
Crystal structures of the starch-binding domain from …...RoGA (Rhizopus oryzae GA) consists of two...
10
Biochem. J. (2008) 416, 27–36 (Printed in Great Britain) doi:10.1042/BJ20080580 27 Crystal structures of the starch-binding domain from Rhizopus oryzae glucoamylase reveal a polysaccharide-binding path Jung-Yu TUNG*, Margaret Dah-Tsyr CHANG†, Wei-I CHOU†, Yen-Yi LIU*, Yi-Hung YEH*, Fan-Yu CHANG†, Shu-Chuan LIN†, Zhen-Liang QIU* and Yuh-Ju SUN* 1 *Institute of Bioinformatics and Structural Biology, National Tsing Hua University, Hsinchu 30013, Taiwan, and †Institute of Molecular and Cellular Biology, National Tsing Hua University, Hsinchu 30013, Taiwan GA (glucoamylase) hydrolyses starch and polysaccharides to β -D- glucose. RoGA (Rhizopus oryzae GA) consists of two functional domains, an N-terminal SBD (starch-binding domain) and a C-terminal catalytic domain, which are connected by an O-glyco- sylated linker. In the present study, the crystal structures of the SBD from RoGA (RoGACBM21) and the complexes with β -cyclodextrin (SBD–β CD) and maltoheptaose (SBD–G7) were determined. Two carbohydrate binding sites, I (Trp 47 ) and II (Tyr 32 ), were resolved and their binding was co-operative. Besides the hydrophobic interaction, two unique polyN loops compris- ing consecutive asparagine residues also participate in the sugar binding. A conformational change in Tyr 32 was observed between unliganded and liganded SBDs. To elucidate the mechanism of polysaccharide binding, a number of mutants were constructed and characterized by a quantitative binding isotherm and Scatchard analysis. A possible binding path for long-chain polysaccharides in RoGACBM21 was proposed. Key words: carbohydrate-binding module, β -cyclodextrin, malto- heptaose, Rhizopus oryzae glucoamylase, starch-binding domain. INTRODUCTION Starch, the primary source of stored energy in plants, is com- posed of amylose and amylopectin. The former consists almost entirely of α-(1,4)-D-glucopyranose units; however, a few α- 1,6 branches and linked phosphate groups may be found [1,2]. The latter is composed of α-(1,4)-D-glucose segments connected by approx. 5 % α-1,6 branching sites [3]. Both amylose and amylopectin fold into helical structures [4,5] and are further organized into the semicrystalline granular form. In the crystalline state, amylose forms double helices [6], whereas in solution it is a flexible chain that can adopt a locally helical shape. In animals, glycogen, similar to the starch in plants, is the storage form of glucose. Glycogen has structural similarities to amylopectin, except that the number of branching sites and glucose units are, in general, more than that of amylopectin. GA (glucoamylase), also known as amyloglucosidase or γ - amylase (EC 3.2.1.3), is a biocatalyst capable of hydrolysing α-1,4 and α-1,6 glycosidic linkages in raw starches and related oligosaccharides to produce β -D-glucose [7,8]. The GA from Rhizopus oryzae (RoGA) comprises a C-terminal catalytic domain, classified as a member of the GH15 (glycoside hydrolase family 15) [9], connected to an N-terminal SBD (starch-binding domain) by an O-glycosylated linker [10]. CAZY (http://www. cazy.org/) classification, which is based on primary sequence similarity, groups the SBD of RoGA into CBMs (carbohydrate- binding modules) family 21 (RoGACBM21) [11,12]. To date, 51 CBM families have been classified, and eight of them (families 20, 21, 25, 26, 34, 41, 45 and 48) have starch- binding activity [13]. The dissociation constants, K d , for the bind- ing of SBDs to starch are in the micromolar range [14,15]; however, SBDs associated with GA only appear in two families (CBM20 and CBM21). Previous reports on structure-based molecular modelling and NMR spectroscopy of RoGACBM21 revealed that although these two families share very low sequence identity, they indeed have similar biological function and structural folding [16]. Therefore the most recent evolutionary study on CBM20 and CBM21 proposes that these two SBD families are grouped into a new CBM clan [17]. However, the detailed binding mechanisms between CBM21 and starch/glyco- gen remain unclear. SBDs can be functionally independent of the catalytic domains and have been proposed to increase the hydrolysis of granular starch by disrupting its structure and concentrating the catalytic domains on the surface of starch [14,18–20]. Ligand binding of RoGACBM21 with starch and soluble oligosaccharides was determined by Chou et al. [15]. The K d values were of a similar order of magnitude to that of CBM20 from AnGA (Aspergillus niger GA), but the maximal amount of bound protein (B max ) of RoGACBM21 was 40- to 70-fold higher than that of AnGACBM20 [21,22]. The binding affinity between RoGACBM21 and soluble ligands, β CD (β -cyclodextrin) and G7 (maltoheptaose), was measured as approx. 5 μM [15]. Three-dimensional structures of several starch-binding CBM superfamily members have been reported: CBM20 from AnGA [23], CBM21 from RoGA [16] and Homo sapien phosphat- ase 1 (PDB: 2EEF), CBM25 and CBM26 from Bacillus halodur- ans maltohexaose-forming amylase [24], CBM34 from Thermo- actinomyces vulgaris α-amylase [25], CBM41 from Klebsiella aerogenes pullulanase [26] and CBM48 from Rattus norvegicus AMP-activated protein kinase [27]. In the present study, we determined the crystal structures of apo-SBD and the complexes with β CD (SBD–β CD) and maltoheptaose (SBD–G7). Additionally, a number of RoGACBM21 derivatives containing point mutations in aromatic residues (Y32A, W47A, F58A, Y67A, Y83A, Y93A and Y94A) and hydrophilic residues (N29A, K34A, N50A, E68A, N96A and N101A) were isolated and studied. We describe detailed interactions between RoGACBM21 and two substrates (β CD and G7) and propose a polysaccharide- binding path. Abbreviations used: AnGA, Aspergillus niger GA; CBM, carbohydrate-binding module; βCD, β-cyclodextrin; G7, maltoheptaose; GA, glucoamylase; PEG, poly(ethylene) glycol; RMSD, root mean square deviation; RoGA, Rhizopus oryzae GA; SBD, starch-binding domain. 1 To whom correspondence should be addressed (email [email protected]). c The Authors Journal compilation c 2008 Biochemical Society
Transcript of Crystal structures of the starch-binding domain from …...RoGA (Rhizopus oryzae GA) consists of two...

Biochem. J. (2008) 416, 27–36 (Printed in Great Britain) doi:10.1042/BJ20080580 27
Crystal structures of the starch-binding domain from Rhizopus oryzaeglucoamylase reveal a polysaccharide-binding pathJung-Yu TUNG*, Margaret Dah-Tsyr CHANG†, Wei-I CHOU†, Yen-Yi LIU*, Yi-Hung YEH*, Fan-Yu CHANG†, Shu-Chuan LIN†,Zhen-Liang QIU* and Yuh-Ju SUN*1
*Institute of Bioinformatics and Structural Biology, National Tsing Hua University, Hsinchu 30013, Taiwan, and †Institute of Molecular and Cellular Biology, National Tsing HuaUniversity, Hsinchu 30013, Taiwan
GA (glucoamylase) hydrolyses starch and polysaccharides to β-D-glucose. RoGA (Rhizopus oryzae GA) consists of two functionaldomains, an N-terminal SBD (starch-binding domain) and aC-terminal catalytic domain, which are connected by an O-glyco-sylated linker. In the present study, the crystal structures ofthe SBD from RoGA (RoGACBM21) and the complexes withβ-cyclodextrin (SBD–βCD) and maltoheptaose (SBD–G7) weredetermined. Two carbohydrate binding sites, I (Trp47) and II(Tyr32), were resolved and their binding was co-operative. Besidesthe hydrophobic interaction, two unique polyN loops compris-
ing consecutive asparagine residues also participate in thesugar binding. A conformational change in Tyr32 was observedbetween unliganded and liganded SBDs. To elucidate themechanism of polysaccharide binding, a number of mutants wereconstructed and characterized by a quantitative binding isothermand Scatchard analysis. A possible binding path for long-chainpolysaccharides in RoGACBM21 was proposed.
Key words: carbohydrate-binding module, β-cyclodextrin, malto-heptaose, Rhizopus oryzae glucoamylase, starch-binding domain.
INTRODUCTION
Starch, the primary source of stored energy in plants, is com-posed of amylose and amylopectin. The former consists almostentirely of α-(1,4)-D-glucopyranose units; however, a few α-1,6 branches and linked phosphate groups may be found [1,2].The latter is composed of α-(1,4)-D-glucose segments connectedby approx. 5% α-1,6 branching sites [3]. Both amylose andamylopectin fold into helical structures [4,5] and are furtherorganized into the semicrystalline granular form. In the crystallinestate, amylose forms double helices [6], whereas in solutionit is a flexible chain that can adopt a locally helical shape.In animals, glycogen, similar to the starch in plants, is thestorage form of glucose. Glycogen has structural similaritiesto amylopectin, except that the number of branching sites andglucose units are, in general, more than that of amylopectin.GA (glucoamylase), also known as amyloglucosidase or γ -amylase (EC 3.2.1.3), is a biocatalyst capable of hydrolysingα-1,4 and α-1,6 glycosidic linkages in raw starches and relatedoligosaccharides to produce β-D-glucose [7,8]. The GA fromRhizopus oryzae (RoGA) comprises a C-terminal catalyticdomain, classified as a member of the GH15 (glycoside hydrolasefamily 15) [9], connected to an N-terminal SBD (starch-bindingdomain) by an O-glycosylated linker [10]. CAZY (http://www.cazy.org/) classification, which is based on primary sequencesimilarity, groups the SBD of RoGA into CBMs (carbohydrate-binding modules) family 21 (RoGACBM21) [11,12].
To date, 51 CBM families have been classified, and eight ofthem (families 20, 21, 25, 26, 34, 41, 45 and 48) have starch-binding activity [13]. The dissociation constants, Kd, for the bind-ing of SBDs to starch are in the micromolar range [14,15];however, SBDs associated with GA only appear in two families(CBM20 and CBM21). Previous reports on structure-basedmolecular modelling and NMR spectroscopy of RoGACBM21revealed that although these two families share very low
sequence identity, they indeed have similar biological functionand structural folding [16]. Therefore the most recent evolutionarystudy on CBM20 and CBM21 proposes that these two SBDfamilies are grouped into a new CBM clan [17]. However, thedetailed binding mechanisms between CBM21 and starch/glyco-gen remain unclear.
SBDs can be functionally independent of the catalytic domainsand have been proposed to increase the hydrolysis of granularstarch by disrupting its structure and concentrating the catalyticdomains on the surface of starch [14,18–20]. Ligand bindingof RoGACBM21 with starch and soluble oligosaccharides wasdetermined by Chou et al. [15]. The Kd values were of a similarorder of magnitude to that of CBM20 from AnGA (Aspergillusniger GA), but the maximal amount of bound protein(Bmax) of RoGACBM21 was 40- to 70-fold higher thanthat of AnGACBM20 [21,22]. The binding affinity betweenRoGACBM21 and soluble ligands, βCD (β-cyclodextrin) andG7 (maltoheptaose), was measured as approx. 5 μM [15].
Three-dimensional structures of several starch-binding CBMsuperfamily members have been reported: CBM20 from AnGA[23], CBM21 from RoGA [16] and Homo sapien phosphat-ase 1 (PDB: 2EEF), CBM25 and CBM26 from Bacillus halodur-ans maltohexaose-forming amylase [24], CBM34 from Thermo-actinomyces vulgaris α-amylase [25], CBM41 from Klebsiellaaerogenes pullulanase [26] and CBM48 from Rattus norvegicusAMP-activated protein kinase [27]. In the present study,we determined the crystal structures of apo-SBD and thecomplexes with βCD (SBD–βCD) and maltoheptaose (SBD–G7).Additionally, a number of RoGACBM21 derivatives containingpoint mutations in aromatic residues (Y32A, W47A, F58A,Y67A, Y83A, Y93A and Y94A) and hydrophilic residues (N29A,K34A, N50A, E68A, N96A and N101A) were isolated andstudied. We describe detailed interactions between RoGACBM21and two substrates (βCD and G7) and propose a polysaccharide-binding path.
Abbreviations used: AnGA, Aspergillus niger GA; CBM, carbohydrate-binding module; βCD, β-cyclodextrin; G7, maltoheptaose; GA, glucoamylase;PEG, poly(ethylene) glycol; RMSD, root mean square deviation; RoGA, Rhizopus oryzae GA; SBD, starch-binding domain.
1 To whom correspondence should be addressed (email [email protected]).
c© The Authors Journal compilation c© 2008 Biochemical Society

28 J.-Y. Tung and others
EXPERIMENTAL
Protein expression and purification
Recombinant enzyme expression, purification and the functionalassay for RoGACBM21 have been reported previously [15].However, the quality and quantity of full-length RoGA proteinexpression was poor due to the flexible O-glycosylated linker.Briefly, the DNA fragment encoding RoGACBM21 was clonedinto the pET23a(+) expression vector and overexpressed inEscherichia coli BL21-Gold (DE3) cells (Novagen). The recombi-nant SBD sample was purified by His-BindR affinity columnchromatography (Novagen). The purified RoGACBM21 samplewas dialysed against sodium acetate buffer (50 mM, pH 5.5). Theresulting C-terminally His6-tagged SBD (11.65 kDa) was purifiedby Ni-NTA (Ni2+-nitrilotriacetate) affinity chromatography witha final yield of 5 mg of purified protein per litre of cells.
Site-directed mutagenesis
All RoGACBM21 mutants were generated using the PCR-basedQuikChange® site-directed mutagenesis method (Stratagene) asdescribed [15] with pET-RoGACBM21 as the template, twocomplementary primers containing the desired mutation, andPfu Turbo DNA polymerase (Stratagene). All constructs weretransformed into competent E. coli BL21-Gold (DE3) for proteinexpression.
Quantitative measurement of binding to starch
The starch-binding isotherm was analysed by a saturation-bind-ing assay as reported previously [15]. Wild-type and mutantRoGACBM21 derivatives (100 μl, 5–50 μM) in sodium acetate(50 mM, pH 5.5) were mixed with 0.1 mg of prewashed insolublestarch and incubated at 25 ◦C with gentle stirring for 16 h.After centrifugation at 16000 g for 10 min at 4 ◦C, the proteinconcentration of the supernatant (unbound protein) was deter-mined by the BCA (bicinchoninic acid) protein assay, and theamount of bound protein was calculated from the differencebetween the initial and unbound protein concentrations. The Kd
and Bmax values were determined by fitting to the non-linearregression of the binding isotherms using a standard single-sitebinding model.
Quantitative measurement of binding to soluble carbohydrate
Fluorescence spectrophotometry of the binding of wild-typeor mutant RoGACBM21 to βCD was recorded by measuringchanges in the intrinsic protein fluorescence intensity. Experi-ments were performed in 50 mM sodium acetate (pH 5.5) at25 ◦C using a PerkinElmer LS-55 spectrophotometer. Circular andlinear carbohydrates (2–20 mM) were titrated into RoGACBM21(1 μM, 2 ml), and the fluorescence-emission spectrum wasmonitored at 350 nm with a fixed excitation at 280 nm. Therelative changes in fluorescence intensity were plotted againstthe ligand concentration, and the data were fitted to a simulatedcurve using the appropriate equation for a single binding site.
Crystallization
Crystallization trials were carried out using the hanging-dropvapour-diffusion method. Both protein (1 μl) and reservoir (1 μl)solutions were mixed and equilibrated against a reservoir solution(500 μl) in Linbro plates. Initial crystallization conditions wereobtained using Hampton Research Crystal Screen kits andthen further optimized to obtain diffraction-quality crystals.The concentration of RoGACBM21 used for crystallization was
Table 1 X-ray diffraction data and refinement statistics of apo-SBD and thecomplexes of SBD–βCD and SBD–G7
Crystal apo-SBD SBD–βCD SBD–G7
X-ray diffraction dataWavelength (A) 0.9732 (BL13C1) 0.9762 (BL13C1) 0.9998 (BL13C1)Resolution 1.25 1.8 2.3Space group P212121 P212121 P21
Unit cell (a/b/c) (A) 26.64/33.89/93.39 42.58/42.72/70.06 37.69/110.91/61.16β = 90.72◦
Reflections collected 277182 54409 76379Unique reflections 23978 12310 22511Completeness (%):overall (outmost shell)
98.5 (93.9) 99.1 (94.1) 99.4 (97.4)
I/σ (I): overall (outmostshell)
31.5 (6.7) 41.3 (2.2) 14.3 (2.5)
Rmerge (%): overall(outermost shell)∗
7.0 (29.4) 3.3 (44.5) 7.8 (39.4)
RefinementResolution (A) 30.0–1.25 30.0–1.8 30.0–2.3R (%)† 14.6 22.4 23.6Rfree (%)‡ 16.9 23.8 27.4Used reflections 22701 10806 18953Residues 106 106 424Atoms (non-hydrogen) 836 827 3,308Sugar molecules – One βCD Four G7Atom of sugar molecule – 77 312Zinc atoms – 1 –Sulfate molecules – – 6Water molecules 210 126 316RMSD for bond length (A) 0.01 0.007 0.009RMSD for bond angle (o) 1.38 1.3 1.5
Average B-factorsAll protein atoms (A2) 9.31 29.28 13.46βCD (A2) – 32.25 –G7 (A2) – – 19.53Zinc atom (A2) – 32.90 –Sulfate molecules (A2) – – 30.08Water molecules (A2) 23.00 42.27 23.07Overall (A2) 12.06 31.10 14.8∗Rmerge = � |Ih − <I>|/� Ih, where Ih is the measured intensity and <I> the average
intensity of reflection hkl.†R = �||F obs |−|F calc ||/�|Fobs |, where Fobs is the observed and Fcalc the calculated structure
factor of reflection hkl.‡Rfree is calculated on the ‘test set’ of reflections.
approx. 10 mg/ml. The βCD and G7 were used at a molar ratioof 1:2 (protein/carbohydrate) to form SBD–βCD and SBD–G7complexes respectively. Three SBD crystals grew in differentconditions at 293 K. The apo-SBD crystal was crystallized in25% PEG [poly(ethylene) glycol] 4000, the SBD–βCD crystalwas grown using 18 % PEG 8000 and 0.2 M zinc acetate, andthe SBD–G7 crystals were grown in 30% PEG 8000 and 0.6 Mammonium sulfate.
X-ray data collection
The X-ray diffraction data were collected at beamline BL13C1at the NSRRC (National Synchrotron Radiation Research Center,Taiwan). The data were processed and scaled using the programHKL2000 [28]. The apo-SBD, SBD–βCD and SBD–G7 crystalsdiffracted to 1.25, 1.8 and 2.3 Å (1 Å = 0.1 nm) resolutionrespectively. Both apo-SBD and the SBD–βCD crystals belongto the orthorhombic P212121 space group with one molecule perasymmetric unit and the VM [29] was calculated as 1.81 Å3 · Da−1
and 2.64 Å3 · Da−1 respectively. The SBD–G7 crystal belongs tothe monoclinic P21 space group with four molecules per asymmet-ric unit and the VM [29] was calculated as 2.74 Å3 · Da−1 (Table 1).
c© The Authors Journal compilation c© 2008 Biochemical Society

Crystal structures of the starch-binding domain of R. oryzae glucoamylase 29
Figure 1 Structure-based multiple-sequence alignment of SBDs
The alignment of SBD from R. oryzae GA (RoGACBM21: PDB code 2DJM) with SBDs from Mucor circinelloides (McCBM21), H. sapiens protein phosphatase 1 (HsCBM21: PDB code 2EEF),A. niger glucoamylase (AnGACBM20: PDB code 1AC0), B. halodurans maltohexaose-forming amylase (BhCBM25: PDB code 2C3X and BhCBM26: PDB code 2C3G), T. vulgaris α-amylase(TvAICBM34: PDB code 1UH3), K. aerogenes pullulanase (KaCBM41: PDB code 2FHF) and R. norvegicus AMP-activated protein kinase (RnCBM48: PDB code 1Z0M). Secondary structuralelements of RoGACBM21 are indicated above the alignment as arrows (β-strands). The locations of β-strands of other CBM families are underlined. The aromatic residues (bold and boxed) andpolar residues (bold and shaded) involved in ligand binding in RoGACBM21 are marked. The corresponding residues in other CBM family members are marked similarly. The asparagine residuesof the polyN loops (Asn46–X–Asn48–Asn49–Asn50–X–Asn52 and Asn96–Asn97–Asn98–X–X–Asn101) of RoGACBM21 are labelled with ∗ and Tyr67 and Tyr93 are labelled with �.
Structural determination and refinement
The crystal structures of RoGACBM21 were determined usingthe unliganded solution structure RoGACBM21 (PDB: 2DJM)[16] as a search model by molecular replacement. The molecularreplacement program MOLREP [30] was used for phase deter-mination. Data between 8.0 and 4.0 Å and a Patterson radiusof 20 Å were used to calculate the rotation and translationfunctions. A similar procedure was applied for the three structuresand significant rotation and translation solutions were obtained.Structural model building was carried out using XTALVIEW [31],and the structural refinement was performed by CNS [32] andthe CCP4 program suite [33]. The final statistics of refinementfor apo-SBD and the complexes, SBD–βCD and SBD–G7, aresummarized in Table 1. The co-ordinates of the RoGACBM21structures have been deposited in the PDB as the accession num-bers 2VQ4 (apo-SBD), 2V8L (SBD–βCD) and 2V8M (SBD–G7).
RESULTS AND DISCUSSION
CBM folding topology
Several CBM superfamilies, including CBM20, CBM21,CBM25, CBM26, CBM34, CBM41 and CBM48, contain anSBD in either the N- or C-terminus [24,25,34–40]. These SBDsreveal low sequence identity but a similar immunoglobulin-likefolding topology. RoGACBM21 contains 106 residues, which haslow sequence identity (<15%) with AnGACBM20, as well asthat of other SBD-containing CBM families. A structure-basedmultiple-sequence alignment of SBDs from nine representativeCBM superfamilies is shown in Figure 1. The sequence alignmentwas carried out based on the overall structural folding of eightβ strands of SBDs and the corresponding ligand-binding sites.Two types of SBD topologies, type I and type II [15], show asimilar overall structure by switching the first and last β strands.
Superfamilies CBM20, CBM25, CBM26 and CBM41 belong tothe type I topology, whereas CBM21, CBM34 and CBM48 belongto type II topology [15].
Overall structures
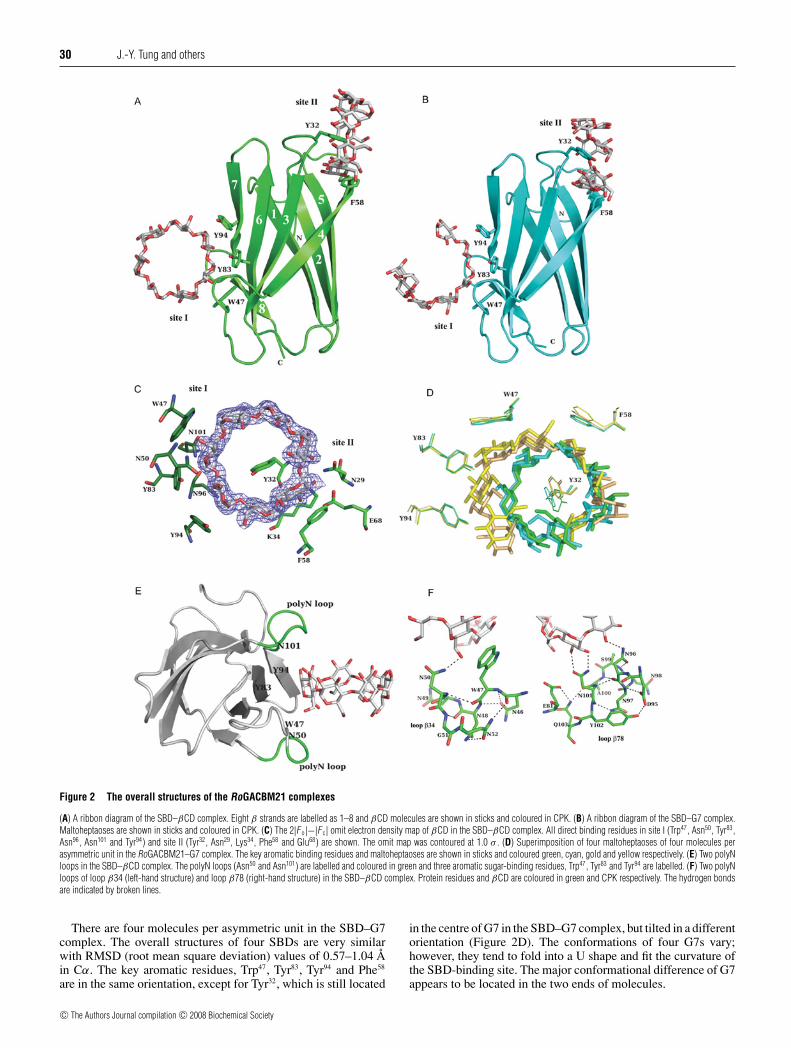
The RoGACBM21 and the SBD–βCD and SBD–G7 complexesshare a similar overall structure, which belongs to a typical β-sandwich fold with an immunoglobulin-like architecture [20] withapproximate dimensions of 28 Å × 30 Å × 42 Å. The β-sand-wich is symmetric and composed of eight β strands, which areantiparallel except β7 and β8. These strands are paired as fourhairpin β strands, β12, β34, β45 and β67/8, and fold as a βbarrel. The β barrel contains a substantial hydrophobic core thatcontributes the major stabilizing force to RoGACBM21. Theoverall structures of SBD–βCD and SBD–G7 are shown (Fig-ures 2A and 2B). Two carbohydrate-binding sites, designated assites I and II, were observed in both SBD–βCD and SBD–G7complexes, in which the sugar ligands, βCD and G7, are locatedat almost diagonal ends of the SBD in a perpendicular orientation.Binding site I is located at Trp47 around loop β34 and site II islocated at Tyr32 around loop β23. The carbohydrate-binding ratiofor both SBD–βCD and SBD–G7 complexes is one sugar ligandper SBD molecule in crystal, in which the SBD shares eachbinding site with the symmetry-related molecule in the crystalsuch that two SBD molecules together hold one sugar ligand.Site I and site II are compatible to bind one sugar molecule co-operatively. In solution, the binding between the SBD and βCD isdetermined to be 1:2 by isothermal titration calorimetry (resultsnot shown). The well-defined electron density map of βCD in theSBD–βCD complex is shown in Figure 2(C). The Kd (∼333 μM)of the αCD complex is much higher than that of βCD and G7complexes [15], because the cyclic ring of the six glucose units ofαCD is too small to fit the essential binding surface of the SBD,such that its binding ability is dramatically reduced.
c© The Authors Journal compilation c© 2008 Biochemical Society
30 J.-Y. Tung and others
Figure 2 The overall structures of the RoGACBM21 complexes
(A) A ribbon diagram of the SBD–βCD complex. Eight β strands are labelled as 1–8 and βCD molecules are shown in sticks and coloured in CPK. (B) A ribbon diagram of the SBD–G7 complex.Maltoheptaoses are shown in sticks and coloured in CPK. (C) The 2|F o|−|F c| omit electron density map of βCD in the SBD–βCD complex. All direct binding residues in site I (Trp47, Asn50, Tyr83,Asn96, Asn101 and Tyr94) and site II (Tyr32, Asn29, Lys34, Phe58 and Glu68) are shown. The omit map was contoured at 1.0 σ . (D) Superimposition of four maltoheptaoses of four molecules perasymmetric unit in the RoGACBM21–G7 complex. The key aromatic binding residues and maltoheptaoses are shown in sticks and coloured green, cyan, gold and yellow respectively. (E) Two polyNloops in the SBD–βCD complex. The polyN loops (Asn50 and Asn101) are labelled and coloured in green and three aromatic sugar-binding residues, Trp47, Tyr83 and Tyr94 are labelled. (F) Two polyNloops of loop β34 (left-hand structure) and loop β78 (right-hand structure) in the SBD–βCD complex. Protein residues and βCD are coloured in green and CPK respectively. The hydrogen bondsare indicated by broken lines.
There are four molecules per asymmetric unit in the SBD–G7complex. The overall structures of four SBDs are very similarwith RMSD (root mean square deviation) values of 0.57–1.04 Åin Cα. The key aromatic residues, Trp47, Tyr83, Tyr94 and Phe58
are in the same orientation, except for Tyr32, which is still located
in the centre of G7 in the SBD–G7 complex, but tilted in a differentorientation (Figure 2D). The conformations of four G7s vary;however, they tend to fold into a U shape and fit the curvature ofthe SBD-binding site. The major conformational difference of G7appears to be located in the two ends of molecules.
c© The Authors Journal compilation c© 2008 Biochemical Society
Crystal structures of the starch-binding domain of R. oryzae glucoamylase 31
Binding sites
Site I mainly comprises three conserved aromatic residues, Trp47,Tyr83 and Tyr94, to form a broad, flat and stable hydrophobicenvironment. The aromatic rings of these residues construct thecurvature of the SBD-binding site and interact with the sugarrings by ideal hydrophobic stacking interactions (Figure 2C).Site I is rather rigid because Tyr83 and Tyr94 are located in βstrands and Trp47 is locked by Asn46 and Asn50. Site II is generallyformed by loops β23 and β45, particularly dominated by twoaromatic residues, Tyr32 and Phe58. Nevertheless, both Tyr32 andPhe58 are in the proximity of the sugar ring where the van derWaals surface and the βCD are closely packed. The sugar ring ofβCD is nearly parallel to the ring of Tyr32, which protrudes intothe non-polar cavity of βCD and forms a hydrogen bond withβCD O27 (Figure 2C). Such unique binding is only observed inthe SBD–βCD complex, where the α-glucan chains of βCD wraparound Tyr32 to stabilize the binding. Similar binding patternswere observed in CBM25, CD glycosyltransferase [41,42] andCBM48, the glycogen-binding domain of AMP-activated proteinkinase [27], in which the corresponding residues in site II areLeu600 and Leu146 respectively. Another key residue in site II isPhe58, of which the hydrophobic phenyl ring forms a flat stackinginteraction with the sugar ring of the glucosyl unit of βCD. Twoplanar rings of Tyr32 and Phe58 pack closely against the van derWaals surface of the βCD molecule and act like a clamp to pickup the βCD.
In addition to the hydrophobic interactions, numeroushydrophilic interactions between the SBD and βCD werefound. In site I, residues Asn50 (ND2-O61:2.7 Å), Asn96 (ND2-O33:2.9 Å) and Asn101 (ND2-O22:2.6 Å; OD1-O32:2.9 Å) formtwo unique polyN loops (Figure 2E) and provide the mainhydrophilic interactions to βCD. In site II, residues Asn29
(NH2-O3:2.8 Å), Lys34 (NZ-O3:2.8 Å and N-O3:3.3 Å) and Glu68
(OE1-O2:2.5 Å and OE2-O3:2.9 Å) supply several hydrogen-bond interactions with βCD. Meanwhile, these residues interactwith each other by hydrogen bonds to provide a tight bindingenvironment for βCD and contribute the forces to stabilize theSBD. The binding area of site II is small and narrow and itprotrudes away from the complex more than site I.
PolyN loop
Besides the hydrophobic interaction, several asparagine residuesfrom β34 and β78 near site I provide additional hydrophilicinteractions for the carbohydrate binding. These two unique polyNloops positioned on the two sides of the sugar molecule interactwith the hydroxy groups to protect the hydrophobic curvature ofthe binding-site groove created by Trp47, Tyr83 and Tyr94 fromexposure to solvent (Figure 2E). The polyN loops consisting ofconsecutive asparagine residues in loop β34 (Asn46, Asn48, Asn49
and Asn50) as well as in loop β78 (Asn96, Asn97, Asn98 and Asn101)(Figure 2F) participate in sugar binding.
The significant interaction networks created in each of thepolyN loops of both complexes stabilize the binding loop andfacilitate sugar binding. In polyN loop β34, residues Asn46
and Asn50 directly interact with the main chains of Trp47 suchthat the aromatic ring of Trp47 forms hydrophobic stacking withthe sugar ring (Figure 2F). Meanwhile, Asn50 and Trp47 lock to-gether to assist the sugar binding by a hydrogen bond (Figure 2F).Furthermore, the geometry of Asn50 is squeezed out byan interaction network, Asn46–Trp47–Asn48–Asn50–Gly51–Asn52–Asn46, and the side chain of Asn50 is forced to interact with thesugar molecule (Figure 2F). Likewise, in polyN loop β78, Asn96
interacts with Ser99, Ala100 and Asn101 by three hydrogen bonds
Table 2 Binding affinity of wild-type and mutant RoGACBM21 for starchand βCD
NB, no binding detected.
Bmax (μmole/g) K d (μM) K d (μM)SBDs for starch for starch for βCD
Wild-type 41.1 +− 1.1 1.4 +− 0.1 5.1 +− 0.7Mutants of aromatic residues
Y32A 23.4 +− 0.7 0.8 +− 0.1 6.0 +− 0.7W47A 23.0 +− 1.3 4.4 +− 0.6 NBF58A 37.5 +− 1.4 4.6 +− 0.5 28.4 +− 3.5Y67A 3.7 +− 0.1 6.5 +− 0.4 10.2 +− 1.4Y83A 6.4 +− 0.1 7.1 +− 0.4 15.4 +− 2.7Y93A 27.8 +− 0.2 2.7 +− 0.1 14.9 +− 1.4Y94A 27.0 +− 1.1 3.5 +− 0.5 12.7 +− 1.4
Mutants of hydrophilic residuesN29A 7.0 +− 0.2 8.1 +− 0.6 6.5 +− 0.9K34A 7.7 +− 0.1 5.2 +− 0.4 22.5 +− 2.5N50A 4.5 +− 0.1 13.6 +− 1.1 7.7 +− 0.9E68A 6.5 +− 0.1 6.8 +− 0.2 22.0 +− 3.0N96A 24.0 +− 0.3 1.6 +− 0.1 23.4 +− 3.0N101A 17.8 +− 0.4 2.9 +− 0.2 25.2 +− 3.3
to build a βCD-binding network (Figure 2F). The orientation ofAsn101 is fixed by Asn96 and Asn97 with two hydrogen bonds,as well as controlled by the adjacent residues, Ala100, Tyr102 andGln103. As a result, Asn101 binds βCD with two hydrogen bonds(Figure 2F). A primary triangle interaction, Asn96:CO–Asn101:N–Asn97:NH2, and a periphery interaction network, Asn96–Ala100–Gln103–Tyr102–Gln95–Asn97, are produced and tight sugar bindingis generated. The orientations of Asn96 and Asn101 are especiallyrestricted and do not reveal favourable geometries in theRamachandran plot.
Some residues in the polyN loops show very restricted geo-metries because these residues have to link one by one and forma series of chain interactions, which not only stabilize the bindingloops, but also confirm the sugar binding. These extra inter-actions from the polyN loops might produce a higher sugar-binding capacity and affinity for RoGACBM21 as compared withthose of other CBM superfamilies. Furthermore, the polyN loopsinteract with two sides of the βCD molecule to assist in turningthe orientation of βCD to form hydrophobic interactions withTrp47, Tyr83 and Tyr94. Interestingly, these characteristic polyNsequences are only observed in RoCBM21 and McCBM21 (whereMc is Mucor circinelloides) (Figure 1) [13,17].
Binding affinity for starch and βCD
To elucidate the mechanism of polysaccharide binding, weexamined the essential residues in the vicinity of the binding sitesaccording to the complex structures. A number of RoGACBM21mutants were generated and characterized by a quantitative bind-ing isotherm and fluorescence spectroscopic analysis (Table 2)[15]. The mutant proteins contained substitutions at the aromaticresidues from site I (W47A, Y83A, and Y94A) and site II (Y32Aand F58A) involved in βCD and G7 binding and two additionaltyrosine residues near sites I and II, Y67A and Y93A. Mutationsat hydrophilic residues in site I (N50A, N96A and N101A) andsite II (N29A, K34A and E68A) involved in direct hydrogen-bondinteractions with ligands were also analysed.
The site I mutants (W47A, Y83A and Y94A) exhibited de-creased binding affinity for starch and βCD when compared withthe Kd values for wild-type RoGACBM21 (Table 2). The βCDbinding cannot be detected in the W47A mutant. Furthermore,
c© The Authors Journal compilation c© 2008 Biochemical Society

32 J.-Y. Tung and others
W47A, Y83A and Y94A showed a reduced Bmax for the bindingcapacity to starch, especially Y83A, possibly because Tyr83 islocated on the inside position of the site I-binding pocket. Site I(W47A) serves as the major carbohydrate-binding site for starch.For the site II mutants (Y32A and F58A), the Kd values of Y32Afor starch and βCD binding were similar to that of wild-typeSBD. In addition, Y32A had less effect on the binding of solubleoligosaccharides. F58A showed an increased Kd for the bindingof starch and had the highest Kd for the binding of βCD amongall of the mutants; however, this mutant exhibited almost no effecton the binding capacity for starch, with a Bmax similar to that ofwild-type SBD. Because Phe58 significantly affected the bindingto βCD and starch, Phe58 might play a key role in site II. TheTrp47- and Tyr32-binding sites were co-operative to sugar binding,because the binding affinity of the single mutant, Y32A or W47A,was partially maintained; however, the binding of the doublemutant (Y32A/W47A) to insoluble starch was almost completelyabolished [15]. The binding roles of Tyr32 and Trp47 can beaccommodated in RoGACBM21 complexes (Figure 2C).
From the structure-based alignment (Figure 1), two additionalinteresting aromatic residues, Tyr67 and Tyr93 were studied. Tyr67
is conserved in most of the SBD-containing CBM superfamiliesand Tyr93 is also conserved in most of the CBM21 superfamilymembers [13,17]. Although Tyr67 and Tyr93 do not directly interactwith sugar in both the SBD–βCD and SBD–G7 complexes(Figure 2), Y67A and Y93A mutants showed apparently lowerBmax and higher Kd values (Table 2). In particular, Y67A had aconsiderably reduced Bmax (3.7 μmol/g), the lowest starch-bindingcapacity of all of the mutants. In the SBD–βCD complex, Tyr67
points into the N-terminal loop, in which the hydroxy group ofTyr67 positions around Pro4–Ser5–Ser6–Ala7 and forms a hydrogenbond with the O atom of Ala7 and stacks with Pro4 in the Nterminus (Figure 3A). The mutant Y67A showed a small Bmax andit might due to the instability of the N terminus. Thus Tyr67 mightplay a role in the N-terminal stabilization. The overall structure ofY67A should be maintained because the binding activity of Y67Ais preserved, and the secondary structure of Y67A examined bycircular dichroism was intact (results not shown). The result inthe present study demonstrates that Tyr67 might play an importantrole in governing the starch-binding capacity for RoGACBM21.
The Kd and Bmax values for mutants with substitutions athydrophilic residues in site I (N50A, N96A and N101A) and siteII (N29A, K34A and E68A) are shown in Table 2. Mutant N50Ahad a reduced Bmax and the highest Kd (∼10-fold higher thanwild-type) for starch binding among all site I and II mutants, andthis may be attributed to the loss of hydrogen-bond interactionsbetween Asn50 and Tyr83. Asn50 might play a major role in thebinding of insoluble polysaccharides and contributes to the overallintegrity of site I. Mutants N96A and N101A (site I) as well asK34A and E68A (site II) exhibited inefficient βCD binding butwith high Kd values due to a loss of hydrogen-bond interactions.Consequently, we suggest that these hydrophilic residues fromsites I and II might play a critical role in the binding of soluble orinsoluble polysaccharides, in either an individual or co-operativemanner.
A continuous polysaccharide-binding path
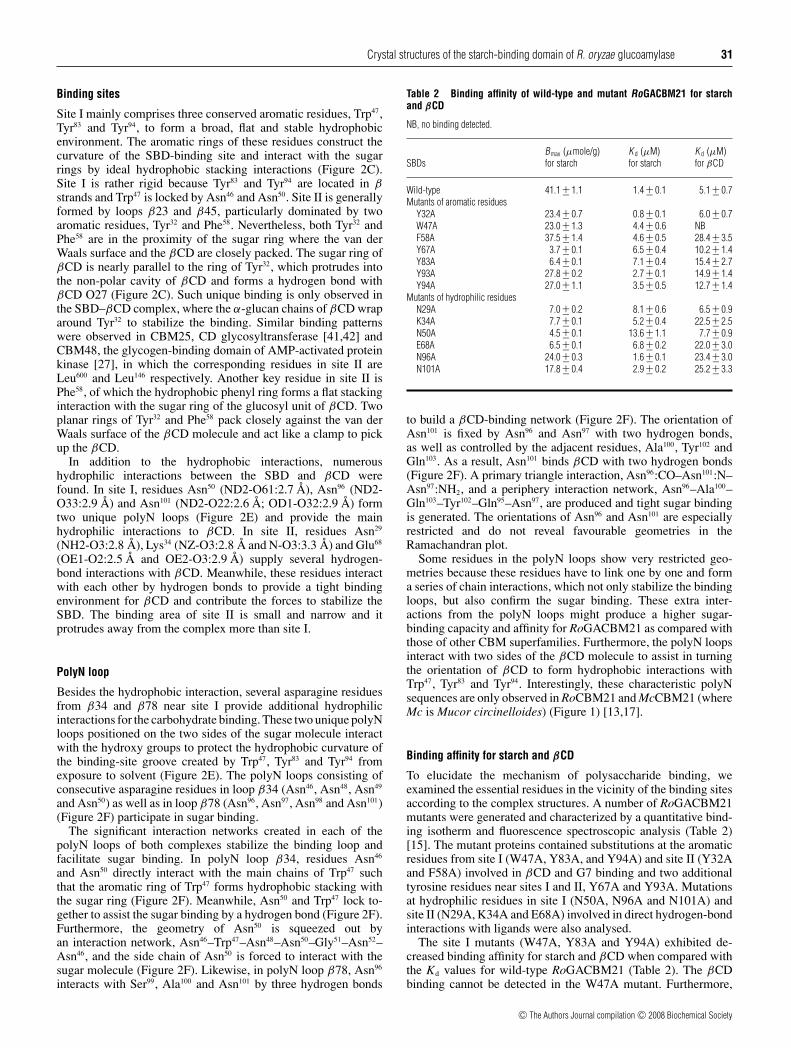
Based on sites I and II, as well as two crucial tyrosine residuesnear sites I and II, Tyr67 and Tyr93, a characteristic connection wasobserved between two carbohydrate-binding sites (Figure 3A).From the surface electrostatic potential distribution of the SBD–βCD complex (Figure 3B), a continuous hydrophobic surface isobserved formed by residues Trp47, Tyr83, Tyr94, Tyr93 and Tyr67.Furthermore, from the solvent-accessible surface of the SBD–
βCD complex (Figure 3C), there are two possible accessiblesurface paths to link site I and site II. One path is throughresidues Asn50, Tyr83, Tyr94, Tyr93 and Tyr67. From the mutationdata (Table 2), Y67A and Y93A mutants showed substantialeffects in the starch-binding assay, Tyr67 and Tyr93 might actas a midpoint to link two binding sites and make a continuousbinding path for longer chain and larger polysaccharides, or evenstarch. The distance of this potential polysaccharide-binding paththrough Trp47 (site I), Tyr93, Tyr67 and Tyr32 (site II) is in a range of45–60 Å with an accessible surface area of approx. 1215 Å2. Theother path is through hydrophilic residues, Asn50, Lys85, Glu87,Lys35 and Lys34, which are consecutive and it might be able tolink sites I and II also. The continuous binding path might beimportant for the raw starch/insoluble polysaccharides, but notfor soluble and smaller carbohydrate molecules, such as βCD andG7. During the initial binding step, sites I and II might act as twopersistent binding markers for soluble/insoluble polysaccharides,after which the continuous polysaccharide-binding path mayassist further binding.
Liganded and unliganded RoGACBM21
The superimposed structures of unliganded and ligandedRoGACBM21 are presented in Figure 3(D). They revealcomparable overall structures, in which RMSD values betweenthe SBD and two complexes, SBD–βCD and SBD–G7, are 0.74and 0.98 Å respectively, and the RMSD value between the twocomplexes is 0.57 Å. The comparison of the residues around thesugar-binding site of unliganded and liganded SBD is shown inFigure 3(E). An apparent conformational change was observedin Tyr32, which formed a hydrogen bond with Asn29 in unligandedSBD; however, the phenyl ring of Tyr32 flipped over and reorientedto form sandwich stacking with the sugar ring upon ligand binding(Figure 3E). Surprisingly, the essential aromatic residues (Trp47,Tyr83 and Tyr94) as well as other ligand-binding residues remainedin the same orientation in unliganded and liganded SBDs. Beforeligand binding, numerous water molecules occupied the sugar-binding site and interacted with several hydrophilic residues, suchas Lys34, Glu68, Asn50, Asn94 and Asn101. However, upon ligandbinding, this water layer was replaced by a sugar molecule andthe interactions were established among the sugar and hydrophilicbinding residues.
These results indicated that the sugar-binding site of SBD israther fixed and stable. As long as the β barrel scaffold is formedand the three key binding positions (Trp47, Tyr83 and Tyr94) canbe preserved, the ligand binding is likely to take place. For linearpolysaccharides, the Kd values of RoGACBM21 decreased withincreasing ligand length and it was worse than that of βCD [15].The Y32A mutant significantly diminished the affinity in G7 buthad no effect in the short linear carbohydrates, such as maltotrioseand maltotetraose [15]. Tyr32 may adjust the orientation of itsphenyl ring to accommodate the conformation of a long ligand.
RoGACBM21 complexes and other CBM superfamilies
The three-dimensional structures of two CBM21 members,apo-RoGACBM21 and apo-HsCBM21 are superimposed inFigure 4(A). They share a similar overall structure except for loopsβ34, β45 and β56. The corresponding residues for ligand bindingresidues in site I (Trp47, Tyr83 and Tyr94) and site II (Tyr32 and Phe58)in the RoGACBM21 complex were also observed in HsCBM21(where Hs is Homo sapiens) in putative site I (Trp72, Ala114 andTrp125) and site II (Phe59 and Tyr82) respectively. These residuesare located in comparable locations except that the orientations
c© The Authors Journal compilation c© 2008 Biochemical Society

Crystal structures of the starch-binding domain of R. oryzae glucoamylase 33
Figure 3 The polysaccharide-binding path
(A) A continuous polysaccharide-binding path in the SBD–βCD complex. Residues Trp47 (site I) and Tyr32 (site II) as well as Tyr67 and Tyr93 are shown as sticks and are coloured in red. βCDsare shown as sticks and coloured in CPK. (B) The surface electrostatic potential of the SBD–βCD complex displayed using the program PyMOL (DeLano Scientific; http://pymol.sourceforge.net/)with the negative potentials (red) and positive potentials (blue). Residues Trp47, Tyr83, Tyr94 (site I) and Tyr32 (site II) as well as Tyr67 and Tyr93 are labelled. (C) The solvent-accessible surface ofthe SBD–βCD complex. The residues on the solvent-accessible surface were calculated by GPSS (http://gpss.mcsg.anl.gov/). Two possible polysaccharide-binding paths to link site I and siteII. Residues Trp47, Tyr83, Tyr94, Tyr93, Tyr67 and Tyr32, as well as residues Asn50, Lys85, Glu87, Lys35 and Lys34 are labelled. Residues Glu95 and Asn98 on the polyN loop β78 as well as residuesAla7 and Gln10 around the N-terminus are labelled. (D) Structural superimposition of unliganded and liganded RoGACBM21s. The structural differences are coloured as unliganded RoGACBM21(gold) and SBD–βCD (green) and SBD–G7 (cyan) complexes. Molecules βCD (green) and G7 (cyan) are coloured in complexes. (E) The sugar-binding site of unliganded (gold) and the ligandedRoGACBM21s, SBD–βCD (green) and SBD–G7 (cyan). Molecules βCD (green) and G7 (cyan) are coloured in complexes. The key sugar-binding residues Asn29, Tyr32, Trp47, Phe58, Tyr83 and Tyr94
are drawn as sticks. The hydrogen bonds are indicated by broken lines.
of the side chains vary. We suggest that HsCBM21 might possessbinding characteristics similar to that of RoGACBM21.
AnGA, a GA from Aspergillus niger [23], shares the sameglycoside hydrolase characteristic as RoGA. However, the SBDsof both proteins, AnGACBM20 and RoGACBM21, fold withdifferent topologies [15] and link with the catalytic domainsthrough their N- and C-terminal ends respectively [23,43].The three-dimensional structures of the two SBDs could be
superimposed by switching the first and the last β strands; thestructural superimposition is presented in Figure 4(B). MostCBM superfamilies contain one sugar-binding site; however,RoGACBM21 and AnGACBM20 complexes contain two bindingsites [24–26,40]. RoGACBM21 binds co-operatively to sugar,whereas AnGACBM20 exhibits independent sugar binding [22].Even though the sugar-binding location is similar in both proteins,several structural differences were noted (Figure 4B), particularly
c© The Authors Journal compilation c© 2008 Biochemical Society

34 J.-Y. Tung and others
Figure 4 Structural comparison of RoGACBM21 with other CBM superfamilies
(A) Structural superimposition of unliganded RoGACBM21 (PDB code: 2VQ4, green) and unliganded HsCBM21 (PDB code: 2EEF, orange). Residues of site I (Trp47, Tyr83 and Tyr94) and site II (Tyr32
and Phe58) in RoGACBM21 and residues of site I (Trp72, Ala114 and Trp125) and site II (Phe59 and Tyr82) in HsCBM21 are shown as sticks and labelled. (B) Structural superimposition of RoGACBM21(PDB code: 2V8L, green) and AnGACBM20 (PDB code: 1AC0, pink) complexes with βCD. Three main structural differences (loops β34, β45 and β78) are shown. The key residues (Trp47, Tyr94
and Phe58) in RoGACBM21 as well as the corresponding residues (Trp543, Trp590 and Tyr556) in AnGACBM20 are shown. To clearly examine the structural differences, the βCD molecules are notshown. (C) Structural superimposition of RoGACBM21 (PDB code: 2V8L, green) and TvAICBM34 (PDB code: 1UH4, blue) complexes. The key residues of site I (Trp47, Tyr83 and Tyr94) and site II(Tyr32 and Phe58) in RoGACBM21 as well as the corresponding residues of site I (Trp51, Tyr89 and Tyr119) and site II (Trp65 and Trp77) in TvAICBM34 are shown as sticks. The βCD (green) of theRoGACBM21 complex and the maltotridecaose (blue) of the TvAICBM34 complex are shown as sticks.
in loops β34, β45 and β78 around the sugar-binding regions,which are in completely different conformations. These distinctloops provide the unique sugar binding for RoGACBM21and AnGACBM20.
The two binding sites of AnGACBM20 and RoGACBM21are structurally and functionally different and the correspondingresidues in AnGACBM20 are Trp543 and Trp590 for site I and Tyr527
and Tyr556 respectively. In AnGACBM20 [23], site I (Trp543) acts
c© The Authors Journal compilation c© 2008 Biochemical Society

Crystal structures of the starch-binding domain of R. oryzae glucoamylase 35
as the initial recognition site for starch, whereas site II (Tyr527) iscapable of recognizing a range of starch strand orientations. SiteI has a larger surface area, undergoes a conformational changeupon sugar binding, and is proposed to act as a more specificsite to lock the ligand into place. However, from the structuralresults and functional assay data (Table 2) for the RoGACBM21complexes, we assume that site I (Trp47) is the essential bindingsite and site II (Tyr32) plays an auxiliary role as a recognition sitebecause a conformational change and the protruded orientationof Tyr32 was clearly observed between liganded and unligandedSBDs (Figure 3E). The residues corresponding to Trp47 inmost SBD superfamilies are highly conserved, except forBhCBM26 (where Bh is B. halodurans) (Figure 1). Site I revealsa larger and broader binding surface, and undergoes furtherconformational changes upon βCD binding.
TvAI, an α-amylase from T. vulgaris [25] that catalyses thehydrolysis of α-D-(1,4)-glucoside linkages in starch to releasethe α-anomer, shows different hydrolase characteristics withRoGA. However, both SBDs belong to the N-terminal starch-binding CBM superfamily and fold with the same type II topology[25,43]. Although the sequence identity and similarity betweenthese two molecules are only 16 and 36% respectively, their struc-tures superimposed quite well, especially the β strands. Thestructural superimposition of RoGACBM21 and TvAICBM34 isshown in Figure 4(C) with an RMSD of 1.6 Å in Cα. Nevertheless,structural differences were observed in loop regions, for exampleloops β34 and β78 (Figure 4C). Two sugar-binding sites, site-NA and site-N, found in TvAICBM34 [25] correspond to site Iand site II respectively in RoGACBM21. Both sugar-bindingsites were in similar orientations but two sugar molecules werein a perpendicular orientation in both proteins. Meanwhile, thefunctional roles of the two sugar-binding sites in both proteinswere comparable: site I/site-NA binds sugar specifically and siteII/site-N helps the enzyme approach starch by recognizing thestarch surface.
There is a carbohydrate-binding curvature observed in severalCBM families, such as TvAICBM34 [25] and BhCBM25 [24]complexes to provide the main protein–carbohydrate interactionsby several key residues, for example Trp51, Tyr89 and Tyr119
in TvAICBM34 and His26, Trp34 and Trp74 in BhCBM25. Thisbinding platform was also formed in RoGACBM21 by thecorresponding residues, Trp47, Tyr83 and Tyr94. These key residueshave a similar location in each protein structure, and theiraromatic rings hold the sugar molecules by hydrophobic stackinginteractions. Within this binding platform, the most conservedresidue is Trp47 in RoGACBM21, corresponding to Trp74 inBhCBM25 and Tyr89 in TvAICBM34. This binding curvaturecould swing around the substrate to create a tight binding pocketand induce at least three glucose units of sugar molecules to bebent in a segmented shape for binding.
Conclusions
The crystal structures of unliganded and liganded RoGACBM21(SBD–βCD and SBD–G7) were determined at 1.25, 1.8 and 2.3 Åresolutions respectively. Two carbohydrate-binding sites, I andII, were found on the surface of SBD, of which site I is a flatand broad hydrophobic-binding region created by the aromaticresidues Trp47, Tyr83 and Tyr94, and site II is a protruded andnarrow binding environment formed by Tyr32 and Phe58. Site Iserves as the major sugar-binding site by forming an essentialbinding curvature for at least three glucose units. Site II may playan auxiliary role for recognizing the long-chain polysaccharides.A significant interaction network, which was created in thepolyN loops unique to RoGACBM21, stabilizes the loops
and confirms the sugar binding. This might produce a higherbinding capacity for RoGACBM21 compared with other CBMsuperfamilies. An especially stable binding environment in SBDwas suggested to accommodate a variety of sugars with differentconformations and lengths. From the structural determination andthe quantitative binding assay of RoGACBM21, a possible long-chain insoluble polysaccharide-binding path was suggested, inwhich the polysaccharide is bound by the specific binding sites Iand II as well as a continuous binding surface with Tyr67 and Tyr93
between two sites.
This work was supported by grants from the National Science Council of Taiwan (NSC96-2311-B-007-014 and NTHU 96N2424E1 to Y.-J. S. and NSC 96-2627-B-007-003, toM. D. T. C.). This work was partially supported by a Council of Agriculture grant, NationalScience and Technology Programme for Agricultural Biotechnology (97AS-1.2.1.-ST-a5, to M. D. T. C.). The RoGACBM21 clone was kindly provided by Mr Chia-Chin Sheu(Simpson Biotech, Taoyan, Taiwan). The X-ray diffraction data were carried out at theNSRRC (National Synchrotron Radiation Research Center, Taiwan).
REFERENCES
1 Buleon, A., Colonna, P., Planchot, V. and Ball, S. (1998) Starch granules: structure andbiosynthesis. Int. J. Biol. Macromol. 23, 85–112
2 Hoover, R. (2001) Composition, molecular structure, and physicochemical properties oftuber and root starches: a review. Carbohydr. Polymers 45, 253–276
3 Parker, R. and Ring, S. G. (2001) Aspects of the physical chemistry of starch.J. Cereal Sci. 34, 1–17
4 Gessler, K., Uson, I., Takaha, T., Krauss, N., Smith, S. M., Okada, S., Sheldrick, G. M. andSaenger, W. (1999) V-Amylose at atomic resolution: X-ray structure of a cycloamylosewith 26 glucose residues (cyclomaltohexaicosaose). Proc. Natl. Acad. Sci. U.S.A. 96,4246–4251
5 Smith, A. M., Denyer, K. and Martin, C. R. (1995) What controls the amount and structureof starch in storage organs? Plant Physiol. 107, 673–677
6 Imberty, A., Chanzy, H., Perez, S., Buleon, A. and Tran, V. (1988) The double-helicalnature of the crystalline part of A-starch. J. Mol. Biol. 201, 365–378
7 Coutinho, P. M. and Reilly, P. J. (1997) Glucoamylase structural, functional, andevolutionary relationships. Proteins 29, 334–347
8 Sauer, J., Sigurskjold, B. W., Christensen, U., Frandsen, T. P., Mirgorodskaya, E.,Harrison, M., Roepstorff, P. and Svensson, B. (2000) Glucoamylase: structure/functionrelationships, and protein engineering. Biochim. Biophys. Acta 1543, 275–293
9 Henrissat, B. (1991) A classification of glycosyl hydrolases based on amino acidsequence similarities. Biochem. J. 280, 309–316
10 Lin, S. C., Liu, W. T., Liu, S. H., Chou, W. I., Hsiung, B. K., Lin, I. P., Sheu, C. C. andChang, M. D. T. (2007) Role of the linker region in the expression of Rhizopus oryzaeglucoamylase. BMC Biochem. 8, 9
11 Coutinho, P. M. and Henrissat, B. (1999) The modular structure of cellulases and othercarbohydrate-active enzymes: an integrated database approach. In Genetics, Biochemistryand Ecology of Cellulose Degradation (Ohmiya, K., Hayashi, K., Sakka, K., Kobayashi, Y.,Karita, S. and and Kimura, T., eds.), pp. 15–23, Uni Publishers Co., Tokyo
12 Hall, J., Black, G. W., Ferreira, L. M., Millward-Sadler, S. J., Ali, B. R., Hazlewood, G. P.and Gilbert, H. J. (1995) The non-catalytic cellulose-binding domain of a novel cellulasefrom Pseudomonas fluorescens subsp. cellulosa is important for the efficient hydrolysisof Avicel. Biochem. J. 309, 749–756
13 Machovic, M. and Janecek, S. (2006) Starch-binding domains in the post-genome era.Cell Mol. Life Sci. 63, 2710–2724
14 Giardina, T., Gunning, A. P., Juge, N., Faulds, C. B., Furniss, C. S., Svensson, B., Morris,V. J. and Williamson, G. (2001) Both binding sites of the starch-binding domain ofAspergillus niger glucoamylase are essential for inducing a conformational change inamylose. J. Mol. Biol. 313, 1149–1159
15 Chou, W. I., Pai, T. W., Liu, S. H., Hsiung, B. K. and Chang, M. D. T. (2006) The family 21carbohydrate-binding module of glucoamylase from Rhizopus oryzae consists of twosites playing distinct roles in ligand binding. Biochem. J. 396, 469–477
16 Liu, Y. N., Lai, Y. T., Chou, W. I., Chang, M. D. T. and Lyu, P. C. (2007) Solution structureof family 21 carbohydrate-binding module from Rhizopus oryzae glucoamylase.Biochem. J. 403, 21–30
17 Machovic, M., Svensson, B., MacGregor, E. A. and Janecek, S. (2005) A new clan of CBMfamilies based on bioinformatics of starch-binding domains from families CBM20 andCBM21. FEBS J. 272, 5497–5513
18 Southall, S. M., Simpson, P. J., Gilbert, H. J., Williamson, G. and Williamson, M. P.(1999) The starch binding domain from glucoamylase disrupts the structure of starch.FEBS Lett. 447, 58–60
c© The Authors Journal compilation c© 2008 Biochemical Society

36 J.-Y. Tung and others
19 Bolam, D. N., Ciruela, A., McQueen-Mason, S., Simpson, P., Williamson, M. P., Rixon,J. E., Boraston, A., Hazlewood, G. P. and Gilbert, H. J. (1998) Pseudomonascellulose-binding domains mediate their effects by increasing enzyme substrateproximity. Biochem. J. 331, 775–781
20 Boraston, A. B., Bolam, D. N., Gilbert, H. J. and Davies, G. J. (2004) Carbohydrate-bindingmodules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781
21 Belshaw, N. J. and Williamson, G. (1990) Production and purification of a granular starchbinding domain of glucoamylase 1 from Aspergillus niger. FEBS Lett. 269, 350–353
22 Paldi, T., Levy, I. and Shoseyov, O. (2003) Glucoamylase starch-binding domain ofAspergillus niger B1: molecular cloning and functional characterization. Biochem. J.372, 905–910
23 Sorimachi, K., Le Gal-Coeffet, M.-F., Williamson, G., Archer, D. B. and Williamson, M. P.(1997) Solution structure of the granular starch binding domain of Aspergillus nigerglucoamylase bound to β-cyclodextrin. Structure 5, 647–661
24 Boraston, A. B., Healey, M., Klassen, J., Ficko-Blean, E., Lammerts van Bueren, A. andLaw, V. (2006) A structural and functional analysis of α-glucan recognition by family 25and 26 carbohydrate-binding modules reveals a conserved mode of starch recognition.J. Biol. Chem. 281, 587–598
25 Abe, A., Tonozuka, T., Sakano, Y. and Kamitori, S. (2004) Complex structures ofThermoactinomyces vulgaris R-47 α-amylase 1 with malto-oligosaccharides demonstratethe role of domain N acting as a starch-binding domain. J. Mol. Biol. 335, 811–822
26 Mikami, B., Iwamoto, H., Malle, D., Yoon, H.-J., Demirkan-Sarikaya, E., Mezaki, Y. andKatsuya, Y. (2006) Crystal Structure of Pullulanase: Evidence for Parallel Binding ofOligosaccharides in the Active Site. J. Mol. Biol. 359, 690–707
27 Polekhina, G., Gupta, A., van Denderen, B. J., Feil, S. C., Kemp, B. E., Stapleton, D. andParker, M. W. (2005) Structural basis for glycogen recognition by AMP-activated proteinkinase. Structure 13, 1453–1462
28 Otwinowski, Z. and Minor, W. (1997) Processing of X-ray diffraction data collected inoscillation mode. Methods Enzymol. 276, 307–326
29 Matthews, B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–49730 Vagin, A. and Teplyakov, A. (1997) MOLREP: an automated program for molecular
replacement. J. Appl. Crystallogr. 30, 1022–102531 McRee, D. E. (1999) XtalView/Xfit: a versatile program for manipulating atomic coordinate
and electron density. J. Struct. Biol. 125, 156–16532 Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve,
R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S. et al. (1998) Crystallographyand NMR system: anew software suite for macromolecular structure determination.Acta Crystallogr. D Biol. Crystallogr. 54, 905–921
33 Collaborative (1994) The CCP4 suite: programs for protein crystallography.Acta Crystallogr. D Biol. Crystallogr. 50, 760–763
34 Kamitori, S., Kondo, S., Okuyama, K., Yokota, T., Shimura, Y., Tonozuka, T. and Sakano, Y.(1999) Crystal structure of Thermoactinomyces vulgaris R-47 α-amylase II (TVAII)hydrolyzing cyclodextrins and pullulan at 2.6 A resolution. J. Mol. Biol. 287,907–921
35 Knegtel, R. M., Wind, R. D., Rozeboom, H. J., Kalk, K. H., Buitelaar, R. M.,Dijkhuizen, L. and Dijkstra, B. W. (1996) Crystal structure at 2.3 A resolution andrevised nucleotide sequence of the thermostable cyclodextrin glycosyltransferasefrom Thermoanaerobacterium thermosulfurigenes EM1. J. Mol. Biol. 256,611–622
36 Mikami, B., Adachi, M., Kage, T., Sarikaya, E., Nanmori, T., Shinke, R. and Utsumi, S.(1999) Structure of raw starch-digesting Bacillus cereus β-amylase complexed withmaltose. Biochemistry 38, 7050–7061
37 Oyama, T., Kusunoki, M., Kishimoto, Y., Takasaki, Y. and Nitta, Y. (1999) Crystal structureof β-amylase from Bacillus cereus var. mycoides at 2.2 A resolution. J. Biochem. (Tokyo)125, 1120–1130
38 Sorimachi, K., Jacks, A. J., Le Gal-Coeffet, M. F., Williamson, G., Archer, D. B. andWilliamson, M. P. (1996) Solution structure of the granular starch binding domain ofglucoamylase from Aspergillus niger by nuclear magnetic resonance spectroscopy.J. Mol. Biol. 259, 970–987
39 Lawson, C. L., van Montfort, R., Strokopytov, B., Rozeboom, H. J., Kalk, K. H., de Vries,G. E., Penninga, D., Dijkhuizen, L. and Dijkstra, B. W. (1994) Nucleotide sequence andX-ray structure of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 in amaltose-dependent crystal form. J. Mol. Biol. 236, 590–600
40 Feese, M. D., Kato, Y., Tamada, T., Kato, M., Komeda, T., Miura, Y., Hirose, M., Hondo, K.,Kobayashi, K. and Kuroki, R. (2000) Crystal structure of glycosyltrehalosetrehalohydrolase from the hyperthermophilic archaeum Sulfolobus solfataricus.J. Mol. Biol. 301, 451–464
41 Penninga, D., van der Veen, B. A., Knegtel, R. M., van Hijum, S. A., Rozeboom, H. J.,Kalk, K. H., Dijkstra, B. W. and Dijkhuizen, L. (1996) The raw starch binding domain ofcyclodextrin glycosyltransferase from Bacillus circulans strain 251. J. Biol. Chem. 271,32777–32784
42 Schmidt, A. K., Cottaz, S., Driguez, H. and Schulz, G. E. (1998) Structure of cyclodextringlycosyltransferase complexed with a derivative of its main product β-cyclodextrin.Biochemistry 37, 5909–5915
43 Rodriguez-Sanoja, R., Oviedo, N. and Sanchez, S. (2005) Microbial starch-bindingdomain. Curr. Opin. Microbiol. 8, 260–267
Received 18 March 2008/18 June 2008; accepted 26 June 2008Published as BJ Immediate Publication 26 June 2008, doi:10.1042/BJ20080580
c© The Authors Journal compilation c© 2008 Biochemical Society


















