
CURSO GMP ANEXO I PROCESO ESTÉRIL - Qualipharma · PROCESOS PRODUCTIVOS ESTÉRILES . CURSO GMP...
38
CURSO GMP ANEXO I PROCESO ESTÉRIL PROCESOS PRODUCTIVOS ESTÉRILES – SIMULACIÓN DEL PROCESO ASÉPTICO
Transcript of CURSO GMP ANEXO I PROCESO ESTÉRIL - Qualipharma · PROCESOS PRODUCTIVOS ESTÉRILES . CURSO GMP...

CURSO GMP ANEXO I
PROCESO ESTÉRILPROCESOS PRODUCTIVOS ESTÉRILES – SIMULACIÓN DEL PROCESO
ASÉPTICO

SUMARIO
1. Visión general
2. Procesos productivos estériles
3. Simulación del Proceso Aséptico

CURSO GMP ANEXO I
PROCESO ESTÉRIL
VISIÓN GENERAL
1.
CURSO GMP ANEXO I: PROCESO ESTÉRIL
VISIÓN GENERAL
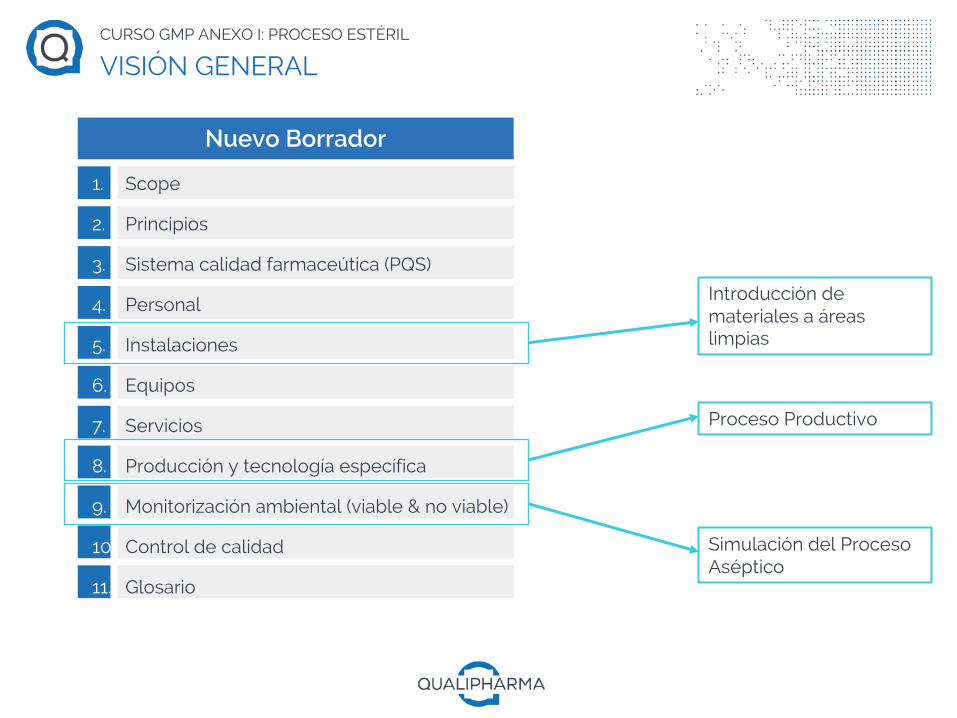
1. Scope
2. Principios
3. Sistema calidad farmaceútica (PQS)
4. Personal
5. Instalaciones
6. Equipos
7. Servicios
8. Producción y tecnología específica
9. Monitorización ambiental (viable & no viable)
10. Control de calidad
11. Glosario
Nuevo Borrador
Proceso Productivo
Simulación del Proceso Aséptico
Introducción de materiales a áreas limpias

CURSO GMP ANEXO I
PROCESO ESTÉRIL
PROCESOS PRODUCTIVOS ESTÉRILES
1.
CURSO GMP ANEXO I: PROCESO ESTÉRIL
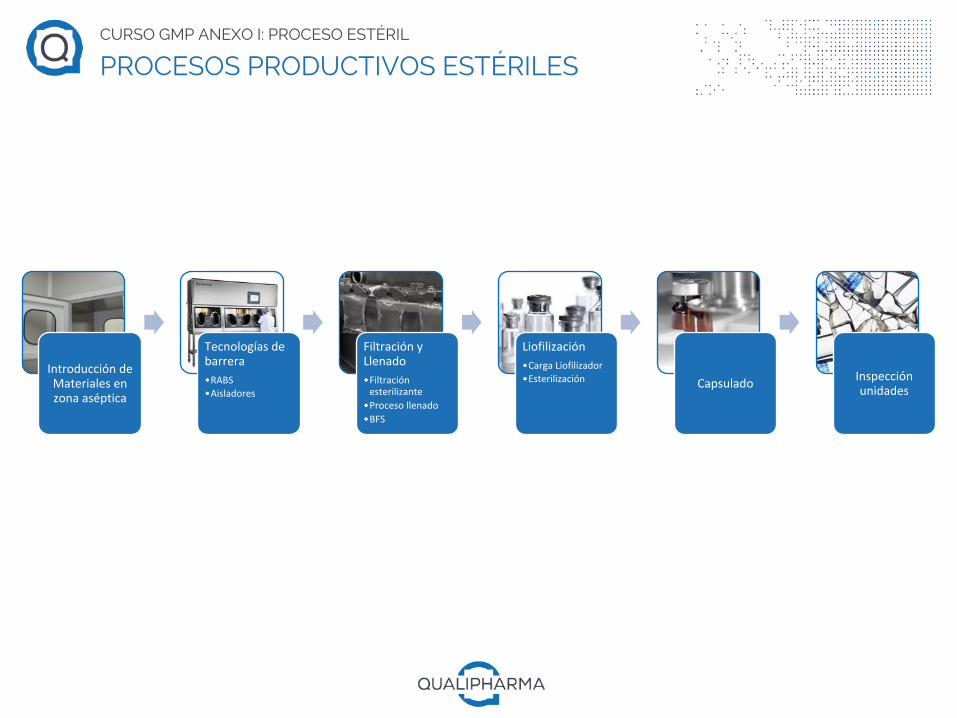
IntroduccióndeMaterialesenzonaaséptica
Tecnologíasdebarrera•RABS•Aisladores
FiltraciónyLlenado•Filtraciónesterilizante
•Procesollenado•BFS
Liofilización•CargaLiofilizador•Esterilización Capsulado Inspección
unidades
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INTRODUCCIÓN DE MATERIAL EN ZONA ASÉPTICA
Debe evitarse el uso de pass-through sin aire activo filtrado.
Sólo los materiales incluidos en una lista cualificada pueden acceder a grado A/B.
Debe asegurarse el mantenimiento de grado A cuando se transfieren materiales de B a A.
Estrategia de sanitización y monitorización de airlocks de materiales será parte del CCS
Los riesgos del trasiego de materiales de zona no clasificada a grado C debe basarse en los
principios de Gestión de Riesgos
ANEXO 1 (5.9.b)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
SAS de materiales con filtración HEPA / ULPA con la clasificación de la sala a la que se accede.
Cualificación y Aprobación de una lista de materiales autorizados en A/B.
Validación del sistema de descontaminación.
Análisis de Riesgos para el paso de materiales de No Clasificado a C.
PROCESOS PRODUCTIVOS ESTÉRILES
INTRODUCCIÓN DE MATERIAL EN ZONA ASÉPTICA

CURSO GMP ANEXO I: PROCESO ESTÉRIL
MATERIALES QUE PUEDAN DESPRENDER PARTÍCULAS
Pasa de
“Deberá minimizarse la presencia”
a
“No deberían estar permitidas”
ANEXO 1 (5.6)
PROCESOS PRODUCTIVOS ESTÉRILES
INTRODUCCIÓN DE MATERIAL EN ZONA ASÉPTICA

CURSO GMP ANEXO I: PROCESO ESTÉRIL
Cuando sea posible, se considerará el uso de equipos tales como RABS, Aisladores o sistemas
cerrados para reducir intervenciones en grado A.
También se considerará la automatización de procesos.
Se debe minimizar el contacto directo con los viales durante la operación de capsulado.
TECNOLOGÍASDEBARRERA
TECNOLOGÍAS DE BARRERA
PROCESOS PRODUCTIVOS ESTÉRILES
ANEXO 1 (8.9)
ANEXO 1 (8.24 – 8.25)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
FILTRACIÓN ESTERILIZANTE
La filtración esterilizante final debe realizarse lo más cerca posible del punto de llenado y, a ser posible, una segunda filtración esterilizante inmediatamente antes del llenado.
La selección de los filtros (incluyendo filtros de gases y de venteo) debe estar justificada.
Se especifican los parámetros que deben validarse para filtros de producto.
FILTRACIÓN ESTERILIZANTE
ANEXO 1 (8.78 – 8.89)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INTEGRIDAD DE FILTRO
Debe ser posible realizarla en el sitio antes de la filtración.
PUPSIT – Integridad antes de uso después de esterilización.
En caso de filtraciones en línea, se debe hacer el test de integridad a todos los filtros esterilizantes antes y después de uso.
FILTRACIÓN ESTERILIZANTE
ANEXO 1 (8.78 – 8.89)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
El llenado se realiza en clasificación:
• Productos con esterilización terminal: Grado C (si no existe riesgo inusual).
• Llenado aséptico: Grado A à Siempre que sea posible con tecnologías de barrera para mantener al operario alejado de la zona crítica.
LLENADO
ANEXO 1 (8.5, 8.10)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
VERTICAL FORM-FILL-SEAL BLOW-FILL-SEAL
LLENADO FORM-FILL-SEAL
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
LLENADO BLOW-FILL-SEAL (BFS)
La técnica BFS realiza en un proceso contínuo:
• Formado del envase primario a partir de un granulado plástico
• Llenado
• Termosellado del envase
El diseño del equipo y los controles a realizar deben basarse en los principios de Gestión de Riesgos.
LLENADO BLOW-FILL-SEAL
ANEXO 1 (8.93 – 8.104)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
LLENADO BLOW-FILL-SEAL (BFS)
Los equipos BFS que cuenten con una ducha de aire grado A se instalarán en al menos grado C (personal con vestimenta A/B) à Grado D si hay esterilización terminal del producto.
Para el control medioambiental se deben tener en cuenta los flujos generados por el propio funcionamiento del equipo.
Se debe prevenir la contaminación por partículas y microbiana de los polímeros plásticos mediante el diseño, mantenimiento y control de los sistemas de almacenamiento y distribución.
Las intervenciones que supongan detener el llenado y/o soplado (y en su caso reesterilización del equipo), deben estar definidas en el SOP de llenado y se deben incluir en la Simulación del proceso aséptico (APS).
LLENADO BLOW-FILL-SEAL
ANEXO 1 (8.93 – 8.104)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
LIOFILIZACIÓN
Los equipos y procesos de liofilización deben asegurar el mantenimiento de la esterilidad hasta elcierre completo de los viales.
Las medidas de control requeridas durante el proceso de liofilización se determinarán en el CCS.
Como novedades se incluyen:
• Esterilización del liofilizador antes de cada carga.
• Monitorización periódica de la integridad del liofilizador
LIOFILIZACIÓN
ANEXO 1 (8.105 – 8.111)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
CARGA DEL LIOFILIZADOR
La distribución de la carga en el liofilizador debe estar especificada y documentada.
El transporte de viales dosificados al liofilizador debe realizarse bajo Grado A (desaparecen las bandejas selladas en B).
Si los viales no se taponan completamente antes de abrir el liofilizador, se deben mantener en Grado A hasta el cierre.
LIOFILIZACIÓN
ANEXO 1 (8.105 – 8.111)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
Otros apartados dedicados a otras tecnologías:
SISTEMAS CERRADOS (8.112 – 8.116)
Para los sistemas cerrados, hace hincapié en la esterilidad de las superficies internas, conexiones
asépticas e integridad del sistema.
SISTEMAS DE UN SOLO USO (Single use Systems – SUS) (8.117 – 8.123)
Para los sistemas de un solo uso, se reconocen riesgos específicos:
• Interacción del material con el producto (adsorción, lixiviables y extraíbles).
• Aumento de operaciones manuales.
• Complejidad del test de integridad pre-uso de los filtros esterilizantes.
• Integridad de los sistemas.
OTRAS TECNOLOGÍAS
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
En caso de que el capsulado se realice como proceso limpio en grado A, los viales sin tapón odesplazados se deben rechazar antes del capsulado.
Debe haber implementado un método automatizado (validado) para la detección de la altura deltapón.
CAPSULADO
ANEXO 1 (8.22 – 8.24)
PROCESOS PRODUCTIVOS ESTÉRILES
Deben utilizarse estudios de ingreso microbiano (o métodoalternativo) para determinar el desplazamiento aceptable de laaltura del tapón.
Cuando se requiera la intervención humana en el capsulado, sedebe utilizar la tecnología apropiada para evitar el contacto directocon los viales y minimizar la contaminación microbiana

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INSPECCIÓN 100%
Todos los contenedores llenos de productos parenterales deben inspeccionarse individualmente para
detectar contaminación externa u otros defectos:
• La clasificación y criticidad de los defectos se determinará mediante herramientas de Gestión de
Riesgos para la Calidad.
• Los lotes con niveles inusuales de defectos deben investigarse y considerar el rechazo parcial o
total del lote.
• Se debe generar y mantener una biblioteca de defectos à herramienta para capacitación del
personal.
• Si se identifican defectos críticos en muestreo posterior à Fallo del proceso de inspección.
INSPECCIÓN 100%
ANEXO 1 (8.26)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INSPECCIÓN MANUAL
Cuando la inspección es manual, debe realizarse en condiciones adecuadas y controladas de
iluminación y fondo.
• La velocidad/ritmo de la inspección deben validarse.
• Los inspectores deben someterse a una cualificación al menos anual, teniendo en cuenta los
peores escenarios (ej. Tiempo de inspección, velocidad de línea, tamaño del envase, fatiga del
operario).
• Los descansos deben ser frecuentes.
INSPECCIÓN 100%
ANEXO 1 (8.27)
PROCESOS PRODUCTIVOS ESTÉRILES

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INSPECCIÓN AUTOMATIZADA
En métodos de inspección automatizados, el
proceso debe validarse para detectar
defectos conocidos con una sensibilidad igual
o mejor a la inspección manual.
El funcionamiento del equipo de inspección
se debe verificar antes de la puesta en
marcha y en intervalos regulares.
INSPECCIÓN 100%
ANEXO 1 (8.28 – 8.29)
PROCESOS PRODUCTIVOS ESTÉRILES
REGISTRO DE DEFECTOS
Los resultados de la inspección deben
registrarse:
• Tendencias de tipos y niveles de defectos.
• Investigar tendencias adversas y nuevos
tipos de defectos (incluir en la investigación
impacto al producto en el mercado).

CURSO GMP ANEXO I
PROCESO ESTÉRIL
Simulación del Proceso Aséptico(APS)
2.

CURSO GMP ANEXO I: PROCESO ESTÉRIL
6 cláusulas
Anexo 1 (en vigor desde el 2009 )
16 cláusulas
Borrador Anexo 1
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
• Deja atrás el término Media Fill à Simulación del
Proceso Aséptico.
• Incluido en el capítulo de Monitorización.
• APS como condición para liberación de producto y
realización de investigaciones y estudios de
tendencia.
• Cambio en el criterio de aceptación.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.34 – 9.49)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
NUEVO ENFOQUE
Es una verificación periódica de la efectividad de los controles para el proceso aséptico.
El plan de APS se debe basar en estrategia de Análisis de Riesgos.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.34 – 9.49)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
Uso de medio nutritivo y/o placebo à
justificar la selección
Cuando las características del proceso
puedan interferir en la viabilidad de la
contaminación microbiológica, se deben
plantear y justificar alternativas (enfriamiento,
liofilización, materiales sólidos, etc.).
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.34)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
APS debe incluir todas las fases críticas de fabricación, específicamente:
• Todas las actividades asépticas que se lleven a cabo después de la esterilización de materiales.
• Si la formulación no es filtrable, todas las fases asépticas adicionales se deben incluir en el estudio.
• Si la fabricación se realiza en condiciones anaerobias estrictas, se debe realizar el APS
adicionalmente con un medio de cultivo anaerobio.
• En el caso de polvo estéril, se debe sustituir por otro material (líquido o semisólido).
• Considerar los procesos de mezcla, molienda, fraccionamiento, etc.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.35)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
LIOFILIZACION
• Considerar el proceso aséptico completo: llenado, transporte, carga del liofilizador, simular la liofilización, aeración de la cámara, taponado, descarga y capsulado.
• La simulación de la liofilización debe replicar el proceso, excepto la congelación y sublimación, incluyendo vacío parcial de la cámara y duración del ciclo.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.35)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
INTERVENCIONES
• Deben incluirse intervenciones rutinarias y en condiciones de peorcaso, considerando:
o Intervenciones inherentes al proceso en la frecuencia máximaadmitida.
o Intervenciones correctivas en el número máximo en que sepodrían producir.
• Debe existir un listado aprobado de intervenciones permitidas
• El listado debe actualizarse siempre que sea necesario para querepresente las actividades reales.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.36 – 9.37)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
USO DE HERRAMIENTAS QRM PARA REALIZAR EL PLAN DE APS PARA DEFINIR
• Identificación de las condiciones peor caso, cubriendo todas las variables y su impacto microbiológico en el proceso.
• Determinar tamaños y materiales a utilizar (configuración envase/cierre).
• Volumen de llenado: suficiente para asegurar el contacto del medio con todas las superficies.
• Holding times máximos permitidos.
• Asegurar la detección de cualquier contaminación.
• Sustitución de gases inertes por aire (¿condiciones anaerobias?).
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.38)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
• Tiempo de duración del llenado (duración máxima del llenado).
• Simulación de interrupciones y mantenimiento de clase A.
• Requisitos y consideraciones especiales de los procesos manuales.
• Fabricación por campañas (riesgos asociados al inicio y final de campaña).
• Riesgos y aspectos relacionados con tecnologías de barrera (RABS, aislador, BFS…).
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.38)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
FRECUENCIA Y REVALIDACIÓN DEL PROCESO
• Validación Inicial: generalmente 3 simulaciones consecutivas satisfactorias.
• Revalidación (3 APS) tras variaciones significativas del HVAC, equipos,cambios en el proceso, nuevos turnos, cierre de instalación, periodos deinactividad, relocalización de líneas.
• Repetición 2 veces al año, aproximadamente cada 6 meses por proceso ylínea de llenado.
• Repetición anual para cada operario.
EN CASO DE LLENADO MANUAL
• Repetición 2 veces al año, por proceso, envase, equipos, operarios.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.40 – 9.41)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
UNIDADES Y RESULTADOS
• El número de unidades debe ser el suficiente para simular todas las actividades representativasen el APS.
• Debe estar justificado y documentado en le Sistema de Calidad Farmacéutico (PQS).
• Si el número de unidades en rutina es menor de 5.000 unidades, el número de unidades del APSdebe ser al menos el mismo.
• Objetivo = Crecimiento 0.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.42 – 9.43)
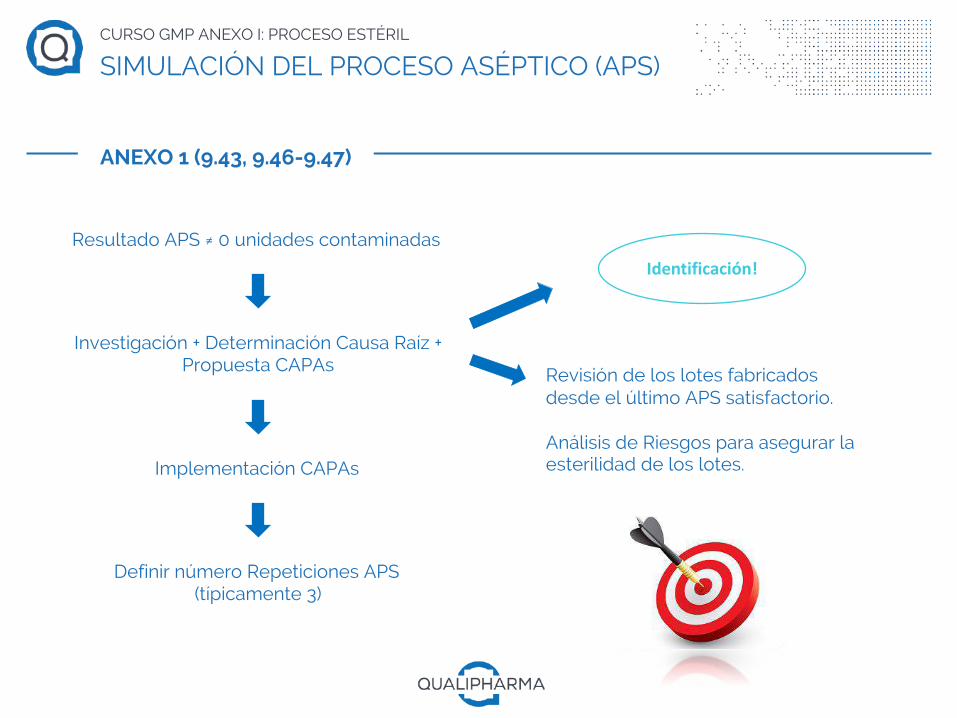
CURSO GMP ANEXO I: PROCESO ESTÉRIL
Resultado APS ≠ 0 unidades contaminadas
Investigación + Determinación Causa Raíz + Propuesta CAPAs
Implementación CAPAs
Definir número Repeticiones APS(típicamente 3)
Revisión de los lotes fabricados desde el último APS satisfactorio.
Análisis de Riesgos para asegurar la esterilidad de los lotes.
Identificación!
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.43, 9.46-9.47)

CURSO GMP ANEXO I: PROCESO ESTÉRIL
DETALLES DE LA INCUBACIÓN
• Las unidades de APS deben ser agitadas, rotadas o invertidas antes de la incubación.
• Se deben incubar todas las unidades, incluidas las defectuosas (defectos cosméticos, controles de peso, … ), siempre que la integridad no esté comprometida.
• Debe documentarse la reconciliación de unidades.
• En caso de descarte de unidades, éstas deben ser coherentes con las unidades retiradas en rutina.
• Las unidades deben incubarse en contenedores transparentes.
• Las condiciones y duración de incubación deben ser justificadas para el proceso y medio de cultivo seleccionado.
SIMULACIÓN DEL PROCESO ASÉPTICO (APS)
ANEXO 1 (9.44 - 9.45, 9.49)

¡MUCHAS GRACIAS!
CURSO GMP ANEXO IPROCESO ASÉPTICO


















