
Creutzfelt-Jacob Disease (CJD) A transmissible spongiform encephalopathy or “prion disease”
28
Creutzfelt-Jacob Disease (CJD) A transmissible spongiform encephalopathy or “prion disease”
-
Upload
maximilian-mclaughlin -
Category
Documents
-
view
218 -
download
2
Transcript of Creutzfelt-Jacob Disease (CJD) A transmissible spongiform encephalopathy or “prion disease”

Creutzfelt-Jacob Disease(CJD)
A transmissible spongiform encephalopathy or “prion disease”

CJD and TSEs• Creutzfelt-Jacob disease (CJD) is one of a small group of disease classified
as transmissible spongiform encephalopathies (TSEs)
• The leading hypothesis is that TSEs are not transmitted by microbes or viruses, but are spread by specific misshaped proteins, given the name prion proteins

CJD and TSEs
• At present, only 6 human man diseases are thought to be caused by prion proteins: Creutzfelt-Jacob disease, new variant CJD (vCJD), Gertmann-Straussler-Scheinker syndrome, fatal familial insomnia, kuru, and possibly Alpers' disease.

CJD and TSEs
• There are up to 25 prion diseases that have been identified in animals, including scrapie (sheep), bovine spongiform encephalopathy (cattle), and chronic wasting disease (deer & elk).

History of CJD and prion diseases5th Century BC: • Hippocrates described disease with symptoms similar to TSE that was
occurring in sheep and cattle4th and 5th Century AD:• Similar disease was recorded by the Roman Renatus1755:• Great Britain’s House of Commons discussed the epidemic of scrapie in
sheep1759 :• There were unsupported claims scrapie might be contagious

History of CJD and prion diseasesEarly 20th Century: • Despite earlier failures in attempting to show transmission of scrapie, it was
finally proven through the intraocular injection of infected nervous tissue from diseased sheep, to healthy sheep, which then developed the disease.
• First instance showing transmissibility of TSEs.1921: • The first described case of disease that became known at Creutzfeldt-Jakob
disease1950s and 60s• The disease kuru was described and was shown to be transmitted via
cannibalism. • Scrapie-Iike lesions were found the brains of kuru victims.• Kuru and CJD shown to be transmissable to chimpanzees.

History of CJD and prion diseases• 1980s: Stanley Prusiner put forth the hypothesis that that a protein was
responsible for the transfer of the disease
• 1988 : Neuropathology, bovine spongiform encephalopathy (BSE), AKA mad cow disease was described in cows
• Late 1980s the prevalence of BSE in British cattle caused concerns that BSE might be transferable to humans. BSE believed to result from scrapie jumping species to cows
• 1990s: a new disease similar to CJD and kuru called “new variant CJD” was found in humans who had been exposed to BSE infected cattle or their products.

Case Study:Patient had a surgical procedure in 1971, at age 38.• Determined to be the most likely source of the infection, after CJD diagnosis • Occurred before prion hypothesis or before it was believed the infectious agent
was resistant to sterilization techniques
Long incubation period• The malformed prion protein induces refolding of normal proteins like itself• Slow process at first (exponential growth)• Much longer replication times than microbes• It takes time to build up the levels of mis-folded prion protein in neural tissue
CJD first suspected 1.5 months after first consulting physicians (11 weeks after first symptoms)
CJD diagnosis at 2 monthsDiagnostic Tests
1. Electroencephalograph2. Cerebrospinal fluid analysis for 14-3-3 protein3. MRI of the brain

Case Study:
Final stages of disease progression (months 5 &6)• Profound dementia• Loss of speech, nearly complete inability to move• Treatment focused on comfort measures, symptom management, sedation, and
prevention of opportunistic infections in hospital• Death, age 68, occurred 25 weeks after first seeking medical help for symptoms
(31 weeks after first signs and symptoms)• Cause of death was pneumonia due to inability to clear lungs
Infection of others?• Because of knowledge of CJT being passed by prions, appropriate precautions
were taken clinically and post mortem, so it is unlikely that others were infected after the acute stage of the disease. Transmissibility is believed to be low during incubation period. The patient was not a blood or tissue donor before or after onset of symptoms, which would have been been a risk for transmission.

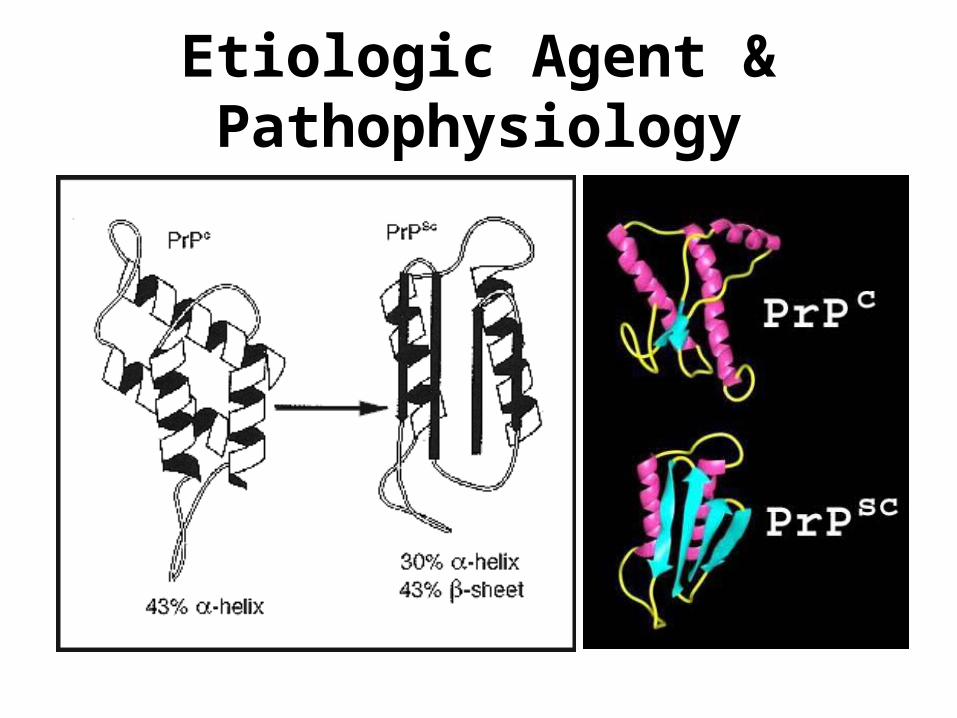
Etiologic Agent & Pathophysiology• The infectious agent in Creutzfeldt-Jacob disease
(CJD) is a mis-folded protein. This malformed protein has fewer alpha-helices and more beta-sheets than the normal protein.
Exposure to the malformed prion protein, can induce correctly folded proteins of the same type, to re-fold into the mutant form of the protein.
Etiologic Agent & Pathophysiology

Etiologic Agent & Pathophysiology• The disease state results from build-up of the malformed protein in
neural tissues which kills neural cells.
• Incubation period can range from months to decades.
• The death of the nerve cells and glial cells is the cause of all signs and symptoms of CJD and other TSE diseases.
• Most instances of CJD are sporadic, occur in people with no known risk factors or gene mutations – ‘unknown source ‘
• Some cases of CJD are transmitted by exposure to prion-contaminated instruments, tissues, serum, hormones, etc, from individuals with CJD - ‘known or suspected source’

The Leading Hypothesis
Protein-only hypothesis• The protein is believed to be the sole infectious agent. • It induces its own replication by causing
conformational (folding) changes in normal proteins

The 2 Competing HypothesesMulti-component hypotheses• Proposes prion protein is the infectious agent for the disease• Also proposes that more than just the mutated prion protein is
required – cofactors also• Cofactors may include lipids and nucleic acids (but no genomic
information contributes to the disease)• Cofactors may form part of the prion or serve as catalysts for the
refolding of normal proteins
Viral hypothesis• Postulates that an infectious virus causes the disease • Support for this hypothesis is waning• Mostly a holdover of the long-held belief that a virus must be
responsible for this type of disease

Related Human Prion Diseases• There are 2 inherited human prion diseases, fatal familial insomnia
and Gertmann-Straussler-Scheinker syndrome. These diseases are caused by the inheritance of mutations in the PRNP gene from a parent. The mutation results in a prion protein being made which folds incorrectly without exposure to malformed proteins.
• There are believed to be cases of the hereditary TSEs that resulted from random point mutations in the PRNP gene, rather than being present in the genes of a parent.
• The human disease new variant CJD (vCJD) is caused by exposure to cows (or their products) that were infected with the prion disease bovine spongiform encephalopathy (BSE) commonly known as mad cow disease.
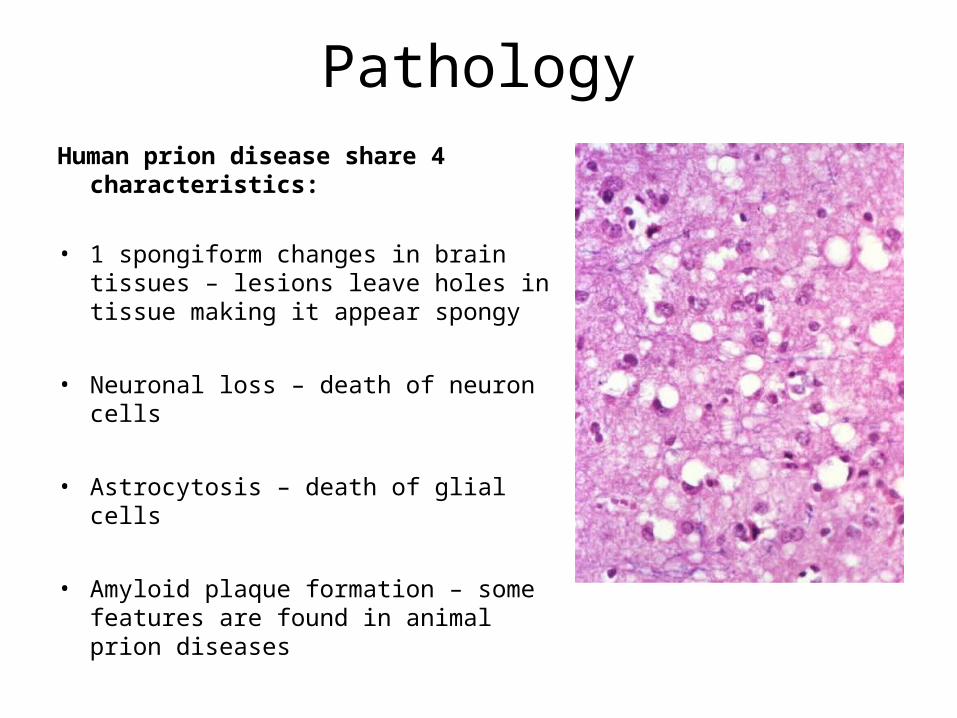
PathologyHuman prion disease share 4 characteristics:
• 1 spongiform changes in brain tissues – lesions leave holes in tissue making it appear spongy
• Neuronal loss – death of neuron cells
• Astrocytosis – death of glial cells
• Amyloid plaque formation – some features are found in animal prion diseases

Clinical Signs and Symptoms of CJD• Personality changes• Psychiatric problems (depression)• Lack of coordination and control• Ataxia• Involuntary jerky movements• Unusual sensations• Insomnia• Confusion• Memory problems
Final stages of the disease:• Dementia• Loss of ability to speak• Loss of ability to move• Eventual death

“Virulence Factors”
While not true virulence factors as we see in microbial diseases, there are several characteristics of all prion diseases which facilitate their transmission.
• Long incubation periods (months to decades)
• No signs or symptoms during incubation
• So far, no way of detecting the mutated prion protein in living individuals– Examinations of brain tissue after death is only sure way to confirm CJD
and other TSEs– Research is working on promising technology which may solve this

“Virulence Factors”• Documented Zoonotic infection of animal TSE diseases to humans
– Scrapie may be the origin of BSE– BSE is the causative prion for new variant CJD– Chronic wasting disease (deer & elk) is being watched closely to see if it jumps to
humans. Hunter are given advise to reduce any risk from game animals they harvest
• Prion proteins are highly stable – very resistant to usual sterilization procedures– Boiling, autoclave, cooking and other heat sterilization methods don’t denature the
prion protein– Irradiation dose not denature the prion protein– Special proteases, bleach, acid baths followed by autoclaving seem effective– We don’t know how long mutated prion proteins persist on surfaces or in
contaminated tissues.– Some TSE may be transmissible thru aerosols (tiny droplets)

Modes of Transmission
• CJD is mostly a sporadic disease. No mode of transmission is proven to account for common form of the disease.
• New variant CJD (nvCJD)is believed to occur in humans as a result of exposure to bovine spongiform encephalopathy
• A hereditary form of CJD (fCJD) occurs as a result of an inherited defective gene that promotes the misfolding of the prion protein with no exposure to malformed proteins
• Exposure or consumption of tissues from animals with any TSE is considered a risk factor
• Nervous tissue (brain and spinal cord) have highest concentration of prion proteins

Diagnostic Procedures
• Electroencephalograph• Cerebrospinal fluid analysis for 14-3-3 protein• MRI of the brain• Brain Biopsy• Autopsy, post mortem

Prevention Measures
• Feeding of rendered mammal proteins to mammals in human food chain (sheep, goats, cows, farmed deer, elk, etc) has been banned in most developed countries
• Brain and spinal cord are separated from carcass early in butchering process
• Hunters in CWD affected areas are advised to bone out meat and avoid contact of meat with any nervous tissue.
• Elimination of cannibalistic funerary practices in New Guinea contributed to the end of kuru epidemic

Treatment of CJD
• No known cure, therapy or vaccine for TSE diseases
• Treatment of the disease is primarily limited to treatment of symptoms and palliative care
• Study of kuru suggests there may have been resistance factors in some individuals in the population
• Active research in all avenues of treatment is ongoing

Prevalence of CJD• 102 cases in Oregon (1991-2009): an avg. of 5.7 per year)• About 1 case per million people, per year, in the US of the
common (sporadic) form of CJD • About 1 case per million people, per year, world wide

Why is this an important disease to study?
• CJD and all other known prion diseases are fatal• Evidence suggests that prion diseases can jump species• Humans have contact with most of these species• BSE is still occasionally discovered – we eat lots of beef• Because of long incubation period, epidemics could
become wide spread, before signs of disease start appearing
• The Prion hypothesis was one of the most significant and controversial biological proposals in the modern era of biology

References:Articles, books:Belay, E. (1999). Transmissible spongiform encephelopathies in humans. Annual Review of Microbiology, 53,
238-314. Retrieved from http://www.cdc.gov/ncidod/dvrd/prions/resources/BelayE_Annu_Rev_Microbio.pdf
Cowan, M., & Bunn, J. (2013). Microbiology fundamentals: A clinical approach. (pp. 477-478). New York: McGraw-Hill.
Webpages:CJD (creutzfeldt-jakob disease, classic). (n.d.). Retrieved from http://www.cdc.gov/ncidod/dvrd/cjd/index.htm Creutzfeldt–jakob disease. (n.d.). Retrieved from http://en.wikipedia.org/wiki/Creutzfeldt-Jakob_diseaseOregon Health Authorty. (2009, Sept 28). Oregon reprted deaths fromm creutzfeldt-jacob disease, 1991-
present. Retrieved from http://public.health.oregon.gov/DiseasesConditions/DiseasesAZ/bse/Documents/cjdeath.pdf
Prion. (n.d.). Retrieved from http://en.wikipedia.org/wiki/PrionPrion diseases. (n.d.). Retrieved from http://www.cdc.gov/ncidod/dvrd/prions/Transmissible spongiform encephalopathy. (n.d.). Retrieved from
http://en.wikipedia.org/wiki/Transmissible_spongiform_encephalopathyvCJD (variant creutzfeldt-jakob disease). (n.d.). Retrieved from
http://www.cdc.gov/ncidod/dvrd/vcjd/index.htm

Photos, Illustrations:Deyo, S., & Deyo, H. (Producer). (2001). CJD USA map. [Print Photo]. Retrieved from http://www.millennium-
ark.net/News_Files/Newsletters/News010113/News010113C.html(n.d.). Major regions of the human brain affected by tse. [Web Graphic]. Retrieved from
http://whyfiles.org/193prion/3.html (n.d.). Normal to abnormal protein. [Web Graphic]. Retrieved from
http://memory.ucsf.edu/cjd/overview/biology/proteins/multiple/cause(n.d.). Plaques from creutzfeldt–jakob disease in the brain.. [Web Photo]. Retrieved from
http://thedallasgeek.com/2010/07/31/creutzfeldt–jakob-disease-cjd/(n.d.). Prion theory. [Web Photo]. Retrieved from http://lawrencekok.blogspot.com/2011/04/ib-biology-
microbes-proteins-and-prions.html(n.d.). Prion graphic, mayo foundation. [Web Graphic]. Retrieved from
http://thedallasgeek.files.wordpress.com/2010/07/r7_prion.jpg(n.d.). Prions, image 1 and image 2. [Web Graphic]. Retrieved from
http://www.stanford.edu/group/virus/prion/2004anderson/index.html(n.d.). Deer with chronic wasting disease (cwd). [Web Photo]. Retrieved from
http://www.prwatch.org/news/2012/05/11500/media-coverage-mad-cow-usda-calls-misleading-columbia-journalism-review-calls-san
(n.d.). sheep. [Web Photo]. Retrieved from http://upload.wikimedia.org/wikipedia/commons/3/3d/Take_ours!.jpg
(n.d.). madcow2. [Web Photo]. Retrieved from http://www.topsecretwriters.com/2011/11/mad-cow-disease-bse/madcow2/

NOTICE• This PowerPoint document is the work product and intellectual property of Ken Irwin.
• Presenting this document, or any portion of it, as your work constitutes academic dishonesty.
• Use, in whole or in part, without written permission is prohibited.


















