
Copyright by Wei-Cheng Lu 2013
140
Copyright by Wei-Cheng Lu 2013
Transcript of Copyright by Wei-Cheng Lu 2013

Copyright
by
Wei-Cheng Lu
2013

The Dissertation Committee for Wei-Cheng Lu Certifies that this is the approved
version of the following dissertation:
EVOLVED ENZYMES FOR CANCER THERAPEUTICS AND
ORTHOGONAL SYSTEMS
Committee:
Andrew D. Ellington, Supervisor
George Georgiou
Walter Fast
Christian Whitman
Hal Alper

EVOLVED ENZYMES FOR CANCER THERAPEUTICS AND
ORTHOGONAL SYSTEMS
by
Wei-Cheng Lu, B.S.; M.S.
Dissertation
Presented to the Faculty of the Graduate School of
The University of Texas at Austin
in Partial Fulfillment
of the Requirements
for the Degree of
Doctor of Philosophy
The University of Texas at Austin
August 2013

Dedication
To my mom and dad, and my brother and sister
For your love and support.

Acknowledgement
I would like to first thank my advisor, Andy Ellington. He gave me a lot of time
and freedom to try and learn in his lab. You need to be very independent to survive in his
lab and I survive.
I would also like to thank my committee members, especially George Georgiou
for giving me an opportunity to join the CGL group. Working in the therapeutics field is
my favorite. I really enjoy in this work. Also, I am grateful to Dr. Fast, Dr. Whitman
and Dr. Alper for giving me lots of feedback to my dissertation and work.
I would like to thank members in the Ellington lab. Thank Matt to guide me in
the beginning and help me a lot in BirA work. I would like to thanks Christien, Supriya,
Amrita. You guys give me warm hugs all the time. Thank Adam, Jared, Tony and
Bingling. You made this lab fun and it is important to have fun in the lab. Thank
Michelle, Arti, Paulina and Jorge. You help me not only on experiments but also on
many other works. Thank Randy, Alex and Xi. You are not just postdocs but also role
models. You teach us how to fight with Andy and expose his weakness to us.
Thank Kaiyin, Reilin, Kengming, Yating and Li. You are my family at Austin.
Finally, I would like to thank my parents. Thank for your sacrifice and support.
In the past several years, I felt upset in most of time and even wanted to give up in
the middle of this journey. Thank all of you to encourage and support me from the
beginning to the end.

vi
Evolved Enzymes for Cancer Therapeutics and Orthogonal System
Wei-Cheng Lu, Ph.D.
The University of Texas at Austin, 2013
Supervisor: Andrew D. Ellington
Directed evolution has been explored for a long time. Various ideas, methods,
have been shown to be feasible and successful in the enzyme field. We were interested in
evolving enzymes for applications. Therefore, we evolved human cystathionine gamma-
lyase (hCGL) and E. coli biotin ligase for therapeutic and biotechnology applications.
Wild-type human cystathionine gamma-lyase does not have any methionine-
degrading activity, unlike the high methionine-degrading abilities of bacterial methionine
gamma-lyase (MGL) found in Pseudomonas putida. The ability to engineer hCGL to
breakdown methionine can be a potential cancer treatment by targeting the methionine-
dependent cancer cells. However, the methionine-degrading activity of previously
engineered hCGL has only shown 1% activity compared to MGL, too low to be useful in
practical cancer therapeutics. By using a combination of protein design and phylogenetic
analysis, we further evolved hCGL to achieve a higher methionine-degrading activity,
with one variant displaying as much as 7% activity compared to bacterial MGL, making
it a more likely candidate in cancer treatment.
In addition, it has been shown that new orthogonal pairs of biotin protein ligase
and biotin have many biotechnology applications. Therefore, we have developed
selection scheme for directing the evolution of E. coli biotin protein ligase (BPL, gene:

vii
BirA) via in vitro compartmentalization, and have altered the substrate specificity of BPL
towards the utilization of the biotin analogue desthiobiotin. Following just 6 rounds of
selection and amplification several variants that demonstrated higher activity with
desthiobiotin were identified. The best variants from Round 6, BirA6-40 and BirA6-47,
showed 17-fold and 10-fold higher activity, respectively, their abilities to use
desthiobiotin as a substrate. Further characterization of BirA6-40 and the single
substitution variant BirAM157T revealed that they had 2- to 3-fold higher kcat values for
desthiobiotin, and 3- to 4-fold higher KM values. The kcat/KM values for these enzymes
were around 0.7-fold that of BirAwt. It is interesting the selections did not lower the KM
for desthiobiotin and actually led to a less efficient enzyme. This is an example of how
“you get what you select for”. Because peptide:DNA conjugates were distributed such
that there was on average one template or less per emulsion compartment there was
selection only for the catalytic rate (kcat) of desthiobiotinylation and not for turnover.
Given these conditions, it might be anticipated that the peptide substrate, rather than
desthiobiotin, should be bound better by the winning variants, and in fact BirA6-40 showed
a reduced KM value for BAP.

viii
Table of Contents
List of Tables ......................................................................................................... xi
List of Figures ....................................................................................................... xii
List of Figures ....................................................................................................... xii
Chapter 1: Directed enzyme evolution: In vitro selection of protein for therapeutic
and biotechnology applications.......................................................................1
In vitro selection of proteins via emulsion compartments ..............................1
Directed evolution of proteins in emulsions ..........................................4
Optimization of binding................................................................6
Optimization of catalysis ..............................................................7
Technical issues in emulsion selections: the oil:aqueous phases.9
Technical issues in emulsion selections: in vitro transcription and
translation...........................................................................10
Technical issues in emulsion selections: template recovery and
amplification ......................................................................12
The future of emulsion selections: double-bagging............................13
The future of emulsion selections: selection for fecundity........15
The future of emulsion selections: synthetic circuits.................17
Advantages and disadvantages ............................................................17
Therapeutic target: human cystathionine gamma-lyase................................18
Cancer and amino acid-dependence.....................................................18
Methionine-dependent cell activities ...................................................20
Pre-clinical trials and Clinical trials.....................................................21
Biotechnology target: E.coli biotin protein ligase ........................................23
Streptavidin:biotin................................................................................23
Applications of streptavidin:biotin orthogonal pair.............................23
Biotinylation reaction...........................................................................24
Function of E.coli biotin protein ligase................................................24
References.....................................................................................................27

ix
Chapter 2: Engineering human cystathionine gamma-lyase to degrade methionine for
cancer treatment ............................................................................................34
Introduction...................................................................................................34
Methionine gamma-lyase.....................................................................34
Cystathionine gamma-lyase .................................................................35
Dr. Stone and Olga Paley’s work.........................................................36
Results...........................................................................................................40
Phylogenetic analysis of MGLs and CGLs:.........................................40
Pilot experiment (test 11 selected positions from phylogenetic analysis)
.....................................................................................................41
Combinatorial library (11 positions from phylogenetic analysis) .......47
Comprehensive phylogenetic analysis of CGLs and MGLs................51
Substrate promiscuity...........................................................................57
Phylogenetic analysis on positions 59, 119 and 339 of hCGL ............59
Discussions ...................................................................................................62
Advantages and disadvantages ............................................................62
Other targets.........................................................................................65
Amino acid-depletion enzymes...................................................65
Antibody-directed enzyme prodrug therapy (ADEPT)...............65
Materials and methods ..................................................................................67
Site-directed mutagenesis ....................................................................67
Combinatorial library construction ......................................................67
96-well plate screening ........................................................................68
Kinetic analysis using MBTH..............................................................69
References.....................................................................................................70
Chapter 3: Directed evolution of the substrate specificity of E.coli biotin ligase 72
Introduction...................................................................................................72
Results and discussions.................................................................................75
Development of a scheme for BirA directed evolution .......................75
Library construction.............................................................................78
Selection for DTB utilization...............................................................80

x
Comparing DTB-utilizing variants ......................................................82
Kinetic characterizations of biotin ligase variants ...............................85
Evolutionary paths ...............................................................................88
Materials and Methods..................................................................................95
Library preparation ..............................................................................95
Cross-linking BAP with DNA .............................................................95
In vitro compartmentalization selection...............................................96
Gel-shift assay......................................................................................96
BPL protein purification ......................................................................97
Pyrophosphate detection ......................................................................97
References.....................................................................................................99
Chapter 4: Demonstration of cooperation and co-evolution of synthetic operon102
Introduction.................................................................................................102
Results and Conclusions .............................................................................105
Construction scheme..........................................................................105
Selection scheme................................................................................106
Operons are functional .......................................................................107
Mock selection ...................................................................................109
The selection of the representative pool (4 operons) .........................110
Non-specific DNA binding of anti-His antibody agarose beads .......111
References...................................................................................................119
Bibliography ........................................................................................................120
Vita…...................................................................................................................126

xi
List of Tables
Table 2-1: Kinetic analysis of Pseudomonas putida methionine gamma-lyase,
human cystathionine gamma-lyase and engineered human
cystathionine gamma-lyase (hCGL-NLV). ...................................38
Table 2-2: List of kinetic values for 22 hCGL variants in methionine
breakdown .......................................................................................45
Table 2-4: List of kinetic values of hCGL variants in methionine breakdown
...........................................................................................................56
Table 2-5: Substrate specificities of hCGL-NLV and hCGL-NLV+RGS+LM.
...........................................................................................................58
Table 2-6: Summary of engineered hCGL variants in methionine breakdown.
...........................................................................................................61
Table 3-1: List of substitutions of isolated biotin ligase variants from Round 6
pool ...................................................................................................82
Table 3-2: Substitution distribution of selected BirA pool (Round 6) by Next-
Generation sequencing ...................................................................91

xii
List of Figures
Figure 1-1: Display technologies for the directed evolution of proteins. .........3
Figure 1-2: In vitro compartmentalized selection for HaeIII methylase variants
.............................................................................................................5
Figure 1-3: In vitro compartmentalized selection for functional streptavidin
variants...............................................................................................8
Figure 1-4: Selections in double emulsions. ......................................................15
Figure 2-1: Enzyme reaction of methionine gamma-lyase. .............................35
Figure 2-2: Enzyme reaction of cystathionine gamma-lyase. .........................36
Figure 2-3: Structure comparison of human cystathionine gamma-lyase (PDB:
3COG) and Trichomoas vaginalis (PDB: 1E5E) with inhibitor
propargylglycine (PAG) .................................................................37
Figure 2-4: Evaluation of PEGylated hCGL-NLV in athymic mice bearing
LAN-1 xenografts............................................................................39
Figure 2-5: Phylogenetic analysis of cystathionine gamma-lyases and methionine
gamma-lyases...................................................................................41
Figure 2-6: Phylogenetic analysis of the carboxyl terminals of 5 eukaryotic
cystathionine gamma-lyases and 6 bacterial methionine gamma-
lyases.................................................................................................43
Figure 2-7: Kinetic analysis of engineered hCGL variants in methionine
breakdown .......................................................................................46
Figure 2-8: 96-well plate screening process ......................................................48
Figure 2-9: Combinatorial library screening ...................................................49

xiii
Figure 2-10:Kinetic analysis of hCGL-NLV and hCGL-NLV+RGS in
methionine breakdown ...................................................................50
Table 2-3: List of kinetic values of 20 hCGL variants in methionine
breakdown. 20 hCGL variants were purified for detailed kinetic
analysis .............................................................................................53
Figure 2-11:Kinetic analysis of hCGL-NLV+RGS+LM in methionine
breakdown .......................................................................................54
Figure 2-12: Methionine and similar molecules. ...............................................58
Figure 2-13: Phylogenetic analysis at positions 59, 119 and 339 .....................60
Figure 2-14:Kinetic analysis of hCGL-NLV+RGS+LM and hCGL-
IAV+RGS+LM in methionine breakdown. ..................................61
Figure 3-1: Biotinylation reaction .....................................................................73
Figure 3-2: Selection scheme of directed evolution of BPL using IVC ..........76
Figure 3-3: Mock selection. Before the real selection, a mock selection was
performed to validate the selection scheme ..................................78
Figure 3-4: Two libraries construction. ............................................................80
Figure 3-5: The selection process of BPL library 1 and BPL library 2. .........81
Figure 3-6: Activity assay of isolated variants (gel-shift assay). .....................84
Figure 3-7: Kinetic characterization of BirAwt, BirA6-40 and BirAM157T86
Figure 3-8: Activity assay of variants with single reintroduced substitutions (gel-
shift assay)........................................................................................89
Figure 3-9: Mapping Met-157 onto the structure of E.coli biotin ligase with
biotin. E.coli biotin ligase is shown here with biotin (1HXD) ...93
Figure 4-1: Structure of Lac operon................................................................103
Figure 4-2: Synthetic operons ..........................................................................104

xiv
Figure 4-3: A Representative synthetic operon pool ......................................106
Figure 4-4: Selection scheme of synthetic operons using IVC .......................107
Figure 4-5: Protein expression of synthetic operons (western blot) .............109
Figure 4-6: Mock selection. ...............................................................................110
Figure 4-7: One round of real selection ...........................................................111
Figure 4-8: Dissection of the selection ..............................................................114
Figure 4-9: One round of selection with new wash buffer ............................115
Figure 4-10:Different capture approach..........................................................116

1
Chapter 1: Directed enzyme evolution: In vitro selection of protein for
therapeutic and biotechnology applications
IN VITRO SELECTION OF PROTEINS VIA EMULSION COMPARTMENTS
The directed evolution of proteins has been used to generate enzymes with a wide
variety of kinetic and physical properties (Griffiths and Tawfik 2000; Bloom and Arnold
2009; Romero and Arnold 2009). In the past, directed evolution of proteins has typically
been carried out in the context of cells. This is both because the production of proteins
requires the complex and difficult-to-maintain translation apparatus, and because the
cellular membrane provides a convenient means of separating genotypes and phenotypes
from one another. However, such in vivo selections have a number of disadvantages.
Manipulating live organisms is time- and labor-consuming, and it is very difficult to
control selection conditions and stringencies within a cell. Quite often these experiments
yield survival of the organism by an otherwise unanticipated route, rather than selection
for a particular protein property. In addition, it is difficult to work outside the well-
known boundary conditions of living systems; for example, selecting for proteins that
operate at extremes of pH or that utilize unnatural amino acids. Also, it has been found
that overexpression of up to 51% of endogenous E.coli ORFs can cause severe growth
defects, while 77% of these ORFs cause growth inhibition (Kitagawa, Ara et al. 2005).
These results strongly argue that in vivo selection experiments for many different
functions might disfavor truly active protein variants. Finally, the population sizes that
can be examined in cells are inherently limited by the gross inefficiencies of
transformation or transfection. In order to overcome these difficulties in vitro systems for
directed evolution were developed.

2
In vitro selections use much the same principles for molecular evolution as in vivo
selections. A pool of heritable information is generated, parsed via function, and
selectively amplified. There are various ways to achieve these three steps with proteins
in vitro, including ribosome display, mRNA display and CIS display (Lipovsek and
Pluckthun 2004; Ullman, Frigotto et al. 2011) (Figure 1-1). For example, ribosome
display involves the in vitro translation of mRNAs that lack a stop codon and that
therefore couple the nascent peptide indirectly to the mRNA via the stalled ribosome.
Ribosome display was first demonstrated by Jozef Hanes and Andreas Plückthun and
used for selection of a single-chain antibody fragment (scFv) that bound hemagglutinin.
In five rounds of purely in vitro selection, the anti-hemagglutinin scFv was enriched from
the starting population in which it was diluted by an anti-β-lactam scFv by 108-fold
(Hanes and Pluckthun 1997; Zahnd, Amstutz et al. 2007). mRNA display is similar to
ribosome display but relies on the formation of a covalent link between a puromycin-
containing mRNA and the nascent translated peptide. In a model selection, a mRNA
fusion with Myc peptide epitope could be enriched 20-40 fold from the mixed population
by immunoprecipitation (Roberts and Szostak 1997). The Szostak group also used this
system for the de novo identification of ATP-binding motifs from a completely random-
sequence library. They found that one functional protein could be recovered from a
sequence space that spanned 1011 different proteins (Keefe and Szostak 2001; Lipovsek
and Pluckthun 2004). In contrast to mRNA display, CIS display relies on a linkage (non-
covalent or covalent) between the template DNA and the nascent translated peptide
instead of linkage of the mRNA to the nascent peptide. The McGregor group has used
RepA, a DNA replication initiator, to link the DNA template and nascent peptide
(Odegrip, Coomber et al. 2004). CIS display can also be engineered to provide a covalent

3
linkage to the DNA template via the replication initiator protein, P2A (Reiersen, Lobersli
et al. 2005).
Figure 1-1: Display technologies for the directed evolution of proteins. Ribosome
display, mRNA display and CIS display provide different means of linking
genotype with phenotype. For ribosome display, mRNA sequences are
engineered without stop codons and the ribosome stalls, leaving the mRNA
and the nascent peptide linked to one another via the ribosome. In mRNA
display, puromycin is covalently conjugated to mRNA, and couples itself to
a newly translated peptide, providing a direct covalent linkage between
mRNA and peptide. For CIS diplay, the RepA (or other DNA binding)
protein binds a specified sequence on a DNA template, such as the ori
sequence. Peptides fused to RepA can bind to their templates, a non-
covalent linkage similar to ribosome display.
The general advantages of these methods relative to in vivo selection are larger
library sizes and ease of manipulation. However, it can be difficult to fully control the
selection environment, especially given that many of the components necessary for
translation or enzymatic activity are diffusible. This is why it has proven useful to try to
both control expression and restrict diffusion within emulsions. Water-in-oil emulsions
can contain 1010
compartments and are stable at 25o C for 24 hours with no obvious
change in the distribution of compartment sizes (Tawfik and Griffiths 1998). This

4
technology has enabled the revolution in NextGen DNA sequencing (Margulies, Egholm
et al. 2005), but has also proved to be of great utility for the directed evolution of
proteins.
Directed evolution of proteins in emulsions
Directed evolution via in vitro compartmentalization (IVC) was first
demonstrated in a seminal paper by Dan Tawfik and Andrew Griffiths (Tawfik and
Griffiths 1998). First, these authors showed that functional dihydrofolate reductase
(DHFR) and HaeIII methyltransferase could be produced via in vitro transcription and
translation in compartments and still retain over 60% activity compared to proteins
produced in nonemulsified reactions. Functional HaeIII methyltransferases could then
individually feedback on the survival of their own templates. In a population of folA
(encoded DHFR) mixed with M.HaeIII (encoded methyltransferase HaeIII), methylated
M.HaeIII genes were protected from HaeIII endonuclease digestion and could be
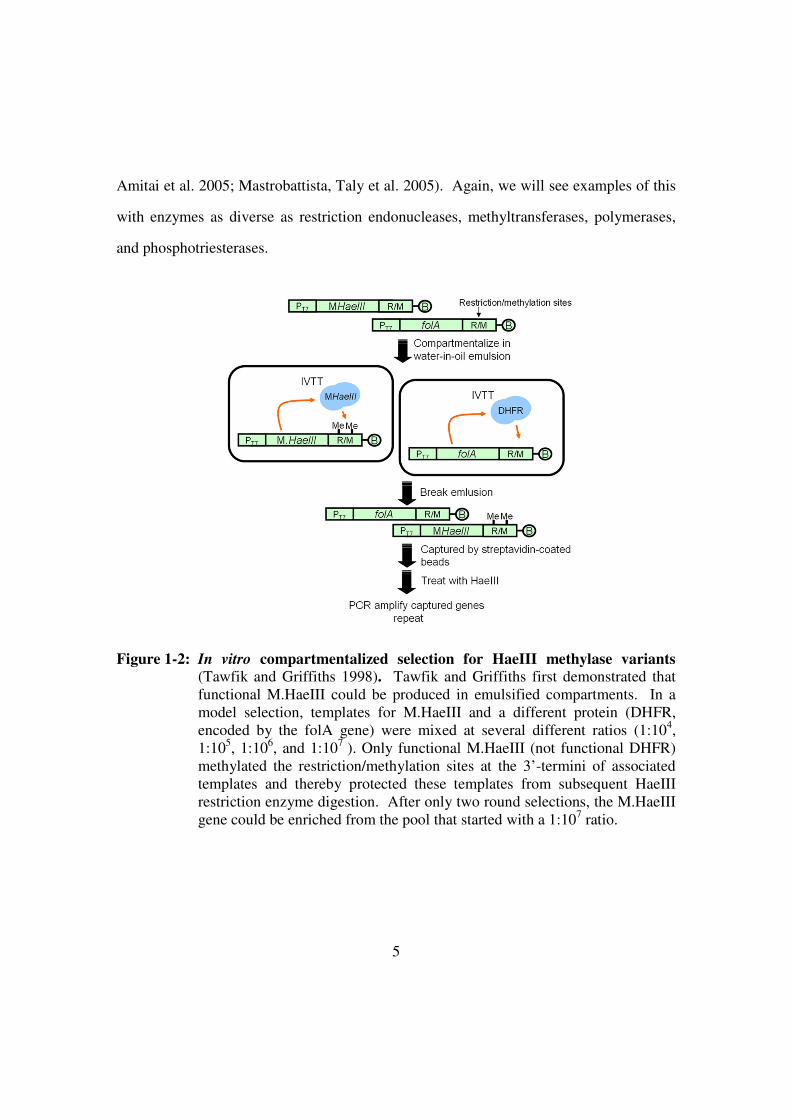
enriched from a pool by a factor of 107-fold after only 2 rounds of selection (Figure 1-2).
For all selections, some manner of genotype-phenotype linkage is necessary.
There are a surprising number of ways to link the genotype and phenotype during IVC
selections. For example, noncovalent linkage by the co-scalled STABLE system (Doi
and Yanagawa 1999), zinc finger protein binding to DNA templates (Sepp and Choo
2005) and covalent linkage by a HaeIII methyltransferase fusion protein (Bertschinger
and Neri 2004) and SNAP-tag (Stein, Sielaff et al. 2007) have been used for the selection
of binding proteins. We will examine a number of these systems below. For enzymes,
fluorescent substrates and FACS have been used to confine genotype and phenotype
either on the surface of beads or in a double emulsion (Bernath, Hai et al. 2004; Aharoni,
5
Amitai et al. 2005; Mastrobattista, Taly et al. 2005). Again, we will see examples of this
with enzymes as diverse as restriction endonucleases, methyltransferases, polymerases,
and phosphotriesterases.
Figure 1-2: In vitro compartmentalized selection for HaeIII methylase variants
(Tawfik and Griffiths 1998). Tawfik and Griffiths first demonstrated that
functional M.HaeIII could be produced in emulsified compartments. In a
model selection, templates for M.HaeIII and a different protein (DHFR,
encoded by the folA gene) were mixed at several different ratios (1:104,
1:105, 1:10
6, and 1:10
7 ). Only functional M.HaeIII (not functional DHFR)
methylated the restriction/methylation sites at the 3’-termini of associated
templates and thereby protected these templates from subsequent HaeIII
restriction enzyme digestion. After only two round selections, the M.HaeIII
gene could be enriched from the pool that started with a 1:107 ratio.

6
Optimization of binding
The methods that Tawfik and Griffiths developed can be generalized to changing
the substrate specificities of a variety of binding proteins. For example, the Ghadessy
group used an anti-p53 antibody to isolate p53 variants that bound to a double-stranded
DNA containing a so-called low response element. When introduced into cells, these
variants could also better mediate transactivation of genes controlled by low response
elements (Fen, Coomber et al. 2007). Similarly, Yu Chen et al. demonstrated that they
could carry out selections with a fusion between a RNA-binding protein and a DNA-
binding zinc finger. The RNA-binding protein could be evolved to recognize an
immobilized target RNA, while the DNA-binding zinc finger bound to the template
DNA; in essence the dual protein served as a bridge between the RNA target and the
DNA template. In this way, these authors demonstrated that the method should be able to
change the recognition specificity of RNA binding protein (Chen, Mandic et al. 2008).
Finally, Levy and co-workers carried out an emulsion-based selection to alter the binding
specificity of streptavidin. The gene encoding streptavidin was modified with the biotin
analogue desthiobiotin, and streptavidin variants that were able also able to bind their
own DNA templates were further isolated via binding of an anti-His antibody to a His tag
on the streptavidin (Figure 1-3). Interestingly, selected variants still bound biotin quite
well, but had greatly reduced off-rates and longer dissociation half-times for
desthiobiotin. Nonetheless, the specificity differences were great enough that the wild-
type and mutant proteins could be used in differential labeling schemes (Levy and
Ellington 2008).

7
Optimization of catalysis
The specificities of enzymes can also be altered by directed evolution in
emulsions. The Griffiths group has used in vitro compartmentalization and directed
evolution to change the recognition specificity of the M.HaeIII methyltransferase, and
identified enzyme variants that could methylate AGCC rather than the canonical
sequence GGCC. Moreover, the mutant methylase shows higher catalytic activity with
AGCC compared to wild-type M.HaeIII with the canonical sequence (Cohen, Tawfik et
al. 2004). Similarly, Nobuhide et al. have shown that it is feasible to use IVC to select
for restriction endonuclease activity. These authors introduced a FokI restriction site into
the template for the gene, and recovered cleaved DNA templates by ligation to to dUTP-
biotin (Doi, Kumadaki et al. 2004).
Enzymes other than those used as molecular biology reagents can also be
optimized. Griffths and Tawfik demonstrated that IVC and FACS could be combined for
the directed evolution of phosphotriesterases (Griffiths and Tawfik 2003). Genotype and
phenotype were linked by immobilizing both genes and translated proteins on
microbeads. The microbeads were then re-emulsified with enzymatic substrates. Finally,
the products were also captured on the microbeads and further detected via an anti-
product antibody binding and FACS. It should be noted that this method selects not only
for catalysis, but for multiple turnover catalysis, and the kcat of the phosphotriesterase was
improved by 63-fold. While library size is limited relative to other emulsion methods (
FACS only selects 107 clones per hour) this combined system can nonetheless be adapted
to the evolution of catalysts that are not readily engineered by other in vitro and in vivo
methods.

8
Figure 1-3: In vitro compartmentalized selection for functional streptavidin
variants. Biotinylated DNA templates encoding streptavidin variants were
compartmentalized in water-in-oil emulsions containing in vitro
transcription and translation reagents. Functional streptavidin variants
translated in compartments could bind to their encoding DNA templates.
After breaking the emulsion, all streptavidin variants were isolated via an
encoded His tag and the genes for the functional streptavidin variants were
recovered by PCR.
Additionally, IVC has been used not only for evolving proteins but also for
evolving RNA. Levy and co-workers developed an IVC selection for multiple turnover
RNA catalysts (Levy, Griswold et al. 2005). In this selection, microbeads that
immobilized a gene pool and one half of a RNA ligase substrate were emulsified with the

9
components of an in vitro transcription reaction and a fluorescent, second half of a RNA
ligase substrate. Transcribed, active ribozyme variants could then link the fluorescent
substrate to the microbeads containing their own templates. The most active ligase
variants were sorted following breaking of the emulsion by FACS, and the templates
were amplified in vitro and re-immobilized for additional rounds of selection and
amplification.
Technical issues in emulsion selections: the oil:aqueous phases
In vitro compartments are typically made of two phases, an aqueous phase and an
oil-surfactants phase. The aqueous phase contains genotype and what are reagents for
expression of phenotype expression. In the case of proteins, this will typically be
transcription and translation, but for ribozymes only transcription is required. The
function of oil-surfactant phase is to produce artificial membranes separating each
aqueous compartment and confining each genotype and phenotype. By stirring the
aqueous phase with the oil-surfactant phase, typically upwards of 1010
compartments can
be generated in a 1 mL emulsion.
The composition of the oil-surfactant phase is a critical issue, in part because the
hydrophobic phase or molecules therein can inhibit complex biochemical processes such
as translation, and in part because the interface can denature newly produced proteins
(Miller, Bernath et al. 2006). In order to stabilize emulsions to heat thermal cycling,
0.05% Triton X-100 or ABIL EM 90 can be added; the latter can prevent compartment
leakage over 35 thermal cycles (Williams, Peisajovich et al. 2006). In addition, unlike
other surfactants ABIL EM 90 does not lead to the rapid inhibition of rabbit reticulocyte
lysates (Ghadessy and Holliger 2004). Because of the idiosyncracies associated with

10
systems and proteins in emulsions, it is typically wise to test different oil-surfactant
phases with different aqueous phases (lysates, below) prior to carrying out a directed
evolution experiment (Davidson, Dlugosz et al. 2009). For example, an expressed
protein may have acceptable yields, but may not be necessarily active due to inhibition by
components of the emulsion reaction (e.g., surfactants).
Technical issues in emulsion selections: in vitro transcription and translation
There are a variety of options for how to produce proteins in emulsion. An E.coli
lysate is generally used as the aqueous phase for most of IVTT-IVC systems. However,
E.coli lysates cannot support post-translational modifications on eukaryotic proteins
while at least rabbit reticulocyte lysates appear to be able to. Moreover, it is apparent that
different proteins translate more or less well in different lysates, perhaps due to the
different surfactants that are available, as alluded to above (Tawfik and Griffiths 1998;
Griffiths and Tawfik 2003; Yonezawa, Doi et al. 2003; Ghadessy and Holliger 2004;
Chen, Mandic et al. 2008). Therefore, wheat germ or rabbit reticulocyte lysates have also
been adapted to emulsion methods.
In our experience, there are a wide variety of variables that impact the efficiency
of expression in these different lysates, and the complexity of the systems makes it
difficult to predict in advance which lysate or conditions may work best for which
emulsion selection. For example, template DNA purification is important for translation
yield in lysates. In our hands, phenol-extracted DNA seems to be better than DNA
purified via spin columns which is in turn better than DNA purified on gels (Davidson,
Dlugosz et al. 2009). A recent selection model for oxygen resistant [FeFe] hydrogenase
overcame expression inefficiencies by using emulsion PCR to increase the amount of

11
template DNA on beads, ultimately generating larger amounts of protein in each
compartment (Stapleton and Swartz 2010).
One commonality between good expression systems seems to be the absence or
inhibition of nucleases. Nucleases in lysates can quickly degrade DNA construct
(genotype) and thereby reduce both the phenotype expression and the recovery of a
functional genotype. In our experience, 1 ng of 2.3kb DNA could not be recovered from
E. coli lysate after 2 hours incubation at 30o C. Adding a non-specific carrier such as
salmon sperm DNA may help, as will making modifications to the DNA template, such
as biotinylation or the use of phosphorothiolated nucleotides (Takei, Kadomatsu et al.
2002). Also, it has been found that Gam protein, a bacteriophage λ protein, can inhibit
exonuclease RecBCD in lysates (Sitaraman, Esposito et al. 2004). A PCR-derived linear
template was protected by Gam against purified RecBCD for up to 4 hours, leading to an
increase in protein production. However, since there are other many other nucleases in
E.coli lysates (such as ExoIII, VIII, and EndoIV) it may be wise into the future to use a
fully defined, recombinant translation system (PURE) to avoid DNA degradation
(Shimizu, Kanamori et al. 2005). In order to enhance RNA production from those
mRNAs that are made, changes in translation initiation can be introduced. For example,
the EMCV IRES has been shown to increase protein expression in rabbit reticulocyte
lysates (Bochkov and Palmenberg 2006). In at least one instance, a construct with an
EMCV IRES increased luciferase expression 4- to 5-fold in rabbit reticulocyte lysates
relative to constructs with Kozak sequences. For similar experiments in wheat germ
extracts, the TMV IRES can be used (Yonezawa, Doi et al. 2003).
Once a protein is produced, there is no guarantee that it will be active, since the
emulsion interface is quite different than a phospholipid interface. In many instances,
proteins likely denature at the interface, even in the presence of emulsifying agents.

12
Once again, it may be necessary to screen emulsifiers to find those that are most useful
for functional expression of a given protein. Adding a bulk protein such as BSA or
negative charged surfactants such as sodium deoxycholate (Tawfik and Griffiths 1998)
can also prevent translated proteins from being trapped at the oil-water interface
(Ghadessy and Holliger 2004). The use of chaperones may also prove helpful in
avoiding interface-mediated denaturation. Parent et al. have found that GroEL- and
GroES-overexpression in lysates can rescue destabilized, temperature-sensitive
bacteriophage P22 coat protein assembly (Parent, Ranaghan et al. 2004). Similarly,
Tokuriki and Tawfik have demonstrated that overexpression of GroEL/GroES helps
enzymes to accumulate mutations that would otherwise be destabilizing, which in turn
allows more pathways to be followed during the evolution of new substrate specificities
(Parent, Ranaghan et al. 2004; Tokuriki and Tawfik 2009). Along these lines, the Tawfik
group has shown that a small library that is first allowed to accumulate neutral mutations
is a better starting point for the evolution of serum paraoxonases (PON1) with different
substrate specificities than a library centered on the wild-type enzyme (Amitai, Gupta et
al. 2007; Gupta and Tawfik 2008). This is likely because mutations that are nominally
'neutral' can actually improve the stability of the wild-type enzyme to denaturation. The
use of such libraries would be a clear advantage in emulsion selections, as well.
Technical issues in emulsion selections: template recovery and amplification
Following the actual selection, it’s important to be able to efficiently recover
functional variants from the pool. Diethyl ether is typically used to break the emulsions
(Miller, Bernath et al. 2006), although other organic solvents such as chloroform and
hexane can also be used (Davidson, Dlugosz et al. 2009). In our experience, there are

13
two potential choke points for recovery. First, it is increasingly difficult to recover longer
genes, especially if you are recovering mRNA rather than DNA. This is likely related to
the aforementioned problem of nucleases in lysates. In addition, there is a related issue.
With low recovery, the possibilities for accumulating additional mutations or
amplification artifacts are proportionately greater, because larger numbers of
amplification cycles are required to generate material for additional rounds of emulsion-
based selection. This can actually lead to a situation not unlike the evolutionary
conundrum known as Muller's Ratchet, in which the mutation rate is sufficiently high and
the recovery rate is sufficiently low that the functionality of the population actually
decreases during the course of a selection, irrespective of stringency.
A secondary issue with respect to the recovery and amplification of mRNAs is
that secondary structure may impede reverse transcription. We have attempted to
circumvent this problem by creating synthetic genes whose mRNA transcripts will have
greatly reduced structure-forming potential.
The future of emulsion selections: double-bagging
It has proven possible to emulsify an emulsion, creating an aqueous suspension of
oil drops that in turn surround aqueous contents. First, an aqueous phase is mixed with
an oil-surfactant phase containing, for example, cholesterol, Span 60, and decane to
generate the water-in-oil emulsion (Figure 1-4). Subsequently, this first emulsion
reaction is further mixed with PBS containing 0.5% Tween 20 and the
aqueous:oil:aqueous compartments are extruded through a 8 µm pore-size membrane
(Miller, Bernath et al. 2006). Double emulsions are particularly useful for looking at
otherwise diffusible products or at individual cells (Bernath, Hai et al. 2004). For

14
example, the Griffiths group has evolved the classic E. coli Ebg enzyme to have greater
β-galactosidase activity (Mastrobattista, Taly et al. 2005). Enzyme variants were
translated in individual compartments, as with single emulsions, but the fluorescent
substrate of the reaction, fluorescein di-β-D-galactopyranoside, was then kept with an
individual template and enzyme variant by a second emulsion. The enzyme variants that
produced the greatest amount of fluorescent product (fluorescein) were separated by
FACS. Eight selected variants showed 300-fold higher kcat/KM values relative to the
unevolved Ebg enzyme.
Bacteria can also be ensconced within double emulsions. This is useful because
in vitro lysates are often inefficient at protein production, and because folding within the
bacteria may prevent the denaturation problems cited above. The Tawfik group has used
this technique to screen individual bacteria that express variants of serum paraoxonase
(PON1). One great advantage of using bacteria as production vehicles is that they can
generate from 104-10
5 enzyme molecules per bacterium per compartment, whereas in
vitro transcription and translation yield only about 10-102
molecules per compartment.
Turnover of a fluorescent substrate and FACS resulted in the identification of enzymes
that were 100-fold improved in thiolactonase activity. Serum paraoxonase can also
hydrolyze organophosphates at a low rate, and in a subsequent experiments variants were
identified that had 105-fold improved activity (kcat/KM) against a coumarin derivative of
Sp-cyclosarinhigher (Gupta, Goldsmith et al. 2011).

15
Figure 1-4: Selections in double emulsions (Bernath, Hai et al. 2004; Miller, Bernath et
al. 2006). A double emulsion further encapsulates water-in-oil emulsions
within an additional aqueous phase. The internal aqueous phase still
contains the in vitro transcription and translation reagents necessary for
protein expression. Surfactants such as Span 60 or Tween 20 provide a
boundary between the internal aqueous phase and the outer oil phase. These
surfactants also help to stabilize encapsulation in a second aqueous phase,
creating an environment akin to a lipid-coated cell. Those ribozyme or
protein templates that generate the most fluorescent products within the first
aqueous layer can be sieved from the multitude of cell-like compartments by
FACS.
The future of emulsion selections: selection for fecundity
While turnover of substrates is a useful way to select for the activity of many
enzymes, self-amplification can be used to select for polymerase function. The Holliger
group has developed a system they term “compartmentalized self-replication” (CSR) for
polymerase selections. They first express Taq DNA polymerase in cells, and then
compartmentalize the cells in emulsions with amplification reagents. Thermocycling is
also carried out directly in emulsions, with the first denaturation step lysing the bacteria.

16
Those polymerase variants that are most active will better amplify their own genes
(Ghadessy, Ong et al. 2001). DNA polymerases with higher thermostabilities (the
isolated Taq clone has 11-fold longer half-life at 97.5oC than the wild type Taq enzyme)
and that are more resistant to the DNA polymerase inhibitor, heparin (130-fold better
than wild type) have been evolved. The same strategy has also been used to expand the
substrate specificity of Taq DNA polymerase. One mutant has been isolated that can
actually use both dNTP and NTP equally well, while another has been shown to use 2’-
substituted nucleotides (Ong, Loakes et al. 2006). The Holliger group has now worked
on evolving polymerases to further expand substrate utilization to primers with a
hydrophobic base analog (Loakes, Gallego et al. 2009) and to the nucleotides 5-
nitroindole, 5-nitroindole-3-carboxamide, FITC-12-dATP(Ghadessy, Ramsay et al.
2004), biotin-16-dUTP (Ghadessy, Ramsay et al. 2004), Cy3-dCTP, and Cy5-dCTP
(Ramsay, Jemth et al. 2010).
Additionally, it is worth mentioning selective gene amplification (SGA) (Kelly
and Griffiths 2007), a method that further mimics natural selection. As an example,
active or inactive phosphotriesterase genes (OPD) and a substrate were immobilized on
streptavidin-coated microbeads. The two different microbead populations (active and
inactive) were emulsified with an in vitro transcription and translation reaction.
Following expression and breaking of the emulsion, conversion of the substrate to a
product was detected by the addition of anti-product antibodies coupled to primers
necessary for the amplification of the original gene. When the microbeads were re-
emulsified with a PCR reaction mixture those genes that originally yielded active
phosphotriesterase variants were preferentially amplified, and could participate in
subsequent rounds of selection and amplification. This method might be adapted to a
wider variety of reactions, depending on the availability of good anti-product antibodies.

17
The future of emulsion selections: synthetic circuits
While individual proteins and enzymes can be expressed in emulsions, it would
be interesting and useful to increase the complexity of selection schemes by having
multiple steps in a synthetic circuit feedback on survival. For example, the Holliger
group demonstrated that CSR could be adapted to select not only polymerases, but also
the nucleoside diphosphate kinase (NDK). In this selection scheme, functional NDK
molecules provide enough dNTPs for DNA polymerase to function and ultimately
amplify the DNA templates encoding functional NDKs. We are similarly working on
demonstration of protein cooperation in a synthetic operon. The desthiobiotin-utilizing
streptavidin variant (Levy and Ellington 2008) that was previously selected has been
combined with a selected biotin ligase (BirA) variant that can also utilize desthiobiotin.
By adding a peptide substrate to the DNA template encoding the operon, it may be
possible to simultaneously select for desthiobiotin addition and capture.
Advantages and disadvantages
Emulsion-based selections have several advantages over competing methods.
They allow selection for multiple turnover reactions, unlike ribosome display, mRNA-
peptide display and CIS display, which primarily select for binding. By avoiding the
need for cells, toxic substrates or unnatural substrates can be used in selections.
Emulsion selections also provide much greater control over the selection process. For
example, by iteratively lowering substrate concentrations, variants with improved KM
values can be selected; by reducing reaction times or adding competitive substrates,
variants with improved kcat values can be isolated. One of the greatest advantages of
emulsion methods is that larger library sizes can be sieved, but this advantage remains
largely unrealized because of the many disadvantages that attend the method.

18
As we have already seen, some of the primary problems with emulsion selections
have to do with the robustness of protein production. Beyond this consideration, there is
considerable variability between emulsion reactions, both from batch to batch and even
within batches (not all emulsion bubbles are of equal volume). In current methods, stir
bars (Yonezawa, Doi et al. 2003), homogenizers (Agresti, Kelly et al. 2005), or
microcapillary devices (Utada, Lorenceau et al. 2005) are used to produce emulsions.
While the first two methods are generally more variable, higher stirring speed or
homogenization may impair enzyme activity (Miller, Bernath et al. 2006). Fortunately,
the general drive for emulsion PCR has led to the development of devices for sorting
emulsions. For example, instruments made by the company RainDance are able to
generate consistent, stable emulsion droplets at a rate of 107 droplets/hour, and the
droplets can potentially not only be sorted, but also merged. Into the future such devices
should help to further standardize selection conditions, and may enable the step-wise
addition of substrates and other small molecules (Kintses, van Vliet et al. 2010).
THERAPEUTIC TARGET: HUMAN CYSTATHIONINE GAMMA-LYASE
Cancer and amino acid-dependence
Cancer has become a leading cause of human death and around one million
people have out of a variety cancers in the United of States. In the current state, cancers
are tough diseases to treat as is it is difficult to preferentially kill cancer cells without
hurting normal cells in the treatment. Conventional chemotherapy is toxic to cancer cells
but also has adverse effects on normal cells. Therefore, cancer cell-specific treatments
have been pursued as a better method.

19
In cancer biology research, it has been found that cancer cells exhibit different
metabolism compared to normal cells (Kroemer and Pouyssegur 2008). It has been
shown that some types of cancer are dependent on particular amino acids because they
are not able to synthesize these amino acids themselves. They must acquire them from
extracellular sources to maintain cell activities such as protein synthesis. Thus this
resource dependent might be a target for preferential killing of cancer cells.
In cancer research, it has been discovered that several amino acids are important
to maintain cancer cell growth such as asparagine, tyrosine, phenylalanine, glutamine,
leucine and methionine. Without these amino acids in culture media, some cancer cell
lines could not survive and stop growing in vitro. Hepatocellular carcinoma (HCCs),
pancreatic tumor and melanomas require exogenous L-arginine (Gong, Zolzer et al. 2000;
Ensor, Holtsberg et al. 2002; Bowles, Kim et al. 2008; Hernandez, Morrow et al. 2010).
Childhood acute lymphoblastic leukemia (ALL), ovarian carcinomas and non-Hodgkin
lymphomas need L-asparagine from extracellular sources (Cooney and Handschumacher
1970; Lorenzi, Llamas et al. 2008; Cantor, Panayiotou et al. 2012). Some cell lines of
neuroblastomas and glioblastomas and breast, lung, colon, kidney carcinomas have been
found to be methionine-dependent (Hoffman 1985; Breillout, Antoine et al. 1990;
Poirson-Bichat, Goncalves et al. 2000; Kokkinakis, Hoffman et al. 2001).
We are interested in methionine-dependent cancer cells for two major reasons. In
the past research, methionine-depletion has been shown to be a promising way to inhibit
tumor growth in vitro and in tumor-bearing animals. (Breillout, Hadida et al. 1987; Guo,
Lishko et al. 1993; Guo, Herrera et al. 1993; Poirson-Bichat, Gonfalone et al. 1997).
Moreover, synergistic effect of methionine-depleting total parenteral nutrition (lacking
methionine and cysteine) with 5-fluorouracil (5-FU) has been reported in tumor-bearing
rats and in clinical trails with gastrointestinal tract cancers (Goseki, Yamazaki et al.

20
1995). Therefore, methionine depletion in serum became our goal to pursue for cancer
treatment.
It is not clear why some cancer cells are sensitive to methionine deprivation. In
past studies, it has been reported that some methionine-dependent cancer cells associate
with a loss of function of methionine synthase, methylthioadenosine phosphorylase
(MTAP) or methylenetetrahydrofolate reductase (MTHFR). These enzymes are
important in the pathway of methionine and homecysteine metabolism (Cellarier,
Durando et al. 2003). Cancer cells would not be able to synthesize methionine
themselves with the loss of function of these enzymes. Therefore, acquiring methionine
from the environment would be the major source for methionine-dependent cancer cells.
Generally, there are two ways to reduce the serum methionine levels: nutritional
deprivation and enzymatic deprivation. The first approach is to provide a methionine-
free diet (Methionine-, Homocysteine-) to lower methionine absorbed in the body. The
second approach is to implement methionine gamma-lyase to degrade absorbed
methionine. By depleting methionine from the environment and therefore, methionine-
dependent cancer cells would not acquire enough methionine could not survive in low
concentration methionine condition.
Methionine-dependent cell activities
Methionine is involved in four major cell activities. First, methionine is one of 20
essential amino acids for all protein synthesis in cells. Without methionine, the synthesis
of all required proteins would be interrupted. Second, methionine is important for
production of polyamines (spermine and spermidine) which are associated with nuclear
and cell division activities (Thomas and Thomas 2001). Third, methionine is involved in

21
the transsulfuration pathway which generates glutathione. Glutathione is an important
antioxidant and can protect cell from oxidative stress (Anderson 1998). Fourth,
methionine provides the methyl group for methylation of DNA and other molecules.
Without methionine, all above cell activities would be interrupted and cancer cells would
be arrested at S-G2 phase of cell cycle (Pavillard et al., 2006) .
Pre-clinical trials and Clinical trials:
It has been reported that some pre-clinical trails have shown efficacy of
methionine deprivation on tumor cells (Breillout, Hadida et al. 1987; Guo, Lishko et al.
1993; Guo, Herrera et al. 1993; Poirson-Bichat, Gonfalone et al. 1997). Purified
methionine gamma-lyase from Pseudomonas putida was used to treat nude mice with
rodent or human tumors. There was no apparent toxicity of the treatment and the growth
of tumors were greatly slowed (Tan, Xu et al. 1996). Furthermore, recombinant
Psuedomonas putida methionine gamma-lase has been used on primates to test the
immunogenicity. However, it has been shown that re-challenge with recombinant
enzyme resulted in anaphylactic shock and death of one primate (Yang, Wang et al.
2004). This result indicated the antigenicity of bacterial enzymes would be a major
hurdle for practical treatment of patients. Recently, PEGylated methionine gamma-lyase
has been generated. PEGlyated therapeutic enzymes have been shown to have greater
resistance to degradation by proteases, longer half-life in vivo, lower clearance and less
immunogenicity (Veronese 2001; Harris and Chess 2003; Tan, Xu et al. 2010).
PEGylated methionine gamma-lyase is one option to reduce antigenicity in practical
treatment.

22
Besides pre-clinical trails, several clinical trails have been reported with positive
results. Methionine-depleting total parenteral nutrition (lacking methionine and cysteine)
displayed synergistic effects with 5-fluorouracil (5-FU) on patients with gastrointestinal
tract cancers (Goseki, Yamazaki et al. 1995). Cystemustine treatment combined one-day
methionine-free diet in patients with metastatic melanoma or recurrent glioma was well
tolerated in toxicity and nutritional status (phase II clinical trail) (Thivat, Farges et al.
2009). Moreover, purified endotoxin-free methionine gamma-lyase from Pseudomonas
putida has been used to test side effects on patients with advanced breast cancers. The
approach significantly lowered methionine concentration in serum (~0.1 uM) and did not
cause any signs of side effects (pilot phase I clinical trail) (Tan, Zavala et al. 1996; Tan,
Zavala et al. 1997).
Taken together, methionine-depletion is a promising approach to specifically
target methionine-dependent cancer cells. Besides methionine-depleting total parenteral
nutrition, an active enzyme with methionine-degrading activity is a powerful option for
this purpose. The hurdle is there is no human enzyme showing methionine-degrading
activity that would be non-immunogenic. Therefore the question is “How could we avoid
immunogenicity using a bacterial methionine gamma-lyase?” or “How do we engineer a
human enzyme to acquire methionine-degrading activity?” Interestingly, recent work has
shown an engineered human cystathionine gamma-lyase is able to degrade methionine
(Stone, Paley et al. 2012) and can be a candidate for further improvement. A further
engineering of human cystathionine gamma-lyase to degrade methionine is described in
Chapter 2.

23
BIOTECHNOLOGY TARGET: E.COLI BIOTIN PROTEIN LIGASE
Streptavidin:biotin
The interaction between biotin and streptavidin is the strongest non-covalent
binding in the nature with a Kd of ~10-15
M. Since the interaction of biotin:streptavidin
pair is so tight and specific, it has been widely applied for protein labeling or purification.
However, the single streptavidin:biotin pair is limited in application. Thus people keep
attempting to expand orthogonal pairs to streptavidin:biotin.
Applications of streptavidin:biotin orthogonal pair
Dr. Levy (an former member of The Ellington lab) implemented an IVC system to
evolve the streptavidin for altering specificity to biotin analog, desthiobiotin (Levy and
Ellington 2008). In his selection, he isolated the variants which show similar binding
affinity to desthiobiotin (~10-13
M) and have around 50 times slower off rate compared to
wild type streptavidin. He has demonstrated that two orthorgonal pairs, streptavidin
variant (SAR7-6):desthiobiotin and wild-type streptavidin:biotin could be used for
different fluorescence labeling on slides. Based on this idea, we questioned whether
there are any interesting biotin relevant targets. Thus, we thought E.coli biotin protein
ligase (BPL) might be an interesting target and it might have potential applications to
generate BPL variants in use of desthiobiotin as a new orthogonal pair. We implemented
in vitro compartmentalization (IVC) as a system to evolve E.coli biotin ligase in use of
desthiobiotin. Detailed work is described in Chapter 3.

24
Biotinylation reaction:
Biotin is an essential cofactor for biotin dependent enzymes to transfer a carboxyl
group in several metabolic pathways. In order to attach biotin to biotin dependent
enzymes such as acetyl-CoA carboxylase, biotin protein ligase is required for this
biotinylation reaction. In this biotinylation reaction, there are two steps. First, BPL
generates an intermediate, biotinoyl-5’-AMP by sequentially recruiting biotin and ATP to
their binding sites. Subsequently, BPL transfers the biotin moiety of biotinoyl-5’-AMP to
the specific lysine of biotin carboxyl carrier protein (BCCP) and then forms an amide
linkage between the carboxyl group of biotin and the ɛ-amino group of lysine. (Chapman-
Smith and Cronan 1999; Chapman-Smith and Cronan 1999)
Function of E.coli biotin protein ligase
E.coli BPL is a 35.5 kDa protein and its structure with bound biotin has been
solved (1HXD) (Weaver, Kwon et al. 2001). It is known that there are three domains, an
N-terminal DNA binding domain, a central catalytic domain, and a C-terminal domain
with unknown function. The N-terminal DNA binding domain has been shown to have a
characteristic helix-turn-helix structure which is associated with repression of biotin
synthesis. E.coli BPL is a bifunctional protein which is not only a biotin ligase but also a
transcription repressor of biotin biosynthesis. In the repression process, biotinoyl-5’-
AMP is a co-repressor which can trigger E.coli BPL to form dimer and bind the operator
to suppress the transcription of the biotin operon. The central catalytic domain contains
binding sites for biotin, ATP and the BCCP as these molecules are required substrates for
biotinylation reaction. Moreover, it has been shown that there are three different
conformation states of E.coli BPL in thermodynamic studies. The first conformation

25
change is caused by biotin binding and helps to recruit ATP binding. After generating
biotinoyl-5’-AMP, it goes through another structural alteration which allows BCCP
binding for biotinylation, or forming a dimer as a repressor (Chapman-Smith and Cronan
1999). Although the function of the C-terminal domain is not clear, it has been found
that some mutations within this domain impaired enzymatic activity. It is thought that
the C-terminal domain is required for catalytic activity and facilitates interaction of ATP
and the BCCP (Chapman-Smith, Mulhern et al. 2001). Regarding protein substrates for
E.coli BPL, it has been shown that there is a conserved peptide sequence (MKM) among
different identified protein substrates and the middle lysine is the specific residue for
formation of the amide bond with biotin. In addition, a 14 amino acid peptide, biotin
acceptor protein (BAP) has been isolated from a peptide library (Schatz 1993). This
small peptide tag has been widely used in protein labeling or purification.
Besides applying streptavidin:biotin orthogonal pair for labeling, Ting’s group has
shown that biotin ligases could be good enzymes to perform site-specific labeling on
proteins with biotin analogs. Labeling proteins with biotin analogs would help provide
an additional functional group to further expand the chemical reaction availability on
proteins (Slavoff, Chen et al. 2008). They assayed biotin ligases from several species and
found desthiobiotin azid and cis-propargyl biotin could be labeled on a short peptide by
P. horikoshii BPL and yeast BPL respectively. In addition, they evolved a new peptide
which could only be specifically recognized by yeast BPL and form a new orthogonal
pair, yeast BAP:yeast BPL. (Chen, Choi et al. 2007). These two orthogonal pairs, yeast
BAP:yeast BPL and E.coli BAP:E.coli BPL orthogonal pairs have been shown to label
different fluorescences on surface. Furthermore, they evolved E.coli lipoic acid ligase (a
homolog of E.coli biotin protein ligase) which has similar ligation reaction to BPL.

26
Engineered E.coli lipoic acid ligase is capable of recognizing 7-hydroxycoumarin and can
be used as a site-specific fluorophore ligase (Uttamapinant, White et al. 2010).

27
REFERENCES
Agresti, J.J., Kelly, B.T., Jaschke, A., and Griffiths, A.D. (2005). Selection of ribozymes
that catalyse multiple-turnover Diels-Alder cycloadditions by using in vitro
compartmentalization. Proc Natl Acad Sci U S A 102, 16170-16175.
Aharoni, A., Amitai, G., Bernath, K., Magdassi, S., and Tawfik, D.S. (2005). High-
throughput screening of enzyme libraries: thiolactonases evolved by fluorescence-
activated sorting of single cells in emulsion compartments. Chem Biol 12, 1281-
1289.
Amitai, G., Gupta, R.D., and Tawfik, D.S. (2007). Latent evolutionary potentials under
the neutral mutational drift of an enzyme. HFSP J 1, 67-78.
Anderson, M.E. (1998). Glutathione: an overview of biosynthesis and modulation. Chem
Biol Interact 111-112, 1-14.
Bernath, K., Hai, M., Mastrobattista, E., Griffiths, A.D., Magdassi, S., and Tawfik, D.S.
(2004). In vitro compartmentalization by double emulsions: sorting and gene
enrichment by fluorescence activated cell sorting. Anal Biochem 325, 151-157.
Bertschinger, J., and Neri, D. (2004). Covalent DNA display as a novel tool for directed
evolution of proteins in vitro. Protein Eng Des Sel 17, 699-707.
Bloom, J.D., and Arnold, F.H. (2009). In the light of directed evolution: pathways of
adaptive protein evolution. Proc Natl Acad Sci U S A 106 Suppl 1, 9995-10000.
Bochkov, Y.A., and Palmenberg, A.C. (2006). Translational efficiency of EMCV IRES
in bicistronic vectors is dependent upon IRES sequence and gene location.
Biotechniques 41, 283-284, 286, 288 passim.
Bowles, T.L., Kim, R., Galante, J., Parsons, C.M., Virudachalam, S., Kung, H.J., and
Bold, R.J. (2008). Pancreatic cancer cell lines deficient in argininosuccinate
synthetase are sensitive to arginine deprivation by arginine deiminase. Int J
Cancer 123, 1950-1955.
Breillout, F., Antoine, E., and Poupon, M.F. (1990). Methionine dependency of
malignant tumors: a possible approach for therapy. J Natl Cancer Inst 82, 1628-
1632.
Breillout, F., Hadida, F., Echinard-Garin, P., Lascaux, V., and Poupon, M.F. (1987).
Decreased rat rhabdomyosarcoma pulmonary metastases in response to a low
methionine diet. Anticancer Res 7, 861-867.
Cantor, J.R., Panayiotou, V., Agnello, G., Georgiou, G., and Stone, E.M. (2012).
Engineering reduced-immunogenicity enzymes for amino acid depletion therapy
in cancer. Methods Enzymol 502, 291-319.

28
Cellarier, E., Durando, X., Vasson, M.P., Farges, M.C., Demiden, A., Maurizis, J.C.,
Madelmont, J.C., and Chollet, P. (2003). Methionine dependency and cancer
treatment. Cancer Treat Rev 29, 489-499.
Chapman-Smith, A., and Cronan, J.E., Jr. (1999a). The enzymatic biotinylation of
proteins: a post-translational modification of exceptional specificity. Trends
Biochem Sci 24, 359-363.
Chapman-Smith, A., and Cronan, J.E., Jr. (1999b). Molecular biology of biotin
attachment to proteins. J Nutr 129, 477S-484S.
Chapman-Smith, A., Mulhern, T.D., Whelan, F., Cronan, J.E., Jr., and Wallace, J.C.
(2001). The C-terminal domain of biotin protein ligase from E. coli is required for
catalytic activity. Protein Sci 10, 2608-2617.
Chen, I., Choi, Y.A., and Ting, A.Y. (2007). Phage display evolution of a peptide
substrate for yeast biotin ligase and application to two-color quantum dot labeling
of cell surface proteins. J Am Chem Soc 129, 6619-6625.
Chen, Y., Mandic, J., and Varani, G. (2008). Cell-free selection of RNA-binding proteins
using in vitro compartmentalization. Nucleic Acids Res 36, e128.
Cohen, H.M., Tawfik, D.S., and Griffiths, A.D. (2004). Altering the sequence specificity
of HaeIII methyltransferase by directed evolution using in vitro
compartmentalization. Protein Eng Des Sel 17, 3-11.
Cooney, D.A., and Handschumacher, R.E. (1970). L-asparaginase and L-asparagine
metabolism. Annu Rev Pharmacol 10, 421-440.
Davidson, E.A., Dlugosz, P.J., Levy, M., and Ellington, A.D. (2009). Directed evolution
of proteins in vitro using compartmentalization in emulsions. Curr Protoc Mol
Biol Chapter 24, Unit 24 26.
Doi, N., Kumadaki, S., Oishi, Y., Matsumura, N., and Yanagawa, H. (2004). In vitro
selection of restriction endonucleases by in vitro compartmentalization. Nucleic
Acids Res 32, e95.
Doi, N., and Yanagawa, H. (1999). STABLE: protein-DNA fusion system for screening
of combinatorial protein libraries in vitro. FEBS Lett 457, 227-230.
Ensor, C.M., Holtsberg, F.W., Bomalaski, J.S., and Clark, M.A. (2002). Pegylated
arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and
hepatocellular carcinomas in vitro and in vivo. Cancer Res 62, 5443-5450.
Fen, C.X., Coomber, D.W., Lane, D.P., and Ghadessy, F.J. (2007). Directed evolution of
p53 variants with altered DNA-binding specificities by in vitro
compartmentalization. J Mol Biol 371, 1238-1248.

29
Ghadessy, F.J., and Holliger, P. (2004). A novel emulsion mixture for in vitro
compartmentalization of transcription and translation in the rabbit reticulocyte
system. Protein Eng Des Sel 17, 201-204.
Ghadessy, F.J., Ong, J.L., and Holliger, P. (2001). Directed evolution of polymerase
function by compartmentalized self-replication. Proc Natl Acad Sci U S A 98,
4552-4557.
Ghadessy, F.J., Ramsay, N., Boudsocq, F., Loakes, D., Brown, A., Iwai, S., Vaisman, A.,
Woodgate, R., and Holliger, P. (2004). Generic expansion of the substrate
spectrum of a DNA polymerase by directed evolution. Nat Biotechnol 22, 755-
759.
Gong, H., Zolzer, F., von Recklinghausen, G., Havers, W., and Schweigerer, L. (2000).
Arginine deiminase inhibits proliferation of human leukemia cells more potently
than asparaginase by inducing cell cycle arrest and apoptosis. Leukemia 14, 826-
829.
Goseki, N., Yamazaki, S., Shimojyu, K., Kando, F., Maruyama, M., Endo, M., Koike,
M., and Takahashi, H. (1995). Synergistic effect of methionine-depleting total
parenteral nutrition with 5-fluorouracil on human gastric cancer: a randomized,
prospective clinical trial. Jpn J Cancer Res 86, 484-489.
Griffiths, A.D., and Tawfik, D.S. (2000). Man-made enzymes--from design to in vitro
compartmentalisation. Curr Opin Biotechnol 11, 338-353.
Griffiths, A.D., and Tawfik, D.S. (2003). Directed evolution of an extremely fast
phosphotriesterase by in vitro compartmentalization. EMBO J 22, 24-35.
Guo, H., Lishko, V.K., Herrera, H., Groce, A., Kubota, T., and Hoffman, R.M. (1993a).
Therapeutic tumor-specific cell cycle block induced by methionine starvation in
vivo. Cancer Res 53, 5676-5679.
Guo, H.Y., Herrera, H., Groce, A., and Hoffman, R.M. (1993b). Expression of the
biochemical defect of methionine dependence in fresh patient tumors in primary
histoculture. Cancer Res 53, 2479-2483.
Gupta, R.D., Goldsmith, M., Ashani, Y., Simo, Y., Mullokandov, G., Bar, H., Ben-David,
M., Leader, H., Margalit, R., Silman, I., et al. (2011). Directed evolution of
hydrolases for prevention of G-type nerve agent intoxication. Nat Chem Biol 7,
120-125.
Gupta, R.D., and Tawfik, D.S. (2008). Directed enzyme evolution via small and effective
neutral drift libraries. Nat Methods 5, 939-942.
Hanes, J., and Pluckthun, A. (1997). In vitro selection and evolution of functional
proteins by using ribosome display. Proc Natl Acad Sci U S A 94, 4937-4942.
Harris, J.M., and Chess, R.B. (2003). Effect of pegylation on pharmaceuticals. Nat Rev
Drug Discov 2, 214-221.

30
Hernandez, C.P., Morrow, K., Lopez-Barcons, L.A., Zabaleta, J., Sierra, R., Velasco, C.,
Cole, J., and Rodriguez, P.C. (2010). Pegylated arginase I: a potential therapeutic
approach in T-ALL. Blood 115, 5214-5221.
Hoffman, R.M. (1985). Altered methionine metabolism and transmethylation in cancer.
Anticancer Res 5, 1-30.
Keefe, A.D., and Szostak, J.W. (2001). Functional proteins from a random-sequence
library. Nature 410, 715-718.
Kelly, B.T., and Griffiths, A.D. (2007). Selective gene amplification. Protein Eng Des Sel
20, 577-581.
Kintses, B., van Vliet, L.D., Devenish, S.R., and Hollfelder, F. (2010). Microfluidic
droplets: new integrated workflows for biological experiments. Curr Opin Chem
Biol 14, 548-555.
Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H.,
and Mori, H. (2005). Complete set of ORF clones of Escherichia coli ASKA
library (a complete set of E. coli K-12 ORF archive): unique resources for
biological research. DNA Res 12, 291-299.
Kokkinakis, D.M., Hoffman, R.M., Frenkel, E.P., Wick, J.B., Han, Q., Xu, M., Tan, Y.,
and Schold, S.C. (2001). Synergy between methionine stress and chemotherapy in
the treatment of brain tumor xenografts in athymic mice. Cancer Res 61, 4017-
4023.
Kroemer, G., and Pouyssegur, J. (2008). Tumor cell metabolism: cancer's Achilles' heel.
Cancer Cell 13, 472-482.
Levy, M., and Ellington, A.D. (2008). Directed evolution of streptavidin variants using in
vitro compartmentalization. Chem Biol 15, 979-989.
Levy, M., Griswold, K.E., and Ellington, A.D. (2005). Direct selection of trans-acting
ligase ribozymes by in vitro compartmentalization. RNA 11, 1555-1562.
Lipovsek, D., and Pluckthun, A. (2004). In-vitro protein evolution by ribosome display
and mRNA display. J Immunol Methods 290, 51-67.
Loakes, D., Gallego, J., Pinheiro, V.B., Kool, E.T., and Holliger, P. (2009). Evolving a
polymerase for hydrophobic base analogues. J Am Chem Soc 131, 14827-14837.
Lorenzi, P.L., Llamas, J., Gunsior, M., Ozbun, L., Reinhold, W.C., Varma, S., Ji, H.,
Kim, H., Hutchinson, A.A., Kohn, E.C., et al. (2008). Asparagine synthetase is a
predictive biomarker of L-asparaginase activity in ovarian cancer cell lines. Mol
Cancer Ther 7, 3123-3128.
Margulies, M., Egholm, M., Altman, W.E., Attiya, S., Bader, J.S., Bemben, L.A., Berka,
J., Braverman, M.S., Chen, Y.J., Chen, Z., et al. (2005). Genome sequencing in
microfabricated high-density picolitre reactors. Nature 437, 376-380.

31
Mastrobattista, E., Taly, V., Chanudet, E., Treacy, P., Kelly, B.T., and Griffiths, A.D.
(2005). High-throughput screening of enzyme libraries: in vitro evolution of a
beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem
Biol 12, 1291-1300.
Miller, O.J., Bernath, K., Agresti, J.J., Amitai, G., Kelly, B.T., Mastrobattista, E., Taly,
V., Magdassi, S., Tawfik, D.S., and Griffiths, A.D. (2006). Directed evolution by
in vitro compartmentalization. Nat Methods 3, 561-570.
Odegrip, R., Coomber, D., Eldridge, B., Hederer, R., Kuhlman, P.A., Ullman, C.,
FitzGerald, K., and McGregor, D. (2004). CIS display: In vitro selection of
peptides from libraries of protein-DNA complexes. Proc Natl Acad Sci U S A
101, 2806-2810.
Ong, J.L., Loakes, D., Jaroslawski, S., Too, K., and Holliger, P. (2006). Directed
evolution of DNA polymerase, RNA polymerase and reverse transcriptase activity
in a single polypeptide. J Mol Biol 361, 537-550.
Parent, K.N., Ranaghan, M.J., and Teschke, C.M. (2004). A second-site suppressor of a
folding defect functions via interactions with a chaperone network to improve
folding and assembly in vivo. Mol Microbiol 54, 1036-1050.
Pavillard, V., Nicolaou, A., Double, J.A., and Phillips, R.M. (2006). Methionine
dependence of tumours: a biochemical strategy for optimizing paclitaxel
chemosensitivity in vitro. Biochem Pharmacol 71, 772-778.
Poirson-Bichat, F., Goncalves, R.A., Miccoli, L., Dutrillaux, B., and Poupon, M.F.
(2000). Methionine depletion enhances the antitumoral efficacy of cytotoxic
agents in drug-resistant human tumor xenografts. Clin Cancer Res 6, 643-653.
Poirson-Bichat, F., Gonfalone, G., Bras-Goncalves, R.A., Dutrillaux, B., and Poupon,
M.F. (1997). Growth of methionine-dependent human prostate cancer (PC-3) is
inhibited by ethionine combined with methionine starvation. Br J Cancer 75,
1605-1612.
Ramsay, N., Jemth, A.S., Brown, A., Crampton, N., Dear, P., and Holliger, P. (2010).
CyDNA: synthesis and replication of highly Cy-dye substituted DNA by an
evolved polymerase. J Am Chem Soc 132, 5096-5104.
Reiersen, H., Lobersli, I., Loset, G.A., Hvattum, E., Simonsen, B., Stacy, J.E., McGregor,
D., Fitzgerald, K., Welschof, M., Brekke, O.H., et al. (2005). Covalent antibody
display--an in vitro antibody-DNA library selection system. Nucleic Acids Res
33, e10.
Roberts, R.W., and Szostak, J.W. (1997). RNA-peptide fusions for the in vitro selection
of peptides and proteins. Proc Natl Acad Sci U S A 94, 12297-12302.
Romero, P.A., and Arnold, F.H. (2009). Exploring protein fitness landscapes by directed
evolution. Nat Rev Mol Cell Biol 10, 866-876.

32
Schatz, P.J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-
modifying enzyme: a 13 residue consensus peptide specifies biotinylation in
Escherichia coli. Biotechnology (N Y) 11, 1138-1143.
Sepp, A., and Choo, Y. (2005). Cell-free selection of zinc finger DNA-binding proteins
using in vitro compartmentalization. J Mol Biol 354, 212-219.
Shimizu, Y., Kanamori, T., and Ueda, T. (2005). Protein synthesis by pure translation
systems. Methods 36, 299-304.
Sitaraman, K., Esposito, D., Klarmann, G., Le Grice, S.F., Hartley, J.L., and Chatterjee,
D.K. (2004). A novel cell-free protein synthesis system. J Biotechnol 110, 257-
263.
Slavoff, S.A., Chen, I., Choi, Y.A., and Ting, A.Y. (2008). Expanding the substrate
tolerance of biotin ligase through exploration of enzymes from diverse species. J
Am Chem Soc 130, 1160-1162.
Stapleton, J.A., and Swartz, J.R. (2010). Development of an in vitro
compartmentalization screen for high-throughput directed evolution of [FeFe]
hydrogenases. PLoS One 5, e15275.
Stein, V., Sielaff, I., Johnsson, K., and Hollfelder, F. (2007). A covalent chemical
genotype-phenotype linkage for in vitro protein evolution. Chembiochem 8, 2191-
2194.
Stone, E., Paley, O., Hu, J., Ekerdt, B., Cheung, N.K., and Georgiou, G. (2012). De novo
engineering of a human cystathionine-gamma-lyase for systemic (L)-Methionine
depletion cancer therapy. ACS Chem Biol 7, 1822-1829.
Takei, Y., Kadomatsu, K., Itoh, H., Sato, W., Nakazawa, K., Kubota, S., and Muramatsu,
T. (2002). 5'-,3'-inverted thymidine-modified antisense oligodeoxynucleotide
targeting midkine. Its design and application for cancer therapy. J Biol Chem 277,
23800-23806.
Tan, Y., Xu, M., Guo, H., Sun, X., Kubota, T., and Hoffman, R.M. (1996a). Anticancer
efficacy of methioninase in vivo. Anticancer Res 16, 3931-3936.
Tan, Y., Xu, M., and Hoffman, R.M. (2010). Broad selective efficacy of recombinant
methioninase and polyethylene glycol-modified recombinant methioninase on
cancer cells In Vitro. Anticancer Res 30, 1041-1046.
Tan, Y., Zavala, J., Sr., Han, Q., Xu, M., Sun, X., Tan, X., Magana, R., Geller, J., and
Hoffman, R.M. (1997). Recombinant methioninase infusion reduces the
biochemical endpoint of serum methionine with minimal toxicity in high-stage
cancer patients. Anticancer Res 17, 3857-3860.
Tan, Y., Zavala, J., Sr., Xu, M., Zavala, J., Jr., and Hoffman, R.M. (1996b). Serum
methionine depletion without side effects by methioninase in metastatic breast
cancer patients. Anticancer Res 16, 3937-3942.

33
Tawfik, D.S., and Griffiths, A.D. (1998). Man-made cell-like compartments for
molecular evolution. Nat Biotechnol 16, 652-656.
Thivat, E., Farges, M.C., Bacin, F., D'Incan, M., Mouret-Reynier, M.A., Cellarier, E.,
Madelmont, J.C., Vasson, M.P., Chollet, P., and Durando, X. (2009). Phase II trial
of the association of a methionine-free diet with cystemustine therapy in
melanoma and glioma. Anticancer Res 29, 5235-5240.
Thomas, T., and Thomas, T.J. (2001). Polyamines in cell growth and cell death:
molecular mechanisms and therapeutic applications. Cell Mol Life Sci 58, 244-
258.
Tokuriki, N., and Tawfik, D.S. (2009). Chaperonin overexpression promotes genetic
variation and enzyme evolution. Nature 459, 668-673.
Ullman, C.G., Frigotto, L., and Cooley, R.N. (2011). In vitro methods for peptide display
and their applications. Brief Funct Genomics 10, 125-134.
Utada, A.S., Lorenceau, E., Link, D.R., Kaplan, P.D., Stone, H.A., and Weitz, D.A.
(2005). Monodisperse double emulsions generated from a microcapillary device.
Science 308, 537-541.
Uttamapinant, C., White, K.A., Baruah, H., Thompson, S., Fernandez-Suarez, M.,
Puthenveetil, S., and Ting, A.Y. (2010). A fluorophore ligase for site-specific
protein labeling inside living cells. Proc Natl Acad Sci U S A 107, 10914-10919.
Veronese, F.M. (2001). Peptide and protein PEGylation: a review of problems and
solutions. Biomaterials 22, 405-417.
Weaver, L.H., Kwon, K., Beckett, D., and Matthews, B.W. (2001). Corepressor-induced
organization and assembly of the biotin repressor: a model for allosteric activation
of a transcriptional regulator. Proc Natl Acad Sci U S A 98, 6045-6050.
Williams, R., Peisajovich, S.G., Miller, O.J., Magdassi, S., Tawfik, D.S., and Griffiths,
A.D. (2006). Amplification of complex gene libraries by emulsion PCR. Nat
Methods 3, 545-550.
Yang, Z., Wang, J., Yoshioka, T., Li, B., Lu, Q., Li, S., Sun, X., Tan, Y., Yagi, S.,
Frenkel, E.P., et al. (2004). Pharmacokinetics, methionine depletion, and
antigenicity of recombinant methioninase in primates. Clin Cancer Res 10, 2131-
2138.
Yonezawa, M., Doi, N., Kawahashi, Y., Higashinakagawa, T., and Yanagawa, H. (2003).
DNA display for in vitro selection of diverse peptide libraries. Nucleic Acids Res
31, e118.
Zahnd, C., Amstutz, P., and Pluckthun, A. (2007). Ribosome display: selecting and
evolving proteins in vitro that specifically bind to a target. Nat Methods 4, 269-
279.

34
Chapter 2: Engineering human cystathionine gamma-lyase to degrade
methionine for cancer treatment
INTRODUCTION
Methionine gamma-lyase
Methionine gamma-lyase (EC 4.4.1.11, also called methioninase or MGL) is a
pyridoxal 5’-phosphate (PLP) dependent enzyme and belongs to the gamma-lyase family
which can break a carbon-sulfur bond at the gamma carbon. The substrate for this
enzyme is L-methionine which is degraded through alpha, gamma-elimination to produce
alpha-ketobutyrate, methane thiol and NH3 as products (Figure 2-1). In nature, it is found
in bacteria, plants, fungi and protozoa but not in mammals. It is known that bacteria and
protozoa use MGL to degrade methionine for ATP generation (Inoue, Inagaki et al. 1997;
Ragsdale 2003; Sato and Nozaki 2009). Importantly, MGL has shown promise for use as
an anti-cancer drug. Depletion of methionine in mice with xenografted tumors inhibited
tumor growth (Tan, Sun et al. 1999; Poirson-Bichat, Goncalves et al. 2000; Kokkinakis,
Hoffman et al. 2001) and several clinical trials reported that a methionine-free diet shown
an improved outcome of cancer treatment (Goseki, Yamazaki et al. 1995; Durando,
Thivat et al. 2008; Thivat, Farges et al. 2009). However, bacterial MGL would be
expected to induce strong immune responses in mammals. In fact, it has been shown to
cause anaphylactic shock and death in primates when bacterial MGL was re-challenged
(Yang, Wang et al. 2004). Therefore, it would be better to use a humanized MGL or a
human enzyme which is able to break down methionine for methionine depletion in
cancer treatment.

35
Figure 2-1: Enzyme reaction of methionine gamma-lyase. Methionine gamma-lyase
is a PLP-dependent enzyme and breaks down L-methionine to methanethiol,
NH3 and 2-oxobutanoate (alpha ketobutyrate).
Cystathionine gamma-lyase
Based on this idea, a paralog of MGL, human cystathionine gamma-lyase
(EC 4.4.1.1, also called cystathionase or hCGL) was chosen by Dr. Stone and Olga Paley
(Georgiou Lab, UT Austin) for engineering to acquire methionine-breakdown activity
(Stone, Paley et al. 2012). hCGL shares around 60% amino acid similarity with
Pseudomonas putida MGL and also belongs to the gamma-lyase family. hCGL is PLP-
dependent enzyme that can break the carbon-sulfur bond of cystathionine to produce
cysteine, NH3 and alpha-ketobutyrate (Figure 2-2). The catalytic mechanism of MGL
and hCGL is the same. Moreover, the substrate, cystathionine is similar and larger than
methionine. Thus hCGL was chosen to be engineered in use of methionine as substrates.
The physiological function of hCGL is involved in a transsulfuration pathway which
converts methionine to cysteine (Rao, Drake et al. 1990; Cavuoto and Fenech 2012) and
aside from diet, it is the only way for humans to synthesize cysteine.

36
Figure 2-2: Enzyme reaction of cystathionine gamma-lyase. Cystathionine gamma-
lyase is a PLP-dependent enzyme and break L-cystathionine to cysteine,
NH3 and 2-oxobutanoate by alpha-gamma elimination.
Dr. Stone and Olga Paley’s work (Stone, Paley et al. 2012):
In order to engineer hCGL to break down methionine, Everett Stone and Olga
Paley did a structural comparison of hCGL (PDB: 3COG) (Sun, Collins et al. 2009) and
Trichomonas vaginalis MGL (tMGL, PDB: 1E5E) and found the side chains of Glu-59,
Arg-119 and Glu-339 are important for the cysteine portion of cystathionione (Figure 2-
3). However, the corresponding positions of tMGL are more hydrophobic amino acids,
Ile-55, Ala-116 and Val-337 (Stone, Paley et al. 2012). Based on this observation, they
hypothesized these positions are important for substrate specificity. Thus, they
performed a combinatorial pairwise saturation mutagenesis on Glu-59, Arg-119 and Glu-
339 of hCGL. First, they carried out saturation mutagenesis on Arg-119 and Glu-339 and
obtained a variant with R119L and E339V showing activity in breakdown of methionine.
And then, they used this variant as a template and further optimized the positions of 59
and 119 (fix the E339V) by another round of saturation mutagenesis. Finally, they
obtained a variant, hCGL-NLV (E59N, R119L and E339V) which displayed around 1%
activity in the breakdown of methionine compared to Pseudomonas putida MGL
(pMGL). The kcat and KM of hCGL-NLV are 7.9 s-1
(kcat of pMGL: 20 s-1
) and 14 mM
(KM of pMGL: 0.34 mM) respectively (Table 2-1). Moreover, the engineered hCGL-

37
NLV is more stable than pMGL in pooled human serum at 37oC. The half-lives of hCG-
NLV and pMGL in pooled human serum are t1/2=78 and t1/2=1.9 hours respectively.
Additionally, the PEGylated hCGL-NLV displayed longer t1/2 (30 hours) than
unPEGylated hCGL-NLV (t1/2=1.8 hours) in the blood of injected mice. Importantly,
mice with neuroblastoma xenografts displayed nearly complete cessation of tumor
growth in PEGylated hCGL-NLV treatment (Figure 2-4) (Stone, Paley et al. 2012). In
order to enhance the efficacy for depletion of methionine in practical cancer therapy, a
more active hCGL variant in break of methionine will be needed in the future.
Figure 2-3: Structure comparison of human cystathionine gamma-lyase (PDB:
3COG) and Trichomoas vaginalis (PDB: 1E5E) with inhibitor
propargylglycine (PAG) (Stone, Paley et al. 2012).

38
Table 2-1: Kinetic analysis of Pseudomonas putida methionine gamma-lyase,
human cystathionine gamma-lyase and engineered human cystathionine
gamma-lyase (hCGL-NLV) (Stone, Paley et al. 2012).
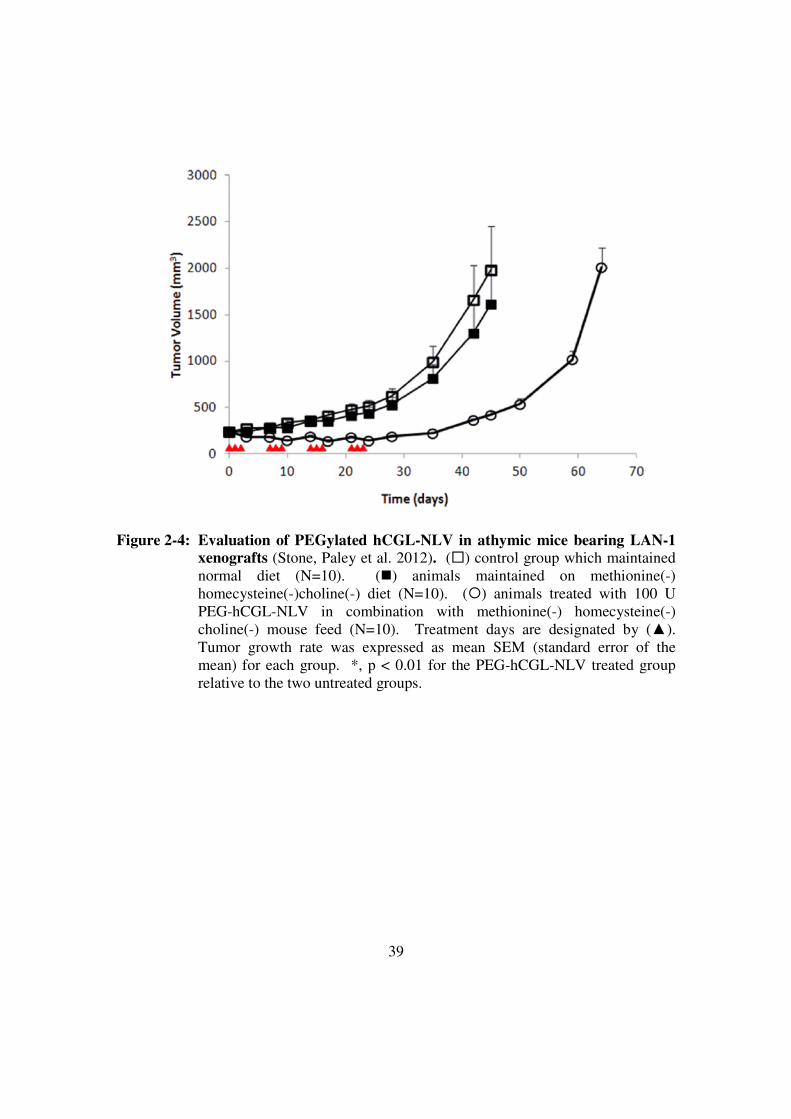
39
Figure 2-4: Evaluation of PEGylated hCGL-NLV in athymic mice bearing LAN-1
xenografts (Stone, Paley et al. 2012). (�) control group which maintained
normal diet (N=10). (�) animals maintained on methionine(-)
homecysteine(-)choline(-) diet (N=10). (�) animals treated with 100 U
PEG-hCGL-NLV in combination with methionine(-) homecysteine(-)
choline(-) mouse feed (N=10). Treatment days are designated by (▲).
Tumor growth rate was expressed as mean SEM (standard error of the
mean) for each group. *, p < 0.01 for the PEG-hCGL-NLV treated group
relative to the two untreated groups.

40
RESULTS
Phylogenetic analysis of MGLs and CGLs:
Besides random mutagenesis, structural comparison, and DNA shuffling,
phylogenetic analysis is an established method of enzyme engineering. The idea is that
enzymes in the same family derive from the same ancestral protein and share similar
protein scaffolds. Evolving to different substrate specificities, these enzymes display
unique conservations at specific amino acid positions and can be further categorized into
different sub-families. For example, both methionine gamma-lyases (MGLs) and
cystathionine gamma-lyases (CGLs) are in the same gamma-lyase family and can be
further categorized into two sub-families showing unique conservations.
MGLs and CGLs are paralogs and both belong to the gamma-lyase family of
proteins. They use the same catalytic mechanism to break the carbon-sulfur bond on the
gamma-carbon of their substrates. The idea of phylogenetic analysis is that “the
differences of conservation in sequence alignment of paralogs may result in the
differences of substrate specificity” (Cole and Gaucher 2011). Theoretically, there are
four situations in the phylogenetic analysis of MGLs and CGLs (Figure 2-5, hypothetical
situations). For Situation 1, the amino acid (S: serine, no difference between CGLs and
MGLs) should be responsible for the properties of gamma-lyase and may be very
important to the catalytic mechanism of gamma lyase. For Situation 2 (CGLs: E, MGLs:
V), 3 (CGLs: S) and 4 (MGLs: V), the conserved amino acids should associate with the
individual sub-family properties. For example, in Situation 3, S (serine) may be
important for substrate specificity of CGLs and V (valine) may be important to the
substrate specificity of MGLs. Substituting S (CGLs: S) with V (MGLs: V), we may be
able to shift the substrate specificity from CGL to MGL (CGLs and MGLs). Since our
goal is to engineer hCGL to acquire the activity of MGLs, we should keep the amino acid

41
(S) as in Situation 1. In addition, the S (CGLs: S) should be substituted with V (MGLs:
V) in the situation 2. In Situation 3, we should use any amino acids of MGLs (A, C, D,
E, T, M, P or Y) to replace S of CGLs and use V to replace amino acid of the CGLs (D,
E, T, M or A) in the Situation 4.
Figure 2-5: Phylogenetic analysis of cystathionine gamma-lyases and methionine
gamma-lyases. (Left) Theoretically, paralogs are from the same precursor
protein. In evolution of different paralogs, they may share the same scaffold
and have different conservations for substrate specificity. (Right) There are
five possible situations for comparison of paralogs.
Pilot experiment (test 11 selected positions from phylogenetic analysis)
The goal is to engineer hCGL to break down methionine. However there is no
strong evidence that phylogenetic analysis will work in this project. Therefore, we
performed a pilot experiment to test the idea.

42
From the solved structure (PDB: 2NMP), hCGL is a tetramer protein with the
active site composed of the C-terminal domain of one monomer and the partial N-
terminal domain of an adjacent monomer. Since the active site is important for substrate
specificity thus we focused on the phylogenetic analysis of the C-terminal domain.
Several eukaryotic cystathionine gamma-lyases (eCGLs: human, cattle, rat, mouse,
zebra) were chosen to compare with several bacterial methionine gamma-lyases (bMGLs:
Ruegeria, Pseudomonas, Ferrimonas, Porphyromonas, Shewanella and Bacillus).
Geneious (sequence analysis software) was used for sequence alignment (Figure 2-6).
K268R, F270C, G273A, C307M, T311G, V314I, G343D, F344A, L350H, I 353S and
V359Y were chosen for site-directed mutagenesis on wild-type hCGL and hCGL-NLV
backbone. These chosen substitutions matched with Situation 2 (Figure 2-5).

43
Figure 2-6: Phylogenetic analysis of the carboxyl terminals of 5 eukaryotic
cystathionine gamma-lyases and 6 bacterial methionine gamma-lyases.
The 11 red arrows indicated conservations chosen as candidates for shifting
substrate specificity.

44
Initially, 11 hCGL mutants (11 aforementioned substitutions) on the wild-type
hCGL backbone were individually screened on a 96-well plate (Figure 2-8, Material and
Method). Unfortunately, none of the 11 mutants showed better activity compared to
hCGL-NLV. However, 2 of the 11 hCGL mutants (11 aforementioned substitutions) on
the hCGL-NLV backbone showed around 2-fold better activity in the breakdown of
methionine compared to hCGL-NLV. These 11 mutants (on hCGL-NLV backbone) were
further purified by Ni-NTA chromatography and characterized to determine the kcat and
KM of methionine breakdown. Interestingly, hCGL-NLV+G and hCGL-NLV+S (T311G,
I353S on hCGL-NLV) both showed around 2-fold better kcat/KM (1.1 and 0.96 s-1
mM-1
)
compared hCGL-NLV (0.56 s-1 mM-1) (Table 2-2). The kcat of hCGL-NLV+G, hCGL-
NLV+S, and hCGL-NLV were 3.1, 4.4, and 7.9 s-1
respectively and KM of them were 2.8,
4.6, and 14 mM respectively (Figure 2-7). Apparently, the KM of hCGL-NLV+G and
hCGL-NLV+S were much improved (3 to 5-fold better) in the breakdown of methionine
compared to hCGL-NLV. Mapping T311 and I353 on the solved structure of wild-type
hCGL (PDB: 2NMP), only I353 is around the active site (similar to N59, L119 and L339)
and T311 is further away from the active site. This indicates that I353S may cooperate
with N59, L119, and L339 to improve the KM in the breakdown of methionine. It is
difficult to explain how T311G affects the KM of the hCGL-NLV+G without solved
structure. hCGL-NLV+R, hCGL-NLV+C and hCGL-NLV+I (K268R, F270C or V314I
on hCGL-NLV backbone) showed similar kcat/KM in the breakdown of methionine
compared to hCGL-NLV.
Furthermore, T311G and I353S were combined on hCGL-NLV backbone to test
for cooperativity. Kinetic assays showed hCGL-NLV+GS (T311G and I353S on hCGL-
NLV) displayed 2.2-fold better kcat/KM value compared to hCGL-NLV.
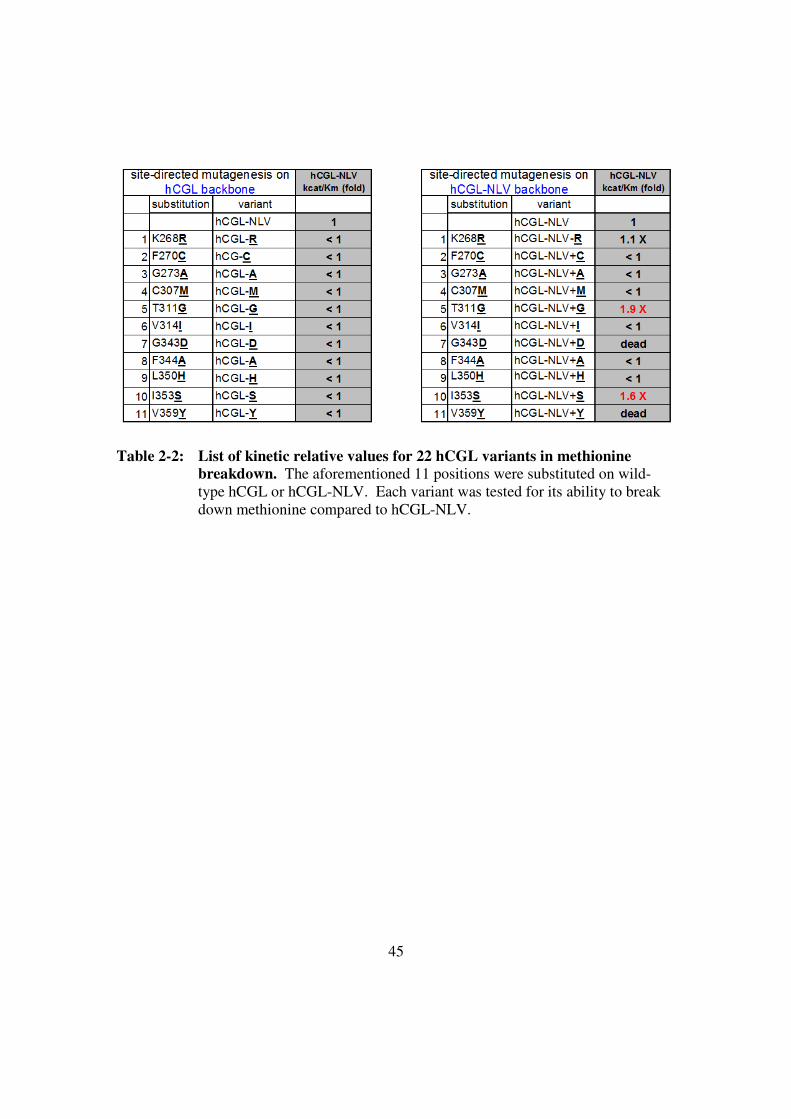
45
Table 2-2: List of kinetic relative values for 22 hCGL variants in methionine
breakdown. The aforementioned 11 positions were substituted on wild-
type hCGL or hCGL-NLV. Each variant was tested for its ability to break
down methionine compared to hCGL-NLV.
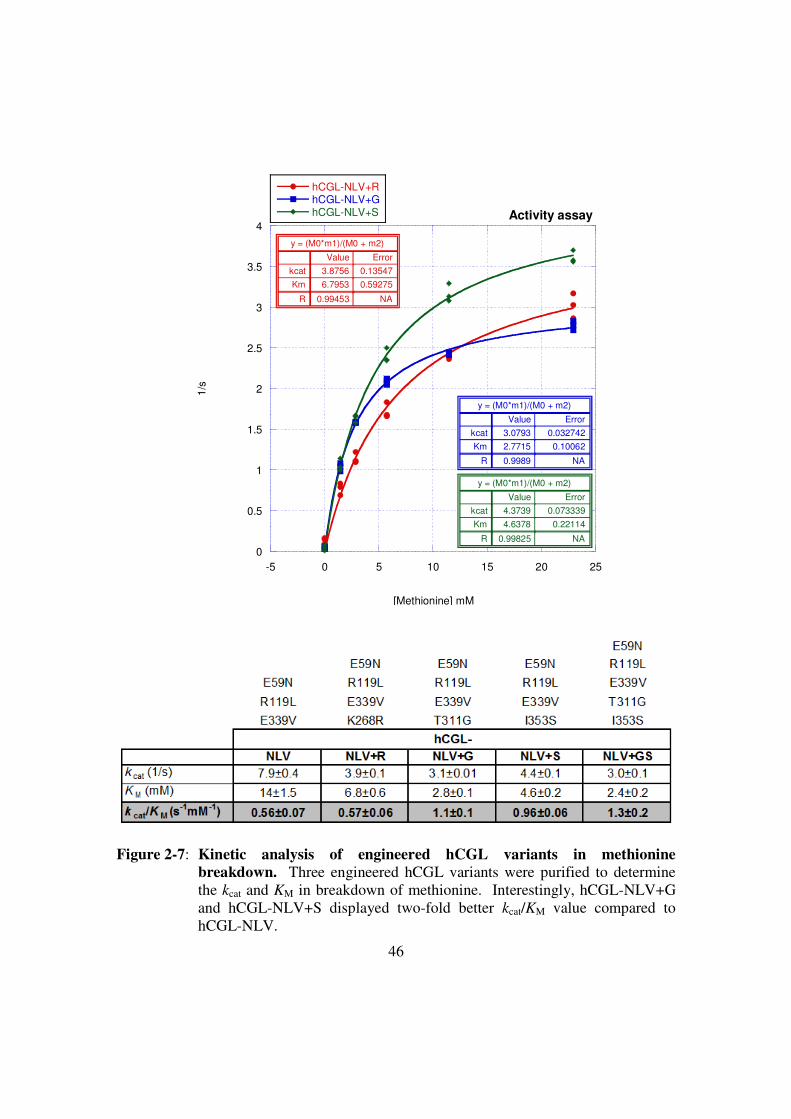
46
0
0.5
1
1.5
2
2.5
3
3.5
4
-5 0 5 10 15 20 25
Activity assay
hCGL-NLV+RhCGL-NLV+GhCGL-NLV+S
1/s
[Methionine] mM
y = (M0*m1)/(M0 + m2)
ErrorValue
0.135473.8756kcat
0.592756.7953Km
NA0.99453R
y = (M0*m1)/(M0 + m2)
ErrorValue
0.0327423.0793kcat
0.100622.7715Km
NA0.9989R
y = (M0*m1)/(M0 + m2)
ErrorValue
0.0733394.3739kcat
0.221144.6378Km
NA0.99825R
Figure 2-7: Kinetic analysis of engineered hCGL variants in methionine
breakdown. Three engineered hCGL variants were purified to determine
the kcat and KM in breakdown of methionine. Interestingly, hCGL-NLV+G
and hCGL-NLV+S displayed two-fold better kcat/KM value compared to
hCGL-NLV.

47
Combinatorial library (11 positions from phylogenetic analysis)
Based on the pilot experiment, it is very possible some of these chosen candidates
(11 aforementioned substitutions) would benefit each other and need to be present at the
same time for cooperation. Therefore a small combinatorial library was generated based
on the 11 aforementioned candidates. Since we did not have a high-throughput screening
method we were not able to screen a library generated from saturation mutagenesis at all
11 positions (2011
variants). Thus, we designed each position to have two amino acid
options (211
=2048 variants) in the combinatorial library to probe either an amino acid
from wild-type hCGL or an amino acid from the phylogenetic analysis of CGLs and
MGLs. We expected the selected amino acids from the phylogenetic analysis of MGLs
and CGLs to be more potent than other amino acids.
This library, “11p-NLV-I” (mutagenesis at 11 positions, on hCGL-NLV
backbone, library I) was constructed by overlapping PCR and verified by sequencing 20
clones. Only 45% (9 clones) contained full-length coding sequences with the desired
substitutions. Others were truncated (premature stop codons) or variants with insertions
or deletions. Theoretically, there are 2048 variants in this library and we screened forty
96-well plates which contained approximately 4000 clones to attempt full coverage.
(Figure 2-8 and 2-9).
Initially, 34 clones displayed higher activity compared to hCGL-NLV in the
screening. The top 10 of these active clones were chosen for further protein purification
and characterization. However, only one variant, P2A11 (plate 2- row A- column 11)
displayed 2.5-fold higher kcat/KM compared to hCGL-NLV in the breakdown of
methionine. The kcat and KM value of P2A11 are 4.5 s-1
and 3.5 mM respectively (Figure
2-10). Sequencing P2A11 showed it contained K268R, T311G and I353S on hCGL-
NLV backbone. Mapping K268 to the solved structure of hCGL (PDB: 2NMP) it is

48
away from the active site and found near the interface of monomers (hCGL is a tetramer)
(Sun, Collins et al. 2009). Perhaps this mutation allows the monomers to juxtapose in a
way that facilitate a more compliant active site. Still, it is difficult to exactly know how it
affects methionine-breakdown activity without a solved structure.
Figure 2-8: 96-well plate screening process. First, a combinatorial library was
transformed to BL21(DE3) competent cells. Individual colonies were used
to inoculate 96-well plates for small scale culture. Second, cells were then
induced for protein expression by IPTG for 2 hours. Induced cells were
lysed by mild detergent and centrifuged to separate soluble protein from cell
debris. The soluble fraction was incubated with methionine for 10 hours.
Finally, the reaction was mixed with MBTH to determine methionine-
degraded products by Absorbance320. Positive variants were further isolated
and purified for detailed kinetic analysis.

49
Figure 2-9: Combinatorial library screening. Green blocks indicate hCG-NLV and
orange blocks indicate hCGL-NLV+G. Pink blocks indicate background.
Yellow and white blocks represent different hCGL variants. Red numbers
are highlighted hCGL variants which may have higher activity in the
breakdown of methionine. In the library screening, we first isolated 34
candidates and then focus on 10 candidates. These 10 candidates were
isolated and further purified for kinetic analysis in detail. Finally, only one
P2A11 (hCGL-NLV+RGS) displayed higher activity in the breakdown of
methionine compared to hCGL-NLV+GS.

50
-1
0
1
2
3
4
5
-10 0 10 20 30 40 50
Activity assay
hCGL-NLVhCGL-NLV+RGS
1/s
[Methionine] mM
y = (m1*M0)/( m2 + M0)
ErrorValue
0.164036.245m1
0.9462815.243m2
NA0.99727R
y = (m1*M0)/( m2 + M0)
ErrorValue
0.091254.5221m1
0.260643.5395m2
NA0.99412R
Figure 2-10:Kinetic analysis of hCGL-NLV and hCGL-NLV+RGS in methionine
breakdown. The kinetic analysis was carried out at 37oC. The kcat/KM of
hCGL-NLV+RGS is approximately 3-fold better than hCGL-NLV.

51
The results of the two experiments indicate the phylogenetic analysis works for
rationally designing proteins. We obtained hCGL-NLV+RGS (K268R, T311G and
I353S) which displayed 3.2-fold better kcat/KM compared to hCGL-NLV in breakdown of
methionine. In addition, it was noted that only one variant was better than hCGL-NLV in
10 isolated clones from screening. Since protein expression could not be normalized by
this screening approach it is possible the other 9 isolated clones expressed more enzymes
and produced false positive signals.
Comprehensive phylogenetic analysis of CGLs and MGLs
Since the previous experiments showed positive results, a comprehensive
phylogenetic analysis was performed to compare full-length eukaryotic CGLs (eCGLs),
bacterial MGLs (bMGLs) and bacterial CGLs (bCGLs). 16 eukaryotic CGLs, 8 bacterial
CGLs and 18 bacterial MGLs were selected for phylogenetic analysis and 67 amino acid
positions were identified for potential mutations. These 67 amino acid candidates not
only included conservations of Situation 2 but also Situation 3 and 4 in phylogenetic
analysis (Figure 2-5). In order to reduce the potential library to a manageable size,
twenty candidates around the active site were chosen for site-directed mutagenesis:
Q49F, P52A, G53E, H55G, S63L, L91M, A92G, T94S, D112T, G116C, W155Y,
T160A, S218G, M222A, Q240K, N241D, P247L, Y317E, E349Q and A357S. These
substitutions were generated individually on the hCGL-NLV+RGS backbone. In order to
avoid the false positive effect in 96-well plate screening, these 20 variants were directly
purified by Ni-NTA column (20 ml bacterial culture) without pre-screening. Since the
initial slope of kinetic assays indicate the approximate kcat/KM values, these variants were
tested at several low methionine concentrations to determine approximate kcat/KM.

52
Variants with G116C, T160A, or P247L substitutions had no enzymatic activity. Variants
with D112T or Q240K substitution expressed poorly and did not produce enough
enzymes for testing. Additionally, variants with Q49F, P52A, G53E, H55G, A92G,
T94S, W155Y, S218G, M222A, Q240K, N241D, E349Q or A357S substitution
displayed similar or worse kcat/KM values compared to hCGL-NLV+RGS. Variants
containing S63L, L91M or Y317E substitution showed improvement compared to
hCGL-NLV+RGS in breakdown of methionine. These three variants were further
analyzed by detailed kinetic analysis to determine kcat and KM values in breakdown of
methionine (Table 2-3). The kcat of hCGL-NLV+RGS+L and hCGL-NLV+RGS+M
were 6.8 and 5.8 s-1, respectively compared to hCGL-NLV+RGS, (4.6 s-1). The KM of
these two variants are 3.3 and 2.8 mM, respectively which are similar to hCGL-
NLV+RGS (2.4 mM). Both variants had slightly better kcat value and similar KM value
compared to hCGL-NLV+RGS in breakdown of methionine and their kcat/KM values
improved only slightly.
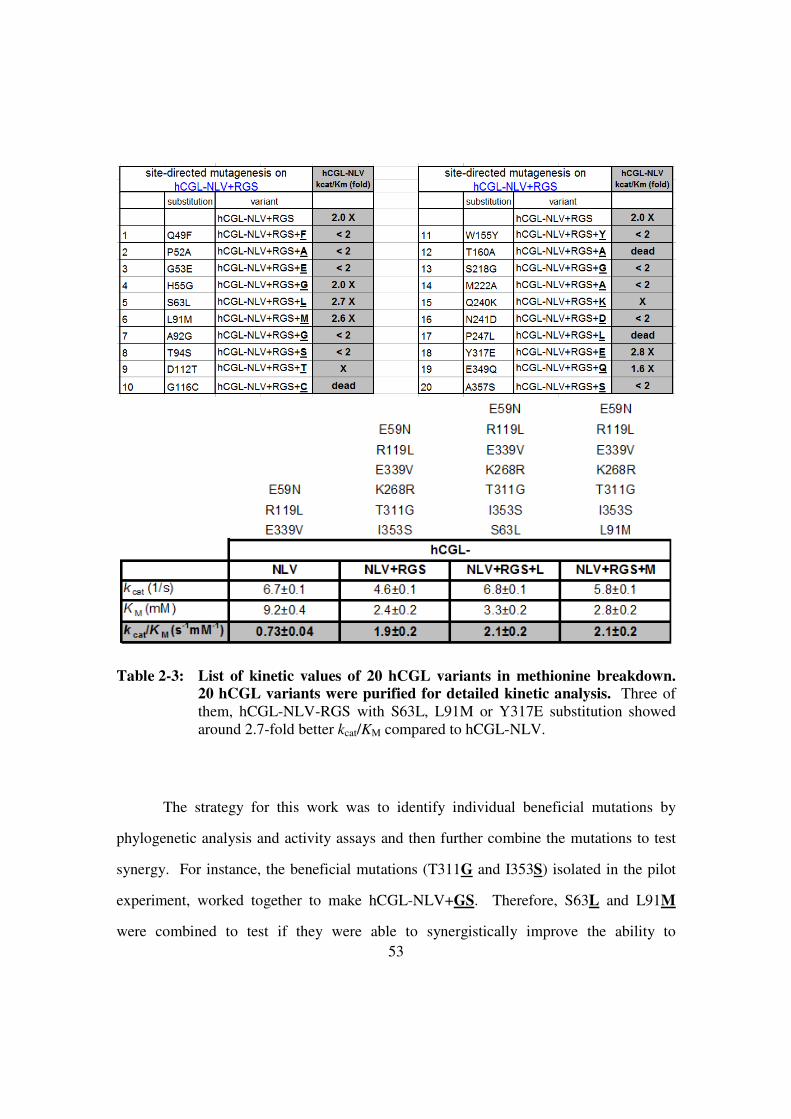
53
Table 2-3: List of kinetic values of 20 hCGL variants in methionine breakdown.
20 hCGL variants were purified for detailed kinetic analysis. Three of
them, hCGL-NLV-RGS with S63L, L91M or Y317E substitution showed
around 2.7-fold better kcat/KM compared to hCGL-NLV.
The strategy for this work was to identify individual beneficial mutations by
phylogenetic analysis and activity assays and then further combine the mutations to test
synergy. For instance, the beneficial mutations (T311G and I353S) isolated in the pilot
experiment, worked together to make hCGL-NLV+GS. Therefore, S63L and L91M
were combined to test if they were able to synergistically improve the ability to

54
methionine breakdown. Surprisingly, hCGL-NLV+RGS+LM did not improve the KM
but significantly changed the kcat value to 2-fold higher compared to hCGL-NLV+RGS in
the breakdown of methionine (Figure 2-11). This variant displayed 5.4-fold better kcat/KM
value compared to the initial hCGL variant, hCGL-NLV. In addition, we further
combined Y317E with hCGL-NLV+RGS+LM. This variant did not show better activity
in the breakdown of methionine. Mapping S63 and L91 on the solved structure of wild-
type hCGL (PDB: 2NMP), these two positions are around active site, indicating S63L
and L91M may work with other active site substitutions such as E59N, R119L, E339V,
T311S.
-2
0
2
4
6
8
-5 0 5 10 15 20
Activity assayhCGL-NLV+RGS+LM
1/s
[Methionine] mM
y = (M0*m1)/(M0+ m2 )
ErrorValue
0.240269.0738m1
0.22692.9115m2
NA0.99451R
Figure 2-11:Kinetic analysis of hCGL-NLV+RGS+LM in methionine breakdown. Since the hCGL-NLV+RGS+L and hCGL-NLV+RGS+M variants showed
higher activity in breakdown of methionine we further combined S63L and
L91M onto hCGL-NLV+RGS. This variant displayed around 5-fold better
kcat/KM value in the breakdown of methionine compared to hCGL-NLV.

55
Compacting all kcat and KM values of isolated variants (Table 2-4), the KM
significantly reduced from 14 mM (hCGL-NLV) to around 3 mM (hCGL-NLV+RGS and
hCGL-NLV+RGS+LM) in use of methionine as substrates. Concurrently, the kcat of
variants dropped from 7.9 s-1 (hCGL-NLV) to 4.6 s-1 (hCGL-NLV+RGS) and then
improved to 9.1 s-1
(hCGL-NLV+RGS+LM). Overall, we further engineered hCGL-
NLV via phylogenetic analysis to yield a better variant, hCGL-NLV+RGS+LM which
displayed around 5% methionine breakdown activity of pMGL (3.1 s-1 mM-1 vs. 59 s-1
mM-1
).

56
Table 2-4: List of kinetic values of hCGL variants in methionine breakdown.

57
Substrate promiscuity
Besides testing the ability to break down methionine, it is also important to test if
hCGL-NLV+RGS+LM is active on other substrates. Is it a methionine-specific enzyme
or a promiscuous enzyme? We tested hCGL-NLV and hCGL-NLV+RGS+LM with L-
methionine, L-cystathionine, L-cysteine and DL-homecysteine (Figure 2-12). Since
hCGL activity is sensitive to changes in salt concentrations these two enzymes were
further purified by size-exclusion chromatography (HiLoad 16/600 Superdex 200 prep
grade, GE Healthcare Life Science) after Ni-NTA column purification. Purified hCGL-
NLV+RGS+LM showed not only 5-fold better kcat/KM value in breakdown of methionine
but also 2-fold better kcat/KM value in use of cysteine compared to hCGL-NLV (7.2 s-
1mM
-1 vs. 2.6 s
-1mM
-1 ) (Table 2-5). Moreover, it displayed 2-fold worse kcat/KM in use
of its original substrate, L-cystathionine, compared to hCGL-NLV (2.1 s-1
mM-1
vs. 1.1
s-1
mM-1
). In use of DL-homocysteine, there was no significant difference between
hCGL-NLV and hCGL-NLV+RGS+LM. In general, hCGL-NLV+RGS+LM seems to be
more like pMGL compared to hCGL-NLV in this engineering process. Additionally,
since L-cysteine and DL-homocysteine are smaller molecules and more similar to L-
methionine than L-cystathionine, it is not surprising that engineered hCGL was active in
use of L-cysteine and DL-homocysteine as in use of methionine (Figure 2-12).
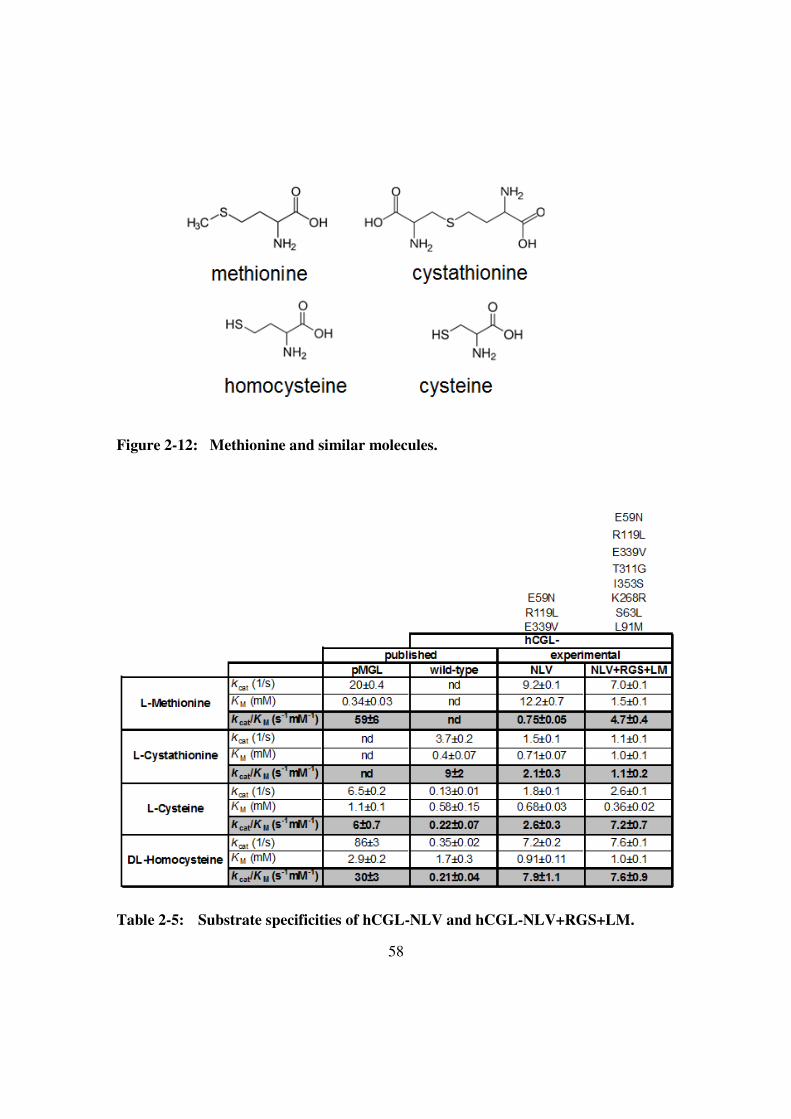
58
Figure 2-12: Methionine and similar molecules.
Table 2-5: Substrate specificities of hCGL-NLV and hCGL-NLV+RGS+LM.

59
Phylogenetic analysis on positions 59, 119 and 339 of hCGL
We further engineered hCGL-NLV based on phylogenetic analysis and found
hCGL-NLV+RGS+LM did improve the activity in breakdown of methionine. We then
questioned if substitutions of E59N, R119L and E339V also matched the phylogenetic
analysis. We revisited positions 59, 119 and 339 which were chosen by structural
analysis and screened from a saturation mutagenized library. Interestingly, we found that
positions 59, 119 and 339 could be potential candidates based on the phylogenetic
analysis (Figure 2-13). The phylogenetic analysis implied isoleucine might be a better
option than asparagine at position 59 and alanine might be more beneficial than leucine at
position 119. Therefore, three variants (hCGL-INV+RGS+LM, hCGL-NAV+RGS+LM
and hCGL-IAV+RGS+LM) were generated to test the activity in methionine breakdown
and surprisingly, hCGL-IAV+RGS+LM showed higher activity in methionine
breakdown compared to hCGL-NLV+RGS+LM. The kcat/KM of hCGL-IAV+RGS+LM
was 5.4 s-1
mM-1
which was 1.5-fold better than the kcat/KM of hCGL-NLV+RGS+LM.
Both hCGL-ILV+RGS+LM and hCGL-NAV+RGS+LM were also better than hCGL-
NLV+RGS+LM though hCGL-IAV+RGS+LM was the best one (Figure 2-14 and Table
2-6).

60
Figure 2-13: Phylogenetic analysis at positions 59, 119 and 339. 16 eukaryotic CGLs,
7 bacterial CGLs and 16 bacterial MGLs were aligned by Geneious
software.
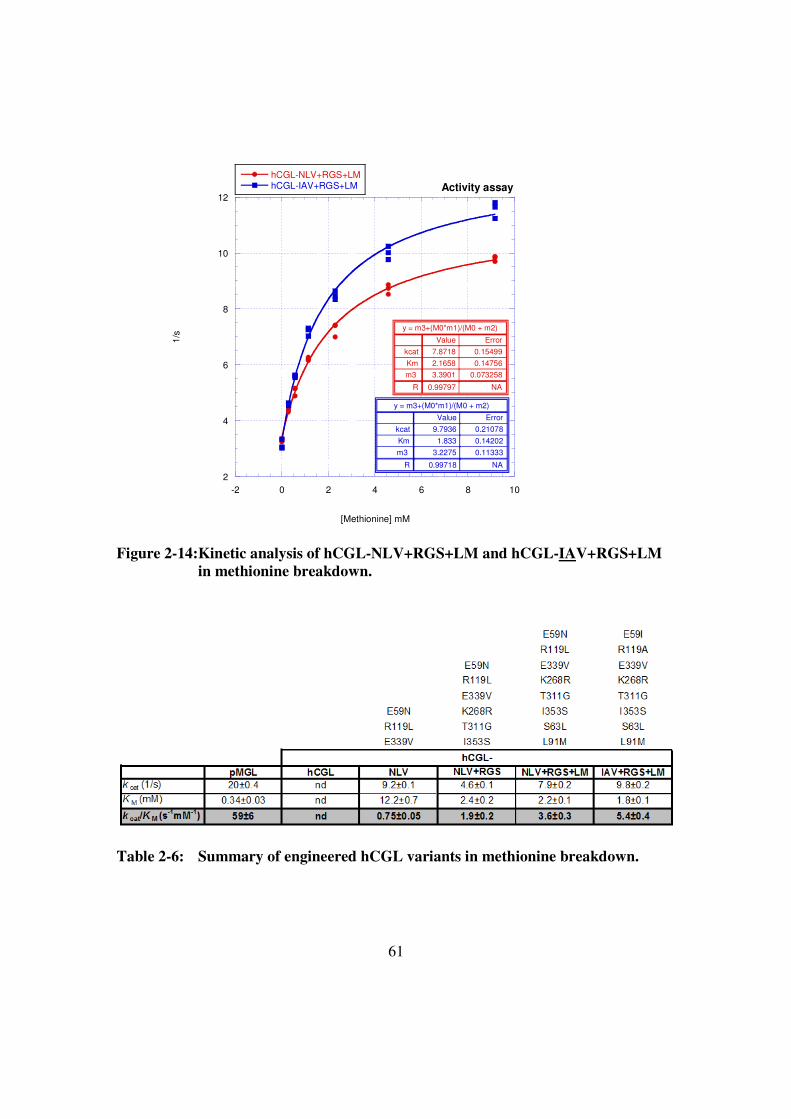
61
2
4
6
8
10
12
-2 0 2 4 6 8 10
Activity assay
hCGL-NLV+RGS+LMhCGL-IAV+RGS+LM
1/s
[Methionine] mM
y = m3+(M0*m1)/(M0 + m2)
ErrorValue
0.154997.8718kcat
0.147562.1658Km
0.0732583.3901m3
NA0.99797R
y = m3+(M0*m1)/(M0 + m2)
ErrorValue
0.210789.7936kcat
0.142021.833Km
0.113333.2275m3
NA0.99718R
Figure 2-14:Kinetic analysis of hCGL-NLV+RGS+LM and hCGL-IAV+RGS+LM
in methionine breakdown.
Table 2-6: Summary of engineered hCGL variants in methionine breakdown.

62
DISCUSSIONS
In past research, Dr. Stone has shown engineered hCGL-NLV displayed 1%
activity in breakdown of methionine compared to pMGL. Moreover, the hCGL variant
was able to inhibit tumor growth of atymic mice with LAN-1 xenografts (Stone, Paley et
al. 2012). In this work, we significantly improved the activity of hCGL variant from 1%
to 7% in breakdown of methionine compared to pMGL. We lowered the KM ten-fold
(from 12.2 mM to 1.8 mM) and restored the kcat (from 9.2 s-1
, 4.5 s-1
to 9.8 s-1
) in
methionine breakdown in the engineering process. Although we only have shown the
improved activity in methionine breakdown in in vitro assays, we are working with others
to perform in vivo analysis for the best hCGL variant (hCGL-IAV+RGS+LM). It will be
very interesting to characterize this variant for stability in pooled serum, IC50, PK and PD
in mice and eventually primates.
Advantages and disadvantages:
Phylogenetic analysis (sequence alignment of paralogs) is a classical method for
defining positions and amino acids which may be important to paralogs in the same
protein family. This simple strategy is based on the idea that “the important amino acids
and positions would be conserved in the evolution”. Since paralogs in the same protein
family are from the same ancient protein, phylogenetic analysis is useful for finding the
“conserved differences” (important positions and amino acids) which may cause the
major different properties among paralogs such as substrate specificity, pH optimum,
temperature optimum, etc. Therefore, knowing these “conserved differences” would be
useful for engineering the properties among paralogs.
This approach is different from directed evolution which starts from a randomly
mutagenized library and then follows several rounds of high-throughput screening. The

63
common directed evolution approach is a short-term evolution and usually finds limited
useful substitutions (positions and amino acids). In order to evolve more functional
substitutions, you will need iterative rounds of mutagenesis and selection. Moreover, you
may need to perform additional saturation mutagenesis to optimize the selected
substitutions. The information (conserved differences) from phylogenetic analysis are
optimized as the result of long-term evolution by nature. By replacing the conserved
differences among related paralogs it is possible to shift certain properties among
paralogs. Based on this idea, we compared bacterial MGLs and eukaryotic CGLs and
found the conserved differences which might define substrate specificity among MGLs
and CGLs. In fact, we did change the substrate specificity of hCGL by implementing this
approach. Moreover, there are still many potential candidates (positions and amino
acids) to tests.
There are several advantages in using of this approach. First, it is easy to perform
phylogenetic analysis compared to structural analysis and there are more sequences than
solved structures in the database. Second, the information (positions and amino acids)
from phylogenetic analysis is more than the information from structural analysis.
Enzyme reaction is dynamic and there are several steps involved in such as substrate
binding, catalysis, conformational change and product release. However, structural
analysis only focuses on the one state-specific interaction (the solved structure is usually
at a specific state such as apo-enzyme or enzyme with substrate analog) and most
interactions are found around the active site. Since phylogenetic analysis shows the
“conserved differences” to specific properties (such as substrate specificities), they may
be involved in substrate binding, catalysis, conformational change or product release.
Moreover, some of the interaction may not be around the active site, such as K268R (at
interface of monomers) and T311G. These key candidates far away from the active site

64
are not easy to identify by structural analysis. Third, phylogenetic analysis could help to
generate a smaller and higher yield library, and identify mutations which may work
synergistically. In this hCGL engineering work, 38 site-directed mutagenized hCGL
mutants and one combinatorial library (2048 variants) were screened for activity in use of
methionine. Surprisingly, 9 out of 38 site-directed mutagenized hCGL mutants are more
active compared to their backbone protein in the breakdown of methionine. In contrast,
only one positive hCGL variant was selected from the combinatorial library.
It seems we have discovered beneficial substitutions from phylogenetic analysis
which have higher possibility to work synergistically such as S63L and L91M. hCGL-
NLV+RGS+LM (5% activity of pMGL) is more active than hCGL-NLV+RGS+L or
hCGL-NLV+RGS+M (both are around 3% activity of pMGL). Another example is
T311G and I353S. Wild-type hCGL with the T311G or I353S substitutions individually
did not show improvement in the breakdown of methionine but hCGL-NLV (E59N,
R119L and E339V) with T311G or I353S substitution displayed synergistic work in the
breakdown of methionine compared to hCGL-NLV.
This approach also has a disadvantage. In order to perform phylogenetic analysis,
you need two groups for comparison. If there is no bacterial MGLs or eukaryotic CGLs
in nature, we would not be able to compare MGLs and CGLs, preventing us finding
conserved difference to shift substrate specificity. The only solution for this situation
would be to perform structural analysis or performing selection or screening from a
randomized pool.
It is very different among phylogenetic analysis, structural analysis and common
directed evolution from a randomized pool. In phylogenetic analysis, you know all
beneficial positions and amino acids which have already been optimized by nature. It is

65
like you have done many rounds of selection and just need to perform recombination to
discover the best combination.
Other targets
Amino acid-depletion enzymes
Besides engineering hCGL, phylogenetic analysis can be applied to similar work
associated with amino acid-depletion enzymes for human cancer treatment such as
asparagine, tyrosine, phenylalanine, glutamine, leucine and cysteine. In addition,
engineered hCGL-NLV+RGS+LM also displays ability in degrading cysteine and can be
used to target cysteine-dependent cancer cells.
Antibody-directed enzyme prodrug therapy (ADEPT)
It has been reported that methionine gamma-lyase could be applied to ADEPT
with selenomethionine for cancer treatment. ADEPT is designed for cancer treatment
and is composed of two parts, non-toxic prodrug and antibody fused enzyme which can
convert the non-toxic prodrug to toxic drug (Xu and McLeod 2001; Zawilska,
Wojcieszak et al. 2013). The idea is to localize the therapeutic enzyme onto cancer cells
with cancer marker targeting antibodies and then systemically administer the prodrug to
be converted in high concentration at tumor site. It has been found that methylselenol
(product of selenomethionine by MGL) inhibited cell growth and arrested cells at G1 and
G2 phase. Subsequently, arrested cells would undergo apoptosis (Zeng, Wu et al. 2009;
Zeng, Briske-Anderson et al. 2012). In order to achieve this goal, the therapeutic enzyme
has to be absent or in low concentration in normal cells. In addition, the enzyme should

66
not induce an immune response, hence it is better to use a human enzyme. Therefore,
engineered hCGL variant with MGL activity can be a better choice than bacterial MGL in
this approach. In addition, the concept of ADEPT is different from amino acid-
deprivation treatments for cancer, and can be used to target different cancer cells with
specific surface markers rather than amino acid-dependent cancer cells.

67
MATERIALS AND METHODS
Site-directed mutagenesis:
We used QuickChange Site-Directed Mutagenesis kit (Agilent Technology) to
generate single site-mutated hCGL variants. All used oligonucleotides were designed
based on the requirement of the kit protocol and ordered from Integrated DNA
Technologies. Mutant strand synthesis reaction (Thermal cycling): 50 ul reaction
included 250 ng oligonucleotide pair (2 X 125 ng), 50-100 ng template plasmid (hCGL
was cloned into pET28a vector), 10 nmole dNTP, 10-fold concentrated Pfu turbo buffer
(Agilent Technology) and 2.5 U Pfu turbo DNA polymerase (Agilent Technology).
Cycling Parameter: one cycle of 95oC for 30 seconds and 16 cycles of 95
oC for 30
seconds, 55oC for 1minutes, 68
oC for 2 minutes. After thermal cycling, reaction would
be treated with 20 unit of Dpn I (New England BioLabs) at 37oC for 1 hour. Finally, 4 ul
of reaction was used for heat-shock transformation on BL21(DE3) competent cells.
Combinatorial library construction:
11 positions were picked from phylogenetic analysis for shifting substrate
specificity and overlapping PCR was used to generate combinatorial library. We used
“DNAWorks” software to design oligonucleotides for combinatorial library. All
oligonucleotides ordered from Integrated DNA Technologies were further purified by
PAGE gel for higher purity. 16 oligonucleotides were designed to cover 336 basepair of
C-terminal of hCGL. Thermal cycling condition for fragment assembly: 50 ul reaction
included 0.1 uM oligonucleotides (final concentration), 20 nmole dNTP, KOD hot start
DNA polymerase buffer (EMD4Biosciences), 1 unit of KOD hot start DNA polymerase
(EMD4Biosciences). The program for thermal cycling: one cycle of 94oC for 2min and

68
24 cycles of 94oC for 30 seconds, 60oC for 30 seconds, 72oC for 1 minutes. 2.5 ul of
assembled reaction would be used for further amplification and amplified product would
be gel extracted for cloning into pET28a vector.
96-well plate screening (Stone, Paley et al. 2012):
Single colonies of E.coli BL21(DE3), containing plasmids encoding either hCGL-
NLV or variant hCGL enzymes were picked into 96-well culture plates containing 75 ul
of TB media/well and 50 ug/ml kanamycine. Cells were grown at 37oC on a plate shaker
to an OD600 of ~0.8-1, cooled to 25oC, whereupon an additional 75 ul of media containing
50 ug/ml kanamycine, and 2 mM IPTG were added and incubation with shaking was
continued for 2 hours at 25oC. Subsequently, 100 ul of culture/well were transferred to a
fresh 96 well plate (assay plate). The assay plates were centrifuged (10 minutes X 3500
g) to pellet the cells, the media was removed, and the cells were chemically lysed by
addition of 50 ul/well of B-PER protein extraction reagent (Pierce) and mixing for 5
minutes on a plate shaker. Then 20 ul of 5 mM L-methionine at pH7.3 was added to the
lysates and incubated at 37oC for approximately 12 hours. The alpha-keto acid reaction
product is then derivatized by addition of 146 ul of MBTH solution to 54 ul of reaction
and heated for 40 minutes at 50oC. The absorbance at 320 nm was determined
spectrophotometrically using a microtiter plate reader. Variants exhibiting high
absorbance values and indicative of production can thus be identified and selected for
further characterization.

69
Kinetic analysis using MBTH (Stone, Paley et al. 2012):
Kinetic analyses were performed as described elsewhere (5) to measure
ketobutyrate production by reaction with 3-methyl-2-benzothiazolone hydrazone
hydrochloride (MBTH) to generate a chromophore with a λmax of ~320 nm. Eppendorf
tubes (1.5 ml) containing 220 µL of substrate in a 100 mM sodium phosphate buffer, 1
mM EDTA (pH 7.3), and 10 uM PLP were incubated at 37°C in a heat block. Reactions
were started by adding 20.3 µL of enzyme solution and quenched with 26.7 µL of 50%
trichloroacetic acid after a set length of time (without shaking). Reactions and blanks
were then mixed with 733 µL of MBTH solution (2.2 ml: 1.6 ml of 1M acetate buffer pH
5.0 and 0.6 ml of 0.1% MBTH in same) and incubated at 50°C for 40 minutes. After
cooling, the samples were transferred to cuvettes and the A320 nm was determined. The
assay was shown to be linear between 0 - 320 µM α-ketobutyrate with a lower detection
limit of 15 µM. One unit of MGL activity was defined as the amount of enzyme that
produced 1 µmol of α-ketobutyrate per minute at infinite concentration of L-methionine.
We also followed enzyme reactions in a continuous assay by detection of product
methane-thiol with 5,5'-Dithiobis(2-nitrobenzoic acid) (DTNB) (2) using 96-well plates
and a microtiter plate reader set at 405 nm.

70
REFERENCES
Cavuoto, P., and Fenech, M.F. (2012). A review of methionine dependency and the role
of methionine restriction in cancer growth control and life-span extension. Cancer
Treat Rev 38, 726-736.
Cole, M.F., and Gaucher, E.A. (2011). Exploiting models of molecular evolution to
efficiently direct protein engineering. J Mol Evol 72, 193-203.
Durando, X., Thivat, E., Farges, M.C., Cellarier, E., D'Incan, M., Demidem, A., Vasson,
M.P., Barthomeuf, C., and Chollet, P. (2008). Optimal methionine-free diet
duration for nitrourea treatment: a Phase I clinical trial. Nutr Cancer 60, 23-30.
Goseki, N., Yamazaki, S., Shimojyu, K., Kando, F., Maruyama, M., Endo, M., Koike,
M., and Takahashi, H. (1995). Synergistic effect of methionine-depleting total
parenteral nutrition with 5-fluorouracil on human gastric cancer: a randomized,
prospective clinical trial. Jpn J Cancer Res 86, 484-489.
Inoue, H., Inagaki, K., Eriguchi, S.I., Tamura, T., Esaki, N., Soda, K., and Tanaka, H.
(1997). Molecular characterization of the mde operon involved in L-methionine
catabolism of Pseudomonas putida. J Bacteriol 179, 3956-3962.
Kokkinakis, D.M., Hoffman, R.M., Frenkel, E.P., Wick, J.B., Han, Q., Xu, M., Tan, Y.,
and Schold, S.C. (2001). Synergy between methionine stress and chemotherapy in
the treatment of brain tumor xenografts in athymic mice. Cancer Res 61, 4017-
4023.
Poirson-Bichat, F., Goncalves, R.A., Miccoli, L., Dutrillaux, B., and Poupon, M.F.
(2000). Methionine depletion enhances the antitumoral efficacy of cytotoxic
agents in drug-resistant human tumor xenografts. Clin Cancer Res 6, 643-653.
Ragsdale, S.W. (2003). Pyruvate ferredoxin oxidoreductase and its radical intermediate.
Chem Rev 103, 2333-2346.
Rao, A.M., Drake, M.R., and Stipanuk, M.H. (1990). Role of the transsulfuration
pathway and of gamma-cystathionase activity in the formation of cysteine and
sulfate from methionine in rat hepatocytes. J Nutr 120, 837-845.
Sato, D., and Nozaki, T. (2009). Methionine gamma-lyase: the unique reaction
mechanism, physiological roles, and therapeutic applications against infectious
diseases and cancers. IUBMB Life 61, 1019-1028.
Stone, E., Paley, O., Hu, J., Ekerdt, B., Cheung, N.K., and Georgiou, G. (2012). De novo
engineering of a human cystathionine-gamma-lyase for systemic (L)-Methionine
depletion cancer therapy. ACS Chem Biol 7, 1822-1829.
Sun, Q., Collins, R., Huang, S., Holmberg-Schiavone, L., Anand, G.S., Tan, C.H., van-
den-Berg, S., Deng, L.W., Moore, P.K., Karlberg, T., et al. (2009). Structural

71
basis for the inhibition mechanism of human cystathionine gamma-lyase, an
enzyme responsible for the production of H(2)S. J Biol Chem 284, 3076-3085.
Tan, Y., Sun, X., Xu, M., Tan, X., Sasson, A., Rashidi, B., Han, Q., Wang, X., An, Z.,
Sun, F.X., et al. (1999). Efficacy of recombinant methioninase in combination
with cisplatin on human colon tumors in nude mice. Clin Cancer Res 5, 2157-
2163.
Thivat, E., Farges, M.C., Bacin, F., D'Incan, M., Mouret-Reynier, M.A., Cellarier, E.,
Madelmont, J.C., Vasson, M.P., Chollet, P., and Durando, X. (2009). Phase II trial
of the association of a methionine-free diet with cystemustine therapy in
melanoma and glioma. Anticancer Res 29, 5235-5240.
Xu, G., and McLeod, H.L. (2001). Strategies for enzyme/prodrug cancer therapy. Clin
Cancer Res 7, 3314-3324.
Yang, Z., Wang, J., Yoshioka, T., Li, B., Lu, Q., Li, S., Sun, X., Tan, Y., Yagi, S.,
Frenkel, E.P., et al. (2004). Pharmacokinetics, methionine depletion, and
antigenicity of recombinant methioninase in primates. Clin Cancer Res 10, 2131-
2138.
Zawilska, J.B., Wojcieszak, J., and Olejniczak, A.B. (2013). Prodrugs: a challenge for the
drug development. Pharmacol Rep 65, 1-14.
Zeng, H., Briske-Anderson, M., Wu, M., and Moyer, M.P. (2012). Methylselenol, a
selenium metabolite, plays common and different roles in cancerous colon
HCT116 cell and noncancerous NCM460 colon cell proliferation. Nutr Cancer 64,
128-135.
Zeng, H., Wu, M., and Botnen, J.H. (2009). Methylselenol, a selenium metabolite,
induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated
kinase 1/2 pathway and other cancer signaling genes. J Nutr 139, 1613-1618.

72
Chapter 3: Directed evolution of the substrate specificity of E.coli
biotin ligase
INTRODUCTION
Biotin is an essential cofactor and is covalently conjugated to biotin-dependent
enzymes such as acetyl-CoA carboxylase by biotin protein ligase (BPL). The E.coli BPL
first generates an intermediate, biotinoyl-5’-AMP, and subsequently transfers the biotin
moiety to a specific lysine of in biotin carboxyl carrier protein (BCCP) (Chapman-Smith
and Cronan 1999; Chapman-Smith and Cronan 1999) (Figure 3-1). E.coli BPL is a 35.5
kDa protein and its structure with bound biotin has been solved (1HXD) (Weaver, Kwon
et al. 2001). The central catalytic domain contains binding sites for biotin, ATP, and
BCCP, and undergoes a series of conformational changes in the multistep reaction
(Chapman-Smith and Cronan 1999). Protein substrates for E.coli BPL contain a
conserved sequence (MKM) that directs lysine modification. Alternatively, a 14 amino
acid peptide (biotin acceptor peptide, BAP) isolated from a peptide library (Schatz 1993)
has been widely used as a tag for protein biotinylation and subsequent purification.
By expanding the substrate specificity of biotin ligase, it may prove possible to
generate a wider array of protein-labeling reagents. For example, Ting’s group purified
biotin ligases from several species (human, S.cerevisiae, B. subtilis, P. acnes, L.
mesenteroides, T. cruzi, M. jannaschii and P. horikoshii), tested these BPLs with a
variety of biotin analogs (desthiobiotin azid, cis-propargyl biotin, trans-propargyl biotin,
iminobiotin, diaminobiotin, nitrobenzoxadiazole r-amino butyric acid, iodouracil valeric
acid and thiouracil valeric acid), and found that P. horikoshii BPL and yeast BPL could
label peptides with desthiobiotin azid and cis-propargyl biotin (Slavoff, Chen et al. 2008).

73
The range of proteins that can be biotinylated has also been expanded by using a
promiscuous E.coli biotin ligase variant that releases the reactive intermediate, biotin-5’-
AMP, and subsequently labels any adjacent lysine residue (Choi-Rhee, Schulman et al.
2004; Cronan 2005). Fusion versions of this variant have been used to label and identify
interaction partners in protein complexes (Cronan 2005; Chen, Choi et al. 2007).
BPL should be an excellent target for directed evolution in vitro, in part because
the strength and utility of the biotin:streptavidin linkage. Phage display has previously
been used to evolve a short peptide that was specifically recognized by yeast BPL. The
yeast BPL and peptide substrate proved to be orthogonal to the E. coli BPL and its
substrate, allowing two color site-specific protein labeling on cell surfaces with
fluorescent streptavidin conjugates (Chen, Choi et al. 2007).
Figure 3-1: Biotinylation reaction. The E.coli BPL first generates an intermediate,
biotinoyl-5’-AMP, and subsequently transfers the biotin moiety to a specific
lysine of in biotin carboxyl carrier protein (BCCP).

74
However, phage display can be limited by library size and skewing due to the
impact of different proteins on phage fitness. For example, a fusion protein to the phage
tail spike (pIII) may impact the functionality of the pIII signal peptide and thereby reduce
infectivity. To overcome these limitations, directed evolution has been carried out in
water-in-oil emulsions. By transcribing and translating individual DNA templates within
individual emulsion bubbles very large libraries can be screened or selected for function
(1010 variants / milliliter; (Tawfik and Griffiths 1998; Griffiths and Tawfik 2003;
Bernath, Hai et al. 2004; Cohen, Tawfik et al. 2004; Miller, Bernath et al. 2006). For
example, Griffths and Tawfik have demonstrated that IVC and FACS could be combined
to select phosphotriesterases from libraries of over 3.4 X 107 variants that have a 63-fold
higher kcat value for the degradation of paraoxon, a potent acetylcholinesterase-inhibiting
insecticide (Griffiths and Tawfik 2003; Bernath, Hai et al. 2004; Aharoni, Amitai et al.
2005; Mastrobattista, Taly et al. 2005). It should also prove possible to use similar
methods for the directed evolution of the substrate specificity of E. coli biotin ligase,
especially as we had previously used in vitro compartmentalization to evolve a biotin-
binding protein, streptavidin, with altered affinities for the biotin analog, desthiobiotin
(Levy and Ellington 2008).

75
RESULTS AND DISCUSSIONS
Development of a scheme for BirA directed evolution
Since biotin ligase is a critical enzyme in many organisms, alteration of its
substrate specificity would likely lead to toxic effects, which would contraindicate an in
vivo selection scheme. In vitro selection via phage-display methods is possible but is not
compatible with the eventual use of enzymes in trans, rather than in cis, and does not
afford the same flexibility in reaction conditions as would selections in the context of an
emulsion (Fernandez-Gacio, Uguen et al. 2003).
Therefore, we have pursued the development of a scheme for the directed
evolution of BirA in emulsions (Figure 3-2). Genes for BirA are conjugated to a peptide
tag that is a substrate for the enzyme, and are distributed into emulsion vesicles such that
there is less than one template / vesicle. The BirA genes are transcribed and translated,
and functional variants can label the peptide tags associated with their own genes. Upon
breaking the emulsion, biotinylated genes are captured with streptavidin-coated magnetic
beads, similar to a scheme we previously developed for the directed evolution of
streptavidin function (Levy and Ellington 2008). The captured genes are amplified and
redistributed to emulsion vesicles. Multiple cycles of selection and amplification are
anticipated to sieve the most functional BirA variants from a library.

76
Figure 3-2: Selection scheme of directed evolution of BPL using IVC. There are
three major steps in this selection. First, linear constructs (library) need to
covalently conjugate with biotin acceptor peptide (BAP). Second, the
conjugates and E.coli lysates are mixed with oil-surfactant to generate
individual compartments. Functional variants which can use desthiobiotin as
substrates would attach desthiobiotin to BAP. Finally, the functional
variants could be separated from other nonfunctional variants by
streptavidin-coated magnetic beads and recovered by PCR amplification.
In order to make this scheme work, each step had to be optimized, including
conjugation of peptide tag with the gene template, in vitro transcription and translation,
breaking of the emulsion, and recovery of the DNA template. Each of these
optimizations was undertaken separately, and the results combined into a workable
protocol. First, we use a chemical cross-linking reagent, sulfo-SMCC
(sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate, from Thermo
Scientific) to conjugate the peptide tags with DNA templates, and have optimized the

77
efficiency to 35% to 50%. A number of different in vitro transcription and translation
(IVTT) options were explored, and ultimately we settled on an E.coli lysate (RTS E.coli
HY kit, from Roche). Salmon testes DNA was added to reduce DNA degradation caused
by nucleases in the lysate (Miller, Bernath et al. 2006). The recovery of biotinylated
DNA templates from the lysate was around 80% in the absence of emulsification. The
efficiency of recovery was diminished following emulsification, but optimization of yield
was partially recovered from 30% by breaking the emulsion with hexane to 60% by
breaking the emulsion with ether. The buffer used for capturing the desthiobiotin-
conjugated templates was also a variable, and surprisingly PBS was found to give better
recovery than a suggested high salt buffer. Finally, the choice of DNA polymerase for
template amplification was very important. KOD DNA polymerase was found to have
better processivity on the full-length BirA template than did Pfu and Taq DNA
polymerase, as has previously been observed (Takagi, Nishioka et al. 1997).
In order to demonstrate the viability of this scheme, we initially selected
functional BirA variants relative to a truncated, inactive enzyme (Figure 3-3). The
truncated variant was recovered from emulsions 50-fold less well than the wild-type
gene, suggesting that there might be substantive purification on a per cycle basis in a real
selection. We therefore mixed small amounts of wild-type BirA with the truncated gene,
and selected for ligation function. As shown in (Figure 3-3), when the wild-type gene
was present at only 1 part in ten, it could be quantitatively recovered.

78
Figure 3-3: Mock selection. Before the real selection, a mock selection was
performed to validate the selection scheme. We prepared four different
pools, full-length BirA pool (positive control pool), truncated BirA pool
(negative control pool) and two mixed pools. For two mixed pools, full-
length wild type BirA (functional) was mixed with truncated BirA (non-
functional) in 1:9 and 1:999 ratios. After one round of selection in emulsion,
recovery of the full-length BirA control pool was defined as 100% while
only 2% of the truncated BirA control pool was recovered. Additionally, the
1:9 ratio pool had 11% recovery. Theoretically, we should recover 10%
DNA from the 1:9 mixed pool because there are 10% of 1:9 mixed pool
DNA is full-length BirA which has enzyme activity in the selection. This
verified that the selection scheme could distinguish functional BirA from
non-functional BirA and recover functional BirA from mixed pool.
Library construction
We wanted to show the utility of our emulsion selection by identifying BirA
variants that could efficiently and orthogonally use desthiobiotin in place of biotin. Two
different libraries were constructed, one that targeted residues that were thought to
interact with biotin, and one that contained random mutations introduced by error-prone

79
PCR. For both pools, we added a C-terminal GGGS link and His6 tag at the C-terminal
end for protein detection and purification.
The structure of E.coli biotin ligase-biotin complex has been solved (Wilson,
Shewchuk et al. 1992), and Tyr-132, Gly-204 and Gly-206 of E.coli are close to the
thiophene ring of biotin. Additionally, Gly-117, Lys-183, Gly-186 and Leu-188 of E.coli
biotin ligase are close to the junction of pentanoic acid and thiophene ring in biotin. In
desthiobiotin, there is no thiophene ring and hexanoic acid directly links to uredio ring.
This difference may affect the binding of E.coli biotin ligase to desthiobiotin (Wu and
Wong 2004). Thus we generated a BirA template that was completely randomized at
these 7 positions by substituting codons with NNK (K=G or T), which spans the 20
amino acids contains only 3% stop codons relative to NNN (Braunagel and Little 1997).
Of course, residues other than those abutting the thiophene ring may prove
important for catalysis with desthiobiotin, and we therefore also generated a pool using
traditional error-prone PCR methods (Fromant, Blanquet et al. 1995). The substitution
rate of the randomized pool was around 2.4% / position, or ca. 24 mutations / gene
(Figure 3-4).

80
Figure 3-4: Two libraries construction. There are two libraries were generated. One is
generated by error-prone PCR for random mutagenesis and the substitution
rate is 2.4%. The substitutions would randomly distribute over the entire
coding sequence. The other library which was generated by overlapping
PCR with designed oligonucleotides (containing 7 positions with NNK
codon).
Selection for DTB utilization
The two libraries were initially selected individually for their ability to conjugate
desthiobiotin to the peptide-gene chimera. A high concentration of DTB (200
micromolar) was utilized to help ensure efficient labeling by enzymatic variants. After
several rounds, the concentration of DTB was dropped to 50 micromolar, and the two
pools were combined for the final three rounds of selection. We did not include a
counterselection with biotin, given that such a counterselection previously failed to yield

81
orthogonal streptavidin variants that could utilize desthiobiotin in preference to biotin
(Levy and Ellington 2008). Therefore, the current selection was primarily to expand and
/ or alter the substrate preference of the E. coli BirA (Slavoff, Chen et al. 2008;
Uttamapinant, White et al. 2010).
The pools were cloned after four (just prior to mixing the pools) and six rounds of
selection. Interestingly, only variants from the PCR-mutagenized pool survived the
selection, which may indicate that only relatively subtle structural alterations to the
biotin-binding pocket remain active with desthiobiotin.
Figure 3-5: The selection process of BPL library 1 and BPL library 2.
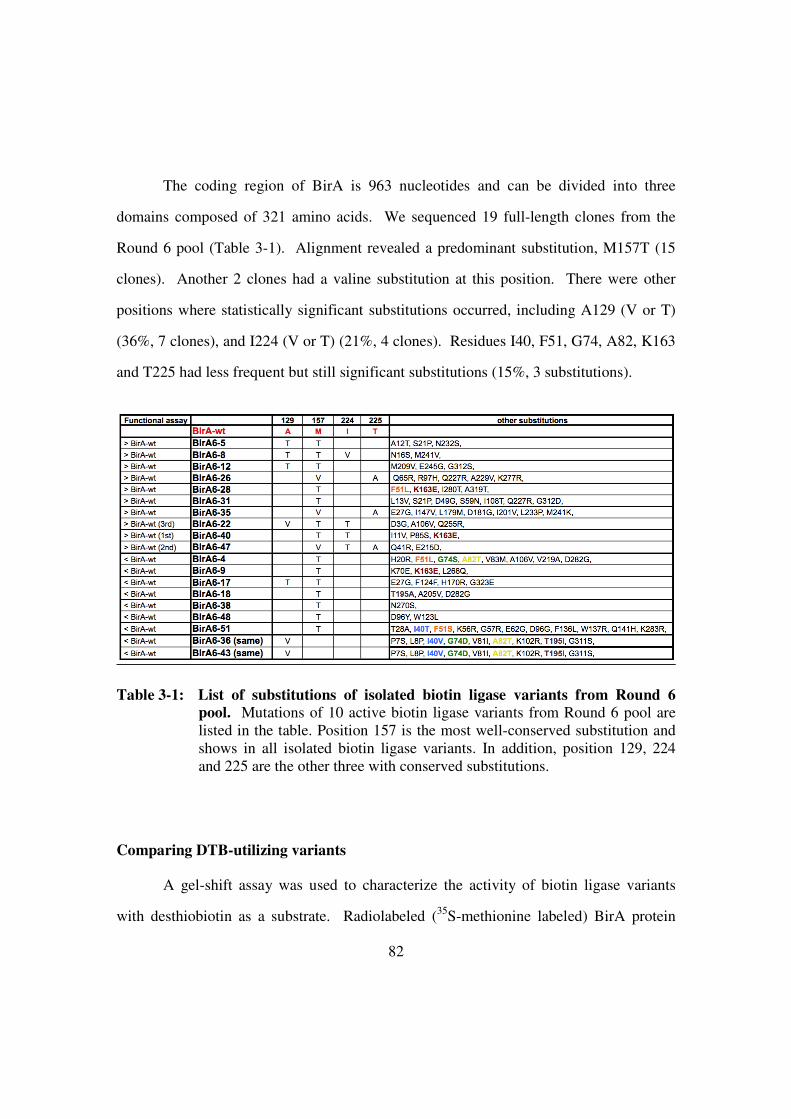
82
The coding region of BirA is 963 nucleotides and can be divided into three
domains composed of 321 amino acids. We sequenced 19 full-length clones from the
Round 6 pool (Table 3-1). Alignment revealed a predominant substitution, M157T (15
clones). Another 2 clones had a valine substitution at this position. There were other
positions where statistically significant substitutions occurred, including A129 (V or T)
(36%, 7 clones), and I224 (V or T) (21%, 4 clones). Residues I40, F51, G74, A82, K163
and T225 had less frequent but still significant substitutions (15%, 3 substitutions).
Table 3-1: List of substitutions of isolated biotin ligase variants from Round 6
pool. Mutations of 10 active biotin ligase variants from Round 6 pool are
listed in the table. Position 157 is the most well-conserved substitution and
shows in all isolated biotin ligase variants. In addition, position 129, 224
and 225 are the other three with conserved substitutions.
Comparing DTB-utilizing variants
A gel-shift assay was used to characterize the activity of biotin ligase variants
with desthiobiotin as a substrate. Radiolabeled (35
S-methionine labeled) BirA protein

83
variants and a peptide-tagged chloramphenicol acetyltransferase were synthesized
separately in lysates. The proteins were then co-incubated in the presence of desthiobiotin
for 8 minutes. After reaction, recombinant streptavidin was added and following gel
electrophoresis a shift in the radioactive CAT band was indicative of BirA activity. The
activity of variants was initially assessed by determining the ratio of shifted signal
(complex amount) to the amount of enzyme present (enzyme amount).
Ten clones showed higher activity than wild-type with desthiobiotin compared to
BirAwt. These 10 most improved BirA variants all contained similar substitutions: M157
to T or V (100%), A129 to T or V (40%), I224 to T or V (40%) and T225 to A (30%)
(Table 3-1). The variants BirA6-22, BirA6-47, and BirA6-40 were best able to utilize
desthiobiotin, and were 8-fold, 10-fold, and 17-fold better than the wild-type in our assay,
respectively (Figure 3-6).
Since the pool was selected for desthiobiotin utilization it was unclear whether it
would retain its ability to utilize biotin as a substrate. We therefore assayed BirA6-22,
BirA6-47, and BirA6-40 using biotin and found that their activities were 0.06-fold, 0.19-
fold, and 0.91-fold that of the wild-type BirA, which would imply 130-fold, 54-fold, and
19.2-fold improvements in the use of desthiobiotin relative to biotin compared with
BirAwt (Figure 3-6). The ability to best utilize desthiobiotin correlated well with the
retention of the ability to utilize biotin, while the greatest orthogonality occurred in
enzymes that had disproportionately lost the ability to utilize biotin. This observation is
consistent with the notion we and others have previously put forward, that enzymes that
are undergoing changes in substrate specificity proceed through a more 'generalist'
intermediate (Khersonsky and Tawfik 2010). That said, when we assayed BirAwt, BirA6-
22, BirA6-22(V106A), BirA6-40, and BirA6-47 with iminobiotin as a substrate none of them
showed easily measured activity. Thus, the remodeling of the active site appeared to
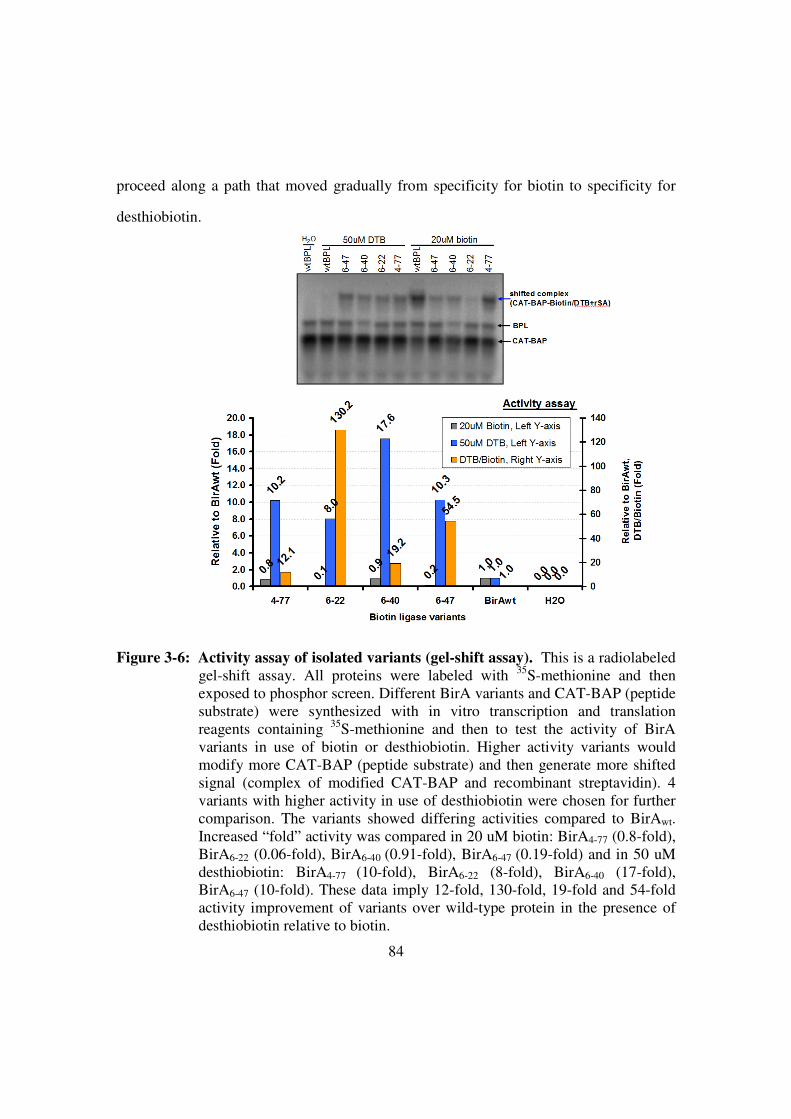
84
proceed along a path that moved gradually from specificity for biotin to specificity for
desthiobiotin.
Figure 3-6: Activity assay of isolated variants (gel-shift assay). This is a radiolabeled
gel-shift assay. All proteins were labeled with 35
S-methionine and then
exposed to phosphor screen. Different BirA variants and CAT-BAP (peptide
substrate) were synthesized with in vitro transcription and translation
reagents containing 35
S-methionine and then to test the activity of BirA
variants in use of biotin or desthiobiotin. Higher activity variants would
modify more CAT-BAP (peptide substrate) and then generate more shifted
signal (complex of modified CAT-BAP and recombinant streptavidin). 4
variants with higher activity in use of desthiobiotin were chosen for further
comparison. The variants showed differing activities compared to BirAwt.
Increased “fold” activity was compared in 20 uM biotin: BirA4-77 (0.8-fold),
BirA6-22 (0.06-fold), BirA6-40 (0.91-fold), BirA6-47 (0.19-fold) and in 50 uM
desthiobiotin: BirA4-77 (10-fold), BirA6-22 (8-fold), BirA6-40 (17-fold),
BirA6-47 (10-fold). These data imply 12-fold, 130-fold, 19-fold and 54-fold
activity improvement of variants over wild-type protein in the presence of
desthiobiotin relative to biotin.

85
The positions of the most frequent substitutions were mapped onto the biotin-
BirA structure (1HXD) (Weaver, Kwon et al. 2001). The substituted position A129 (to T
or V) is near to the biotin-binding loop (amino acids 110 to 128), while the substituted
positions I224 (to V or T) and T225 (to A) are in a different loop (amino 212 to 233) that
is known to change its conformation from disordered to ordered upon biotin-binding
(Weaver, Kwon et al. 2001). Interestingly, the most well-conserved substitution, T157
(V or T) is far from the biotin binding site.
Kinetic characterizations of biotin ligase variants
More detailed kinetic characterizations were carried out with the best
desthiobiotin-utilizing variants. Our assay was based on the release of pyrophosphate in
the first step of the ligase reaction. We initially measured the kcat and KM of BirAwt with
ATP, and found that the measured KM value (0.22 mM) was very similar to the value that
was previously published (0.2 mM). Similarly, the measured kcat, 0.48 s-1
, was close to
similar to the range of values that have appeared (0.17 to 0.44 s-1
) (Chapman-Smith,
Mulhern et al. 2001).
We proceeded to use this method with desthiobiotin. However, since
desthiobiotin is not readily soluble in water it was initially dissolved in dimethyl
sulfoxide (DMSO) and then diluted with water. While it has been shown that DMSO can
impair wild-type biotin ligase activity, we characterized all of the BirA variants using the
a concentration of DMSO (2.4%) that has been shown to leave wild-type enzyme activity
largely intact (90%) (Nah 2009). Surprisingly, the kcat of BirA6-40 and BirAM157T were
found to be 0.73 s-1
and 0.53 s-1
, respectively; 2-fold faster than the kcat of BirAwt with
desthiobiotin (0.29 s-1
). However, the KM of BirA6-40 and BirAM157T for desthiobiotin

86
were 6.7 mM and 4.3 mM, values that are 2- to 3-fold worse than the KM of BirAwt (0.17
mM) (Figure 3-7).
Figure 3-7: Kinetic characterization of BirAwt, BirA6-40and BirAM157T. Purified
BirAwt, BirA6-40 and BirAM157T were characterized by kinetic analysis via
detection of pyrophosphate generated in the biotin ligase reaction. BirA6-40
and BirAM157T show much lower kcat/KM compared to BirAwt in use of
biotin. However, the KM values for BirA6-40 and BirAM157T are too high to
define. Additionally, BirA6-40 and BirAM157T show 0.6-fold and 0.7-fold
kcat/KM in the presence of desthiobiotin as compared to BirAwt. Moreover,
BirA6-40 and BirAM157T have around 2-fold higher kcat value with
desthiobiotin as compared to BirAwt.

87
Selected variants were also characterized for their ability to use biotin. The very
high KM values of both BirA6-40 and BirAM157T for biotin prevented measurement of
individual kinetic constants, but the ratio kcat/KM could be determined. This value was
similar for both biotin ligase variants, around 0.01 s-1 mM-1. This value is roughly 5
orders of magnitude less than that of the wild-type BirA (106 s
-1 M
-1) (Figure 3-7).
The pyrophosphate assay proved limiting for attempting to characterize the
kinetics of the second substrate, the peptide BAP. However, if we consider the gel-shift
assays (Figure 3-6 and 3-8) and kinetic assays (Figure 3-7) for BirAwt and BirA6-40 with
50 uM desthiobiotin, it might be expected that BirAwt should be faster than BirA6-40 with
desthiobiotin, since BirAwt has a larger kcat/KM value. In fact, the result of the gel-shift
assay shows a stronger signal for BirA6-40 (this was consistent across two independent
gel-shift assays, Figure 3-6 and Figure 3-8). Taken together, these results imply that
BirA6-40 has a smaller KM for BAP than does the wild-type enzyme.
The observed changes in the kinetic parameters seem to mirror the particular
features of the emulsion-based selection method. The fact that the amount of
desthiobiotin provided was essentially saturating throughout the selection led to 2-fold
improvements in kcat for desthiobiotin, and may have also allowed an increase in the KM
for desthiobiotin. Conversely, the low peptide concentration in each compartment (1
peptide / compartment, or ca. 0.3 nM) led to selective pressure for improved binding of
the peptide. While it is possible that variants exist that show greatly improved kcat and
KM values for each substrate, such variants were not seen and are apparently rare relative
to variants where only the kinetic parameter actually under selection (kcat) is improved.
In general the stepwise optimization of individual kinetic parameters would seem to be
more productive than attempts to find rare variants that are simultaneously optimized
over a range of kinetic and biophysical parameters.

88
Evolutionary paths
In order to determine how the various prevalent mutations contributed to the
change in substrate specificity, we created variants containing individual amino acid
substitutions based on the three clones with the highest activities. Some 12 single
substitutions were introduced into the wild-type protein: D3G, I11V, Q41R, P85S,
A106V, A129V, M157T, I224T and T225A, K163E, E215D and Q255R, and the
activities of these variants were again assessed using both desthiobiotin and biotin as
substrates (Figure 3-8).
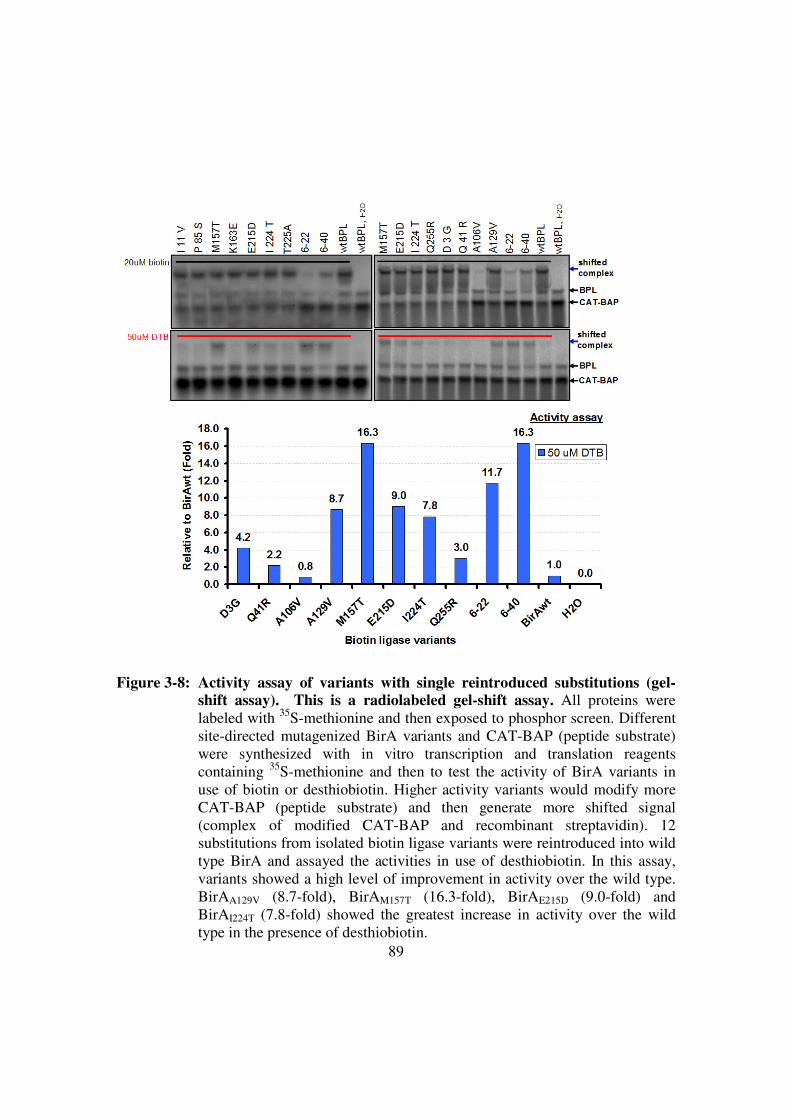
89
Figure 3-8: Activity assay of variants with single reintroduced substitutions (gel-
shift assay). This is a radiolabeled gel-shift assay. All proteins were
labeled with 35
S-methionine and then exposed to phosphor screen. Different
site-directed mutagenized BirA variants and CAT-BAP (peptide substrate)
were synthesized with in vitro transcription and translation reagents
containing 35
S-methionine and then to test the activity of BirA variants in
use of biotin or desthiobiotin. Higher activity variants would modify more
CAT-BAP (peptide substrate) and then generate more shifted signal
(complex of modified CAT-BAP and recombinant streptavidin). 12
substitutions from isolated biotin ligase variants were reintroduced into wild
type BirA and assayed the activities in use of desthiobiotin. In this assay,
variants showed a high level of improvement in activity over the wild type.
BirAA129V (8.7-fold), BirAM157T (16.3-fold), BirAE215D (9.0-fold) and
BirAI224T (7.8-fold) showed the greatest increase in activity over the wild
type in the presence of desthiobiotin.

90
The single, most highly observed mutation (95.7% of the Round 6 pool, Next-Gen
sequencing, Table 3-2), BirAM157T, on its own shows much higher activity with
desthiobiotin (a kcat of 0.53 s-1
and a KM of 4.3 mM with desthiobiotin, compared with a
kcat of 0.73 s-1 and a KM of 6.7 mM for the best isolated BirA variant, BirA6-40) (Figure 3-
7). While the M157T substitution does not directly contact biotin it is close to highly
conserved residues such as Tyr-132, Gly-204, Ala-205 and Gly-206 which in turn are
close to the thiophene ring of biotin (Wu and Wong 2004). Also Met-157 is in proximity
to Gly-186 and Ile-187 that are near the junction of the pentanoic acid and thiophene ring
of biotin (Figure 3-9, left). All of the substitutions at this position are smaller than
methionine (T (59% of the total), V (30%), I (4%) and A (1%)). Interestingly, it has been
shown that Pyrococcus horikoshii biotin ligase can use desthiobiotin-azide (DTB-Az)
(Slavoff, Chen et al. 2008), and the position corresponding to Met-157 in this enzyme is
valine. However, this may not be significant as other biotin ligases from
Bacillus, Leuconostoc, and Methanococcus also have valine at this position and these
were found not to be able to use DTB-Az as a substrate.
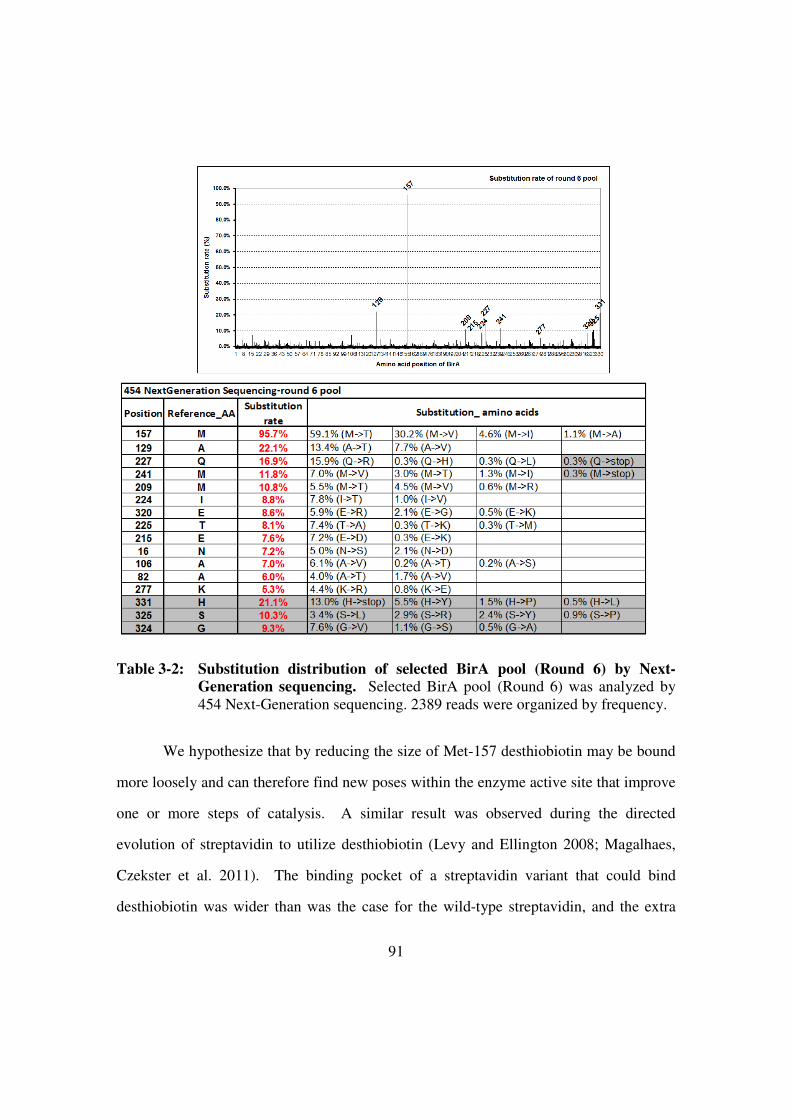
91
Table 3-2: Substitution distribution of selected BirA pool (Round 6) by Next-
Generation sequencing. Selected BirA pool (Round 6) was analyzed by
454 Next-Generation sequencing. 2389 reads were organized by frequency.
We hypothesize that by reducing the size of Met-157 desthiobiotin may be bound
more loosely and can therefore find new poses within the enzyme active site that improve
one or more steps of catalysis. A similar result was observed during the directed
evolution of streptavidin to utilize desthiobiotin (Levy and Ellington 2008; Magalhaes,
Czekster et al. 2011). The binding pocket of a streptavidin variant that could bind
desthiobiotin was wider than was the case for the wild-type streptavidin, and the extra

92
space gave the open thiophene ring more rotational freedom. Thus, although the M157T
does not directly contact to desthiobiotin reducing the size of Met-157 might mean fewer
structural constraints on immediately adjacent residues such as Tyr-132, Gly-204, Ala-
205, Gly-206, Gly-186, and Ile-187, and thus possibly also more rotational freedom for
binding desthiobiotin (Figure 3-9, right).
The next most highly observed substitutions BirAA129V and BirAI224T also show
higher activity with desthiobiotin than the wild-type E.coli biotin ligase, as does
BirAE215D (Figure 3-6). That said, there are clearly multiple paths to high activity clones,
as the best variant, BirA6-40, contains only 2 of the 4 highly observed substitutions, while
the next most active variants contain 3 of the 4 highly observed substitutions.
In contrast, BirAA106V has lost activity with both biotin and desthiobiotin. This
substitution was introduced based on the sequence of BirA6-22. It seems likely that
A106V is a deleterious mutation that randomly arose, and has been compensated for by
the other mutations in BirA6-22. To test this hypothesis, we 'repaired' the A106V
substitution in BirA6-22 and found there was no significant difference compared to BirA6-
22 in the use of biotin or desthiobiotin. While none of the other 9 active variants
originally screened contained substitutions at positions 106, deep sequencing revealed
that 7% of selected variants contained substitutions at this position. Thus, A106V may be
a potentiating or compensatory mutation that improves the fitness variants that proceed
along a specific mutational path. Deep sequencing provided sufficient data to search for
nearby covariations, and we discovered that A106V demonstrated significant covariance
with A129V (p<0.0001), a mutation that appeared 22.1% in Round 6 pool (Rhee, Liu et
al. 2007).

93
Figure 3-9: Mapping Met-157 onto the structure of E.coli biotin ligase with biotin.
E.coli biotin ligase is shown here with biotin (1HXD). Tyr-132 (blue
spheres), Gly-204, Ala-205, Gly-206 (pink spheres), Ile-187, Gly-186
(yellow spheres), Met-157 (left, assorted color spheres) and M157T (right,
assorted color spheres, mutagenized by PyMOL) are labeled on the 1HXD.
Tyr-132, Gly-204, Ala-205, Gly-206, Gly-186 and Ile-187 are close to
biotin’s thiophene ring or to the junction of the pentanoic acid and thiophene
ring in biotin. M157T substitution does not directly interact with biotin but
may affect aforementioned conserved amino acids (blue, pink and yellow
spheres). Substitution of the methionine with threonine or another smaller
amino acid (valine, isoleucine or alanine based Next-Gen sequencing result)
may provide more space for desthiobiotin at binding pocket of enzyme.
The identification of a deleterious substitution in an otherwise active variant is
consistent with the fact that while 10 of the 19 clones from Round 6 had improved
activities, 8 had virtually no activity with desthiobiotin. While this could indicate that the
selection stringency was not sufficiently high, this hypothesis is contradicted by the fact

94
that we were able to recover clones with improved activities. Therefore, it seems more
likely that this result indicates that there is a large mutational burden that occurs as a
result of the amplification process.

95
MATERIALS AND METHODS:
Library preparation:
To begin selection, two biotin protein ligase pools were created. The first was
generated by error-prone PCR for the random mutagenesis of E. coli BirA per Fromant et
al. (Fromant, Blanquet et al. 1995). Buffer conditions were: 10 mM Tris, pH 8.7, 50 mM
KCl, 5 µg/ml BSA, 0.5 µM of each of the two primers, 0.12 mM dATP, 0.10 mM dCTP,
0.55 mM dGTP, 3.85 mM dTTP, 0.5 mM MnCl2, 4.8 mM Cl2, 20 pg of template DNA
(966 bp), and 2 units of Taq DNA polymerase (New England Biolabs). Amplification
was carried out over 25 cycles and amplified product was subsequently gel-extracted then
further mutagenized for another 25 cycles. The second library was generated by
saturation mutagenesis at 7 specific positions. After mutagenesis, the assembled
fragments were overlapped with N-terminal and C-terminal fragments of the E. coli BirA
gene in order to generate a full coding region. Finally, the randomized coding regions
were overlapped with commercial fragments (Roche) containing T7 promoters and
terminators to generate the full linear constructs.
Cross-linking BAP with DNA:
Sulfo-SMCC (Pierce) was used to cross-link the BAP (23 amino acids,
CGGGSGGGSGLNDIFEAQKIEWH, from Bio-Synthesis) and DNA linear constructs
(1.3 kb). First, DNA library was amplified with an oligonucleotide primer containing a
5’-amino group. After this, 2 µg of amplified library DNA was mixed with 10 mM, 500
µl sulfo-SMCC at room temperature for 1 hr. In the next step, activated DNA library was
purified by silicon-member-based DNA purification spin column (Qiagen) and then

96
incubated with BAP overnight at room temperature. Finally, the BAP-library conjugates
were purified by silicon-membrane-based DNA purification spin column again.
In vitro compartmentalization selection (Miller, Bernath et al. 2006; Levy and
Ellington 2008):
The DNA library was added into RTS E. coli lysate (Roche) with 0.5% sodium
deoxycholate. Then it was emulsified with 500 µl of an oil-surfactant mixture containing
mineral oil, 4.5% Span 80, 0.5% Tween 80, and 0.1% Triton X-100 by stirring for 4 min
on ice. Subsequently, the emulsion reactions were incubated at 30 °C for 2 hr. To stop the
reactions, the emulsions were incubated at 90 °C for 15 min. The aqueous phase was
extracted the first time with 900 µl of water-saturated ether in the presence of 500 µl
PBS. It was further extracted two more times with 900 µl water-saturated ether. Excess
ether was removed by vacuum centrifugation for 10 min at room temperature. Free
desthiobiotin in the extracted reaction was removed by silicon-membrane-based DNA
purification (Qiagen). Purified DNA was incubated with streptavidin-coated magnetic
beads for capture. After three PBS washes, captured DNA was recovered by PCR
amplification.
Gel-shift assay:
35S-methionine-labeled BPL and CAT-BAP (peptide substrate) were generated
separately from Roche HY100 E. coli lysate after 30 °C incubation for 3 hr.
Subsequently, BPL and CAT-BAP were mixed together with 4 mM ATP; these solutions
were further mixed with either 20 µM biotin or 50 µM desthiobiotin for 8 min. Reactions
were then terminated by heating the mixtures for 10 min at 95 °C. In order to

97
differentiate the modified CAT-BAP (biotinylated or desthiobiotinylated), 3.8 µmol
recombinant streptavidin was added into the reactions to bind modified CAT-BAP. A
complex of streptavidin and modified CAT-BAP showed a shifted signal on a 4-20%
gradient SDS-PAGE gel. The signals of all proteins were be detected by phosphor
screen.
BPL protein purification (Naganathan and Beckett 2007):
The BirA variants were first subcloned into a pRS.1 plasmid containing a tac
promoter and then transformed into JM109 competent cells for protein expression. Cells
grew in Terrific Broth medium (Sigma-Aldrich) and protein expression was induced by 1
mM IPTG for 5 hr after OD600 reached 0.6-0.9. Soluble protein was separated from
debris by 40,000 g centrifugation for 30 min and further purified using a Ni-NTA column
(Qiagen). The purified BPL was then run through FPLC with HiLoad 16/600 Superdex
75 pg column (GE) for higher purity and buffer exchange. Storage buffer for purified
BPL was composed of 50 mM Tris-HCl, pH 7.5, and 200 mM KCl.
Pyrophosphate detection:
An EnzChek pyrophosphate assay kit (Life Technologies) was used for the
detection of generated pyrophosphate from the biotinylation reaction; steps were
modified as necessary to fit the experiment. In the first step, the biotinylation (or
desthiobiotinylation) reactions (50 mM bicine buffer, pH 8.3, 10 mM Magnesium
oxaloacetate, 2 mM ATP, 70 µM BAP, and 0.5-1 µM enzyme in 50 µl volume) were
carried out at 30 °C (with different concentrations of biotin or desthiobiotin).
Subsequently, the reactions were inactivated at 90 °C for 4 min at different time points.

98
Finally, these terminated reactions were mixed with 50 µl pyrophosphate reagents and
incubated at 22 °C for 1 hr. 95 µl of the final reaction were transferred to a new 96-well
plate and measured for absorbance at 360 nm. Readouts were taken at various time points
to determine the rate of reaction at different concentrations of biotin or desthiobiotin.

99
REFERENCES
Aharoni, A., Amitai, G., Bernath, K., Magdassi, S., and Tawfik, D.S. (2005). High-
throughput screening of enzyme libraries: thiolactonases evolved by fluorescence-
activated sorting of single cells in emulsion compartments. Chem Biol 12, 1281-
1289.
Bernath, K., Hai, M., Mastrobattista, E., Griffiths, A.D., Magdassi, S., and Tawfik, D.S.
(2004). In vitro compartmentalization by double emulsions: sorting and gene
enrichment by fluorescence activated cell sorting. Anal Biochem 325, 151-157.
Braunagel, M., and Little, M. (1997). Construction of a semisynthetic antibody library
using trinucleotide oligos. Nucleic Acids Res 25, 4690-4691.
Chapman-Smith, A., and Cronan, J.E., Jr. (1999a). The enzymatic biotinylation of
proteins: a post-translational modification of exceptional specificity. Trends
Biochem Sci 24, 359-363.
Chapman-Smith, A., and Cronan, J.E., Jr. (1999b). Molecular biology of biotin
attachment to proteins. J Nutr 129, 477S-484S.
Chapman-Smith, A., Mulhern, T.D., Whelan, F., Cronan, J.E., Jr., and Wallace, J.C.
(2001). The C-terminal domain of biotin protein ligase from E. coli is required for
catalytic activity. Protein Sci 10, 2608-2617.
Chen, I., Choi, Y.A., and Ting, A.Y. (2007). Phage display evolution of a peptide
substrate for yeast biotin ligase and application to two-color quantum dot labeling
of cell surface proteins. J Am Chem Soc 129, 6619-6625.
Choi-Rhee, E., Schulman, H., and Cronan, J.E. (2004). Promiscuous protein biotinylation
by Escherichia coli biotin protein ligase. Protein Sci 13, 3043-3050.
Cronan, J.E. (2005). Targeted and proximity-dependent promiscuous protein
biotinylation by a mutant Escherichia coli biotin protein ligase. J Nutr Biochem
16, 416-418.
Fernandez-Gacio, A., Uguen, M., and Fastrez, J. (2003). Phage display as a tool for the
directed evolution of enzymes. Trends Biotechnol 21, 408-414.
Fromant, M., Blanquet, S., and Plateau, P. (1995). Direct random mutagenesis of gene-
sized DNA fragments using polymerase chain reaction. Anal Biochem 224, 347-
353.
Griffiths, A.D., and Tawfik, D.S. (2003). Directed evolution of an extremely fast
phosphotriesterase by in vitro compartmentalization. EMBO J 22, 24-35.
Khersonsky, O., and Tawfik, D.S. (2010). Enzyme promiscuity: a mechanistic and
evolutionary perspective. Annu Rev Biochem 79, 471-505.

100
Levy, M., and Ellington, A.D. (2008). Directed evolution of streptavidin variants using in
vitro compartmentalization. Chem Biol 15, 979-989.
Magalhaes, M.L., Czekster, C.M., Guan, R., Malashkevich, V.N., Almo, S.C., and Levy,
M. (2011). Evolved streptavidin mutants reveal key role of loop residue in high-
affinity binding. Protein Sci 20, 1145-1154.
Mastrobattista, E., Taly, V., Chanudet, E., Treacy, P., Kelly, B.T., and Griffiths, A.D.
(2005). High-throughput screening of enzyme libraries: in vitro evolution of a
beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem
Biol 12, 1291-1300.
Miller, O.J., Bernath, K., Agresti, J.J., Amitai, G., Kelly, B.T., Mastrobattista, E., Taly,
V., Magdassi, S., Tawfik, D.S., and Griffiths, A.D. (2006). Directed evolution by
in vitro compartmentalization. Nat Methods 3, 561-570.
Naganathan, S., and Beckett, D. (2007). Nucleation of an allosteric response via ligand-
induced loop folding. J Mol Biol 373, 96-111.
Nah, B.N.L. (2009). High-throughput assays for biotin protein ligase: A novel antibiotic
target. Master's thesis, 1-145.
Rhee, S.Y., Liu, T.F., Holmes, S.P., and Shafer, R.W. (2007). HIV-1 subtype B protease
and reverse transcriptase amino acid covariation. PLoS Comput Biol 3, e87.
Schatz, P.J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-
modifying enzyme: a 13 residue consensus peptide specifies biotinylation in
Escherichia coli. Biotechnology (N Y) 11, 1138-1143.
Slavoff, S.A., Chen, I., Choi, Y.A., and Ting, A.Y. (2008). Expanding the substrate
tolerance of biotin ligase through exploration of enzymes from diverse species. J
Am Chem Soc 130, 1160-1162.
Takagi, M., Nishioka, M., Kakihara, H., Kitabayashi, M., Inoue, H., Kawakami, B., Oka,
M., and Imanaka, T. (1997). Characterization of DNA polymerase from
Pyrococcus sp. strain KOD1 and its application to PCR. Appl Environ Microbiol
63, 4504-4510.
Uttamapinant, C., White, K.A., Baruah, H., Thompson, S., Fernandez-Suarez, M.,
Puthenveetil, S., and Ting, A.Y. (2010). A fluorophore ligase for site-specific
protein labeling inside living cells. Proc Natl Acad Sci U S A 107, 10914-10919.
Weaver, L.H., Kwon, K., Beckett, D., and Matthews, B.W. (2001). Corepressor-induced
organization and assembly of the biotin repressor: a model for allosteric activation
of a transcriptional regulator. Proc Natl Acad Sci U S A 98, 6045-6050.
Wilson, K.P., Shewchuk, L.M., Brennan, R.G., Otsuka, A.J., and Matthews, B.W. (1992).
Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure
delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci U S A 89,
9257-9261.

101
Wu, S.C., and Wong, S.L. (2004). Development of an enzymatic method for site-specific
incorporation of desthiobiotin to recombinant proteins in vitro. Anal Biochem
331, 340-348.

102
Chapter 4: demonstration of cooperation and co-evolution of synthetic
operon
INTRODUCTION
Circuits and pathways in living organisms are complex, as they are finely tuned
and co-optimized throughout the course of evolution. An operon is a simple form of a
genetic circuit which contains a cluster of genes under the control of a single promoter.
For example, the canonical lac operon contains lacY (beta-lactose permease), lacZ (beta-
galactosidase), and lacA (thiogalactoside acetyltransferase) driven by a single promoter
(Figure 4-1). The lac operon is required for the lactose catabolism and is tightly
controlled to response to the environment stimulus by cooperation of lacY and lacZ. The
beta-lactose permease transfers lactose into cells and then the beta-galactosidase digests
lactose to galactose and glucose (Juers, Matthews et al. 2012). Therefore, bacteria use
simple sugars as carbon source. Although the role of thiogalactoside acetyltransferase is
controversial, it has been hypothesized to play a role in detoxification (Roderick 2005).
Tight regulation of lac operon is based on the availability of lactose. Without
lactose present, the lac I (repressor) is constitutively expressed and binds the operator (O)
which is just downstream of the lac operon promoter (P). The repressor blocks the RNA
polymerase from binding the promoter and transcribing lacY, lacZ and lacA in order to
avoid unnecessary enzyme production (Wilson, Zhan et al. 2007). With lactose present,
the metabolite, allolactose, binds to the repressor which changes its conformation to
allow release from the operator. Once the operator is unbound, RNA polymerase can
bind to the promoter and transcribe lacY, lacZ and lacA to conduct lactose metabolism.
The long evolutionary history of the lac operon has allowed for fine tuning of
individual genes, as well as, the interplay between genes in the operon. The creation of a
synthetic operon with analogous complexity and evolved cooperativity would enable

103
better understanding of the process of natural evolution. Through the course of an in vitro
evolutionary experiment, similar evolutionary development can be observed as evolution
orchestrates the co-optimization of parts (Davidson, Meyer et al. 2012).
Figure 4-1: Structure of Lac operon. The lac operon is composed of three structure
genes, lacY, lacZ and lacA, which encode three different enzymes for
lactose metabolism. Besides three structure genes, it has one promoter (P),
operator (O) and lacI (repressor). The lacI is constitutively expressed to
block the transcription of structure genes by binding operator and prevent
the wasting of enzymes production without lactose present as a carbon
source in the environment. These regulators tightly control the cooperation
of these three structures for lactose metabolism in bacteria.
Biotin ligase and streptavidin are functionally relevant proteins and have been
evolved by our lab (Chapman-Smith and Cronan 1999; Levy and Ellington 2008;
Magalhaes, Czekster et al. 2011) (Figure 4-2). It would be interesting to combine a biotin
ligase (BirA) with streptavidin (SA) under the same T7 promoter to demonstrate
cooperation and co-evolution of synthetic operons. The idea is to demonstrate the
synthetic operon in use of desthiobiotin.
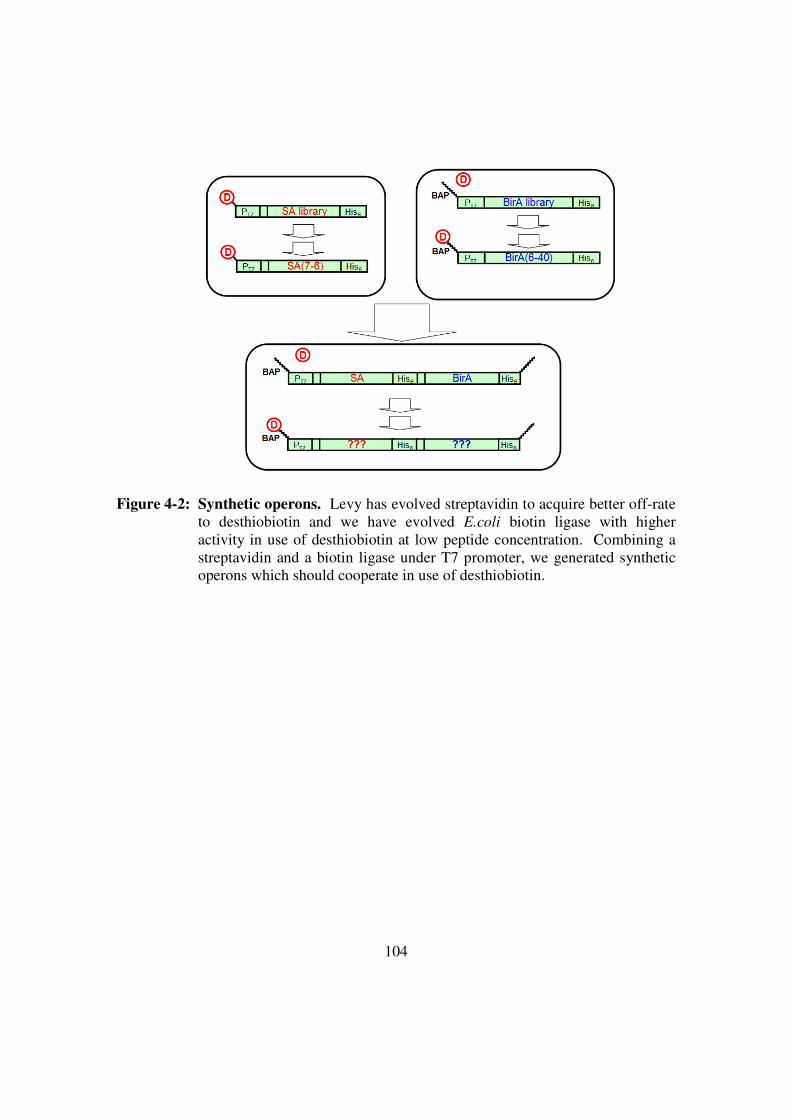
104
Figure 4-2: Synthetic operons. Levy has evolved streptavidin to acquire better off-rate
to desthiobiotin and we have evolved E.coli biotin ligase with higher
activity in use of desthiobiotin at low peptide concentration. Combining a
streptavidin and a biotin ligase under T7 promoter, we generated synthetic
operons which should cooperate in use of desthiobiotin.

105
RESULTS AND CONCLUSIONS
Construction scheme:
In order to demonstrate cooperation and co-evolution of synthetic operons, two
genes (streptavidin and biotin ligase) with two versions (wild-type version optimized for
biotin and evolved version optimized for desthiobiotin) were chosen to generate four
different synthetic operons (Figure 4-3) (Chapman-Smith and Cronan 1999; Levy and
Ellington 2008; Nah 2009; Magalhaes, Czekster et al. 2011). The constructs: [SAwt-
BirAwt], [SAwt-BirA6-40], [SA7-6-BirAwt] and [SA7-6-BirA6-40] were placed under the same
T7 promoter for transcription. Each of these operons also had two separate ribosomal
binding sites for translation and all conjugated with biotin acceptor peptide (BAP) at two
ends (5’ of DNA). BAP is a peptide substrate for biotin ligase (Schatz 1993). These
four different synthetic operons (as a representative pool) were selected in use of
desthiobiotin.

106
Figure 4-3: A Representative synthetic operon pool. Two different streptavidin, wild-
type streptavidin (SAwt), streptavidin variant (SA7-6) and two different biotin
ligases, wild-type BirA (BirAwt) and BirA variant (BirA6-40) were used to
generate four synthetic operons as a representative pool. They were [SAwt-
BirAwt], [SAwt-BirA6-40], [SA7-6-BirAwt] and [SA7-6-BirA6-40]. Each of
synthetic operon has unique restriction site in the middle of construct and
can be differentiated from each other by restriction enzyme digestion.
Selection scheme:
A selection scheme was designed to test these 4 synthetic operons (Figure 4-4).
In order to “survive” in this selection scheme, two genes in the same operon have to
cooperate together in emulsions. First, a representative pool (four operons) is conjugated
to BAP which is a substrate for biotin ligase (BAP:operon conjugates). Then they are
emulsified with E.coli lysates to generate compartments which is less than one conjugate
/ compartment and the conjugates are transcribed and translated in emulsions. A
synthesized biotin ligase recognizes desthiobiotin and then attaches it on BAP of
conjugate. A synthesized streptavidin binds the modified conjugate. After breaking the
emulsions, anti-His antibody agarose beads capture the complex (His-tagged streptavidin
and desthiobiotinylated BAP:operon conjugates). Finally, selected operon recovers by
PCR amplification and analyzes by restriction enzymes. Different synthetic operons
survive in different selection conditions. We would expect that [SAwt-BirAwt] survives
with biotin present and [SA7-6-BirA6-40] survives with desthiobiotin present in the
selection. The selection platform is water-in-oil emulsions and is similar to a scheme we
implemented in Chapter 3 (Figure 4-4) (Griffiths and Tawfik 2003; Miller, Bernath et al.
2006; Levy and Ellington 2008; Davidson, Dlugosz et al. 2009).
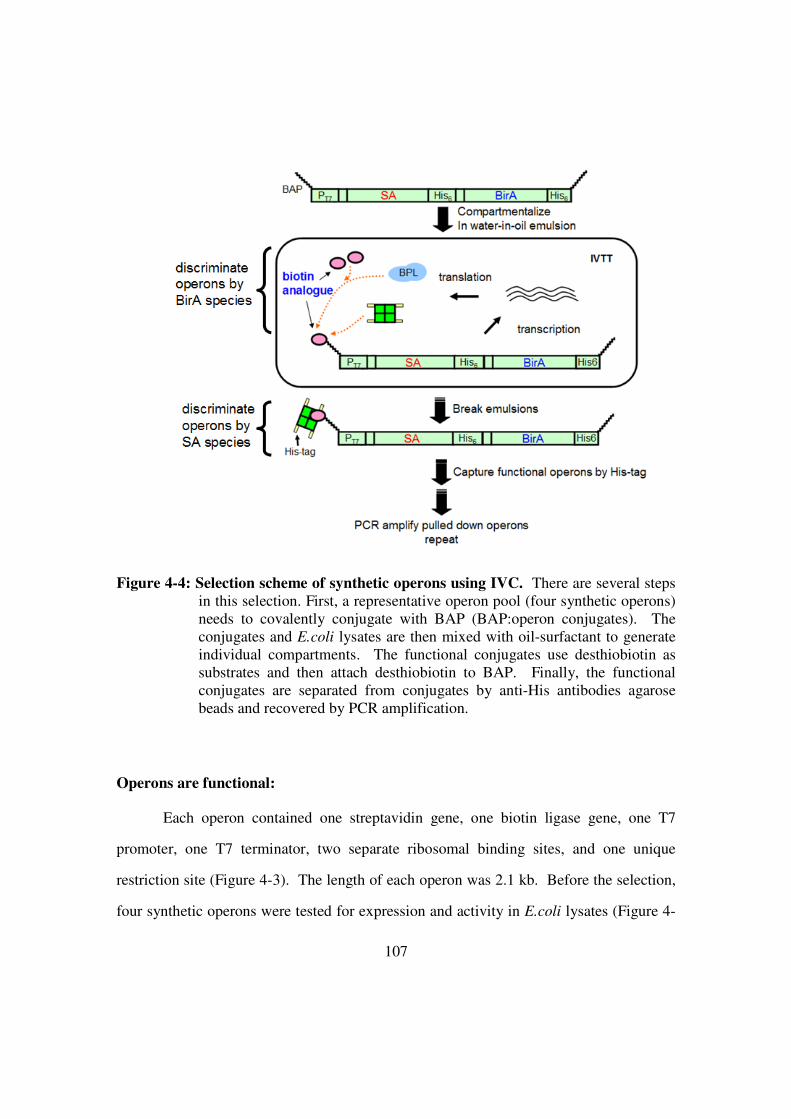
107
Figure 4-4: Selection scheme of synthetic operons using IVC. There are several steps
in this selection. First, a representative operon pool (four synthetic operons)
needs to covalently conjugate with BAP (BAP:operon conjugates). The
conjugates and E.coli lysates are then mixed with oil-surfactant to generate
individual compartments. The functional conjugates use desthiobiotin as
substrates and then attach desthiobiotin to BAP. Finally, the functional
conjugates are separated from conjugates by anti-His antibodies agarose
beads and recovered by PCR amplification.
Operons are functional:
Each operon contained one streptavidin gene, one biotin ligase gene, one T7
promoter, one T7 terminator, two separate ribosomal binding sites, and one unique
restriction site (Figure 4-3). The length of each operon was 2.1 kb. Before the selection,
four synthetic operons were tested for expression and activity in E.coli lysates (Figure 4-

108
5, western blot). Four operons were separately incubated with E.coli lysates at 30oC for 1
hour and then divided into two sets for gel electrophoresis and then western blot. One set
of samples was boiled and the other set of samples was not boiled before gel
electrophoresis (Figure 4-5). Lane1, 2, 3, and 4 are [SAwt-BirAwt], [SAwt-BirA6-40], [SA7-
6-BirAwt] and [SA7-6-BirA6-40], respectively and lane5, 6, 7, and 8 are aforementioned
operons with additional CAT-BAP (Chloramphenicol AcetyltTansferase with Biotin
Acceptor Peptide) DNA as substrates for biotin ligase. The western blot was stained
with alkaline phosphatase conjugated anti-His antibodies (aHis Ab-AP) (left part of
Figure 4-5) to detect synthesized His-tagged biotin ligases and streptavidin and then
further stained with alkaline phosphatase conjugated streptavidin (SA-AP) to detect
biotinylated CAT-BAP (right part of Figure 4-5). The expression of biotin ligases and
streptavidin of four operons were good (left part of Figure 4-5, lane1-4). Moreover, since
the protein samples were not boiled before gel electrophoresis the complex of
biotinylated CAT-BAP and streptavidin was not disrupted. That said, the complex
(shifted signal) was detected by aHis Ab-AP (left part of Figure4-5, lane 5-8). The
biotinylated CAT-BAP (not the complex) was differentiated from the shifted complex by
SA-AP (right part of Figure 4-5, lane 5-8). Taken together, these 4 synthetic operons
expressed and functioned well in the in vitro transcription and translation reaction.

109
Figure 4-5: Protein expression of synthetic operons (western blot). We tested 4
synthetic operons if they were all able to express functional protein in E.coli
lysates. (Left: stained by aHis Ab-AP) Lane1 to lane4 were [SAwt-BirAwt],
[SAwt-BirA6-40], [SA7-6-BirAwt] and [SA7-6-BirA6-40], respectively and all of
them expressed well. Lane5 to lane8 were aforementioned operons with
additional CAT-BAP (substrate of biotin ligase) and displayed different
pattern compared to lane1 to lane4. It indicated CAT-BAP was biotinylated
by biotin ligases and formed shifted complex with streptavidin. (Right:
stained with SA-AP) In addition, CAT-BAP (not complex) was biotinylated
by biotin ligase and was stained by SA-AP.
Mock selection:
In order to demonstrate the feasibility of selection scheme in emulsions, a mock
selection was performed. We generated three pools to test and they were three ratios
(0:1, 1:9 and 1:99) of mixing “non-functional” operon (no BAP-tagged) and “functional”
operon (BAP-tagged) (Figure 4-6). Without BAP-tagged, operon was not recovered in
the selection. Additionally, an Xho I restriction site was introduced into non-functional
operon. Thus it was able to differentiate from functional operon after selection. One
round of selection was performed with 20 uM of what present in the emulsions and we

110
found functional operon in 1:9 pool was enriched (Figure 4-6). This result proved the
selection worked. Although we did not find significant enrichment of functional operon
in the 1:99 pool it probably needed more rounds of selection to achieve it.
Figure 4-6: Mock selection. Non-functional and functional operons were mixed in three
different ratios (0:1, 1:9, and 1:99) for the mock selection. Non-functional
operons were not BAP-tagged and had an Xho restriction site. Thus it was
able to differentiate from functional operons in Xho I restriction enzyme
reaction. After one round of selection, we found functional operons in the
1:9 pool were enriched and proved the feasibility of selection scheme.
The selection of the representative pool (4 operons)
After mock selection, the real selection was implemented in emulsions with 50
uM desthiobiotin present. Four BAP:operon conjugates were added into in vitro
transcription and translation reagents (RTS E.coli HY kit, Roche) and then emulsified to
generate small vesicles with one conjugate / compartment. After one round of selection,
recovered operons were further digested with 4 restriction enzymes (Cla I, Sac I, Sal I or
Xho I) individually for differentiation (Figure 4-7). Comparing the signal intensity of
digested fragments, we were able to find which operon was enriched in the selection.

111
However, there was no enrichment in anyone of 4 operons in the selection (Figure 4-7)
and the result was consistent among several repeats.
Figure 4-7: One round of selection. Four operon conjugates were mixed in the same
amount and competed in the selection with 50 uM desthiobiotin present in
emulsions. After one round of selection, we expected to recover [SA7-6-
BirA6-40] more than the other three. However, there was no difference
among all synthetic operons.
Non-specific DNA binding of anti-His antibody agarose beads
As previously demonstrated, the BirA6-40 is a better enzyme in use of
desthiobiotin compared to BirAwt with 50 uM desthiobiotin present in the selection
(Chapter 3). In addition, the koff of SA7-6 is 50-fold better than SAwt. That indicates we
should have more SA7-6 binding on desthiobiotinylated operons than SAwt after washes
(Levy and Ellington 2008). In order to figure out why the selection did not work, we
dissected the selection into two steps. First, [SAwt-BirA6-40] and [SA7-6-BirA6-40] should
modify themselves more than [SAwt-BirAwt] and [SA7-6-BirAwt] with 50 uM desthiobiotin
present in the selection. Second, after pulling out by anti-His antibody agarose beads and
washes, SA7-6 should bind tighter to modified BAP:operon conjugates than SAwt in
washes. Taken together, we should see [SAwt-BirA6-40] and [SA7-6-BirA6-40] modify

112
themselves more than [SAwt-BirAwt] and [SA7-6-BirAwt] in the first step of selection.
Subsequently, we should see more [SA7-6-BirA6-40] than [SAwt-BirA6-40] after washes.
In order to check the first step, we did PCR cleanup after the selection and then
used streptavidin magnetic beads to capture functional conjugates. Captured conjugates
were then amplified by PCR. Thus, we avoided the interference of synthesized
streptavidin and anti-His antibody agarose beads in the selection. It was clear that we had
lower signals in background than recovered functional conjugates (Figure 4-8 B, lane1
and lane2). Moreover, we used four restriction enzymes to differentiate which operon
used desthiobiotin better than other operons (Figure 4-8 D). The result showed [SAwt-
BirA6-40] and [SA7-6-BirA6-40] used desthiobiotin better than [SAwt-BirAwt] and [SA7-6-
BirAwt]. Therefore, the first step of the selection was not a problem.
Besides using streptavidin magnetic beads, we also performed a selection using
anti-His antibody agarose beads to pull out functional conjugates. However, when we
performed PCR amplification to recover operons from the selection we found high
background signal (untagged operon) compared to BAP:operon conjugates (Figure 4-8 C,
lane5 and lane6). It was consistent across experiments. Since untagged operon should
not be recovered from the selection it was very possible that anti-His antibody agarose
beads had high non-specific DNA binding and covered real signal.
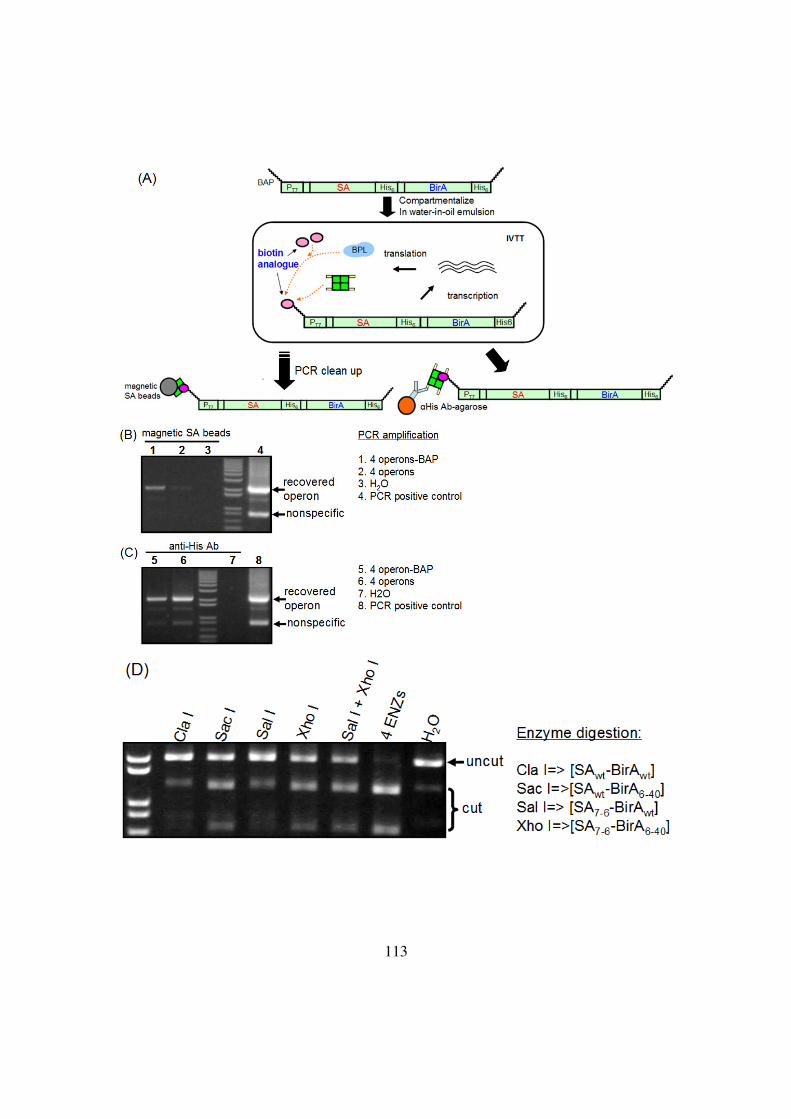
113

114
Figure 4-8: Dissection of the selection. (A) The selection had two steps. First,
synthesized biotin ligase modified BAP of conjugates with desthiobiotin and
then synthesized streptavidin bound to modified conjugates in the
compartment. Second, anti-His antibodies agarose beads were used to pull
out the complex (modified conjugates with streptavidin). And then we
recovered operon by PCR amplification. BirA6-40 has better activity in use
of desthiobiotin and SA7-6 has better off rate in binding to desthiobiotin
compared to their corresponding wild-type protein. Therefore, we should
pull out [SA7-6-BirA6-40] after one round of selection with 50 uM
desthiobiotin present. However, we did not have more [SA7-6-BirA6-40] back
in the selection. (B and C) After breaking emulsions we used streptavidin
magnetic beads or anti-His antibodies agarose beads to pull out modified
conjugates. Then we recovered conjugates with PCR amplification.
Streptavidin magnetic beads avoided the interference of synthesized
streptavidin and anti-His antibody agarose beads in the selection. (B,
streptavidin magnetic beads) The signal of recovered functional operons in
PCR amplification (lane 1) was lower than background (lane2). (C, anti-His
Ab agarose beads) However, the background signal was stronger than
recovered functional conjugates. (D) Further analysis of the recovered
conjugates (B, lane1) using restriction enzymes, we found we had more
[SAwt-BirA6-40] and [SA7-6-BirA6-40] back than the other two at the first step
of selection. It indicated modification of conjugates was not a problem.
The problem should be the non-specific DNA binding to anti-His Ab
agarose beads.
Therefore, we tried different wash buffers and capture approaches to avoid non-
specific DNA binding. The original wash buffer was TBS with Tween 20 (TBST) which
was used in streptavidin selection by Levy (100 mM Tris-HCl [pH7.4], 150 mM NaCl,
0.1% Tween 20) (Levy and Ellington 2008). Therefore, we tested wash buffers with
different salt concentration (150 mM, 300 mM, 600 mM and 1 M NaCl) and there was no
significant improvement in non-specific DNA binding of anti-His Ab agarose beads.
Also, we tried longer wash time, larger volume, more washes and there were no
significant improvement. But blocking anti-His Ab agarose beads with salmon testes
DNA (Sigma) offered minor improvements. The new wash buffer was 100 mM Tri-HCl,
[pH7.4], 150 mM NaCl, 0.1% Tween 20 and washed resin 7 times with 1.2 ml buffer.

115
A selection was performed with 50 uM desthiobiotin present and used pre-
blocked anti-His Ab agarose beads (by salmon testes DNA) for capture. Also, new wash
buffer was applied for washes (100 mM Tri-HCl, [pH7.4], 150 mM NaCl, 0.1% Tween
20 and washed resin 7 times with 1.2 ml buffer). The non-specific binding was reduced
(Figure 4-9, lane 4 and lane 5). We further analyzed the selection (Figure 4-9, lane 5) by
restriction enzymes but still did not see any significant enrichment in any operons. It
indicated that the non-specific DNA binding was still too high compared to enriched
signal.
Figure 4-9: One round of selection with new wash buffer. (top figure) After one
round of real selection it was clear that we lowered the background with
compared to recovered operons (lane4 and lane5). (bottom figure)
However, we still did not see any difference among 4 operons after one
round of real selection.

116
Besides the aforementioned tests, we also tested two beads to avoid non-specific
DNA binding. They were anti-His Ab resin (R&D systems) and cobalt-immobilized
metal affinity chromatography (IMAC) magnetic beads (Invitrogen). Three of them
(including original anti-His Ab from Sigma) were tested (but not in emulsions and also
had no free biotin in the reaction) for non-specific binding to beads (Figure 4-10). The
result showed the original beads were better than others in recovery. By comparing the
signal of untagged operon (lane 1) and biotin-tagged operon (lane 2), the anti-His Ab
agarose beads may be the best one so far.
Also, it should be noted the recovery of biotin tagged operon in this test was
better than in the selection because all operons were biotin-tagged. Furthermore, there
was no free biotin in the reaction. Therefore, there was no competition to binding sites of
synthesized streptavidin and it improved the recovery of biotin-tagged operon in this test.
Figure 4-10:Different capture approach. We suspected the problem was non-specific
DNA binding to agarose beads so we compared different anti-His antibodies
(Sigma or R&D) and Cobalt-IMAC magnetic beads to capture complex
(streptavidin with modified operon). However, the original anti-His
antibodies was the best one compared to others.

117
In fact, besides the nonspecific binding to beads, there were several factors that
also affected the selection. First, the operon (2.1 kb) is longer than the linear construct
(0.7 kb) used in Levy’s work and it has been found the short DNA has less non-specific
binding to beads compared to 2.1 kb linear construct. Second, free desthiobiotin in the
selection also affects the recovery of modified operon because it competes binding sites
of streptavidin. Higher recovery would help to differentiate from background (non-
specific binding). Third, the expression of both biotin ligase and streptavidin in an
emulsion vesicle is not as high as only expression of biotin ligase or streptavidin in an
emulsion vesicle. It would reduce the modification of BAP-tagged operon (by biotin
ligase) and recovery of modified operon (by streptavidin).
Taken together, non-specific binding to beads and the competition effect of free
desthiobiotin are likely the two major factors. Since we would not be able to reduce the
size of operons the best way is to find a better capture approach for the modified operon.
Optimize the use of Co-IMAC magnetic beads would be an option because we already
knew magnetic beads cause much lower background than agarose beads. In the
experiment of “evolving of biotin ligase for desthiobiotin”, the linear construct was 1.3
kb and did not show significant non-specific binding to magnetic beads. Thus the Co-
IMAC magnetic beads might a potential candidate. In addition, the free desthiobiotin in
the selection is 50 uM which is around 1.5 x 105 molecules of free desthiobiotin in an
emulsion vesicle. Although we don’t know the concentration of synthesized streptavidin
in an emulsion vesicle it would be better to reduce free desthiobiotin for the competition
of binding sites. However, reducing desthiobiotin concentration in the reaction may also
affect the activity of biotin ligase. Therefore, these two factors influence the selection
conversely and would be difficult to tune. Although they affect the selection in opposite
ways, the concentration of desthiobiotin in modification reaction is not a limiting step but

118
the concentration of BAP (one BAP-tagged operon in one compartment: 0.3 nM).
Therefore, we would be able to reduce the concentration of free desthiobiotin without
influence the modification reaction and help to recover more modified operons from the
selection.

119
REFERENCES
Chapman-Smith, A., and Cronan, J.E., Jr. (1999). The enzymatic biotinylation of
proteins: a post-translational modification of exceptional specificity. Trends
Biochem Sci 24, 359-363.
Davidson, E.A., Dlugosz, P.J., Levy, M., and Ellington, A.D. (2009). Directed evolution
of proteins in vitro using compartmentalization in emulsions. Curr Protoc Mol
Biol Chapter 24, Unit 24 26.
Davidson, E.A., Meyer, A.J., Ellefson, J.W., Levy, M., and Ellington, A.D. (2012). An in
vitro autogene. ACS Synth Biol 1, 190-196.
Griffiths, A.D., and Tawfik, D.S. (2003). Directed evolution of an extremely fast
phosphotriesterase by in vitro compartmentalization. EMBO J 22, 24-35.
Juers, D.H., Matthews, B.W., and Huber, R.E. (2012). LacZ beta-galactosidase: structure
and function of an enzyme of historical and molecular biological importance.
Protein Sci 21, 1792-1807.
Levy, M., and Ellington, A.D. (2008). Directed evolution of streptavidin variants using in
vitro compartmentalization. Chem Biol 15, 979-989.
Magalhaes, M.L., Czekster, C.M., Guan, R., Malashkevich, V.N., Almo, S.C., and Levy,
M. (2011). Evolved streptavidin mutants reveal key role of loop residue in high-
affinity binding. Protein Sci 20, 1145-1154.
Miller, O.J., Bernath, K., Agresti, J.J., Amitai, G., Kelly, B.T., Mastrobattista, E., Taly,
V., Magdassi, S., Tawfik, D.S., and Griffiths, A.D. (2006). Directed evolution by
in vitro compartmentalization. Nat Methods 3, 561-570.
Nah, B.N.L. (2009). High-throughput assays for biotin protein ligase: A novel antibiotic
target. Master's Thesis, 1-145.
Roderick, S.L. (2005). The lac operon galactoside acetyltransferase. C R Biol 328, 568-
575.
Schatz, P.J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-
modifying enzyme: a 13 residue consensus peptide specifies biotinylation in
Escherichia coli. Biotechnology (N Y) 11, 1138-1143.
Wilson, C.J., Zhan, H., Swint-Kruse, L., and Matthews, K.S. (2007). The lactose
repressor system: paradigms for regulation, allosteric behavior and protein
folding. Cell Mol Life Sci 64, 3-16.

120
Bibliography
Agresti, J.J., Kelly, B.T., Jaschke, A., and Griffiths, A.D. (2005). Selection of ribozymes
that catalyse multiple-turnover Diels-Alder cycloadditions by using in vitro
compartmentalization. Proc Natl Acad Sci U S A 102, 16170-16175.
Amitai, G., Gupta, R.D., and Tawfik, D.S. (2007). Latent evolutionary potentials under
the neutral mutational drift of an enzyme. HFSP J 1, 67-78.
Anderson, M.E. (1998). Glutathione: an overview of biosynthesis and modulation. Chem
Biol Interact 111-112, 1-14.
Bernath, K., Hai, M., Mastrobattista, E., Griffiths, A.D., Magdassi, S., and Tawfik, D.S.
(2004). In vitro compartmentalization by double emulsions: sorting and gene
enrichment by fluorescence activated cell sorting. Anal Biochem 325, 151-157.
Bertschinger, J., and Neri, D. (2004). Covalent DNA display as a novel tool for directed
evolution of proteins in vitro. Protein Eng Des Sel 17, 699-707.
Bochkov, Y.A., and Palmenberg, A.C. (2006). Translational efficiency of EMCV IRES
in bicistronic vectors is dependent upon IRES sequence and gene location.
Biotechniques 41, 283-284, 286, 288 passim.
Braunagel, M., and Little, M. (1997). Construction of a semisynthetic antibody library
using trinucleotide oligos. Nucleic Acids Res 25, 4690-4691.
Cellarier, E., Durando, X., Vasson, M.P., Farges, M.C., Demiden, A., Maurizis, J.C.,
Madelmont, J.C., and Chollet, P. (2003). Methionine dependency and cancer
treatment. Cancer Treat Rev 29, 489-499.
Chapman-Smith, A., and Cronan, J.E., Jr. (1999). The enzymatic biotinylation of
proteins: a post-translational modification of exceptional specificity. Trends
Biochem Sci 24, 359-363.
Chapman-Smith, A., Mulhern, T.D., Whelan, F., Cronan, J.E., Jr., and Wallace, J.C.
(2001). The C-terminal domain of biotin protein ligase from E. coli is required for
catalytic activity. Protein Sci 10, 2608-2617.
Chen, I., Choi, Y.A., and Ting, A.Y. (2007). Phage display evolution of a peptide
substrate for yeast biotin ligase and application to two-color quantum dot labeling
of cell surface proteins. J Am Chem Soc 129, 6619-6625.
Chen, Y., Mandic, J., and Varani, G. (2008). Cell-free selection of RNA-binding proteins
using in vitro compartmentalization. Nucleic Acids Res 36, e128.
Choi-Rhee, E., Schulman, H., and Cronan, J.E. (2004). Promiscuous protein biotinylation
by Escherichia coli biotin protein ligase. Protein Sci 13, 3043-3050.

121
Cronan, J.E. (2005). Targeted and proximity-dependent promiscuous protein
biotinylation by a mutant Escherichia coli biotin protein ligase. J Nutr Biochem
16, 416-418.
Davidson, E.A., Dlugosz, P.J., Levy, M., and Ellington, A.D. (2009). Directed evolution
of proteins in vitro using compartmentalization in emulsions. Curr Protoc Mol
Biol Chapter 24, Unit 24 26.
Davidson, E.A., Meyer, A.J., Ellefson, J.W., Levy, M., and Ellington, A.D. (2012). An in
vitro autogene. ACS Synth Biol 1, 190-196.
Doi, N., Kumadaki, S., Oishi, Y., Matsumura, N., and Yanagawa, H. (2004). In vitro
selection of restriction endonucleases by in vitro compartmentalization. Nucleic
Acids Res 32, e95.
Doi, N., and Yanagawa, H. (1999). STABLE: protein-DNA fusion system for screening
of combinatorial protein libraries in vitro. FEBS Lett 457, 227-230.
Fen, C.X., Coomber, D.W., Lane, D.P., and Ghadessy, F.J. (2007). Directed evolution of
p53 variants with altered DNA-binding specificities by in vitro
compartmentalization. J Mol Biol 371, 1238-1248.
Fernandez-Gacio, A., Uguen, M., and Fastrez, J. (2003). Phage display as a tool for the
directed evolution of enzymes. Trends Biotechnol 21, 408-414.
Fromant, M., Blanquet, S., and Plateau, P. (1995). Direct random mutagenesis of gene-
sized DNA fragments using polymerase chain reaction. Anal Biochem 224, 347-
353.
Ghadessy, F.J., and Holliger, P. (2004). A novel emulsion mixture for in vitro
compartmentalization of transcription and translation in the rabbit reticulocyte
system. Protein Eng Des Sel 17, 201-204.
Ghadessy, F.J., Ong, J.L., and Holliger, P. (2001). Directed evolution of polymerase
function by compartmentalized self-replication. Proc Natl Acad Sci U S A 98,
4552-4557.
Ghadessy, F.J., Ramsay, N., Boudsocq, F., Loakes, D., Brown, A., Iwai, S., Vaisman, A.,
Woodgate, R., and Holliger, P. (2004). Generic expansion of the substrate
spectrum of a DNA polymerase by directed evolution. Nat Biotechnol 22, 755-
759.
Griffiths, A.D., and Tawfik, D.S. (2003). Directed evolution of an extremely fast
phosphotriesterase by in vitro compartmentalization. EMBO J 22, 24-35.
Gupta, R.D., Goldsmith, M., Ashani, Y., Simo, Y., Mullokandov, G., Bar, H., Ben-David,
M., Leader, H., Margalit, R., Silman, I., et al. (2011). Directed evolution of
hydrolases for prevention of G-type nerve agent intoxication. Nat Chem Biol 7,
120-125.

122
Gupta, R.D., and Tawfik, D.S. (2008). Directed enzyme evolution via small and effective
neutral drift libraries. Nat Methods 5, 939-942.
Hanes, J., and Pluckthun, A. (1997). In vitro selection and evolution of functional
proteins by using ribosome display. Proc Natl Acad Sci U S A 94, 4937-4942.
Juers, D.H., Matthews, B.W., and Huber, R.E. (2012). LacZ beta-galactosidase: structure
and function of an enzyme of historical and molecular biological importance.
Protein Sci 21, 1792-1807.
Kelly, B.T., and Griffiths, A.D. (2007). Selective gene amplification. Protein Eng Des Sel
20, 577-581.
Khersonsky, O., and Tawfik, D.S. (2010). Enzyme promiscuity: a mechanistic and
evolutionary perspective. Annu Rev Biochem 79, 471-505.
Kintses, B., van Vliet, L.D., Devenish, S.R., and Hollfelder, F. (2010). Microfluidic
droplets: new integrated workflows for biological experiments. Curr Opin Chem
Biol 14, 548-555.
Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H.,
and Mori, H. (2005). Complete set of ORF clones of Escherichia coli ASKA
library (a complete set of E. coli K-12 ORF archive): unique resources for
biological research. DNA Res 12, 291-299.
Kroemer, G., and Pouyssegur, J. (2008). Tumor cell metabolism: cancer's Achilles' heel.
Cancer Cell 13, 472-482.
Levy, M., and Ellington, A.D. (2008). Directed evolution of streptavidin variants using in
vitro compartmentalization. Chem Biol 15, 979-989.
Levy, M., Griswold, K.E., and Ellington, A.D. (2005). Direct selection of trans-acting
ligase ribozymes by in vitro compartmentalization. RNA 11, 1555-1562.
Lipovsek, D., and Pluckthun, A. (2004). In-vitro protein evolution by ribosome display
and mRNA display. J Immunol Methods 290, 51-67.
Loakes, D., Gallego, J., Pinheiro, V.B., Kool, E.T., and Holliger, P. (2009). Evolving a
polymerase for hydrophobic base analogues. J Am Chem Soc 131, 14827-14837.
Magalhaes, M.L., Czekster, C.M., Guan, R., Malashkevich, V.N., Almo, S.C., and Levy,
M. (2011). Evolved streptavidin mutants reveal key role of loop residue in high-
affinity binding. Protein Sci 20, 1145-1154.
Margulies, M., Egholm, M., Altman, W.E., Attiya, S., Bader, J.S., Bemben, L.A., Berka,
J., Braverman, M.S., Chen, Y.J., Chen, Z., et al. (2005). Genome sequencing in
microfabricated high-density picolitre reactors. Nature 437, 376-380.
Mastrobattista, E., Taly, V., Chanudet, E., Treacy, P., Kelly, B.T., and Griffiths, A.D.
(2005). High-throughput screening of enzyme libraries: in vitro evolution of a

123
beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem
Biol 12, 1291-1300.
Miller, O.J., Bernath, K., Agresti, J.J., Amitai, G., Kelly, B.T., Mastrobattista, E., Taly,
V., Magdassi, S., Tawfik, D.S., and Griffiths, A.D. (2006). Directed evolution by
in vitro compartmentalization. Nat Methods 3, 561-570.
Naganathan, S., and Beckett, D. (2007). Nucleation of an allosteric response via ligand-
induced loop folding. J Mol Biol 373, 96-111.
Nah, B.N.L. (2009). High-throughput assays for biotin protein ligase: A novel antibiotic
target. Master's Thesis, 1-145.
Ong, J.L., Loakes, D., Jaroslawski, S., Too, K., and Holliger, P. (2006). Directed
evolution of DNA polymerase, RNA polymerase and reverse transcriptase activity
in a single polypeptide. J Mol Biol 361, 537-550.
Parent, K.N., Ranaghan, M.J., and Teschke, C.M. (2004). A second-site suppressor of a
folding defect functions via interactions with a chaperone network to improve
folding and assembly in vivo. Mol Microbiol 54, 1036-1050.
Pavillard, V., Nicolaou, A., Double, J.A., and Phillips, R.M. (2006). Methionine
dependence of tumours: a biochemical strategy for optimizing paclitaxel
chemosensitivity in vitro. Biochem Pharmacol 71, 772-778.
Ramsay, N., Jemth, A.S., Brown, A., Crampton, N., Dear, P., and Holliger, P. (2010).
CyDNA: synthesis and replication of highly Cy-dye substituted DNA by an
evolved polymerase. J Am Chem Soc 132, 5096-5104.
Reiersen, H., Lobersli, I., Loset, G.A., Hvattum, E., Simonsen, B., Stacy, J.E., McGregor,
D., Fitzgerald, K., Welschof, M., Brekke, O.H., et al. (2005). Covalent antibody
display--an in vitro antibody-DNA library selection system. Nucleic Acids Res
33, e10.
Rhee, S.Y., Liu, T.F., Holmes, S.P., and Shafer, R.W. (2007). HIV-1 subtype B protease
and reverse transcriptase amino acid covariation. PLoS Comput Biol 3, e87.
Roberts, R.W., and Szostak, J.W. (1997). RNA-peptide fusions for the in vitro selection
of peptides and proteins. Proc Natl Acad Sci U S A 94, 12297-12302.
Roderick, S.L. (2005). The lac operon galactoside acetyltransferase. C R Biol 328, 568-
575.
Schatz, P.J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-
modifying enzyme: a 13 residue consensus peptide specifies biotinylation in
Escherichia coli. Biotechnology (N Y) 11, 1138-1143.
Sepp, A., and Choo, Y. (2005). Cell-free selection of zinc finger DNA-binding proteins
using in vitro compartmentalization. J Mol Biol 354, 212-219.

124
Shimizu, Y., Kanamori, T., and Ueda, T. (2005). Protein synthesis by pure translation
systems. Methods 36, 299-304.
Sitaraman, K., Esposito, D., Klarmann, G., Le Grice, S.F., Hartley, J.L., and Chatterjee,
D.K. (2004). A novel cell-free protein synthesis system. J Biotechnol 110, 257-
263.
Slavoff, S.A., Chen, I., Choi, Y.A., and Ting, A.Y. (2008). Expanding the substrate
tolerance of biotin ligase through exploration of enzymes from diverse species. J
Am Chem Soc 130, 1160-1162.
Stapleton, J.A., and Swartz, J.R. (2010). Development of an in vitro
compartmentalization screen for high-throughput directed evolution of [FeFe]
hydrogenases. PLoS One 5, e15275.
Stein, V., Sielaff, I., Johnsson, K., and Hollfelder, F. (2007). A covalent chemical
genotype-phenotype linkage for in vitro protein evolution. Chembiochem 8, 2191-
2194.
Stone, E., Paley, O., Hu, J., Ekerdt, B., Cheung, N.K., and Georgiou, G. (2012). De novo
engineering of a human cystathionine-gamma-lyase for systemic (L)-Methionine
depletion cancer therapy. ACS Chem Biol 7, 1822-1829.
Takagi, M., Nishioka, M., Kakihara, H., Kitabayashi, M., Inoue, H., Kawakami, B., Oka,
M., and Imanaka, T. (1997). Characterization of DNA polymerase from
Pyrococcus sp. strain KOD1 and its application to PCR. Appl Environ Microbiol
63, 4504-4510.
Takei, Y., Kadomatsu, K., Itoh, H., Sato, W., Nakazawa, K., Kubota, S., and Muramatsu,
T. (2002). 5'-,3'-inverted thymidine-modified antisense oligodeoxynucleotide
targeting midkine. Its design and application for cancer therapy. J Biol Chem 277,
23800-23806.
Tawfik, D.S., and Griffiths, A.D. (1998). Man-made cell-like compartments for
molecular evolution. Nat Biotechnol 16, 652-656.
Thivat, E., Farges, M.C., Bacin, F., D'Incan, M., Mouret-Reynier, M.A., Cellarier, E.,
Madelmont, J.C., Vasson, M.P., Chollet, P., and Durando, X. (2009). Phase II trial
of the association of a methionine-free diet with cystemustine therapy in
melanoma and glioma. Anticancer Res 29, 5235-5240.
Thomas, T., and Thomas, T.J. (2001). Polyamines in cell growth and cell death:
molecular mechanisms and therapeutic applications. Cell Mol Life Sci 58, 244-
258.
Tokuriki, N., and Tawfik, D.S. (2009). Chaperonin overexpression promotes genetic
variation and enzyme evolution. Nature 459, 668-673.
Ullman, C.G., Frigotto, L., and Cooley, R.N. (2011). In vitro methods for peptide display
and their applications. Brief Funct Genomics 10, 125-134.

125
Utada, A.S., Lorenceau, E., Link, D.R., Kaplan, P.D., Stone, H.A., and Weitz, D.A.
(2005). Monodisperse double emulsions generated from a microcapillary device.
Science 308, 537-541.
Uttamapinant, C., White, K.A., Baruah, H., Thompson, S., Fernandez-Suarez, M.,
Puthenveetil, S., and Ting, A.Y. (2010). A fluorophore ligase for site-specific
protein labeling inside living cells. Proc Natl Acad Sci U S A 107, 10914-10919.
Williams, R., Peisajovich, S.G., Miller, O.J., Magdassi, S., Tawfik, D.S., and Griffiths,
A.D. (2006). Amplification of complex gene libraries by emulsion PCR. Nat
Methods 3, 545-550.
Wilson, C.J., Zhan, H., Swint-Kruse, L., and Matthews, K.S. (2007). The lactose
repressor system: paradigms for regulation, allosteric behavior and protein
folding. Cell Mol Life Sci 64, 3-16.
Yonezawa, M., Doi, N., Kawahashi, Y., Higashinakagawa, T., and Yanagawa, H. (2003).
DNA display for in vitro selection of diverse peptide libraries. Nucleic Acids Res
31, e118.
Zahnd, C., Amstutz, P., and Pluckthun, A. (2007). Ribosome display: selecting and
evolving proteins in vitro that specifically bind to a target. Nat Methods 4, 269-
279.

126
Vita
Wei-Cheng Lu was born in Taipei, Taiwan. He entered the Tzu-Chi University
and received his B.S. degree in Laboratory Medicine and Biotechnology. After
mandatory military service he then started work for Institute of Biomedical Sciences,
Academia Sinica at Taiwan. He entered the Cell and Molecular Biology graduate
program at the University of Texas at Austin, under the supervision of Dr. Andrew D.
Ellington. His publications include:
Lu, W. C., Liu, Y. N., Kang, B. B., Chen, J. H. Trans-activation of heparanase promoter
by ETS transcription factor. Oncogene 22: 919-923, 2003
Ru, H. Y., Chen, R. L., Lu, W. C., Chen, J. H. hBUB1 defects in leukemia and
lymphoma cells. Oncogene 21: 4673-4679, 2002
KC Chen, CH Wu, CY Chang, WC Lu, Q Tseng, ZM Prijovich, W Schechinger, YC
Liaw, YL Leu and SR Roffler. Directed evolution of a lysosomal enzyme with
enhanced activity at neutral pH by mammalian cell-surface display. Chem Biol.
15:1277-86, 2008
CP Chen, YT Hsieh, ZM Prijovich, HY Chuang, KC Chen, WC Lu, Q Tseng, YL Leu,
TL Cheng, SR Roffler. ECSTASY, an adjustable membrane-tethered/soluble
protein expression system for the directed evolution of mammalian proteins
Protein Eng Des Sel., 25:367-375, 2012
Lu WC, Ellington AD. In vitro selection of proteins via emulsion compartments.
Methods. 15;60(1):75-80, 2013
Email: [email protected]
This dissertation was typed by the author.


















