
COVALENCY EFFECTS IN MAGNETIC INTERACTIONS
33
HAL Id: jpa-00216628 https://hal.archives-ouvertes.fr/jpa-00216628 Submitted on 1 Jan 1976 HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci- entific research documents, whether they are pub- lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers. L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés. COVALENCY EFFECTS IN MAGNETIC INTERACTIONS B. Tofield To cite this version: B. Tofield. COVALENCY EFFECTS IN MAGNETIC INTERACTIONS. Journal de Physique Col- loques, 1976, 37 (C6), pp.C6-539-C6-570. 10.1051/jphyscol:19766115. jpa-00216628
Transcript of COVALENCY EFFECTS IN MAGNETIC INTERACTIONS

HAL Id: jpa-00216628https://hal.archives-ouvertes.fr/jpa-00216628
Submitted on 1 Jan 1976
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
COVALENCY EFFECTS IN MAGNETICINTERACTIONS
B. Tofield
To cite this version:B. Tofield. COVALENCY EFFECTS IN MAGNETIC INTERACTIONS. Journal de Physique Col-loques, 1976, 37 (C6), pp.C6-539-C6-570. �10.1051/jphyscol:19766115�. �jpa-00216628�

CO VAL ENC Y EFFECTS AND MA GNETIC H YPEISFINE' INTERA C TIONS.
COVALENCY EFFECTS IN MAGNETIC INTERACTIONS
B. C. TOFIELD
Materials Physics Division, A. E. R. E., Harwell, OX11 ORA, U. K.
RBsumB. - On prksente une revue des interactions magnetiques dans le but de dkgager les informations sur la covalence qui peuvent en Stre dkduites.
Les interactions hyperfines sur des ligands, les champs hyperfins dans les mktaux mesurks par spectromktrie Mossbauer et diffracti~n neutronique sont prksentkes en detail, en insistant sur la dernihre approche qui est la plus productive en ce domaine. Les informations dkj& recueillies sont confrontkes aux connaissances en chimie et on essaie de degager les axes intkressants pour de futures etudes.
Abstract. - Magnetic interactions, categorised into ligand based, metal based and neutron diffraction, are reviewed with regard to the information they give on covalency. The ligand hyper- fine interaction, the metal hyperfine field measured by the Mossbauer effect and neutron diffraction measurements are mentioned in some detail, with particular emphasis on the latter as being the most productive experimental technique in this field. The information already gathered is assessed with reference to chemical knowledge, and useful avenues for future investigation are suggested.
1. Introduction. - The ionic model occupies a central position, both in the teaching of solid state inorganic chemistry and in the explanation of many solid state phenomena in the current literature. I t has been used for example to rationalise many thermo- dynamic data and in this context has been lucidly described in text books such as those of Phillips and Williams [I] and Johnson [2]. Such descriptions rely on the concept of the ionic radius, more or less transfe- rable from compound to compound, and the deriva- tion of accurate radii and the determination of the effect on them of factors such as ionic spin, coordina- tion number, bond distortion and so on, has received new impetus with the publication of many accurate crystallographic data in the last few years. The tables of Shannon and Prewitt [3] represented a significant milestone in this endeavour, and stimulated much supplementary activity, detailed in the revised list of Shannon [4]. Such work is inherently empirical in nature, but even in some of the most sophisticated quantitative lattice potential calculations currently performed, to explain [5] , for example the occurence of very short fluorine-fluorine distances in anion-excess fluorites such as Cal -,YxF2 +,, it has not been found necessary to invoke deviations from formal ionic charges.
Nevertheless, there are many indications in thermo- dynamic and structural solid state chemistry of devia- tions from ionic model behaviour, even though self- compensating features of the ionic model do tend to compensate for departures from formal ionic charges. This is discussed by Phillips and Williams [I], who detail the covalent contribution to heats of formation of
B-group metal compounds by comparison with the compounds formed by A-metal ions of similar radii.
More traditional evidence has been the reduction of coordination numbers associated with metals suspected of showing significant covalency, or directed bonding, in their compounds, particularly for B-group metals such as silver, zinc, mercury, and so on. The elastic constants of tetrahedrally bonded materials with zinc blende or wurtzite structure have been related to the degree of covalency by Phillips [6] who showed how the ratio of bending to stretching force constants increased linearly with decrease of ionicity, as determined from the dielectric properties of the crystals. The shear constants reflect the magnitude of covalent bond charge and resistance to bond bending, but unfortunately such data are either not available, or not so straight- forwardly analysed in most situations of interest to the inorganic chemist, where either the symmetry is too low, the unit cell too complicated or the ionicity higher than in the simple tetrahedral semiconductors.
The adoption of structures, particularly layer lattices, not explicable for any combinations of ionic radii is also generally accepted to be evidence for metal-ligand covalency, as is the shrinkage [I, 4, 71 of effective ionic radii for some metals such as Cu', Tli, Pb2+ on bonding to relatively more polarisable ligands (e. g. I- c. f. F- or S2- C. f. 0'-) this effect being intimately related to the thermodynamic features mentioned. However, it is not at all clear what is the amount of covalency relative to other polarization effects such as van de Waals interactions which is necessary to produce such structural manifestations. Although layer lattice compounds such as those of the
Article published online by EDP Sciences and available at http://dx.doi.org/10.1051/jphyscol:19766115

C6-540 B. C. TOFIELD
transition metal dichalcogenides have been much studied lately with regard to the intercalation of metals, e. g. lithium into titanium disulphide [a], and organics, e. g. pyridine into tantalum disulphide [9], there do not seem to be any well established rules for predicting what reduction of the formal charge of the anion by covalency is required before the van der Waals attraction between layers of sulphur, chlorine, or other anions often found in layer lattices, becomes the dominant structure-determining feature. Tetrahedral coordination of the metal atoms, a consequence if covalent interactions are strong, according to Phil- lips [6], is not a feature of many layer lattice compounds.
From structural chemistry also one might enquire about the role and the nature of covalency in the defect behaviour of high oxidation state oxides. Does the occurrence of crystallographic shear [lo], and the elimination of point defects in oxides of Ti, Nb and W, for example, depend at least in part on relatively strong ligand-to-metal n-bonding, permitting edge-sharing or face-sharing of octahedra ? Shear is not observed in isovalent oxides of the post-transition B-group metals such as Sn and Sb although for these, covalency is surely just as important, but is now dominated by ligand- to metal s-bonding. The structures of A+BS+03 compounds have been justified in similar terms [ll]. Some years ago I suggested it would be interesting to study BaPtO, to compare with related phases for earlier transition metals. Now PtlV, d6 low spin, is strongly stabilised by o-covalent interaction in an octahedral environment but BaPtO, does not form as a perovskite. Phases such as Ba4Pt06 are much more easily prepared however and these facts seem to be another structural consequence of strong covalency : in Ba,Pt06 each oxygen linked to Pt is shared only with a barium ion, but in BaPtO, the P ~ o Z - octahedra are not isolated and we can say that the oxygen is too strongly electronegative to be able to donate sufficient electron density to satisfy two pt4+ ions. In other words ~ t0 : - is now effectively an oxy-anion like COO:-, F~o:- and other high oxidation state metal-oxygen solid state complexes.
It is to provide explanations to features such as these and also to be able to quantify predictive che- mistry that the chemist is interested in covalency. In solid state inorganic chemistry and physics this seems to be most easily comprehended from the standpoint of the ionic model with effective charges of ions reduced by some amount from the formal ionic charges. This concept is nicely quantified in the molecular orbital model of covalency, applied below to the case of transition metal ions.
One of the most fertile areas for illustrating effects of covalency has indeed been the solid state compounds of the transition elements [12]. The wide variety of size and charge of ion possible in structure types such as perovskite permits many different regimes of electrical and magnetic behaviour, some of which demand an explanation involving covalency effects. Because many
of these ions have a nonzero spin, associated more or less with a specific d or f orbital, the fact that a covalent interaction between a closed shell anion orbital and an unpaired spin on a metal ion alters the spatial distri- bution of this spin provides a sensitive probe for investi- gating at least a part of the total bonding interaction. It is for this reason that most quantitative, or semi- quantitative data on covalent effects have been obtained by measurement of magnetic interactions, principally by magnetic resonance, Mossbauer spectroscopic or neutron diffraction techniques.
The direct measurement of unpaired spin distribu- tion, either by examining the region near the ligand nucleus, transferred by covalent interactions, using Iigand hyperfine interactions (LHFI), or the total spin distribution by neutron diffraction seems to provide the most direct information. Hyperfine interactions with the metal ion nucleus measured either by ESR or Mossbauer spectroscopy are clearly also very sensitive to d electron covalency, but being modulated by s orbitals, are sometimes more difficult to interpret quantitatively. This review will attempt to assess and compare the information gathered by the various techniques. Many trends are clearly observable ; these are indeed often thought to be satisfactory if agreeing with the type of thermochemical data mentioned. Nevertheless there are inconsistencies, both compared with thermochemical data and in comparing measure- ments of one technique with another, although some- times these are understandable in that different techniques probe different regions of the electron charge cloud. One quickly realises the need for theoreti- cal calculation to supplement the experimental effort, both to interpret the data obtained and to draw attention to defects in the models used for interpreta- tion, although there is much virtue in simple models, especially when used for comparative purposes, unless hopelessly physically unrealistic. Several calculations have seemed to indicate that the molecular orbital model of covalency in transition metal complexes, unlike the ionic crystal field model, might be acceptable on these terms.
The magnetic techniques essentially probe the electron spin distribution, without significantly disturb- ing it, by measuring nuclear interactions or by elastic scattering. It should be mentioned that several other techniques, which do not depend on the presence of unpaired spin, are used to probe covalent effects. These include measurement of charge or momentum distributions by NQR, Mossbauer spectroscopy, X-ray or Compton scattering, and spectroscopic methods, such as ligand field spectroscopy or photoelectron spectroscopy. These various techniques have been compared with those of interest here in a recent review 1131 and although impressive advances have been made in several since the time of writing, will only be mentioned in passing here. It should be said, however, that the information gained is essentially complementary to that from the magnetic methods

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-541
and an aim of theoretical calculations should be to synthesise the various pieces of data gained on parti- cular compounds. Also, this review is competent to deal only with quite simple lattice-type compounds. All the techniques mentioned, with the exception of neutron diffraction, have been widely applied to bond- ing in complexes which in many respects differ only from simpler compounds in the bulkier nature of the ligand. As the difficulties presently encountered even in understanding quite simple solids become more understood the emphasis generally will naturally shift to more complicated chemical situations involving complexes, often trying to simulate biological situa- tions. Although most of the presently obtained knowledge on complexes will not be much mentioned here I will hope to indicate some situations where experiments might usefully be carried out in the near future.
This review will also show an emphasis towards neutron diffraction methods, because of the author's greater familiarity with this area, because it is the least widely appreciated and because, by virture of spin density mapping, it has the greatest promise for provid- ing useful new data. Although I am no longer actively involved in this field I would like to use this occasion to encourage others to perform accurate spin density measurements using the excellent facilities which now exist. The task has hardly begun but the rewards would be great.
A review of the progress to date of the covalency information gained from neutron diffraction was written two years ago [27]. One can justify a further
discussion because new data have been obtained both by neutron diffraction and other techniques, because there is the chance to mention the correlations between magnetic properties and other parameters recently drawn [14], and because there is the possibility of proposing some directions which future study might usefully take.
2. Experimental Data Interpretation. - The expe- rimenter obtains numbers which he must relate to the physical system under investigation. The extraction of covalency information from magnetic measurements has generally been achieved using a simple molecular orbital model. This model was first invoked [15] in this context to account for the discovery of a hyperfine interaction with the chlorine nuclei in 1r~12- and the reduced g-factor in this complex, and it is rather appo- site that the spin density in this complex revealing the presence and symmetry of the spin covalently delocalis- ed onto the chlorine atoms in K,IrCI, has recently been determined. This complex is rather unusual in fact, being based on a third-row transition metal ion bonded to chlorine ; most of the data collected by spin resonance or neutron diffraction methods have been on first row metals' bonded to oxygen or fluorine. Hopefully this situation will change before too long, but we must note that Mossbauer spectroscopy has recently provided some interesting information on covalency involving iodine ligands.
If the positive and negative axes of an octahedron for the x, y and z directions are labelled 1 and 4, 2 and 5, and 3 and 6 respectively, the antibonding orbitals for d-electron bonding are :
1 +3,2-2. = N, (d3.2-.2 - - ,I,(- 2pr3 + 2pz6 + pxt - px4 + py2 - py5) -
Jiz
where
so = 2 < d , I p , > , S s = 2 < d , l s > and S , = 2 < d , I p , > . (8)

C6-542 B. C. TOFIELD
It should be remembered that these wavefunctions describe the antibonding orbitals derived mainly from d electron states in a transition metal complex. If there are no outer d electrons there cgn still be significant covalency in compounds of (formally) ~ i ~ " , Nb5 ", Mo6+ and so on. This will also be the case for o bonding in octahedral compounds with no e, electrons (e. g. Cr3 '(t:,), 1r4+(tZg)). Magnetic measurements will not be sensitive to covalency in these situations, but charge sensitive measurements (e. g. quadrupole coupling constants) will be. Similar remarks apply to dia- magnetic complexes containing antibonding d-elec- trons in, for example, low spin d6 octahedral, or square- planar d8 configuration. Nor will magnetic measure- ments reflect bonding into other than d (or f) states on the metal, except sometimes indirectly. Yet ions such as Cd2+ and zn2+ with a completed d-shell seem to be as covalent in their behaviour as Ni2" and Mn2+ where d-electron covalency is well established by magnetic measurements. The bonding to outer s and p levels occurring for the B-group metals will occur in transi- tion compounds as well, although to a lesser extent because of the lower effective nuclear large. The Mossbauer isomer shift is very sensitive to the partial occupation of outer s orbitals. Sometimes, in favou- rable situations, the experimental data can directly reveal deficiencies in the model. This is the case for spin polarised e, electrons in c r3+ compounds where the model given above predicts no o-bonding effect to be revealed in the magnetic behaviour. For Mn2', on the other hand, where both o and n bonding are measured by neutron diffraction or spin resonance, the conse- quences of spin polarisation cannot be readily disen- tangled [16].
In general, therefore, although one can usefully use the covalency parameters obtained from experiment via the simple MO model to demonstrate trends in a series of compounds, one must be cautious when seeking a true physical interpretation. It is the province of the theoretician to be able to reproduce by calcula- tion the numbers obtained experimentally, and to inform us, both on the approximations involved on the way and their likely consequences, and on the real physical significance of the numbers, i. e. where the models are good or bad and where they need modifying. Such interaction is essential.
The admixture coefficients I,, I, and I,, which may be derived from experiment contain both covalency and overlap information. If y is the admixture parame- ter in a bonding orbital, then to first order
Although normally neglected in qualitative discussions, the treatment of I as purely a measure of bonding may be a rather serious approximation in situations where the metal-ligand admixture and overlap are quite strong. The rather small s admixture parameter observed for transition metal complexes may arise principally from overlap effects.
The complex as proposed is presumed isolated from other magnetic complexes, as occurs in the experimen- tal situation for EPR experiments. It is customery to refer to the fraction of unpaired spin (f) transferred to a single ligand orbital when the metal d orbitals of the appropriate symmetry are singly occupied (d3, d5 high spin for t,, .n electrons, d5 and d8 for ep o electrons). Then
~ b 2 62 Nb2 2 ~ , 2 fu = , fs = - 2, and f = - A' (10) 3 , 4 n '
The relations are not correct for other d states and, in reading the literature one has both to ascertain what symbols are being used and whether f really means f or is used as a measure of 1 in eq. (10) in all cases.
The simple LCAO model assumes that the combining atomic orbital states are radially unchanged. However, the charge transfer occurring will undoubtedly cause modifications of screening effects and for transition metal complexes this is expected to allow an increase in the radial extent of the d orbitals. Such an effect may occur via transfer of charge to outer s and p orbitals also. On the other hand, the Coulombic effect of the other ions in the lattice may cause either an expansion or a contraction. Jorgensen [17] distinguished the expansion of d orbitals via screening, or central field covalency, from the charge transfer occurring via the LCAO molecular orbital model, or symmetry restricted covalency, to account for the nephelauxetic effect. It would appear that these mechanisms are sometimes confused in the literature, but anyway it is not at all clear whether the MO model described is sufficiently close to physical reality to allow such fine distinctions to be meaningful. One must appeal again to calculation. Experimentally, the measurement of neutron form factors may throw light on the radial functions involved, but here the evidence so far is equivocal, which encourages caution in attempts to explain other data such as reduced quadrupole coupling constants on the basis of significantly expanded d functions.
3. The Ligand interactions. - Writing the antibond- ing orbitals in the form
where x,,,, etc. refer to the normalised ligand atomic orbital combinations detailed in eqs (1) to (5), the general behaviour of spin related properties expected in a partially covalent complex may be examined by considering such an antibonding orbital to contain one electron. The moment distribution, previously given by the square of the d wave function, is now given by the square of the molecular orbital. To second order we have :

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-543
+ 2 & A s Xzs Xzpa (13~)
I +,(TI l 2 = dn2(1 - 22) + ( 1 4 ~ )
+ 2(Az S, d; - A, dz x2pn) (14b)
+ 1 2 ~ g p n . (14~)
The spin density can be considered to be in three parts, a term of the form dZ(l - A'), a term of the form A2 x2 and a term containing overlap integrals and products of metal and ligand wavefunctions. The magnitude of the spin density on the ligand is directly proportional to the admixture parameter and thus provides a very direct measurement of the covalency in the complex. The perturbation of the ligand nuclear levels caused by this new magnetic field, the ligand hyperfine interaction LHFI, provided the first direct demonstration of covalency in transition metal complexes [IS], and has since been measured in many different complexes by spin resonance techniques. Recently, measurement of the hyperfine field at '''1 by Mossbauer spectroscopy has also provided covalency information, for example for CrI, [19].
The Hamiltonian for the interaction at the ligand may be written generally as
X = - g N ~ l u H s I (1 5)
where H is the transferred hyperfine field and I the nuclear moment on the ligand. For 19F, I = 3 and many spin resonance data have been obtained on fluoride complexes. For 160, I = 0 and spin resonance data have only been collected on oxides with the fairly recent preparation of materials doped with 170 (I = +).
Where I > + (170, the heavier halides) there will be a quadrupole interaction in addition. Thus to fit the lz9I Mossbauer spectra of CrI, below the Curie temperature the Hamiltonian needed was
where eq is the electric field gradient along the principal (2) axis of the EFG tensor (e is the charge on the proton), Q is the nuclear quadrupole moment and y is the asymmetry parameter. Such an analysis yields H, eq and the angles between the coordinate systems of the EFG and the hyperfine field. The results may be obtained from polycrystalline samples in suitable cases which offers some advantages in comparison with spin resonance results where the hyperfine interaction must be measured as a function of orientation withsingle crystals.
y = 0 in the case that the symmetry is axial, as along a metal-ligand bond in an octahedral complex, the frequently encountered geometry in spin resonance measurements. The spin Hamiltonian used to describe the ligand interaction in magnetic resonance measure- ments is then conveniently expressed in the form :
+ Bf(S, I, + S, I,)
where the first term is the nuclear Zeeman interaction with the applied field. When the ground state of the magnetic ion is an orbital singlet A' and B' may be quite directly related to the contributions from the separate types of orbitals containing unpaired spin, and the dipolar contribution, Ad from the metal unpaired electrons :
A' = As + 2(Ad + A, - An) (18) B' = A, - (A, f A, - A,) (19)
where
and
R is the metal-ligand distance, and the dipolar and contact hyperfine interactions corresponding to a singly occupied p orbital, and s orbital, respectively are :
There is also an isotropic contact term arising from the unpaired ligand p orbitals due to polarisation of core s electrons, which is of the form
where k = 2 g, p, p, < r i 3 > u and K 0.1 [20]. A: is normally much smaller than A, but may be important where, to first order,f, = 0 by symmetry, as in Cr3+, d3. However, calculations to estimate the magnitude of K have only been done for F- ions so there is considerable uncertainty as to the correct values to use for heavier ligands such as I-.
From a measurement of A' and B' by spin resonance of single crystals the difference (f, - f,) andf, may be determined, although where the symmetry is not axial this restriction does not necessarily hold. Thus f, and f,

C6-544 B. C. TOFIELD
could be determined independently for CrI, [19]. Such measurements were comprehensively discussed by Owen and Thornley [20], and as they note, the data may be interpreted at various levels of approximation. Certainly the ligand wavefunctions must be known to provide I $(0) l 2 and < r i 3 > and these will vary with the charge state which will vary with the covalency. This could make a difference of up to 10 % for F - ; for 0'-, and for more complicated ligands uncertain- ties of at least this magnitude are probably inherent for any chosen charge state. The real ionic charges corrected for covalency must be known to permit the correct determination of A, and the R must also be known. For EPR or ENDOR work, this may well be subject to some uncertainty as the magnetic complex is doped into a diamagnetic host. Other problems can arise from competing processes not accounted for in the simple MO model. These can be quite important, but difficult to estimate quantitatively and include contri- butions from inner ligand electrons (e. g. 2p electrons for chlorine [21]), admixture with empty ligand orbits (expected to be particularly important in cases where metal-to-ligand z bonding is occurring), spin polarised transfer to empty metal orbitals (especially noticeable for d3 ions and discussed beIow), and spin-orbit admixture of excited charge-transfer states into the ground state [20].
Once again, we see that a quantitative interpretation requires extensive calculation, but that qualitative interpretation of data, and of trends via the MO model is in order. This is particularly important as comparison can then be made with data collected by other tech- niques such as neutron diffraction and Mossbauer spectroscopy. Although the collection of magnetic resonance LHFI data is an active field still, the complexity of the interpretation for more complex materials, which includes all complexes with poly- atomic ligands, sometimes makes the extraction of quantitative covalency information quite difficult. Fairly recent discussions have been given, for example, for CuCli- and C u ~ r i - (ref. [22]), for proton NMR in RU(NH,):+ (ref. 1231) and for 170 LHFI in 6 3 ~ ~ ( ~ 2 0 ) 2 6 ' (ref. [24]). The determination of spin density distributions by neutron diffraction will be as important an advance in this area as was the measure- ment of covalency parameters by neutron diffraction for simpIe oxides, and fluorides.
The quadrupole interaction is quite complementary to the magnetic hyperfine interaction as it reflects not spin but charge transfer. eq and possibly q can be extracted from the parameters of the spin Hamiltonian. In a crystal,
The Sternheimer antishielding and shielding para- meters y, and R [0 < (1 - R) < 11 describe the induced distortions of the charge cloud of the ligand due to the lattice and due to the valence electrons. R is
quite small (0.2-0.3 often), y, is large and negative (often - lo), and neither is known very precisely. The lattice contribution has often been neglected in interpretation of quadrupole coupling constants as it is often considerably smaller than the valence term. For ligand p electrons in an octahedral complex the valence contributions to q (neglecting the shielding factor) are
4 q = - - < r - 3 5 > for p,
and
If then, we consider bonding to metal d states only, and that A = y (eq. (9)) then a quadrupole interaction arises when there is a net imbalance of electrons (N) in the p, relative to the px and p, orbi- tals [25]. If
then
fQ may be related tof, and f, [20, 261 and the relations have been tabulated [I 3,271.
In general, fQ will measure different combinations of f, and f, (which are fixed relations of A, and A, by eq. (10)) than obtained through the ligand hyperfine interaction although for high spin d5 ions (Mn2+, Fe3+) fQ = f, - f, and for d8 complexes fQ = fo. fQ, relecting charge transfer also provides results on non-magnetic ions and is, for example, 2(f, - f,) and 2 f, for do and low-spin d6 species respectively.
Because of the several approximations involved in this approach, however, any absolute interpretation of fa is highly qualitative, although broad consistency with LHFI-measured spin transfer coefficients is claimed [28, 291 for Mn2+, Fe3 +, Cr3' and Ni2+ in 170-doped MgO.
A more promising approach is possible where the observed eq may be normalised with reference to an atomic species. This is possible for C1, Br and I and in this case, at least if one is considering the same formal charge as in the simple halides, the < r-3 > and (1 - R) terms will disappear if we define
eq (crystal) 'Q = eq (atomic)
The use of fo as an indicator of the bonding in several series of compounds has recently been discussed [13]. Hopefully, for isostructural series of compounds of the relatively covalent transition metal ions or B-group ions, where the lattice contribution is appa- rently small (by comparison with A-group metal compounds), the trends discerned are at least qualita- tively correct.

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-545
A fair range of compounds with the K,IrC16 structure (Fig. 1) have been studied. Here the halide ion is indeed bonded to one metal atom only, as part of an octahedral complex, and the simple relations off , and f, to fQ, mentioned above, are appropriate.
FIG. 1. -The crystal structure of KzIrC16. The only variable positional parameter is that of C1 which may be slightly displaced from (00 a) and equivalent positions to (00z). The structure is related to that of an ordered perovskite K2Ir Cl6 where is a vacancy so that the 1rCIt- octahedra are isolated from one ano- ther. Alternatively it may be viewed as an antifluorite Kz(IrC16).
It would appear that bromides are consistently more covalent than chlorides but fQ is generally very similar for bromides and iodides. This is interesting as other data generally suggest that the reverse if anything might be true (chlorides - bromides and less covalent than iodides). Whether this,reflects neglect of essential factors, such as overlap effects, in the model, or different mechanisms of bonding does not seem to be clear at the present time. Other trends apparent are a similarity of 4d and 5d covalency for the chlorides and bromides of Pd and Pt, and a greater covalency per ligand for tetrahedrally coordinated compared to octahedrally coordinated snIV and PbIV. This latter result appears to be supported by '19Sn Moss- bzuer isomer shift data [30], apparently indicating also, therefore, a greater total charge transfer in the tetrahedral case. It is interesting to compare these results with the fairly similar conclusions (see below) reached from neutron diffraction experiments on oxides containing octahedrally and tetrahedrally coordinated rnxtal ions, although for the B-group metals the bonding is dominantly by charge transfer to metal outer s and p orbitals and tetrahedral coor- dination encourages favourable sp3 hybridisation. For the transition metals this mechanism is less important as the 4s and 4p orbitals are higher in energy and radially more diffuse.
Mossbauer data for Fe3' oxides indicate significant 4s population (2 0.1 electron) as discussed below and there is also NQR evidence pointing to 4s and 4p involvement in bonding, in particular the C1 quadrupole coupling constant of KMnCl, where for Mn2+ we recall that fQ should equal f, - f,. However for a linear Mn-C1-Mn situation, f, - f, by NMR is roughly zero but fQ is 16.7 % [31] (for CsMgCl,, fQ is 8.1 %).
Finally, by comparing the fQ data for K,MC16 (M = W to Pt) it appears that covalency must increase, as expected, across the 5d series.
Most halides of divalent and trivalent metals do not have the simple geometry considered above and a proper analysis invoking the particular geometry must be made if a comparison of quadrupole data with spin transfer data is required. This has been done [19] recently using Mossbauer data for CrI, which is discussed below. Because iodine is a Moss- bauer nucleus it is however appropriate to mention here the qualitative picture of bonding for iodine compounds which can be presented by combining isomer shift with quadrupole coupling data [32]. A semi-empirical relation relates the isomer shift relative to ZnTe to the 5s and 5p occupations :
6 = ah, -i- bh, + 6, (28)
where h, is the number of 5s holes and h, is the number of 5p holes. a and b will be of opposite sign as the loss of 5p electrons reduces the 5s screening, and de Waard estimated [32] the best values from various compilations with the probable errors to be,
and So = - 0.54 2 0.02 (mm s-I). We recall (eqs (25), (26)) that the sign of q will reflect the dominance of TC or B bonding (negative and positive respectively) and thus if 6 - 6, is plotted against q an indication of the nature of the bonding is displayed (Fig. 2). The straight lines represent bonds with predominant n or a bonding and little hybridisation. For positive 6 - 6,, points between the lines represent mixed TC and a bonding and points below indicate 5s invol- vement in the bonding. It would be encouraging to see the broad features of such a plot reproduced by calculation. For the group valence, KIO,, IF,, etc. a significant 5s contribution is indicated.
We have mentioned the outlines for the interpre- tation of the LHFI only for the case of transition metal ions with orbital singlet ground states. When the ground state is orbitally degenerate spin orbit coupling acts in first order to scramble orbital and spin states which makes the extraction of covalency information considerably more tedious 1331. In general it is not straightforward to extract f, and f, individually and either approximations must be made (e. g. f, = f,) or the ligand hyperfine informa- tion must be combined with other information such
35

FIG. 2. - A plot of isomer shift versus q for 1291 compounds. The straight lines indicate pure a and pure a bonding according to the simple Townes-Dailey model. For positive 6 - 60 points between the lines represent mixed a and a bonding, and negative 6 - 60 indicates 5s involvement in the bonding (from ref. [32]).
as the g-factor reduction. The magnitude of the inter- action may, however, be related to the total bonding interaction so that comparisons of this sort for different ligands and a given metal ion have been made in a few cases. Nevertheless, the great majority of the quantitative information gained so far has been collected on spin only metal ions and except in special cases we will not discuss in any detail spin distribution measurements on systems where the spin-orbit coupling is not quenched to first order. For Co2 + (d7) and Ir4+ (d5 low spin) quite comprehensive discus- sions have been given [34,35].
A similar situation applies to the neutron diffrac- tion case for transition metal species. The treatment for magnetic neutron scattering of combined orbital and spin moment in the most suitable fashion remains a topic of lively theoretical interest (see for example ref. [36] and references therein), but because the effects of covalency are generally small in the first instance any added uncertainty due to unquenched orbital moment makes extraction of bonding para- meters of any significance very difficult indeed, especially as the effects may be quite similar in some cases [37].
4. Metal based interactions. - Several parameters primarily related to metal electron charge or spin distributions are sensitive to covalency, although often in a fairly complex way. These include hyper- fine interactions measurable by ESR or Mossbauer spectroscopy, supertransferred hyperfine interactions, metal isomer shifts and quadrupole splittings obser-
vable in Mossbauer work, particularly of 5 7 ~ e , orbital reduction factors of orbitally degenerate ground state species accessible by ESR or magnetic susceptibility, g-values measurable by ESR where spin-orbit coupling acts in second order, core level splittings observed by photoelectron spectroscopy, and ligand field splittings and electron repulsion parameters measured in electronic spectra.
A reduction of the orbital contribution to the magnetic moment was observed for the complex 1rC12- by Owen and Stevens [18] in the same set of experiments in which the ligand hyperfine interaction was first observed, and an explanation given by Stevens 1151. Effectively, the reduction is caused by the transfer of d electrons to the ligand orbitals by covalency, and the orbital reduction factor is defined :
where I is the orbital angular momentum operator and 1 d > and 1 $ > are free ion d orbitals and molecular orbitals. Orbital reduction factors have been reviewed with reference to EPR [20, 331 and magnetic susceptibility measurements [38, 391. For interactions within t2, .n orbitals
k,, = 1 - 2 f , (30)
and between n and rs orbitals
1 k,, = 1 - - 2 N , N , x
For 1rC1;- the orbital reduction factor [20] gave f , = 8 % via eq. (30), in quite good agreement with the value from the LHFI, although the g-value determination was later revised upwards to 14 % in a more comprehensive analysis [35] involving the effect of excited and charge transfer states. For ions (e. g. dZ, d7 in an octahedral field) whose ground states are mixtures of e, and t,, configurations both k,, and kc, appear in the overall orbital reduction factor and the analysis [34] for COF:- has already been mentioned.
Although it was one of the first experimental para- meters measured that was clearly related to covalent effects, and has been widely referred to in this context, orbital reduction factor analysis has not provided a mass of semi-quantitative covalency data as has the measurement of LHFI, perhaps because the latter, measured on spin only complexes, is more readily analysed. Nevertheless, even for spin only ions, spin-orbit coupling acting in second order causes small g-factor changes from the spin-only value, readily measurable by EPR. It was again early on observed that the small orbital contribution to theSg-factor for d3 and d8 octahedral ions :

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-547
8 h in electronic spectra might be expected to show a Ag = 2.0023 - -
A (32) decrease
was less than expected for the free ions which was explained by Owen [40] in terms of covalency, and this evidence for covalency in transition metal com- plexes has also been widely quoted. A is the ligand field splitting and for the free ion I = + c/2 S, where ( is the spin orbit interaction for one electron. The treatment is quite analogous to the derivation of the orbital reduction factor and was reviewed by Owen and Thornley [20], and calculations per- formed in detail for N~F;- by Misetich and Wat- son [41]. More recently, for V F ~ - , it was shown [42] in a similar treatment that the observed g-factor was consistent with the spin transfer coefficients determined from the LHFI. In the covalent situation
. where k,, is the orbital reduction factor, and
where 5, is the spin-orbit coupling constant for the central d orbit and 5, that for the ligand p orbit. In addition to taking into account the covalently induced spin-orbit coupling on the ligand it is also necessary to consider effects due to coupling with charge transfer states. Such a complete analysis has only apparently been carried through for the ~ i " and V2+ fluoride complexes, and even here, as is the case with the orbital reduction factor, the fairly complex relation describing Ag makes it most useful as a check on covalency parameters determined by measurement of LHFI. It does appear, however, that the analyses carried out do not demand a signi- ficant expansion of the d electron radial functions, although is was originally suggested [43] that such an effect might explain the observed results.
For the most part spin-orbit effects have been interpreted in terms of an apparent reduction of A (eq. (32)) where Ag,,, = 2.0023 - 8 l ' /A. A table of X/A, was given [20] for d3 and d8 ions (Table I). In view of the several factors entering into A', which will not necessarily change similarly from ligand to ligand such data gives a very qualitative estimate of the total covalency and relative magnitudes may not be particularly significant.
Some parameters available from electronic spec- troscopy have been discussed recently 1131. Ligand field splittings are broadly interpretable in terms of the nature of the bonding in a similar fashion to partial isomer shifts 1441 and partial quadrupole splittings [45], although it is difficult to understand some features of the spectrochemical series, such .as the order of the halide ions. The nephelauxetic series, derived from the Coulombic repulsion terms
Spin-orbit coupling reductions (A'/&) ,for d3 and d8 ions
from free ion values according to N4 where N is the normalisation factor (eqs. (6), (7)) and also according to changes in < r-' > for the d orbitals. The nephelauxetic series for Iigands [I] :
I- > Br- > CN- 2 C1- > OH- >
is indeed fairly similar, for example to a table [I] of enthalpy discrepancies between compounds of the similar-sized Ca2+ and Cd2+ :
> NO, > so:- > F- (36)
and the nephelauxetic series for metal ions [I] :
roughly follows expectations of covalency. Also the effect is much weaker for rare earth ion f-f spectra than for transition metal spectra. The detailed mecha- nism underlying such series do not seem to be very well understood, however, and a recent calculation [46] for N~F:- perhaps encourages caution in drawing detailed conclusions on d-electron distributions from the effect.
The hyperfine field at the metal nucleus, observed particularly extensively for the d5 high spin ions Cr+, MnZf and Fe3+ by EPR and by the Mossbauer effect has long been known [47] to be reduced as the covalency of the metal-ligand bond increases (according to the electronegativity difference for example) (Fig. 3). In contrast to the hyperfine fields at ligand nuclei discussed above however, where the core polarisation term is small, for metal ions containing unpaired electrons this term is often dominant and, because of the several factors which can contribute, a detailed interpretation was not forthcoming for a long time. Spin polarised covalently occupied outer 4s orbitals [48] as well as polarised core s orbitals and d electron transfer (the covalent reduction of the metal spin

C6-548 B. C. TOFIELD
FIG. 3. - The hyperfine interaction constant of Mn2+ plotted as a function of a covalency parameter, c, divided by the number of ligands, n. c is derived from the electronegativity difference of Mn and the ligand using the expression of Hannay and Smyth
(from ref. [48]).
reduces the s orbital polarisation) must all be taken into account. Hyperfine interactions and the Moss- bauer isomer shift were comprehensively discussed [49] by Sawatzky and van der Woude recently and we summarise their discussion below.
First of all however, let us mention metal core level splittings observed by photoelectron spectroscopy of transition metal compounds as these should directly reflect the magnitude of the spin and thus covalency effects. The splitting of 3s levels is the result of exchange coupling between the remaining 3s electron and the unpaired 3d electrons and although the magnitude is influenced by other effects also the order should correspond to the d electron covalency (as reflected by the total metal spin).
For Mn2 + the order is MnF, :6.5eV(ref [50]), 6.3eV(ref [51]),
MnCl, : 6.0 eV (ref [51]), MnO : 5.7 eV (ref [50]), 5.5 eV (ref [51]), MnS : 5.3 eV (ref [51]),
MnBr, : 4.8 eV (ref [51]) . (38)
This series is in agreement with most other data which indicate fluorides to be the most ionic salts but it is not clear how the overall order should be interpreted.
For Cr3 + some splittings are [51,52] :
CrF, : 4.2 eV, Cr203 : 4.1 eV, CrC1, : 3.8eV, K,Cr(CN), : 3.6 eV and CrBr, : 3.1 eV . (39)
In particular we note the location of the cyanide complex. The initial measurement [51] found no splitting for this compound, but is was recently shown [52] that this was probably the result of impu- rity effects. It was pointed out [52] that the splitting of 3.6 eV is in accordance with the hyperfine interactions at the Cr nucleus which are typical of Cr3' compounds.
The discussion [49] of the Mossbauer isomer shift, hyperfine and supertransferred hyperfine inter- actions has rather elegantly related these three effects for high-spin Fe3' oxides in particular and also permitted quantitative contact with covalency para- meters determined by neutron diffraction. The dis- cussion was pursued via an LCAO molecular orbital model with all the effects of covalency and overlap being incorporated into the ligand orbitals, i. e. the bonding orbitals, instead of primarily via the antibonding orbitals (eq. (1)-(5)) normally used to interpret LHFI and neutron diffraction. It is helpful to follow this interpretation. The metal wavefunctions in this approach are unchanged. We note that the transfer parameters b of ref. [49] are synonymous with y (eq. (9)) and B, is related to y as .f is to A. A2 is synonymous with f (this was a convention adopted by Hubbard and Marshall [53] in discussing the effects of covalency in neutron scattering).
The problem then is to calculate the charge density at the nucleus, ]$(0) 12, to calibrate the isomer shift, and the spin density at the nucleus to calibrate the hyperfine field. The isomer shift (IS) may be written
where ARIR is the fractional nuclear radius change on excitation, not known from other studies, cc is the calibration constant for the IS, and S 1$(0) l 2 is the change in charge density at the nucleus from source to absorber. The hyperfine interaction Hamil- tonian is given by eq. (15). The contact field at the nucleus is from ey. (15) and (23)
where 1$,(0) l 2 is the spin density at the nucleus. Both IS and Hco, involve only s orbitals so, separating up and down spin orbitals, for the isomer shift
is needed and for the hyperfine field
For high spin Fe3+ compounds other contributions to the hyperfine field may be neglected.
The metal and Iigand wavefunctions are made orthogonal and the effects of covalency included via the LCAO molecular orbital model discussed above. All the modifications to free ion wavefunctions being

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-549
lumped into the ligand wavefunction, this has the
We note that the covalency parameter b is inversely proportional to the energy difference between the ground, and excited states with one electron trans- ferred from ligands to metal, and a dependence on the electronegativity difference is observed for the hyperfine field as mentioned above (Fig. 3) and also, where other factors are constant, for the isomer shift (Fig. 4). For these high spin iron halides the decreasing IS with decreasing ligand electronegativity indicates that ligand-to-metal 4s donation is probably dominant. However an opposite trend is observed for example for low spin divalent iron dichalcogenides (F. W. D. Woodhams, personal communication) and in such a case ligand-to-metal 3d bonding is presu- mably giving the larger effect.
general form : 1.4
I = N i ( Ii - S S i j C j + Z:bi.q;
where $, is the free ion ligand wavefunction, p j a 1.2 filled metal wavefunction and q i an empty metal wavefunction. The second term describes the orthogon- m
\
alisation and the third the covalency. The normalisa- E E - tion constant is given by 4 1.0- U
FIG. 4. -The isomer shift of divalent iron halides plotted against the average electronegativity of the ligands (fromref. 149491).
- FeSiF6.6H 0 ' ~ e ~
x FeCt2+LH20 FeCI2-2H20
- FeCI2-H20 FeCI,
FeBr2
Fe12
Using wavefunctions of this type with the metal functions unchanged, parameters such as those of interest here are then the sum of the metal free ion term and the contribution arising from (eq. (42)). For the charge and spin densities at the nucleus where only the ligand a,, p orbital combination is of the appropriate symmetry, and the only metal orbitals directly involved are the 1s to 3s filled core orbitals and the initially empty 4s orbitals, then we have :
and
The first terms are the free ion values and the second an3* = anid + anid = 2 ~ ; 2 ~ , 2 + terms have an overlap distortion contribution from the orthogonalisation procedure and a 4s covalency + 3NL2 S: + 2~; 'b ,2 + 3Ni2b,2. (47) term. smaller correction terms involving ligand orbitals have been omitted. In oxides at least the first terms are dominant in both but will scale accord- ing to the d electron populations.
For the isomer shift this can be expressed as :
Thus comparing eqs (44) and (46), (47) the opposing effects of ligand to-metal transfer to 3d and 4s orbitals are seen. On the other hand metal-to-ligand back- handing will decrease the 3d shielding and for 57Fe, decrease the IS. The similarity of the IS of F~"(cN)~-
3 and F~"'(CN);- was interpreted 1541 as arising from 2 C I cpns(0) l 2 = C - 1-8(an3d)
n = 1 (46) the transfer of the extra ferrous ion electron on to the
ligands by n back-bonding. where C is the free ion density at the nucleus and If the 3d or 4s orbital populations are known an3, is the change in the total d electron population. or can be calculated, the calibration constant a The constant of proportionality was fixed [49] from of eq. (40) can be determined. This would make the Hartree-Fock calculations for the free ions Fe3+ isomer shift a very useful parameter for investigating and ~ e ~ + . covalency particularly for those valencies and spin

C6-550 B. C. TOFIELD
states where there is a significant spread of values, and especially in conjunction with d orbital population evidence from other measurements. Nevertheless, although there has been much effort directed towards this end in the last few years, the several contributing influences, as exemplified in the examples mentioned, will encourage circumspection in drawing detailed conclusions about bonding.
In suitable circumstances both 3d and 4s occup- ations may be extracted from hyperfine field data as discussed by Sawatzky and van der Woude [49]. This was done for a few different situations but most elegantly for the rare earth orthoferrites [55] where, in addition, comparison of the d orbital population could be made with values from neutron diffraction data determined [56] by myself a few years ago. The procedure adopted is one which allows the determi- nation of the bond-angle independent portion of the supertransferred hyperfine field, STHF, part of the total hyperfine field. The 3d and 4s orbital populations can then be extracted using catalogued wavefunctions.
The first term of eq. (45) will be scaled by the change in spin of the 3d electrons arising from overlap and covalency. For Fe3+, with the up spin d states full and the down states empty, the up spin occupation will decrease due to charge transfer and increase due to overlap, with the opposite being the case for the down spin occupation. The net change in spin is the difference of the changes in up and down spin popu- lations :
central cation s orbitals by ligand orbitals unpaired by transfer into unoccupied 3d orbitals of the neigh- bouring cations. The latter is dominated by p to d transfer and for antiferromagnets such as the rare earth orthoferrites, where one spin state is full and the other empty of d electrons, the orbitals on the ligands relatively depopulated by transfer to the outer cations will be of up spin if these Fe3+ ions have down spin. This means that the overlap distortion of the central Fe3' down spin s orbitals will be slightly greater than of the up spin orbitals, so that the down spin density at the central cation nucleus will be the greater.
The reduction in the negative, up spin overlap distortion part of the second term of eq. (45), which gives rise to the STHF is determined by the bonds from the ligands to the surrounding cations and may be expected [55] to depend on spin transfer in a similar kind of way to superexchange. We will not discuss the relation between covalency, exchange interactions, and magnetic ordering temperatures in any detail here because, although aspects of this relation are quite clear, the process can only be to rationalise qualitatively magnetic ordering temper- atures using known covalency parameters and not the other way round. The subject was well discussed by Owen and Thornley [20], and more recently, for example by Sawatzky et a1 1571 who also consider the STHF and superexchange in an ion like Cr3+ where the e, metal orbitals are initially empty but exchange split. We will just note for reference, however, that the magnetic ordering temperatures of KMnF,, KCOF, and KNiF, (88 K, 125 K and 275 K respec- tively), scale well with the covalency parameters determined by spin resonance and neutron diffrac- tion [20]. For these perovskites an 1800 interaction
- 2 Nb2 bb2 - 3 3:' b121 (48) will be dominant. where Boekema et al. [55] treated -the STHF in the ortho-
ferrites by reference to the superexchange reflected NJ,, = (1 - s$ ,J -~ /~ in the magnetic ordering temperatures, noting that
N:,, = (1 + 2 b,,, So,, + b:,,)a112. (49) Jiuo a f ~ f ~ (neglecting bonding) and J90 a 2 f&. If the magnetic ordering temperature, TN, is pro-
Thus the free ion contribution to the hyperfine portional to J, thenin this approximation field is
Hfree [(So - Ahy)/.yo]
where H,,,, has been calculated to be - 630 kOe. The second term in (eq. (45)) comprises contribu-
tions from overlap distortion of the iron core orbitals and the differential charge transfer to the spin polarised 4s orbitals, of which the latter is dominant [49] and would give a field of - 420 kOe for a full 4s shell.
Finally, in concentrated magnetic materials such as the orthoferrites there is a small correction to the second term of eq. (45). This additional, super- transferred, hyperfine field (STHF) arises from the neighbouring Fe3 + ions. For neighbouring ions with spin, a supertransferred hyperfine field at a central ion arises from ov'erlap distortion of the
where 8 is the Fe-0-Fe bond angle. Such a relation is applicable to the orthoferrites because 8 varies through the series as the rare earth ions get smaller and the ~ e 0 : - octahedra rotate more to acdommodate this reduction in size, but the Fe-0 bond lengths do not change significantly. Fe-0 covalency is assumed to be unchanged, although it is not altogether clear why this should be so, and appears to be reflected in a constant IS [55] and similar moment reductions for LaFeO, and YFeO, [56]. Perhaps any small perturbation by the smaller, more polarising heavier rare earths is more or less cancelled by the decrease

COVALENCY EFFECTS M MAGNETIC INTERACTIONS C6-551
in 8 bringing a second oxygen o orbital more into play.
Writing TN = TN (900) + ( TN (1800) - TN (900) ) x x c0s2 8 (51)
extrapolation of the observed linear curve of TN against cos2 8 gave [55] f, If, = 5.3, in reasonable agreement with 4from neutron and LHFI data (below). The general expression for the STHF correc- tion is obtained by considering ligand a orbital overlap with the central cation s orbitals, the neglect of .n interaction being justified by the data just discussed. Because 8 # 1800 the ligand orbital required will be a mixture of the o and n bonding orbitals used to bond to the outer Fe3+
p, = p, cos 8 - p, sin 8 (52)
and the supertransferred spin density at the central Fe3 + nucleus takes the form,
where I is the number of nearest neighbour irons. Thus there are three contributions to the observed
hyperfine field :
where HA,, is H,,,,(S, - AS)/&, Hco, arises from the second terms of eq. (45) and HsTHF comes from eq. (53).
The angular dependent term involved in HsTHF may be rewritten as { (f, - f,) cos2 8 + f, } and thus this treatment predicts a linear variation also of H, as well as of TN, with cos2 6' which is indeed observed (Fig. 5). Using the relative magnitudes of fa and f, determined from the Ntel temperature variation an angular independent part of HsTHF involving only f, in eq. (53) could be determined from this plot and thus the magnitude of (~f',,, + Hco,) found as well. As already mentioned H,,,, and Hco, had been calculated for a fully ionic Fe" ion (- 630 kOe) and for a full 4s shell (420 kOe) so that, using free ion wave functions and an average Fe-0 distance of 2.01 1 A, the various overlap integrals and normalisation constants were determined and the 3d and 4s covalency parameters scaled to fit the measured fields. The values found were [55] f, = 0.144, f, = 0.027 and 4s occupation 0.10 electrons. .fa and f, are somewhat higher than found from neutron diffrac- tion and spin resonance work (see below).
In view of the difficulties involved in extracting such numbers from the Mossbauer data such agreement must be viewed as very encouraging. Although the orthoferrite series is fairly ideal for such a study the angular range which is accessible (- 150 in total) is quite limited and it would also be interesting to know if the effect of direct metal-to-metal transfer on the
FIG. 5. - The hyperfine field at 57Fe measured by the M O S ~
bauer effect for the rare earth orthoferrites plotted against (COS~ where 0 is the Fe-0-Fe bond angle. This plot was used to derive information on the supertransferred hyperline field and thus on the covalency parameters of the ~ e 0 2 - clusters
(from ref. [49]).
estimated STHF has been estimated. It would appear that in some cases [58] such a direct transfer may be significant. Nevertheless, if 57Fe Mossbauer data can be used to provide quantitative covalency data in a complementary manner to neutron diffraction and LHFI this is a significant step forward although a thorough analysis as was carried out for the ortho- ferrites will always be needed. For example, it has been shown recently that the isomorphous replacement 2 02- = F- + N3- may sometimes be made. It has not been possible to study nitrides at all by the other methods mentioned here, although one presumes the covalency order will be F- < 02- < N3-. But Mossbauer data of the high spin Fe3+ fluoronitride Fe,N,F3, which has the cubic bixybite (Fe, Mn),03, structure with apparently random F- and N3- ions have recently been published 1591 and the hyperfine fields at the two metal sites (505 kOe and 475 kOe) are much less than observed for Fe3+ fluorides. Deter- mination of the covalency for F- and, particularly, for N3- by the methods used for the Fe3' oxides would be very encouraging. A relatively new method of determining STHF uses the Perturbed Angular Correla- tion of y-rays (PAC) technique, described [60] recently for lllCdm in fluorides containing transition metal ions. So far, the analysis does not seem te have proceeded sufficiently to achieve consistency with neutron diffrac- tion and spin resonance data.
57Fe quadrupole interactions have not been men- tioned in this discussion. As is the case with the hyper-

C6-552 B. C . TOFIELD
fine field, an analysis is more complicated than for the normalised to unity. The form factor of the magnetic ligand quadrupole interactions discussed above, moment distribution as expressed in eq. (13) gives, although in principle, such interactions should reflect at Q = 0, the central field covalency through the < r-3 > term. For divalent iron compounds, where the valence efg will f,(o) = 1 - n,2 - a," be present in first order, partial quadrupole splittings [45] seem to provide analogous qualitative
fb(0) = 0 (510
information to partial isomer shifts 1441 and the spectro- f,(o) = 2: + A: - .
chemical series, as mentioned above. However, because in any particular compound, the quadrupole splitting depends on the crystal field splitting and the spin orbit coupling as well as on central field and charge transfer covalency, a detailed analysis to provide information on either of the latter effects does not seem to be easily achieved at the present time [61, 13, 491.
5. Neutron diffraction investigations. - Boekema et al. [55], in discussing the comparison of covalency parameters determined from Mossbauer data with neutron values indicated that the expression used in determining covalency parameters from moment reductions observed by neutron diffraction was obtained by assuming 2 = y (eq. (9)). The first terms of eqs. (13) or (14) used to estimate A or f from moment reductions observed by neutron scattering, certainly do not provide the same moment reduction as given by Boekema et al. (eq. 48). eqs. (13) and (14) may however be rearranged to :
I+,,(r) l2 = d:(1 + 2 A, S, + 2 As Ss - 2: - A,") -
By putting b = y = A - S and neglecting spin polarisation we see that eq. (48) and the first terms of eqs. (55) and (56) are now in agreement, and that the true moment reduction associated with the metal d function is indeed somewhat less than given by the (1 - A2) term of eqs. (13a) and (14a), because of the effect of overlap terms in AS. When considering the effective magnetic moment observed in the scattering from an antiferromagnet such as LaFeO,, however, the spatial extent of the magnetic moment distribution, which is reflected in the angular dependence of the scattered intensity, must be considered.
This is given by the magnetic form factor, which in the spin only case may be simply written as
where D(r) is the normalised spin density, Q is the scattering vector (1 Q I = 4 n sin O/A), and f (Q) is
and analogously for eq. (14). In other words, the only contribution to magnetic scattering at Q = 0 comes from the first terms of eqs. (13) and (14) containing no overlap terms of the form AS, and the ligand spin term. Moreover, in antiferromagnets of the orthoferrite type where each Iigand is symmetrically surrounded by metals with opposite spin (shown in one dimension - . in figure 6) the l&nd moment disappears so that only
FIG. 6 . - A diagram showing the cancelling of the covalently induced ligand moment in a simple antiferrornagnet. For clarity
this is shown in one dimension only.
the metal term in fact is present at Q = 0. The various overlap terms represented in the second term of eq. (13) peak at sin 0/A - 0.2-0.3. As the magnetic intensities used to determine accurate magnetic moments have generally been measured at sin 011 - 0.1 it has been assumed that this overlap term will be small and that to a good approximation the observed moment reduc- tion is given by A rather than by A(l - S) . Of course the product of the spin and form factor is measured at any particular' Q and, in the absence of other informa- tion concerning the detailed shape of the form factor, the free ion shape has been assumed. This is given by the dotted line in figure 7 where the different contri- butions to the form factor are detailed. The moment reductions expected for high spin ions in octahedral coordination are given in Table 11, together with covalency parameters determined from the LHFI for spin only ions.
As is shown, the inclusion of the overlap terms will cause an expansion of the form factor (the covalent form factor of figure 7), relative to the free ion curve, if unchanged radial functions are assumed. A radial expansion of the d wavefunction will, on the other hand, cause a contraction from the free ion curve. The free ion curves for most commonly occuring first row transition metal valence states have been calculated [62].
The first antiferromagnetic form factor to be determined was that of Ni2+ in NiO, by single crystal diffractometry [63]. The expansion predicted was indeed observed, or rather, the expansion and reduced

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-553
FIG. 7. - A schematic representation showing the effect of covalency on the magnetic form factor according to the simple molecular orbital theory of ref. [53]. The metal, overlap and ligand contributions according to eqs. (13) and (58) are shown as dotted lines, and the combined paramagnetic form factor is also indicated. The metal spin form factor has the same shape as the free ion form factor and for convenience in this diagram the effect of the covalent moment reduction is shown as a reduction in the magnitude of the form factor (in the text the usual convention is followed that f(Q) is always normalised to unity). In an anti- ferromagnet the ligand moment is quenched, but above Q % 20 nm-1 the antiferromagnetic and paramagnetic form factors are very similar. The covalent form factor is slightly expanded relative to the metal curve because of the effect of overlap, but at Q x 12 nm-1 (sin 0/1 - 0.1) where magnetic reflections are generally measured to estimate the magnetic moment, the assumption of the free ion form factor is a fairly
good approximation.
moment observed were subsequently explained by Hubbard and Marshall [53] who first presented the molecular orbital interpretation of the effects of cova- lency in neutron scattering reproduced here. Quite good agreement was obtained [53] with the experi- mental curve when the effects of the small orbital moment and the spin polarisation of the inner shells were included.
More recently, the magnetic moment of Ni2' in NiO was measured by Fender et al. [64] using a poly- crystalline sample by comparison of the first magnetic reflection ($*$) with adjacent nuclear reflections [(I 11) and [2OO)J. The use of the real form factor for this compound, measured by Alperin [63] and the cal- culated 1621 free ion form factor provided an opportu- nity to assess the error involved in the normal approxi- mation. The actual value of the form factor at the magnetic (444) reflection is 0.937 whereas the free ion calculated value is 0.914. The observed moments will scale inversely (S,/S, = f,lfi) and thus will differ by less than 3 %. However, because the covalency para-
TABLE II
Moment reductions for antiferromagnets (octahedral complex)
Covalency Number of parameter electrons Moment reduction from LHFI - - -
1 ( g e ; 4 fn ---
2 (t;g e,O) 4fn ---
3 (ti, e;) 4fn f z
meters are obtained from the quite small difference between the measured and the expected spin, this small effect on the observed moment has a much larger effect on the covalency parameters, which change from fu + f, = 3.8(2) % using the Alperin form factor to 3.1(2) % using the free ion form factor, a difference of 18 %. Even this difference, however, which reflects a part of the negative overlap contribution to the moment reduction (eq. (55a)) is not qualitatively significant.
When we come to examine the data for Fe3+, although the moment reduction given in ref. [56] is increased slightly when a revised value for the scattering length of germanium (which was used to calibrate the intensity of the first magnetic reflection (orthorhombic (001)+ (101) at sin O/il=O.ll)) is used (0.8193 (ref. [65]) c. f. 0.84) the new value off, + 2 fn(l 1.4 $. 1.2 %) [27] is considerably less than the value found by Boekema et al. (20 %) [55]. As we have shown, the form factor at Q = 0 is reduced by the magnitude of il only, not including overlap, and at sin 012 - 0.1 a small part of the overlap moment of eqs. (13b) or (14b) will apply as discussed for NiO. This term itself is somewhat smaller, because of the negative two-centre overlap terms, than the adjustment needed to bring (l3a) or (14a) into line with eqs. (48), and (55a) and (56a), so that a correction similar to that found for NiO might be expected (bringing f, + 2 f, from 11.4 _+ 1.2 % to -- 13.7 %). As mentioned below, there is no evi-

C6-554 B. C. TOFIELD
dence for any significant change in shape of the form factor for octahedral coordination by oxygen and thus there would in fact seem to be a discrepancy between the Mossbauer and neutron values for.fa + 2 f,. Nevertheless the agreement is sufficiently close that the overall result should certainly be regarded as a success. I think it would be worthwhile re-examining the ortho- ferrites with neutrons using the more precise technique of total profile refinement [66] to provide a better estimate for the overall error of the covalency sum, but only slightly higher covalency has been found more recently for octahedrally coordinated Fe3+ in BiFeO, and Sr2Fe205 (see below). We might note also that had not a correction for the zero point spin deviation been applied in the neutron case (< S, > was considered to be 2.5 % less than the total spin of 3 according to the calculation of Davis [67]) the value of (f, + 2 f,) would have been about 16 + 1 %, really quite close to the Mossbauer value. The correct value of the zero point spin deviation seems to provide one of the larger uncertainties in the neutron experiments.
Thus we see that, subject to the approximations discussed above powder neutron diffraction data can give results directly comparable with those obtained from the measurement of the LHFI. But combina- tion of the two techniques allows further insight for d3 and d5 ions. For the latter the values off, and (fa -- f,) are normally obtained from the LHFI whereas neutron diffraction measures the sum (f, + 2 f, +A), so that both fa and f, may be obtained if both types of experi- ment are done. For d3 ions comparison of covalency parameters determined by the two types of method reveals dramatically the presence of spin polarisation of the e, orbitals.
Data for d3, d5 and dB ions are given in Tables III- VI. These provide some clear trends and some puzzling features.
The neutron data are consistent within them- selves. Thus we see the increase in covalency sum for Mn2+ from MnO through MnTe (MnO (3.6 %), MnS (7.0 %), MnSe (7.5 %), MnTe (9.8 %)) which parallels the increase for Co2+ halides measured by EPR ($(fa + f,) = 2.4 %, 5 %, 5,3 % and 7.5 % for F-, C1-, Br- and I- ligands respectively [34,68]). The similarity between S and Se is parallel by that between C1 and Br. Also the covalency in MnSe, is very similar to that in ~ n ~ e . The expected increase in covalency from divalent Mn2+ to trivalent Fe3+ d5 oxides and fluorides is observed (thus, fa f 2 f, + f, = 3.6 % for MnO and - 13 % for Fe3+ oxides). For the iron oxides the several values now determined are grati- fyingly consistent. There does not seem to be any trend revealed with Fe3+-0'-average distance ; although at face value there is an increase in covalency with an increase in distance this is very unlikely to be a real effect.
The revolution in diffraction analysis of poly- crystaIline samples since the introduction of profile analysis [66] should be mentioned. In the first measure- ments on MnO, NiO [64], LaFeO, [56], etc., one was constrained to use quite simple materials to avoid problems of overlapping reflections. More recent refinements, such as those of Sr2Fe,05 (ref. [69]) have allowed the determination of several positional para- meters as well as the moments of more than one type of atom. In this particular example, which was undertaken principally to assist investigation of the structural and thermodynamic behaviour in the SrFeO, ., -, ., system a choice could also be made between three possible
Spin transfer coeficients for Ni2+
I Fluoride ! Oxide I Chloride I Host and method
(Bond length (A))
KNiF3 (neutrons) [I ]
2.004
( Host and
Covalency method I (Bonsygth
fa + f s = 2.6(1.8) % NiO (neutrons) 141
2.084 fa = 3.8 P/d fs = 0.54 % MgO
(ENDOR) [5] 2.106
fa = 3.1 O/d fs = 0.53 %
1 I Host and Covalency method
[I] HUTCHINGS, M. T. and GUGGENHEIM, H. J., J. Phys. C 3 (1970) 1303. [2] SHULMAN, R. G. and SUGANO, S., Phys. Rev. 130 (1963) 506. [3] HALL, T. P. P., HAYES, W., STEVENSON, R. W. H. and WILKENS, J., J. Chem. Phys. 38 (1963) 1977. [4] FENDER, B. E. F., JACOBSON, A. J. and WEDGWOOD, F. A., J. Chem. Phys. 48 (1968) 990. [5] FREUND, P., J. Phys. C 7 (1974) L 33. [6] RINNEBERG H., HAAS, H. and HARTMANN H., J. Chem. Phys. 50 (1969) 3064.

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-555
Spin transfer coeficients for MnZS
[1] NATHANS, R., ALPERIN, H. A.,PICKART, S. 3. and BROWN, P. J., J. Appi. Phys. 34(1963) 1182. [2] HALL, T. P. P., HAYES, W., STEVENSON, R. W. H. and WILKENS, J., J. Chem. Phys. 38 (1963) 1977. [3] WALKER, M. B. and STEVENSON, R. W. H., PYOC. Phys. Soc. 87 (1966) 35. [4] JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F., J. Phys. C6 (1973) 1615. [5] FREUND, P., OWEN, J. and HANN, B. F., J. Phys. C 6 (1973) L 139. [6] RINNEBERG, H. and HARTMANN,.H., J. Chem. Phys. 52 (1970) 5814. [7] TSAY, F. D. and HELMHOLZ, L., J. Chem. Phys. 50 (1969) 2642. 181 FENDER, B. E. F., JACOBSON, A. J. and WEDGWOOD, F. A., J. Chem. Phys. 48 (1968) 990. [9] JACOBSON, A. J. and FENDER, B. E. F., J. Chem. Phys. 52 (1970) 4563.
[lo] FENDER, B. E. F. and COFFIN, P. S., unpublished data (1971).
space groups. The profile is shown in figure 8. In the experiment on BiFeO, (ref. [70]) it was shown that conventional profile analysis using a medium flux reactor was as satisfactory a means of determining the magnetic moment as the use of polarization analysis [71] to separate the magnetic and nuclear scattering using a high-flux reactor. Except for NiO 1641 discussed above and Tb,Fe,O,, [72] where the Y,Fe,O,, form factors for octahedrally and tetra- hedrally coordinated Fe3 + determined [73] by pola- rised neutrons were used, these powder studies have used free ion form factors to estimate moment reduc- tions. It should be added that although profile analysis can handle successfully quite complicated nuclear unit cells it is essential that the magnetic moments be determined as accurately as possible because of the fact that the covalency parameters are given by the quite small difference between the expected and observed magnetic moments. Where the magnetic cell is complicated by spiral or non-collinear ordering, or when angles between spin orientations and crystallo-. graphic axes must be determined thus cannot generally be done.
The data indicate fluorides to be the most ionic compounds studied, in line with most other evidence. This is seen in neutron work, for example in the increase in covalency sum from FeF, (6.2 %) to Fe3+ oxides (- 13 %), and from CrF, (3.0 %) to LaCrO, (4.4 %). The neutron diffraction studies seem to indicate similar covalency for MnO and MnF,, however (3.3 % and 3.6 % respectively) but this may well reflect the relative inaccuracy of the determination for MnF,. This value was obtained from an elegant polarised beam experiment 1741 utilising the particular symmetry of the rutile lattice which did indeed directly reveal the presence of unquenched moment on the fluoride ions, but the final value for the covalency sum may be only qualitatively comparable with those determined from the magnitude of the metal-based moment such as that for MnO [75]. It would be quite valuable to perform a conventional powder study on MnF, and RbMnF, to compare with the other powder data. The difference between the relatively ionic divalent oxides and fluorides will only be small and therefore, remembering that the covalency itself is determined as a small difference between observed and expected moments,
Fluoride Chloride/Chalcogenides Oxide
Host and method
( ~ o n d length (A))
Host and method
(Bond length ('9)
CsMnCls (NMR) 161
K4CdC16 (ESR) [71
2.63A a-MnS
(neutrons) [8] 2.601 a-MnSe
(neutrons) [91 2.732 MnSel
(neutrons) [91 2.71
MnTe (neutrons) [lo]
Host and method
(Bond length (A))
Covalency Covalency
--
f, - f, = 0.0 f , = 0.57 % ) ClI
fa - fn = 0.6 % f , = 0.50 Cl I1
fa -fn = 4.3 % f , = 0.4 %.
f, + 2f, + fs = 7.0(3) %
f, + 2fn +f, = 7.5(3) %
f, -I- 2fn + fs = 7.8(1.1) %
f, + 2f, + f , = 9.W) %
Covalency
f, + 2f, + fs = 3.6(5) %
f u -fn = 0.8(6) % f , = 0.8 %
MnO MnFz f, + 2 fn + fs = 3.3 % (neutron) [I I
2.119 RbMnF3
(NMR) PI 2.120
f, = 0.5 fa = 1.1(5) % fn = 0.8(5) o/d
KMgFs (ESR) [21
(neutrons) 141 2.187
f u -fn = 0.3 % 1 Mgo f , = 0.52 o/d
I
f, - fn = 0.3 f, = 0.55
(ENDOR) [5] 2.106
f, = 0.8 % f u = 1.5(6) % f n = 0.7(4) %
!!-

C6-556 B. C. TOFIELD
FIG. 8. - The powder neutron diffraction pattern for SrzFezOs from which the magnetic moments of octahedrally and tetrahe- drally coordinated iron were determined. The refinement was carried out by a least squares fitting to the measured profile (after ref. [66]). Small circles are the experimental points and the continuous line passes through the caIculated points. The small vertical lines are the calculated hkl positions and the bottom trace is the difference between observed and calculated
intensities.
quite difficult to measure. But there seems to be a difference, at least judging from the neutron data, for Ni2+ between KNiF, (2.6 %) and NiO (3.8 %) observed in very carefully performed experiments [64, 761. The error for KNiF, reflects the authors' uncer- tainty in the magnitude of the zero point spin correc- tion to be applied rather than experimental error. Both experiments used the experimental form factor of Alperin [63]. It must be admitted however that the EPR and NMR data show the covalency of Ni2+ in KMgF, and KNiF, to be the same as found by neutrons in NiO.
Discrepancies are more clearly apparent when we examine the results on dilute systems (EPR) and concentrated systems (NMR, neutrons) for anions other than fluoride (the data for Mn2+ in RbMnF, and KMgF,, Cr3+ in K2NaCrF6 and KMgF,, and at least fairly closely for Ni2+ in KNiF, and KMgF, are consistent). We notice, in particular the differences for Ni2' in NiO (f, + f, = 3.8 %) and MgO (9.2 %) and Mn2+ in CsMnC1, (f, - f, = 0.6 %) and K,CdC16 (4.3 %). If the EPR measurements over- estimate the covalency relative to the undiluted salts, then the combination of neutron and ENDOR data taken to determine individual f, and f, for Mn2+, ~e~ + and Cr3+ oxides might be unreliable, and lead to a too-large ratio of fcFz. The case for Ni2+-0'- has been qualitatively discussed [27], but the reasons for these differences do not seem well understood. Clearly 1 7 0 NMR data for NiO and neutron data for halides
such as the perovskites KMnCI, and KNiCl, would help to claify the experimental situation.
Because of this uncertainty it becomes quite difficult to determine an order of ligand covalency from these data if indeed there is such an order which can be transferred from one ion to another, except to be confident that fluoride ion is the most ionic. For example for Cr3+ in MgO, ENDOR gives f, - f, = - 7.1 %, in good agreement with that for CrBr, (-- 7.5 %) determined by NMR, and for Ni2+, the ENDOR value for fa is greater in MgO (8.5 %) than that determined by NMR for CsNiC1, (7.3 %). The neutron determination for NiO (fa+f,=3.8 %) on the other hand indicates much less covalency than for CsNiCl, (f, + f , = 7.9 %).
One can examine the data from other techniques. From 3s splittings (see above) we have the orders :
From Ca!Cd AH deviations [l] :
From spin orbit coupling reductions (Table I)
From the nephelauxetic effect

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-557
Spin transfer coeficients for Fe3+
Host and method Covalency Host and method 1 (Bond length (A)) 1 (Bond length (A)) Covalency I
KMgF3 (ESR) [ll
1.994 FeF3
(neutrons) [2] 1.92
fs = 0.8 % fa = 4.1(6) % fz = 0.7(3) %
Fluoride
MgO (ENDOR) [3]
2.106 LaFeO 3
(neutrons) [4] 2.006
YFeO 3
(neutrons) [4] 2.015
Tb3FesOl2 (neutrons) [7]
2.023 BiFeO3
(neutrons) [5] 2.027
Sr~Fe205 (neutrons) [6]
2.058
Oxide
[I] HALL, T. P. P., HAYES, W., STEVENSON, R. W. H. and WILKENS, J., J. Chem. Phys. 38 (1963) 1977. [2] JACOBSON, A. J., MCBRIDE, L. and FENDER, B. E. F., J. Phys. C7, (1974) 783. [3] FREUND, P., OWEN, J. and HANN, B. F., J. Phys. C 6 (1973) L 139. [4] TOFIELD, B. C. and FENDER, B. E. F., J. Phys Chem. Solids, 31 (1970) 2741. [5] JACOBSON, A. J. and FENDER, B. E. F., J. Phys. C 8 (1975) 844. [6] GREAVES, C., JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F., Acta Cryst. B 31 (1975) 641. [7] Fu~ss, H., BASSI, G., BONNET, M. and DELAPALME, A., Solid State Commun. 18 (1976) 557.
I
From the isomer shift total covalent energy. It is interesting that the order
Fez+ (ref. [14]) : F - > 0 2 - >Cl->I- >S2- >Se2-.
From the metal hyperfine interaction
Mn2+ (ref. [48]) :
F- > 02- ,- Cl- > S2- Se2- > Te2-
and from comparative unit cell volumes for Mn2+, Fez +, Co2 + and Ni2+ (ref. [14])
F- > 02- > c1- > S2 > I - .
The atomic electronegativities (Pauling) are 1771
F (3.98) > 0 (3.44) > Cl(3.16) > Br (2.96) > I(2.66) > S (2.58) > Se (2.55).
The discussion of the isomer shift and the hyperfine field has shown how several factors can contribute to any one type of observation and there is no reason why these should remain in the same ratio as either metal or ligand are varied. Nevertheless, one might presume that the covalency contraction parameter R, of Shan- non and Vincent [14] defined as the ratio of the unit cell volume of a transition metal compound M, X, to that of an ionic compound M& X, (where M' = Mg in ref. [14]) might be a relatively useful guide to the
found parallels that- of the atomic ele~trone~ativities as do those of the isomer shift and hyperfine interaction and it seems likely that the order of d electron covalency determined by neutrons will not be dissimilar. We have noted already the similarity observed by neutrons for S2- and Se2- and by EPR for C1- and Br- which reflects the electronegativity sequence. It is by no means clear, however, that we will be able to invoke significant differences in metal-ligand covalency between oxides and chlorides, for example, to explain the frequent presence of layer structures among the latter, which may rather just reflect the different oxidation states, ionic radii and coordination numbers.
There has been some questioning concerning the relative covalency of Mn2+ and ~ i " . This was discussed more fully elsewhere [27] and had three strands of argument : that fluorides and oxides could not have equal covalencies, that fa for Mn2 +, being less than for Ni2 + was anomalously low, and that the form factor for ~ n ' + was contracted relative to the free ion value so reflecting an expansion of the electrons. The first of these points was discussed above and the second is a result of the different electron configurations. Although fa is certainly less for Mn2+ than for Ni2+, the total covalency for the oxides is very similar for the

two ions (f, + 2 f, + f, = 3.6 % and 3.8 % respecti- vely where f, = 0 for Ni2+). The difference in f, arises because for Ni2+ the t2, n antibonding levels are full so that charge transfer to the d levels can only occur by o bonding. We noted above that the magnetic ordering temperatures of KMnF,, KCoF, and KNiF, corre- lated well with the observed o covalency parameters. Similarly, although the total charge transfer might be similar for Mn2+ and Ni2+ oxides and fluorides the covalent bond energy will be greater for Ni2+, where all the d electron interaction is via o bonding, which correlates with other evidence indicating that Mn2 + is the more ionic.
Of course there is no reason why for all other metals, oxides should be nearly as ionic as fluorides, or for all other ligands the total covalency should be similar for Mn2+ and Ni2+. It is topically of interest that the difference in Ag+ location in silver beta alumina [78] relative to the NaC location in sodium beta alumina [79] may be the result of a covalent interaction between silver and oxygen which helps to stabilise the former in the anti-Beevers-Ross position, and in line with this there is a large R, contraction from Ag-F to Ag-0 [7] which reflects a significantly greater covalency for the oxide than for the fluoride. For other ligands of Mn2+ and Ni2+ recent lZ9I Mossbauer data [80] for the di-iodides show an ionic character in the order v2+ N ~ n ' + > ~ e " > cd2' > co2+ > ~ i ' + . The ionicities were obtained from 5 p ~ orbital popula- tions derived from the isomer shift and quadrupole coupling constant in an analogous manner to that described for CrI, below.
The original data indicating an expanded Mn2+ form factor were collected [81] before the effects of covalency on the magnetic moment were realised, and could therefore have arisen from a scaling error, and also extended out only to I Q I -- 5.0 A w l . A more recent, careful examination [75] of the form factor for polycrystalline MnO out to I Q I = 6.75 showed the,form factor to follow the free ion curve quite closely. An expansion arising from overlap effects was certainly not observed as for NiO [63], which may reflect an expansion of the t,, orbitals in MnO as calculated [82] for M~F;-, although it would appear that this, calculation claimed agreement with the original form factor determination. A survey of form factors so far determined gives some empirical support to this possibility (see below). A proper re-determina- tion of the Mn2+ form factor by single crystal methods is certainly called for however.
In fact this should be quite straightforwardly achieved for several compounds such as MnO, MnS and MnS, by single crystal diffractometry because of the high spin of Mn2+. Also, Mn2+ has a high spin configuration in some organometallic compounds such as dicyclopentadienyl manganese which should make a very interesting study.
For d3 ions, especially Cr3 + for which there is most
data, effects of spin polarisation are clearly revealed. Although spin polarisation effects may be most impor- tant for d5 ions they are difficult to observe because both o and n covalency contribute to the observed quantities. For Cr3+ however, the simple MO model outlined predicts only a n-bonding contribution to the moment reduction and LHFI so that neutrons and resonance techniques should both measure f, as they do fa for Ni2+. However, the moment reduction in CrF,, and indeed in LaCrO, yielded a smaller f, then found for Cr3+ in KMgF, or K2NaCrF, by ESR and NMR. It was realised [56] that the presence of spin polariza- tion in the e, orbitals will leave a net down spin in the Iigand p, orbitals, increasing the magnitude off, - f,, and an up spin on the metal e, orbitals so reducing the observed moment, which would explain the observed behaviour. Combination of the resonance and neutron data allows an estimate to be made off, and the spin polarisation-induced fa, and f, is surprisingly large, even for the fluoride (Table VI).
Unfortunately there are no neutron data for V2+, but from the EPR study of V2+ in KMgF, [42] fa was estimated, by comparison with f, and the calculation for Mn2+ of Hubbard et al. [16], to be only 0.3 k0.2 % (with f, = 2.6 + 0.3 %). Clearly a neutron determina- tion of the covalency for V2+ would be very useful and this might now be possible as the preparation of pure phases of the cubic V2+ perovskites KVF, and RbVF, has been recently described [83]. The high NCel temperatures found [83] for antiferromagnetic ordering (120 K and 130 K respectively) indicate rather strong antiferromagnetic interactions of similar magnitude to those in KMnF, and KCoF,. This is not at all what is expected for d3 configuration ions interacting through fluorine at 1800, unless there is a significant spin-polarisation induced spin in the e, orbitals. From the g-value found [42] for V2+ in KMgF, the average o-bonding admixture was esti- mated to give an fa of about 2-2.5 % (where now f, is independent of spin polarisation and is a measure of A,, eq. (10)). In such a case the total charge transfer away from V2+ to the six ligands would be about 0.7 e compared with about 0.2 e for Mn2+ and Ni2+ fluorides. This will apparently be at variance with the indications from some data, for example the quadru- pole coupling and isomer shifts in the transition metal di-iodides [80] and the nephelauxetic effect, that V2+ is similarly ionic to Mn2+ and Ni2+, unless in fact the greater part of the observed [42] fa - f, is due to the spin polarisation-induced fa term, itself reflecting a fairly large proportion of the total o bonding interac- tion, and which will explain the large 1800 super- exchange interaction. In such a situation, with the spin-polarisation-ind~fced fa being greater than f,, an increased rather than a decreased moment would be observed in a neutron scattering experiment.
For CrI, (Table VI, ref. [19]) the covalency para- meters were determined by measurement of the ligand hyperfine interaction in the ferromagnetic phase by

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS
TABLE VI
Spin transfer coeficients for V2+, Cr3+, ~ n ~ +
Host and method
(Bond length (A))
KMgF3 (ENDOR) [I]
1.994
/ Host and I Covalency method Covalency -I Host and
method
CrF3 (neutrons) [4]
1.90 fa = - 2.3(4) % f, = 2.6(4) %.
- .. .
f, = - 3.2(9) % fn = 3.9(6) %
CrBr 3
(NMR) C71 CrI 3
(Mossbauer) 171
- ~~
LaCr03 (neutrons) [6] - 1.98
Mn4+
Covalency
fs = 0.10 % f, - fn = - 9.2 %
f, + 2 fn +f, = 7.6(16)%
fu = - 3.6(8) % f, = 5.6(8) O/d
f, + 2f, +f, =3.0(8) %
fa + 2 f, + f, = 4.4(12) %
[I] DAVIES, J. J., SMITH, S. R. P., OWEN, J. and HANN, B. F., J. Phys. C 5 (1972) 245. [2] HALL, T. P. P., HAYES, W., STEVENSON, R. W. H. and WILKENS, J., J. Chem. Phys. 38 (1963) 1977. [3] SHULMAN, R. G. and KNOX, K., Phys. Rev. Lett. 4 (1960) 603. [4] JACOBSON, A. J., MCBRIDE, L. and FENDER, B. E. F., J. Phys. C 7 (1974) 783. [5] FREUND, P. OWEN, J. and HANN, B. F., J. Phys. C 6 (1973) L 139. 161 TOFIELD, B. C. and FENDER, B. E. F., J. Phys. Chem. Solids. 31 (1970) 2741. [7] SANCHEZ, J. P., DJERMOUNI, B., FRIEDT, J. M. and SHENOY, G. K., Hyperfine Interactions 1 (1976) 313. [8] HELMHOLZ, L., GUZZO, A. V. and SANDERS, R. N., J. Chern. Phys. 35 (1961) 1349.
CaMnOr (neutrons) [6] - 1.87
lz9I Mossbauer spectroscopy. The data given in the Table were obtained by analysing the hyperfine field components along the Cr-I bonds (eq. (17)), but, by combining the hyperfine field data with the isomer shift and quadrupole coupling constant, individual values of f, and fn could in fact be estimated. In Cr13 each Cr has two nearest neighbour iodides and the symmetry axis of the bonds is not parallel to the crystallographic c-axis so that y # 0. The coordinate system is shown in figure 9 and hybridised orbitals were set up
$4 = 1C/p~ FIG. 9. - The local environment of iodine in CrI3 showing the two nearest-neighbour chromium atoms, the crystal axis and the
where $2 and $3 are 0 orbitals directed along the bonds coordinate system used in the interpretation of the 1291 M ~ s s - and $, is a 7~ orbital. The orbital populations are 2, bauer data (after ref. [19]).

(1 -t i,), (1 + i,) and (1 + i,) respectively, where i, and i, are the ionicities of the a and n bonds. Then adding up the populations of the constituent 5s and 5p orbitals gives
h, = 2 s(1 - i,) (60)
h, = 3 - 2 i, - i, - 2s(l - i,)
so that from eq. (28)
6 = 21.45 s(i, - 1) - 1.5(2 i, + i,) + 3.96 (61)
using values for a and b of - 9.2 and + 1.5 respec- tively.
Nx = 1 + i, + 2 s(1 - i,) , Ny = 1 + i, and N, = 1 + i, (62)
so that from eqs (25), (26) and (27)
e2qQ = [i, - i, + s(1 - i,)] e2qQ ,,,, ,, . (63)
The quadrupole coupling constant is
From the experimental 6, eq and 'I, the values of s, i, and i, were found to be 0.07,0.62 and 0.64 respectively.
The ligand hyperfine field along the x, y and z axes was known in the magnetically ordered phase from the magnitude and orientation of H. From the spin direc- tion Hy = 0. From Hz and Hx the unpaired spins, a in $, (= $,) and b in $, could be estimated and hence f, and f,. Remembering that < 3 cos2 0 - 1 >, along the i direction is 415 for a pi orbital and - 215 for pj and p, orbitals, then the p orbital contributions
2 4 along x and z are - (a-4 as- b) and - (as- a+ b) res-
5 5 pectively. Using the value of s (0.07) determined from the analysis above T, gave
f, - f, = - 12.8 % agrees quite well with the value in table V. Unfortunately, the more symmetrical coor- dination of the three metal atoms around each iodine in the di-iodides did not permit [80] the determination of f, or f,.
Although the great majority of neutron diffraction covalency determinations have been concerned with octahedrally coordinated transition metal ions, there have been some recent studies on tetrahedrally coor- dinated oxide ions. d2, d5 and d7 tetrahedral ions have spin only ground states and Fe3', Ma2' (d5) and Co2+ (d7) have been investigated. Although for tetra- hedral coordination the t, and e crystal field states do
not correspond separately to a and n antibonding levels in an analogous manner to the octahedral case, it is often assumed that the 7~ bonding contribution to the upper t2 levels is negligible, in which case the distinction can be made. It is also instructive to compare the total moment reduction in the two coordinations to give a relative assessment of the total covalencies. As we men- tioned above, NQR and Mijssbauer evidence indicates that the covalency per bond is the greater for tetra- hedrally coordinated SnIV and ~ b ' " halides.
For tetrahedral d5 ions
and for tetrahedral d7 ions
where the second equality assumes the t, and e states to be a and n antibonding levels respectively. A list of moment reductions observed is given in table VII with selected octahedral cases for comparison. The three Fe3 + -02- examples indicate that the total d electron covalency is roughly similar for both coordinations and therefore the covalency per bond is greater for the tetrahedral situation. The form factors were measur- ed [73] by polarised neutrons for both types of iron in ferrimagnetic yttrium iron garnet (YIG, Y,Fe,O,,) (Fig. 10) by Fourier transforming the measured magnetic scattering amplitudes,
where z is a reciprocal lattice vector and v, the unit cell volume, and integrating the moment associated with each site to give < S >. The form factors were then obtained from the measured valves of < S > f (Q). The octahedral form factor follows quite closely the Hartree-Fock free ion curve but the tetrahedral form factor is considerably contracted. Also, by observing non zero magnetic scattering amplitudes for some reflections to which only oxygen spin density could contribute, the presence of covalent spin density on oxygen, the result of the different bonding characteris- tics of the two types of iron, was directly observed. These results were reproduced in an ab initio spin polarised calculation [84] where a larger radial extent of the spin densities compared to the free ion was found, although it was pointed out that different exchange approximations caused significant changes in these. The YIG form factors were used in the profile refine- ment [72] of the terbium iron garnet data, while for Sr,Fe,O, the Hartree-Fock free ion form factor was used [69]. A contracted tetrahedral site form factor would decrease the tetrahedral moment somewhat.
The other compounds studied were the CoZf oxide spinels Co,O,, CoRhzO, and MnRh204 [85]. Spinels

COVALENCY EFFECTS IN MAGNETIC INTERACrrIONS
NiO [5]
TABLE VII
Magnetic moments and spin transfer in Tetrahedral Systems
Octahedral moment (Bond distance, A)
Tetrahedral moment (Bond distance, A)
[I] BONNET, M., DELAPALME, A., TCHEOU, F. and FUESS, H., Znt. Congr. Magnetism, Moscow, 1973, Vol. IV p. 351 (1974 ), [2] Fuess, H., BASSI, G., BONNET, M. and DELAPALME, A., SolidState Commun. 18 (1976) 557. [3] GREAVES, C., JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F., Acta Cryst. B 31 (1975) 641. [4] INFANTE, C. E., D. Phil Thesis, Oxford (1975). [ 5 ] FENDER, B. E. F., ~ACOBSON, A. J. and WEDGWOOD, F. A., J. Chem. Phys. 48 (1968) 990.
FIG. 10. -The octahedral and tetrahedral form factors for Fe3+ in Y3Fe5012 (taken from the data of ref. [73]). The experimental points are denoted by + and . for the octahedral and tetrahedral cases respectively. The smooth curve is the free ion form factor. The octahedral form factor follows this closely but the tetrahedral form factor is significantly contracted.
are not generally suitable materials for this type of investigation because of the rather complicated magne- tic structures often observed, and the problems of knowing the exact oxidation states and site occupan- cies of the metal ions. But in these three normal spinels the structure refinements indicate [85] that the Co2+ d7 ions are effectively totally located in the tetrahedral A site. Also, because the octahedral B site is occupied by a non-magnetic ion (low-spin d6 Co3' or Rh3+) the only exchange path possible (A-A) gives rise to simple antiferromagnetic ordering. The NCel tempera- tures (about 40 K, 27 K and 20 K respectively) are sufficiently high that magnetic saturation should be achievable at easily accessible temperatures (2-4 K).
In Co304 the co2'-0 bond length at room tempe- rature is considerably shorter (1.9404(4) A [85]) than estimated from the ionic radii of zn2+ and Co2+ (0.74 A and 0.72 A respectively [4]) and the Zn-0 distance in ZnCo204 (2.03 A). This indicates a consi- derable covalent contraction of the cobalt-oxygen bond, and is supported by the significant super- transferred hyperfine interaction observed at the Co3 +
site [86]. The Co-0 bond length in CoRh204 is longer (1.989(1) A at room temperature [85]), and close to that expected. It was, therefore, surprising that a previous study of Co,04 had determined a rather high moment of 3.26 ,uB [87].

The Roth value [87] of the magnetic moment seems to have been the result of an incorrect value for the cobalt scattering length (0.232 x lo-'' cm). This was refined at room temperature in Infante's investiga- tion [85] and the value found (0.248(1) x 10-I' cm), 7 % higher than used by Roth, is in agreement with other more recent determinations. It was found that the moment reduction in Co304 was indeed somewhat greater than in CoRh,04, but that both were quite similar to that found in NiO [64]. To compare the total covalency for the two cases, however, the moment reductions must be multiplied by the number of unpair- ed electrons so that the total charge transfer for the tetrahedral Co2+-oxygen situation would seem to be somewhat greater than for octahedral NiO, with, therefore, a considerably greater covalency per bond. As mentioned above, such an effect of greater total covalency in the tetrahedral case seems to be less pronounced for Fe3+, although the charge transfer per bond is certainly greater for the tetrahedral oxide coordination. The rather large value of the moment reduction for Mn2" found in MnRh,04 (as great as for Fe3') is puzzling. There does not seem to be any inversion or oxidation state changes in this compound but the explanation might perhaps lie [85] in spin canting, intrinsic spin disorder or errors in the zero point spin correction. Considerable care was taken in the sample preparations, for either incomplete reaction or decomposition to metallic rhodium is very easily achieved. In the cobalt spinel refinements the effect on the overall R-value of expanding or contracting the Co2" form factor was investigated. Although this technique will not give a precise determination, in both cases a broad minimum was indicated for an expansion of 5-10 %, while the agreement became sharply worse for a contraction from the free ion curve. 'It might perhaps be interesting to continue these studies to encompass the analogous sulphide spinels MnRh,S,, CoRh,S4, and MnSc2S4. Another tetrahedral material of some interest is the high d5 co4+ oxide Ba,Co04.
The means and experimental techniques of detecting the effects of covalency by neutron scattering were recently discussed [27] and we have mentioned most of the data collected from experiments on polycrystalline samples. Ideally, of course, one wishes to obtain a complete spin density map which entails a form factor determination using single crystals. This may be done by conventional diffractometry on antiferromagnets or, more accurately, using polarised neutrons in which case one needs a ferromagnetic or paramagnetic sample (or exceptionally an antiferromagnetic material), in which the spins can be aligned perpendicular to the scattering vector by an external field. In some cases the spin covalently transferred to non-metal atoms can be seen directly by observing a magnetic component for reflections to which only the non-magnetic atoms give a nuclear contribution. Experiments of all these types have already been mentioned (NiO [63] MnF, [74], Y3Fe,0,, [73]). One can indeed separate magnetic
from nuclear intensity in antiferromagnetic or para- magnetic materials using polarization analysis [71] but the heavy loss of intensity with such instruments as currently designed is a severe disadvantage. The most attractive and useful area for further study must surely be paramagnetic complexes where the spins can be aligned and the form factor measured with pola- rised neutrons. An experiment of this type, on K2NaCrF6 [88], shows the power of the technique. We will now mention some of the rather few form factor determinations for non-metallic systems so far carried out, in the general order 3d, 4d and 5d transi- tion metals, rare earths and actinides.
FIG. 11. - Fourier transform of the magnetic scattering ampli- tudes of K2NaCrF6 in the (001) plane through a chromium site. Contour units are in 0.01 p ~ / A 3 . The crystal structure of KzNaCrFs is similar to that of K2IrCI6 (Fig. 1) in that the CrFi- clusters are isolated from one another, but there are sodium ions in alternate octahedral sites instead of vacancies. The tzg shape of the electron density around the chromium is clearly seen, although because of the finite resolution of the map the nodes are washed out, and also spin density covalently
transferred to the fluorines (after ref. [88]).
Spin density covalently transferred to the fluorine was observed in K,NaCrF6 (Fig. 1 I), and the t,, shape of the metal d electron spin density was seen. Wedgwood showed quite clearly that the data were not consistent with a single value of A,. The spherical and aspherical parts of the form factor were separated and the value of A, which gave the best fit to the sphe- rical part (A, = 0.30, f, - 2 %) was lower than for the aspherical part (A, = 0.48, f, - 5 %). As the spherical form factor is particularly sensitive to the metal spin, except at low Q, but the aspherical form factor to the ligand spin density these data seem to confirm the deductions from the powder data discussed above that there is a considerable spin-polarised o spin density, which adds to the n density observed on the ligands and reduces the moment reduction on the metal. Wedgwood used free ion wavefunctions for Cr3" and F- to calculate form factors to compare with the

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-563
experimental ones and a significant change in the Cr3+ radial function was not apparently indicated. It was pointed out that the Fourier transformed spin density, although qualitatively revealing was only of limited value in analysing the data because termination errors were of similar magnitude to the expected effects of covalency. The most satisfactory method of analysis for an experiment of this type would be to perform a complete form factor calculation of the type done for Y3Fe5012 [84], although the interpretation then loses the simplicity offered by the MO model. Of the few spin polarised calculations done for Cr3+ complexes, one for K2NaCrF6 1891 gives quite good agreement with the experiment and Table V (fa = - 2.19 % and f, = 2.55 %), but another for c~F;- and CrC1:- gives a dominant spin polarized f, contribu- tion (c~F;- : fa = - 4.8 %, f, = 1.0 %, crC1;- : f, = - 10.2 %, f, = 0;3 %) [go], which would seem to be difficult to reconcile with the results for K2NaCrF6, CrF, and CrI, (Table VI). It is clearly just as necessary to have experimental data to check on the calculations as vice versa.
d3 complexes seem to be well suited to a study of several aspects of chemical bonding and should receive priority in the neutron investigations. In view of the considerable spin,polarised o spin density, apparently of comparable magnitude to f,, found for the fluorides and oxides, it will be extremely interesting to look at the effect of varying the nature of the ligand bonded to Cr3+. In particular, H,O and NH3 are thought to be dominantly o bonders and thus covalently trans- ferred spin density should arise more or less totally from spin-polarisation effects without competing ligand-to-metal n bonding. CN-, on the other hand, is high in the spectrochemical series and breaks Hund's rule for d4, d5, d6 and d7 systems. This is not a problem for the d3 configuration of course and thus the possible effects of metal-to-ligand n bonding, thought to be important along with ligand-to-metal o donation in the bonding of CN- and revealed in the isomer shift of iron cyanide complexes [49, 541, could be conveniently investigated by neutrons in the c ~ ( c N ) ~ - complex. It would be especially interesting if analysis by X-ray diffraction of the charge distribution in the bond regions of such complexes were done in conjunction with the spin density determinations.
Spin pairing is also an almost universal feature of 4d and 5d transition metal complexes and once again d3 systems seem to be best suited to compare the bonding in the first row with the later transition elements. Although it is often stated that 4d and 5d ions are more covalent than 3d ions there has been very little detailed comparison of particular configurations in the same oxidation state. The basis for the hypothesis seems primarily to be the ligand field splittings but these are not totally reliable quantitative guides to covalency and the nephelauxetic effect does not indicate any substantial covalency differences between, for example, Mo3+ and Cr3+, and Rh3+ and Co3+. The greater
spatial extent of the 4d and 5d radial functions entails a more sharply dropping form factor for these ions than for 3d ions but the recent determination [91] of the form factor of K,IrCl, (S = 3) shows that this is not an insuperable problem. MoF, has a similar magnetic and nuclear structure to CrF, [92] and could be studied by conventional diffractometry, and as a powder even. Several simple complexes of first and later row metals are directly comparable and could be studied by polarised beam techniques (e. g. ~e'"C1;- and ~ n ' ~ C l 2 -, and the corresponding fluorides).
CoS, is a low-spin d7 ( t i ed) metallic ferromagnet. The metallic behaviour presumably arises from the broadening of the quarter-filled eg level implying considerable Co-S covalency 1121, and a delocalisation path Co-S-S-Co. The molecular nature of the x Z - groups in the pyrites structure has been demonstrated for ~ e ; - by Mossbauer spectroscopy by the observa- tion of a large quadrupole splitting for 25Te [93]. With CoS, however, one is in two types of regime which are not properly accounted for by the LCAO MO model. First of all the large anion-anion interac- tion in sulphides leads to a wide valence band (5-8 eV [57]) situated only just below the d levels (as shown in photoemission experiments [94, 95]), so that the anion-cation mixing will be dependent on the k vector of the anion band state [57], and covalency and superexchange effects may well be transmitted beyond the nearest or next-nearest neighbours, and secondly, the delocalisation of the d states destroys the localised moment on the metal as the band width increases (see for example, the discussion for d2 ions in perov- skites 1961). The form factor was measured by the polarised beam [97] and there was no evidence for any
COBALT
NUCLEUS I0101 ---3
FIG. 12. - Fourier transform of the magnetic scattering ampli- tudes of CoSz in the (001) plane showing aspherical eg character
(after ref. [97]).

C6-564 B. C. TOFIELD
significant negative magnetic moment arising from 4s states delocalised in the region between the cobalt atoms as was found in the case of the ferromagnetic transition metals, but the shape of the form factor was very like that expected for an ionic Co2+ situation. In spite of the metallic conductivity the dominant magnetic moment is localised on the Co2+ atoms and this localised moment has an e, character (Fig. 12). A small magnetic moment distributed around the sulphur atoms was directly observed by measuring [98] the flipping ratios for reflections to which the Co atoms did not contribute. This was observed to be also aspherical and the larger component could be rationalised on the basis of bonding between the Co2+ eg arbitals and the 3p, arbitals of the s$- group (Fig. 13). This study
FIG. 14. -The KzCuF4 crystal structure shown as an ideal tetragonal K2NiF4 type. The infinite perovskite like sheets along
the a-axis are separated along c by KF sheets.
0 : su l f u r nucleus
:cobalt nucleus
FIG. 13. - Schematic overlap picture between the 3p, and 3p, orbitals of the sulphur atoms shown in projection and the 3d-eg orbitals of the cobalt atoms in the (001) plane of CoS2. The 3p, orbital overlaps the 3d-e, orbitals while the 3p, does not to any great degree and this is in accord with the observation of an aspherical magnetic moment covalently transferred to the sulphur atoms. The S2 molecular axis is along the [111] direction so that the sulphur atoms are not actually located on the (001)
plane (after ref. [98]).
is thus quite significant as it would seem that in spite of the electron delocalisation implied by the metallic conductivity some at least of the magnetic properties can still be interpreted using a localised electron pic- ture. If crystals could be prepared it would be interest- ing to perform this type'of study on other narrpw band metallic ferromagnets, such as the perovskite oxide SrCoO, [99] where in this case the CoIY ion is low spin so that the t2, levels must be delocalised.
K2CuF4 is one of the few known ferromagn2tic insulators and has the K,NiF4 structure (Fig. 14). As originally determined it appeared that there was a compression of the CUF;- octahedra along the crystal- lographic c-axis. This would have been a unique situa- tion, as in all other cases studied the Jahn-Teller distortion around the d9 Cu2+ ion takes the form of a
tetragonal elongation. The reasons for this do not seem to have been well explained but may be involved with covalent bond energetics ; certainly for systems where a Jahn-Teller distortion should arise out of a n antibonding electron configuration the distortions observed are generally small or non-existent as is the case for K,IrCl, mentioned below. Khomskii and Kugel showed [101], however, that such a compression was unlikely on the basis of the probable superexchange paths and proposed a situation where the octahedra had tetragonal elongations along alternate crystallo- graphic a axes (Fig. 15). This leads to a doubling of the a axis and crystallographic superlattice reflections are indeed observed [102], and another consequence is that the dXZ-y2 orbital containing the unpaired electron is similarly alternately oriented (Fig. 15). This has recently been confirmed by neutron diffraction [I031 by measurement of the magnetic structure factors of the superlattice reflections below T, (6.25 K) using the polarised beam with the field along c whereby the aspherical spin density associated with the distortion was revealed. The observed magnetic scattering ampli- tude at any reciprocal lattice vector is a function of the difference of the form factors associated with the two orientations and thus no magnetc scattering was observed for the 4 (1 10) reflection. Agreement was only obtained at lower sin 911 values, where the ligand moment is particularly significant (Fig. 7), if the effect of covalency was included. Only the square planar inter- action between C U ~ + ~ ~ ~ - ~ ~ orbital and four F- ions affects the spin distribution (with two distances of c and two of s, figure 15) and the values off, used in all
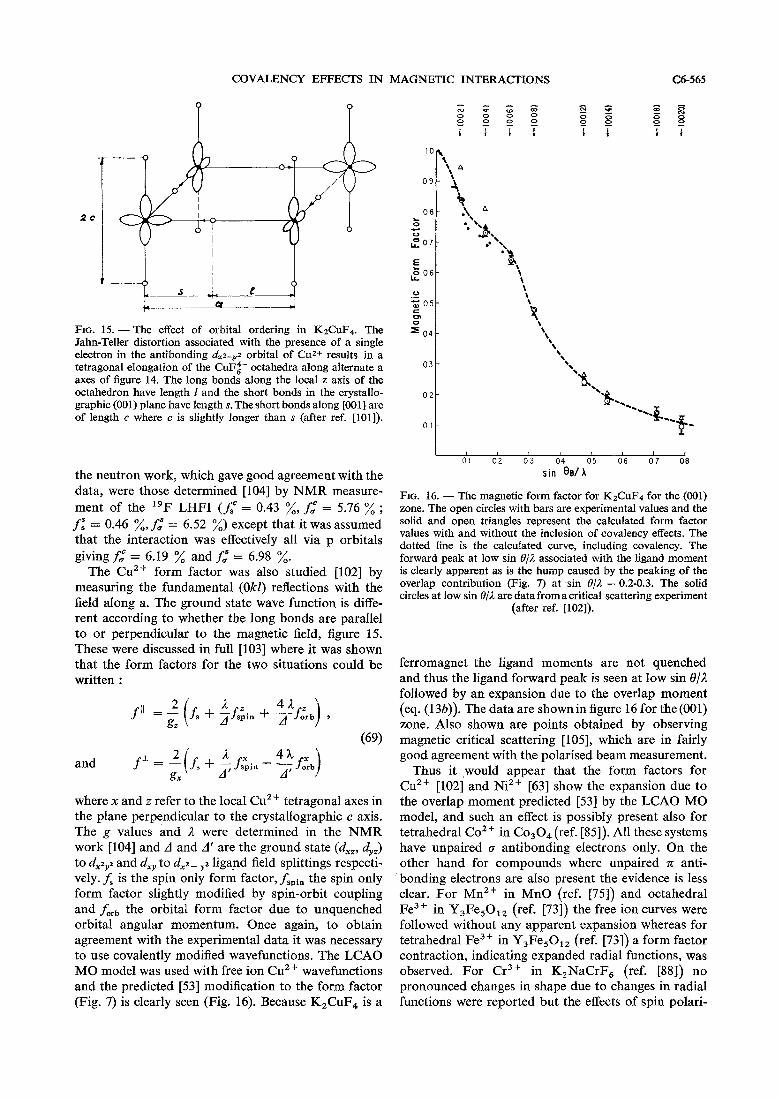
COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-565
FIG. 15. -The effect of orbital ordering in K2CuF4. The Jahn-Teller distortion associated with the presence of a single electron in the antibonding dZ2-@2 orbital of C U ~ + results in a tetragonal elongation of the C U F ~ - octahedra along alternate a axes of figure 14. The long bonds along the local z axis of the octahedron have length I and the short bonds in the crystallo- graphic (001) plane have length s. The short bonds along [OOl] are of length c where c is slightly longer than s (after ref. [loll).
the neutron work, which gave good agreement with the data, were those determined [I041 by NMR measure- ment of the 19F LHFI (&' = 0.43 %, f,' = 5.76 % ; f," = 0.46 %, f," = 6.52 %) except that it was assumed that the interaction was effectively all via p orbitals giving f,' = 6.19 % and f: = 6.98 %.
The Cu2+ form factor was also studied [I021 by measuring the fundamental (Okl) reflections with the field along a. The ground state wave function is diffe- rent according to whether the long bonds are parallel to or perpendicular to the magnetic field, figure 15. These were discussed in full [I031 where it was shown that the form factors for the two situations could be written :
1 2 and f =-(f s d- T f & i n - - f z b ) 4 h gx A A'
where x and z refer to the local Cu2+ tetragonal axes in the plane perpendicular to the crystallographic c axis. The g values and 1 were determined in the NMR work [104] and A and A' are the ground state (d,,, d,,) to dX2,2 and d,, to dX2- ,2 ligand field splittings respecti- vely. f, is the spin only form factor, fSpi, the spin only form factor slightly modified by spin-orbit coupling and f,, the orbital form factor due to unquenched orbital angular momentum. Once again, to obtain agreement with the experimental data it was necessary to use covalently modified wavefunctions. The LCAO MO model was used with free ion Cu2+ wavefunctions and the predicted [53] modification to the form factor (Fig. 7) is clearly seen (Fig. 16). Because K2CuF4 is a
L I
01 0 2 0 3 04 05 06 0 7 08
s in ee/X
FIG. 16. - The magnetic form factor for K2CuF4 for the (001) zone. The open circles with bars are experimental values and the solid and open triangles represent the calculated form factor values with and without the inclusion of covalency effects. The dotted line is the calcufated curve, including covalency. The forward peak at low sin 611 associated with the ligand moment is clearly apparent as is the hump caused by the peaking of the overlap contribution (Fig. 7) at sin 611 = 0.2-0.3. The solid circles at low sin 011 are data froma critical scattering experiment
(after ref. 11021).
ferromagnet the ligand moments are not quenched and thus the ligand forward peak is seen at low sin 8 / A followed by an expansion due to the overlap moment (eq. (13b)). The data are shownin figure 16 for the (001) zone. Also shown are points obtained by observing magnetic critical scattering [105], which are in fairly good agreement with the polarised beam measurement.
Thus it ,would appear that the form factors for Cu2+ [I021 and ~ i ~ + [63] show the expansion due to the overlap moment predicted [53] by the LCAO MO model, and such an effect is possibly present also for tetrahedral Co2+ in Co304 (ref. [85]). All these systems have unpaired a antibonding electrons only. On the other hand for compounds where unpaired .n anti- bonding electrons are also present the evidence is less clear. For Mn2+ in MnO (ref. [75]) and octahedral Fe3 + in Y,Fe,O,, (ref. [73]) the free ion curves were followed without any apparent expansion whereas for tetrahedral Fe3+ in Y,Fe,O,, (ref. [73]) a form factor contraction, indicating expanded radial functions, was observed. For Cr3+ in K,NaCrF, (ref. [88]) no pronounced changes in shape due to changes in radial functions were reported but the effects of spin polari-

sation were apparently observed. Thus there is some experimental evidence in support of the expansion of the t,, n antibonding electrons proposed by Freeman and Ellis [82] but the interpretation is possibly confused by the presence of spin polarisation effects. More accurate form factor determinations for several diffe- rent metal ligand combinations are needed to provide the data needed to be able to pin down the various effects.
d4 ions are in some respects analogous to d9 ions like Cu2+, in having one hole in a half-filled shell. Rb,CrC14 is also a ferromagnet based on the K,NiF4 structure [106], although the detailed crystallographic structure is not yet known, and would make an ideal candidate for study especially in view of the coordina- tion by chlorine. The other well-characterized d4 ion, Mn3+, has not been investigated at all except in an early study 11071 of LaMnO, where no moment reduc- tion was observed. This seems odd in view of the results for Cr3 +, Fe3 +, Mn2+ and Mn4' oxides and should be re-investigated. Ferromagnetic SrCoO, [99] was mentioned above. d4 Fe4+ in SrFeO, has a high spin configuration and there is an antiferromagnetic transi- tion below 134 K [108]. It was originally hoped [I091 to determine the covalency by measuring a magnetic moment reduction, but the lack of a Jahn-Teller distortion [I081 and the magnetic behaviour indicates that the e, electron is delocalised and the magne- tic structure is in fact a spiral propagating along [Ill], [110]. Difficulties with sample preparation meant that our interest turned more towards the crystallographic nature of the non-stoichiometry in this compound, although the covalency of octahedral and tetrahedral Fe3+ in Sr,Fe,O, [69] was one outcome of this study, and a new phase containing 50 % Fe3 + and 50 % Fe4+, Sr4Fe401 was discovered [1 1 I]. Mossbauer data on this compound collected at room temperature by Barry Dale at Harwell has shown that the Fe3+ is probably half octahedrally and half tetra- hedrally coordinated as in Sr,Fe,O,, while the ~ e ~ + spectrum shows a quadrupole splitting, in contrast to SrFeO, [log]. This might arise just from lattice effects but it is also possible that the dilution with localised electron Fe3 ' has localised the Fe4+ e, electron so that the covalency of all three species might perhaps be investigated in this compound, either by neutrons, or via the Mossbauer hyperfine interactions.
We might note here also that it would be very interesting to have neutron investigations of spin distri- butions in compounds with iron bonded to sulphur, with relevance in particular to biological systems. By analogy with MnS (Table 111) and from the other properties of such compounds the covalent effects are likely to be significant.
The first direct measurement of covalent delocalisa- tion of spin was the observation of LHFI in the 1r~1:- complex [18]. The total moment density in K,IrC16 has recently been revealed by the measurement of magnetic scattering amplitudes below the antiferro-
magnetic ordering transition (3.05 K) using single crystal diffractometry [91]. The crystal structure of K,IrCl, (Fig. 1) is that of a defect double-perovskite K,(OIr)C16 with vacancies in half the octahedral sites, or alternatively it may be viewed as on antifluorite K2(IrC16). The Ir4+ ion is low-spin d5 with one hole in the n antibonding t,, state and below the Nkel tempe- rature the spins are arranged so that the magnetic unit cell is twice the volume of the crystallographic cell, being doubled along the spin (2) axis. There does not appear to be any crystallographic distortion or Jahn-Teller splitting and an isotropic g-factor was observed in K,Pt(1r)Cl6 [21]. The total multiplicity of the 2T ground state (2 L + 1) (2 S + I), is six and spin-orbit coupling splits this into a spin doublet ground state and an upper quartet. The antiferromagnetic ordering will further define a singlet ground state which may be written [15, 911 as
where
If the molecular orbitals (eq. (3), (4), (5)) are written in place of d,,, etc. and the contributions to the various ligands added it is found that the up and down spin moments cancel on the ligands on the x and y axes leaving the total transferred spin only on the two ligands along z (the spin axis). Thus a cylindrical moment density is predicted and the Fourier inversion of the form factor data in the (xOz) plane shows just this effect (Fig. 17). The ligand moment is not cancelled
(001) K21rCie MAGNETIZATION DENSITY (101)
FIG. 17. - Fourier transform of themagnetic scattering ampli- tudes of KzTrCl6 in the (xOz) plane showing the large elongation of spin density in the spin direction (z axis) associated with the covalent transfer of spin to the z axis chlorines (after ref. [91]).

COVALENCY EFFECTS IN MAGNETIC INTERACTIONS C6-567
because of antiferromagnetism (c. f. Fig. 6) in this case because of the indirect Ir-el-C1-Ir exchange path. Also, the large unit cell means that there are reflections in the low sin 8!1 region where the effect of the ligand moment is particularly pronounced (Fig. 7). Experi- ments such as this seem a more profitable means of directly observing the ligand moment than attempting to measure the low angle paramagnetic diffuse scatter- ing [112,27]. A calculation with 30 % of the total spin transferred to the z-axis C1 atoms (using the LFHI result that about 5 % of the Ir spin is lost for each ligand) gave quite a good fit to the form factor data and thus it seems that the two techniques are in agreement. The neutron experiment was a particularly impressive one in the light of the low moment of Ir4+ and the low density of magnetic ions which meant that the magnetic intensities were some four orders of magnitude smaller than the nuclear reflections. It would be interesting to investigate other compounds in this series, particularly K2ReC1, (Re4+, d3) now that it has been shown that such experiments are possible on a high-flux reactor.
In contrast to the results for d-series transition metal ions, it appears that covalency effects are not very important for compounds of the rare earth and actinide metals, at least in regard to the interpretation of neutron diffraction data. Thorough studies of Tb(OH), [I131 and Gd203 [114] have been made using the polarised beam and in both cases zero moment was found on the oxygen sites. A study of gadolinium metal 11151 had revealed the effect of spin polarised 6s conduction band spin density and that the calculated non-relativistic free ion form factor [116] did not adequately describe the data. Good agreement was obtained with a calculated Gd3 + form factor including relativistic effects [I 171 for the inner electrons. These have the effect of increasing the shielding of the 4f elec- trons so causing a slight expansion relative to the non- relativistic case, and thus a slight contraction in the form factor. The fact that such a subtle effect could be established is perhaps one of the most impressive demonstrations of the power of neutron diffraction techniques to determine precise spin density distribu- tions. A similar effect was found for Tb3+ in Tb(OH), 11131.
The initial studies using polarization analysis [71] indicated that it might be a very useful means of deter- mining form factors by examination of paramagnetic diffuse scattering. Application of the technique to Gd20, at room temperature [115] surprisingly appeared to indicate that the form factor in this case was better described by the non-relativistic curve, but a more thorc~lgh examination [114] using both unpolarized ncutrons and the conventional polarized beam mode has shown that even though the magnetic ordering temperature is as low as 1.6 K, spin correla- tions, which will affect the paramagnetic scattering, still exist at 300 K. After correction for this effect there was no convincing evidence to support a difference in
the 4f form factor between Gd metal and Gd20?, and either of the two form factors fitted the Gd203 data equally well (Fig. 18). It was concluded that measurement of the paramagnetic scattering is not a good method of obtaining form factor data in concen- trated paramagnets unless an independent study of the magnitude of spin correlations is made. The latter study on Gd203 also allowed the susceptibilities of the two crystallographically independent Gd3+ ions to be measured. The discovery of their different depen- dence on temperature below 10 K permitted the expla- nation of apparent anomalies in the magnetic suscepti- bility data which indicate only the average suscepti- bility, and demonstrated the power of the polarized beam technique to investigate complicated para- magnets.
o 0. t a2 0.3 a4 a5 sin B / X
FIG. 18. - The calculated and experimental form factors for Gd metal and Gd2O3. The Gd metal form factor clearly follows the DiraoFock calculated curve which includes relativistic effects rather than the non relativisitic Hartree-Fock curve. The Gd203 data fits either of the two form factors equally well
(after ref. [I 141).
One effect of the increase of covalency with oxidation state of transition metal ions is to induce a change from localised electron to collective electron behaviour so that, for example, stoichiometric d5 ~ e ~ + oxides are generally good insulators, but SrFeO, containing Fe4+ is a metallic conductor [108]. The itinerant electron in this case is contained in the o antibonding e, orbital and is delocalised by virtue of the covalent Fe-0-Fe interaction rather than via direct metal-metal interac- tions. As the oxidation state is increased further of course, a localised electron state is again reached where now there are isolated oxyanion complexes as in BaFeO, and SrFeO,. The high lattice energy of the perovskite structure, responsible for stabilising Fe4+ in SrFeO,, is also able to stabilise high oxidation state cations of rare earth cations so that Ce4+, Pr4+ and Tb4+ are readily prepared in air in the perovskites BaCeO,, etc. [118], whereas the preparations of the binary oxides of Tb4+ and pr4+ require unusual condi- tions. Tb4+ is isoelectronic with Gd3', having a half-

'OFIELD
filled f shell with a spin-only ground state, but BaTbO, has, for insulating rare earth compounds, a relatively high antiferromagnetic-ordering temperature of 36 K and the same simple spin configuration as the rare earth-transition metal perovskites such as LaCrO, and LaFeO,. This fact may well indicate either a certain amount of f electron covalency or effects of spin polarisation, involving the occupied 5s25p6 outer shell or covalently occupied 5d orbitals.
Because of the high spin of the Tb4+ ion and the relatively simple magnetic and nuclear structures it proved possible to measure < S > f ( Q ) out to sin 8/A -- 0.5 from polycrystalline data [I191 (single crystals have not been prepared). This could not be done quite as accurately as for MnO [75], because of the more complicated nuclear structure of BaTbO,, and there was also no calculated free ion form factor for Tb4+ for comparison. Nevertheless, from the calculated Hartree-Fock curves for EU'+ and Gd3 + [I161 an estimate of the Tb4+ form factor was made for the lowest angle reflection (sin 812 = 0.10), where any error should not be too significant. As expected the form factor for the other seven reflections measured appears to be expanded relative to those for the lower valent ions (Fig. 19). The effective
FIG. 19. - The form factor for Tb4+ in BaTbOs. The experi- mental points have been normalised to the value of the Tb4+ form factor at the lowest angle reflection (IC = Q = 1.27) estimated by extrapolation from the non-relativistic EuZ+ and Gd3+ theoretical free ion curves. The higher angle points indi- cate a slightly expanded form factor compared to Gd3+ and Euz+, consistent with a contracted 4f spin density for the higher- valent Tb4+ ion. The data from two separate determinations are
shown (after ref. [119]).
magnetic moment of 6.66 ) 0.04 p, is reduced from the expected value, but at least half of this is probably caused by zero point spin deviation effects, and the remaining reduction of 2-3 % might be reduced by N 1 % if a relativistic form factor had been used Although, as for Mn2+ in the transition metal series, a small moment reduction does not imply a non- negligible covalency, because to compare charge transfers the moment reduction must be multiplied by
the number of unpaired electrons, it is clear that even for this tetravdent rare earth ion, covalency effects on the 4f electrons are not large, and are probably less than for the most ionic 3d ions such as Mn2+. The Nee1 temperature of BaTbO,, although high for rare earth ions, is considerably less than that of MnO (120 K). The isolation of the 4f electrons by the outer 5s' 5p6 filled shell means therefore that the formation of higher oxidation state rare earth ions is limited by the oxidisability of the anion without any intermediate range of oxidation states showing considerable cova- lency as found for transition metal ions. Only in situations of high lattice energy favouring tetravalent ion formation, such as the BaMO, perovskites, can such ions be stabilised, and we have provided evidence for the formation of Nd4+ also in this environ- ment [120].
The relatively poorer shielding of the 5f electrons in actinide compounds leads to chemical properties more analogous to the 3d transition series and more indications of covalency effects in the magnetic proper- ties might be expected, although, because of the larger effect of the environment, the crystal fields are much larger than in rare earth ions and the magnetic descrip- tion of the actinide ions is complicated by the similarity of the crystal field and the spin orbit coupling. Never- theless, the presence of unquenched moment on ligand atoms observed by polarised neutrons should still be a sensitive technique of looking for covalency effects but in studies on US [121], a ferromagnet, and the paramagnetic phase of UO, [122], a semiconductor with antiferromagnetic ordering below 30.8 K, no such moment was observed. Handling problems have limited this type of study to uranium compounds and unfortunately the uranyl ion U02,+, where a covalent interaction presumably contributes to the structure, is diamagnetic. Compounds containing the neptunyl, N ~ o ~ + or plutonyl PUO$+ ions (fl and f2 respectively) would therefore be most suitable for a study of cova- lent effects by neutron diffraction, but handling pro- blems might direct effort instead to compounds containing the pentavalent UO: ion.
5. Summary. - Various magnetic measurements which provide information on covalency have been mentioned. Until recently most information has been provided by measurement of the ligand hyperfine interaction by spin resonance methods, but in the last few years neutron diffraction has provided a considerable body of complementary data especially for octahedrally coordinated oxides and fluorides. Many trends have been revealed but many areas of uncertainty remain, and more data for other ligands and other coordinations are needed, although interest- ing comparisons between octahedrally and tetra- hedrally coordinated oxides have been made. The measurement of accurate magnetic form factors by neutron diffraction provides the most complete deter-

COVALENCY EFFECTS f N MAGNETIC INTERACTIONS C6-569
mination possible ofspin density distribution and hence of covalency effects. The few experiments so far done have been reviewed but this field has not been much explored so far, and some fruitful areas for future investigation have been mentioned, where d3 configu- ration ions in particular stand out. The presence of high flux polarised beam diffractometers on both sides of the Atlantic will, hopefully, permit the collection of much more data in the near future. Approaches to the extraction of covalency information from 5 7 ~ e isomer shift and byperfine field data are promising, and although few results are known, reasonable agreement with neutron data has been found from the analysis of
hyperfine fields of ferric oxides. If reliable covalency parameters can be extracted from hyperfine field data this will be very important, but the most interesting recent development in Mossbauer spectroscopy is the measurement of the LHFI for '''I, a ligand largely neglected in spin resonance and neutron studies. The covalency parameters found for CrI, were nicely complementary to data on oxides and fluorides from other techniques, and the advantage of LHFI measure- ment in a complicated molecule, where the bonding of one particular atom may be studied in suitable cases has been utilised in a study of Fe (111) bis-dithio- carbamato iodides [123].
References
[I] PHILLIPS, C. S. G. and WILLIAMS, R. J. P., Inorganic Chemistry (Clarendon Press, Oxford) 1965.
[:!I JOHNSON, D. A., Some Thermodynamic Aspects of Inor- ganic Chemistry (Cambridge University Press, Cam- bridge) 1968.
[3] SHANNON, R. D. and PREWITT, C. T., Acta Cryst. B 25 (1969) 925.
[4] SHANNON, R. D., Acta Cryst. A 32 (1976) 751. [5] CATLOW, C. R. A., J. Phys. C 9 (1976) 1859. [6] PHILLIPS, J. C., Bonds and Bands in Semiconductors
(Academic Press, New York and London) 1973. [7] SHANNON, R. D. and GUMERMAN, P. S., J. Inorg. Nucl.
Chem. 38 (1976) 699. [8] WHITT~NGHAM, M. S., Science, 192 (1976) 1126. 191 GAMBLE, F. R., OSIECKI, J. H. and DISALVO, F. J., J.
Chem. Phys. 55 (1971) 3525. [lo] ANDERSON, J. S., in Surface and Defect Properties of
Solids ed. M . W. Roberts and J. M. Thomas (Specialist Periodical Reports) The Chemical Society, London 1972, Vol. 1, p. 1.
[ l l ] GOODENOUGH, J. B. and KAFALAS, J. A., J. Solid State Chem. 6 (1973) 493.
[12] GOODENOUGH, J. B., Magnetism and the Chemical Bor?d (Interscience, New York, 1963), J. Appl. Phys. 37 (1966) 1415.
[13] TOFIELD, B. C., Progress in Inorganic Chemistry, 20 (1976) 153.
[14] SHANNON, R. D. and VINCENT, H., Structure and Bonding 19 (1974) 1.
[15] STEVENS, K. W. H., Proc. R. SOC. A 219 (1953) 542. [I61 HUBBARD, J., RIMMER, D. E. and HOPGOOD, F. R. A.,
Proc. Phys. Soc. 88 (1966) 13. [17] J~RGENSEN, C. K., .4bsorption Spectra and Chemical
Bonding in Complexes (Pergamon Press, Oxford) 1962. [IS] OWEN, J. and STEVENS, K. W. H., Nature 171 (1953) 836. [19] SANCHEZ, J. P., DJERMOUNI, B., FRIEDT, J. M. and SHE-
NOY, G. K., Hyperfine Interactions, 1 (1976) 313. [20] OWEN, J. and THORNLEY, J. H. M., Rep. Prog. Phys. 29
(1966) 675. [21] CIPOLLINI, E., OWEN, J., THORNLEY, J. H. M. and WII~D-
SOR, C. G., PYOC. Phys. Soc. 79 (1962) 1083. [22] CHOW, C., CHANG, K. and WILLETT, R. D., J. Chem.
Phys. 59 (1973) 2629. [23] WAYSBORT, D. and NAVON, G., J. Chem. Phys. 59 (1973)
5583. [24] GETZ, D. and SILVER, B. L., J. Chem. Phys. 61 (1974)
630. [25] TOWNES, C. H. and DAILEY, B. P., J. Chem. Phys. 17
(1949) 782. [26] BERSOHN, R. G. and SHULMAN, R. G., J. Chem. Phys.
45 (1966) 2298.
[27] TOFIELD, B. C., Structure and Bonding 21 (1975) 1. [28] FREUND, P., OWEN, J. and HANN, B. F., J. Phys. C 6
(1973) L 139. 1291 FREUND, P., J. Phys. C 7 (1974) L 33. [30] PARISH, R. V. and PLATT, R. H., J. Chem. Soc. A (1969)
2145. [31] RINNEBERG, H. and HARTMANN, H., J. Chem. Phys. 52
(1970) 5814. [32] DE WAARD, H., MiiSSbauer Efect Data Index Eds.
J. G. Stevens and V. E. Stevens, Plenum (New York and London : 1975) p. 447.
[33] ARRAGAM, A. and BLEANEY, B. Electron Paramagnetic Resonance of Transition Metal Ions (Clarendon Press, Oxford) 1970, p. 784.
1341 THORNLEY, J. H. M., WINDSOR, C. G. and OWEN, J., Proc. R. Soc. A 284 (1965) 252.
[35] THORNLEY, J. H. M., J. Phys. C 1 (1968) 1024. [36] BALCAR, E., J. Phys. C 8 (1975) 1581. [37] BALCAR, E., LOVESEY, S. W. and WEDGWOOD, F. A.,
J. Phys. C 6 (1973) 3746. 1381 GERLOCH, M. and MILLER, J. R., Pmg. Inorg. Chem. 10
(1968) 1. [39] BASU, C., MAJUMDAR, D. and GHOSH, U. S., Phys. Stat.
Solidi (b) 53 (1972) 71 1. [40] OWEN, J., Pmc. R. SOC. A 227 (1955) 183. [41] MISETICH, A. A. and WATSON, R. E., Phys. Rev. 143
(1966) 335. 1421 DAVIES, J. J., SMITH, S. R. P., OWEN, J. and HANN, B. F.,
J. Phys. C 5 (1972) 245. 1431 MARSHALL, W. and STUART, R., Phys. Rev. 123 (1961)
2048. 1441 BANCROFT, G. M., MAYS, M. J. and PRATER, B. E.,
J. Chem. Soc. A, (1970) 956. [45] BANCROFT, G. M., Coord. Chem. Rev. 11 (1973) 247. [46] WACHTERS, A. J. H. and NIEUWPOORT, W. C., Phys. Rev.
B 5 (1972) 4291. [47] VAN WIERINGEN, J. S., Disc. Far. Soc. 19 (1955) 118. [48 SIMLNEK, E. and MULLER, K. A., J. Phys. Chem. Solids,
31 (1970) 1027. [49] SAWATZKY, G. A. and VAN DER WOUDE, F., J. Physique,
Colloq. 12 (1974) C 6-47. [50] FADLEY, C. S. and SHIRLEY, D. A., Phys. Rev. A 2 (1970)
1109. [51] CARVER, J. C., SCHWEITZER, G. K. and CARLSON, T. A.,
J. Chem. Phys. 57 (1972) 973. 1521 ORCHARD, A. F., STOCCO, G. C. and THORNTON, G.,
J. Chem. Soc. Far. Trans I1 72 (1976) 1045. [53] HUBBARD, J. and MARSHALL, W., Proc. Phys. Soc. 86
(1965) 561. [54] SHULMAN, R. G. and SUGANO, S., J. Chem. Phys. 42
(1965) 39.

[55] BOEKEMA, C., VAN DER WOUDE, F. and SA~ATZKY, G. A., Znt. J. Magnetism 3 (1972) 341.
[56] TOFIELD, B. C. and FENDER, B. E. F., J. Phys. Chem. Solids 31 (1970) 2741.
[57] SAWATZKY, G. A., GEERTSMA, W. and HAAS, C. Commu- nicated to the author.
[58] EVANS, B. J. and SWARTZENDRUBER, L. J., Phys. Rev. B 6 (1976) 223.
[59] MENIL, F., PEZAT, M. and TANGUY, B., Comptes Rendus C 281 (1975) 849.
[60] RINNEBERG, H. and SHIRLEY, D. A., Phys. Rev. B 11 (1975) 248.
[61] DALE, B. W., J. Phys. C 4 (1971) 2705. [62] WATSON, R. E. and FREEMAN, A. J., Acta Cryst. 14 (1961)
27. [63] ALPERIN, H. A., J. Phys. Soc. Jap. Suppl. B 111, 17 (1962)
12. [64] FENDER, B. E. F., JACOBSON, A. J. and WEDGWOOD, F. A.,
J. Chem. Phys. 48 (1968) 990. [65] SCHNEIDER, C. S., Acta Cryst. A 32 (1976) 375. [66] RIETVELD, H. M., J. Appl. Cryst. 2 (1969) 65. [67] DAVIS, H. H., Phys. Rev. 120 (1960) 789. [68] WINDSOR, C. G., THORNLEY, J. H. M., GRIFFITHS, J. H. E.
and OWEN, J., Proc. Phys. Soc. 80 (1962) 803. [69] GREAVES, C., JACOBSON, A. J., TOFIELD, B. C. and FEN-
DER, B. E. F., Acta Cryst. B 31 (1975) 641. [70] JACOBSON, A. J. and FENDER, B. E. F., J. Phys. C 8 (1975)
844 [71] MOON, R. M., RISTE, T. and KOEHLER, W. C., Phys.
Rev. 181 (1969) 920. [72] Fu~ss , H., BASSI, G., BONNET, M. and DELAPALME, A.,
Solid State Commun. 18 (1976) 557. 1731 BONNET, M., DELAPALME, A., TCHEOU, F. and Fu~ss, H.,
Znt. Cong. Magnetism Moscow, 1973, Vol. IV, p. 351 (1974).
[74] NATHANS, R., ALPERIN,. H. A., PICKART, S. J. and BROWN, P. J., J. Appl. Phys. 34 (1963) 1182.
[75] JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F., J. Phys. C 6 (1973) 1615.
[76] HUTCHINGS, M. T. and GUGGENHEIM, H. J., J. Phys. C 3 (1970) 1303.
[77] COTTON, F. A. and WILKINSON, G., Advanced Inorganic Chemistry, 3rd Edition, John Wiley Interscience, New York and London (1972).
[78] ROTH, W. L., J. Solid State Chem. 4 (1972) 60. [79] PETERS, C. R., BEITMAN, M., MOORE, J. W. and
GLICK, M. D., Acta Cryst. B 27 (1971) 1826. [80] FRIEDT, J. M., SANCHEZ, J. P. and SHENOY, G. K., J.
Chem. Phys., to be published. [81] HASTINGS, J. M., ELLIOTT, N. and CORLISS, L. M., Phys.
Rev. 115 (1959) 13. [82] FREEMAN, A. J. and ELLIS, D. E., Phys. Rev. Letts. 24
(1970) 516. [83] CROS, C., FEURER, R. and POURCHARD, M., Mat. Res.
Bull. 11 (1976) 117. 1841 BYROM, E., FREEMAN, A. J. and ELLIS, D. E., Proc. 1974,
AIP Magnetism Conference. [85] INFANTE, C: E., D. Phil Thesis, University of Oxford,
1975. [86] KAMIMURA, H., J. Phys. SOC. Japan 21 (1966) 484. [87] ROTH, W. L., J. Phys. Chem. Solids 25 (1964) 1. [88] WEDGWOOD, F. A., Proc. R. Soc. A 349 (1976) 447. [89] BROWN, R. D. and BURTON, P. G. Theoretica Chimica
Acta 18 (1970) 309. [90] LARSSON, S. and CONNOLLY, J. W. D., J. Chem. Phys.
60 (1974) 1514.
[91] LYNN, J. W., SHIRANE, G. and BLUME, M., Phys. Rev. Lett. 37 (1976) 154.
[92] WILKINSON, M. K., WOLLAN, E. O., CHILD, H. R. and CABLE, J. W., Phys. Rev. 121 (1961) 74.
[93] PASTERNAK, M., in Mossbauer Spectroscopy and its Applications, Vienna, 1971 (I. A. E. A., Vienna) 1972, p. 197.
[94] LI, E. K., JOHNSON, K. H., EASTMAN, D. E. and FREEOUF, J. L., Phys. Rev. Lett. 32 (1974) 470.
[95] WILLIAMS, P. M. and SHEPHERD, R. F., J. Phys. C 6 (1973) L 36.
[96] GOODENOUGH, J. B., LONGO, J. M. and KAFALAS, J. A., Mat. Res. Bull. 3 (1968) 471.
[97] OSHAWA, A., YAMAGUCHI, Y., WATANABE, H. and ITOH, H., J. Phys. Soc. Japan 40 (1976) 986.
[98] OSHAWA, A., YAMAGUCHI, Y., WATANABE, H. and ITOH, H., J. Phys. Soc. Japan 40 (1976) 992.
[99] TAKEDA, T. and WATANABE, H., J. Phys. Soc. Japan 33 (1972) 973.
[loo] KNOX, K., J. Chem. Phys. 30 (1959) 991. [loll KHOMSKII, D. I. and KUGEL, K. I., Solid State Commun.
13 (1973) 763. [I021 AKIMITSU, J. and ITO, Y., J. Phys. Soc. Japan 40 (1976)
1621. [I031 ITO, Y. and AKIMITSU, J., J. Phys. Soc. Japan 40 (1976)
1333. [I041 YAMADA, I., J. Phys. Soc. Japan 33 (1972) 979. [I051 HIRAKAWA, K. and IKEDA, H., Phys. Rev. Lett. 33 (1974)
374. [I061 GREGSON, A. K., DAY, P., LEECH, D. H., FAIR, M. J.
and GARDNER, W. E., J. Chem. Soc. (Dalton) (1975) p. 1306.
[I071 NATHANS, R., WILL, G. and Cox, D. E., Proc. Intern. Conf. Magnetism, Nottingham, 1964, p. 327.
[I081 GALLAGHER, P. K., MACCHESNEY, J. B. and BUCHA- NAN, D. N. E, J. Chem. Php . 41 (1964) 2129.
[I091 TOFIELD, B. C., D. Phil. Thesis, Oxford, 1969. [I101 TAKEDA, T., YAMAGUCHI, Y. and WATANABE, H., J.
Phys. Soc. Japan 33 (1972) 967. [ I l l ] TOFIELD, B. C., GREAVES, C. and FENDER, B. E. F., Mat.
Res. Bull. 10 (1975) 737. [I121 TOFIELD, B. C. and FENDER, B. E. F., J. Phys. C 4 (1971)
1279. [I131 BRUN, T. 0. and LANDER, G. H., Phys. Rev. B 9 (1974)
3003. [I141 MOON, R. M. and KOEHLER, W. C., Phys. Rev. B 11 (1975)
1609. [I151 MOON, R. M., KOEHLER, W. C., CABLE, J. W. and
CHILD, H. R., Phys. Rev. B 5 (1972) 997. [I161 BLUME, M., FREEMAN, A. J. and WATSON, R. E., J. Chem.
Phys. 37 (1962) 1245. [I171 FREEMAN, A. J. and DESCLAUX, J. P., Znt. J. Magnetism 3
(1972) 311. [I181 JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F.,
Acta Cryst. B 28 (1972) 956. [I191 TOFIELD, B. C., JACOBSON, A. J. and FENDER, B. E. F.,
J . Phys. C 5 (1972) 2887. [I201 JACOBSON, A. J., TOFIELD, B. C. and FENDER, B. E. F.,
Proc. 10th Rare Earth Research Conference, Carefree, Arizona (1973) p. 194 (CONF-730402-P 1, USAEC Technical Information Center).
[I211 WEDGWOOD, F. A., J. Phys. C 5 (1972) 2427. [I221 LANDER, G. H., FABER, Jr., J., FREEMAN, A. J. and DES-
CLAUX, J. P., Phys. Rev. R 13 (1976) 1177. [I231 PETRIDIS, D., SIMOPOULOS, A., KOSTIKAS, A. and PAS-
TERNAK, M. Communicated to the author.












![Interfacial magnetic coupling between Fe nanoparticles in Fe ...magnetic moment [10] and magnetic interactions [11–13]. Concerning interparticle magnetic interactions, it has been](https://static.fdocuments.net/doc/165x107/60eea974519ccd0158590d85/interfacial-magnetic-coupling-between-fe-nanoparticles-in-fe-magnetic-moment.jpg)





