
concept of biosimilars
51
CONCEPT OF BIOSIMILARS KIRANKAUR OBEROI, FIRST YEAR M.PHARM(QUALITY ASSURANCE)
-
Upload
kkoberoi -
Category
Health & Medicine
-
view
158 -
download
2
Transcript of concept of biosimilars

CONCEPT OF BIOSIMILARS
KIRANKAUR OBEROI,
FIRST YEAR M.PHARM(QUALITY ASSURANCE)

OVERVIEW
Introduction
Definition
Biologics vs. small molecules
Science behind biologics
Regulations
Approval pathways
Commercial aspects
Role of pharmacist
Conclusion
References

Introduction Biopharmaceuticals are well established in biomedicine and
have opened new therapy options particularly in disease areas
where previously no, or only insufficient, therapies were
available. Some 165 biopharmaceutical products have gained
approval.
“A protein or nucleic acid based pharmaceutical substance
used for therapeutic or in vivo diagnostic purposes, which is
produced by means other than direct extraction from a native
(non-engineered) biological source.”
A generic term for a biomolecule (e.g., proteins),including anti
bodies and nucleic acids and antisense oligonucleotides,which
are produced in a transgenic organism—
e.g., mice, livestock, fish, or plant—
and used as a therapeutic agent.

The first recombinant protein drugs, like Eli Lilly’s insulin
(developed by Genentech, Inc.), were launched in the
1980’s.
The patent and regulatory data protection periods for the
first and second waves of biopharmaceuticals based on
recombinant proteins have started to expire, opening the
way for other manufacturers to place follow-on products to
the market as this has occurred since many years for
conventional medicines containing small-molecule drug
substances.
There are fundamental differences between conventional
small-molecule based drugs and biopharmaceuticals. This
has led to the adoption of distinct legal and regulatory
frameworks for follow-on products to biopharmaceuticals
(‘‘biosimilars”) in various parts of the world.
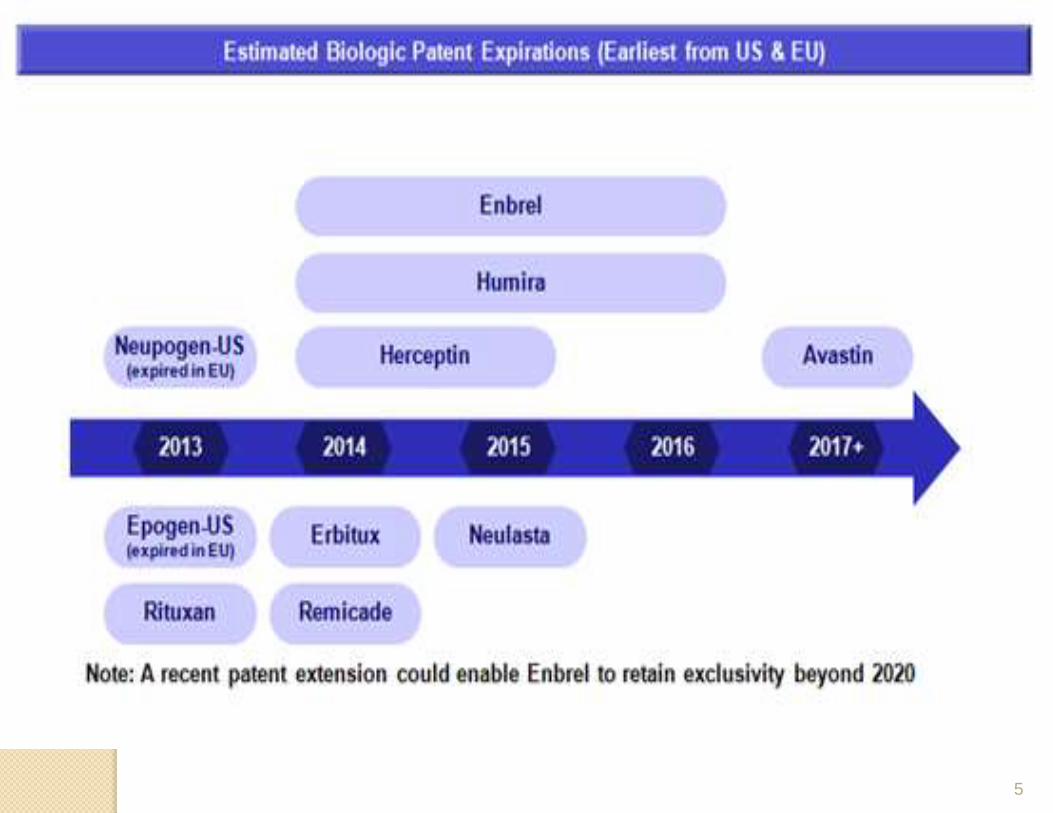
5

Definition Several terms are used in various countries for
‘‘intended copy” products to biopharmaceuticals
(e.g., Biosimilars, follow-on biologicals, follow-on
protein products, subsequent-entry biologicals,
similar biological medicinal products). In this review,
Biosimilars are defined as biological medicinal
products which are:
similar in terms of quality, safety and efficacy to an
already licensed, well-established reference
medicinal product, and
marketed by an independent applicant following
expiry of patent and regulatory data/market
exclusivity periods of the reference product.
There are many differences between
biopharmaceuticals and the small molecule drugs.

Definition1)As per EMA: A similar biological or
'biosimilar' medicine is a biological medicine
that is similar to another biological medicine
that has already been authorized for use.
Biological medicines are medicines that are
made by or derived from a biological source,
such as a bacterium or yeast. They can consist
of relatively small molecules such as human
insulin or erythropoietin, or complex
molecules such as monoclonal antibodies.
2)As per WHO: A biotherapeutic product which
is similar in terms of quality, safety and
efficacy to an already licensed reference
biotherapeutic product. 7

Definition3) ) As per US FDA: “Biological Product” in the
Public Health Service Act (PHS Act) now includes
“protein”: a virus, therapeutic serum, toxin, antitoxin,
vaccine, blood, blood component or derivative,
allergenic product, protein (except any chemically
synthesized polypeptide), or analogous product
applicable to the prevention, treatment, or cure of a
disease or condition of human beings
8
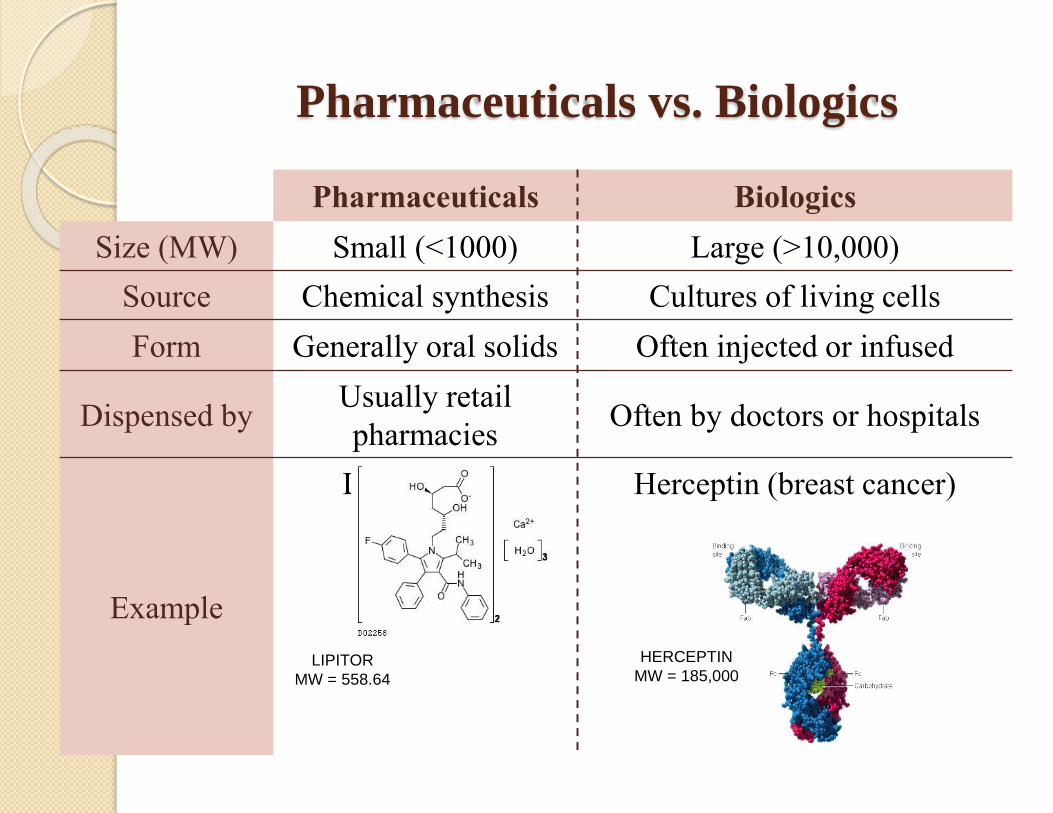
Pharmaceuticals vs. Biologics
Pharmaceuticals Biologics
Size (MW) Small (<1000) Large (>10,000)
Source Chemical synthesis Cultures of living cells
Form Generally oral solids Often injected or infused
Dispensed byUsually retail
pharmaciesOften by doctors or hospitals
Example
Lipitor (anti-
cholesterol)
Herceptin (breast cancer)
HERCEPTIN
MW = 185,000LIPITOR
MW = 558.64

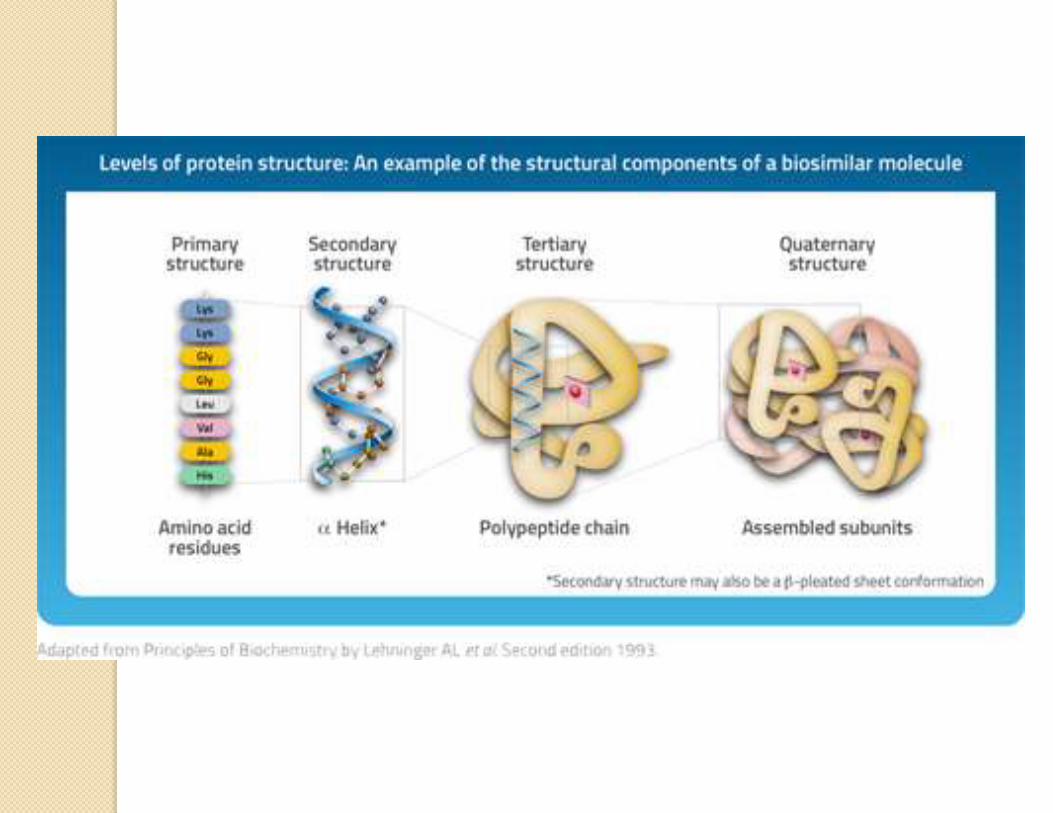
Science of biologics
COMPLEXITY OF
PROTEIN MOLECULE:
Size – Proteins have
molecular weights from
about 10,000 up to more
than 200,000 Daltons, so
typically they are 100- to
1000-fold larger molecules.
Structure-To possess
biological activity, proteins
have to adopt the correct
three-dimensionally folded
secondary, tertiary, and
quaternary structures.
Stability – Proteins are
inherently unstable molecules,
and may structurally be
damaged by heat, prolonged
storage, denaturants, organic
solvents, oxygen, pH changes,
and by other factors, leading to
reduction or complete loss of
biological activity.
Micro heterogeneity– No
protein product will leave the
producing cell and the
manufacturing process as
predicted theoretically based on
the encoding DNA sequence
alone. Proteins are modified
both biologically by the
producing cell as well as by the
process conditions.
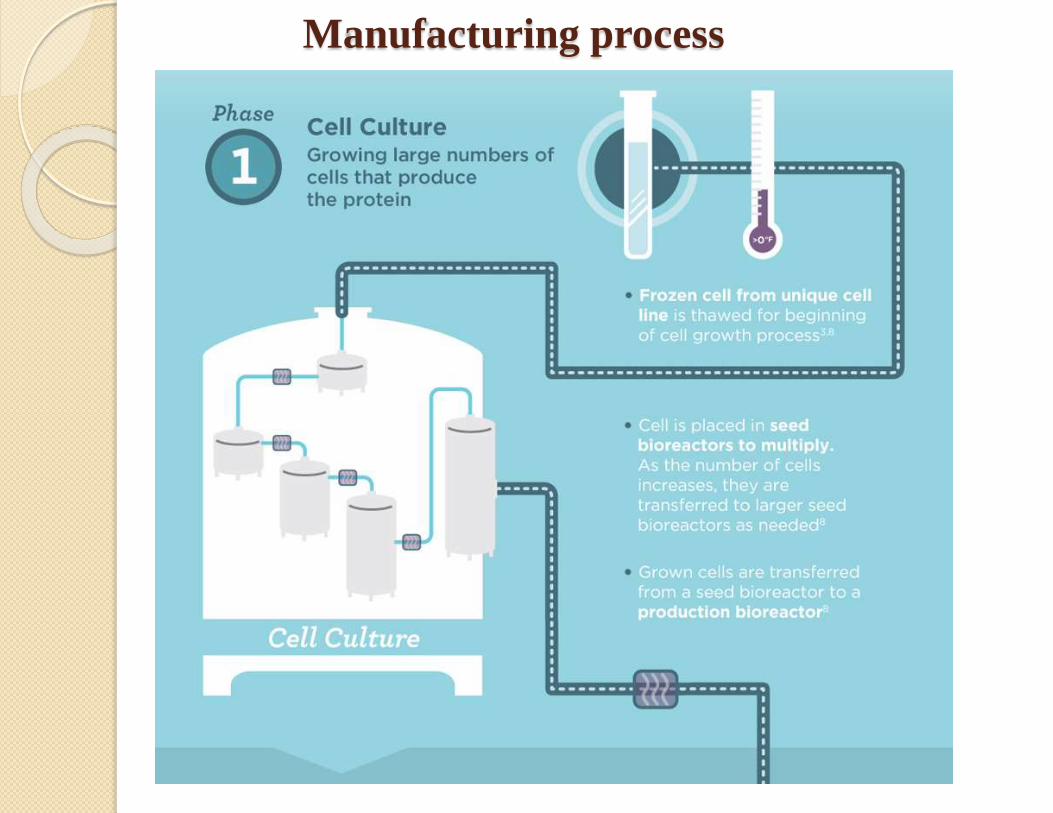
Manufacturing process
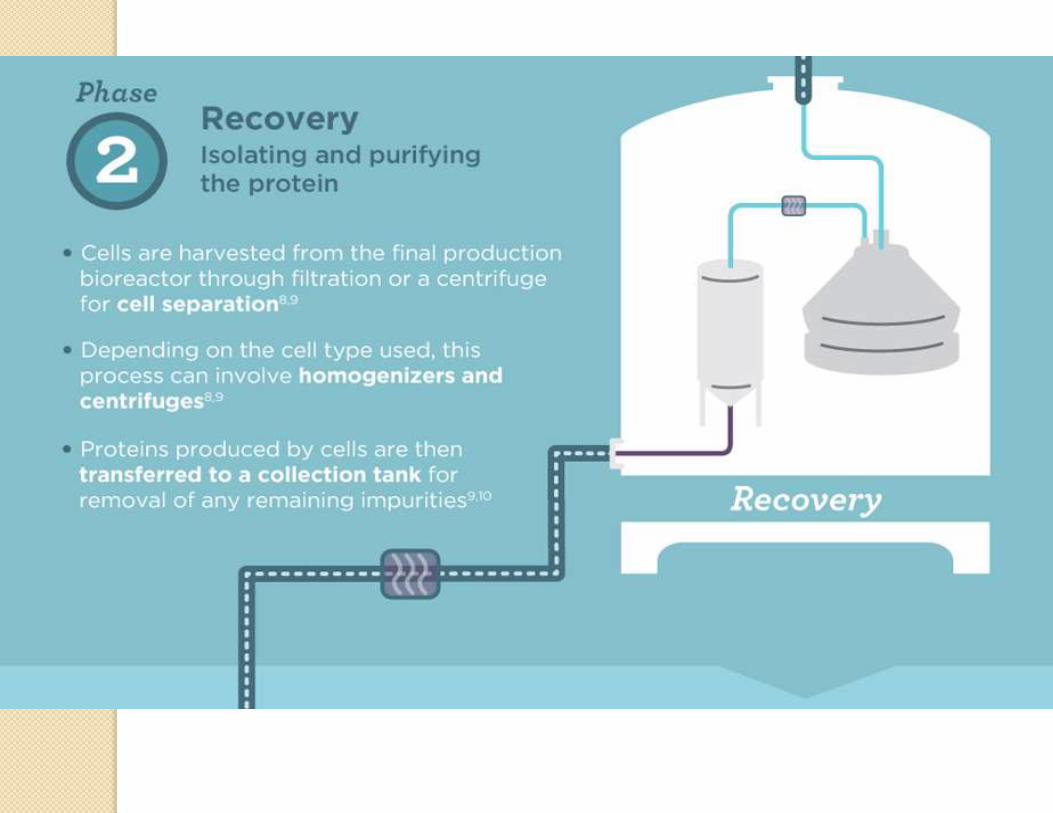
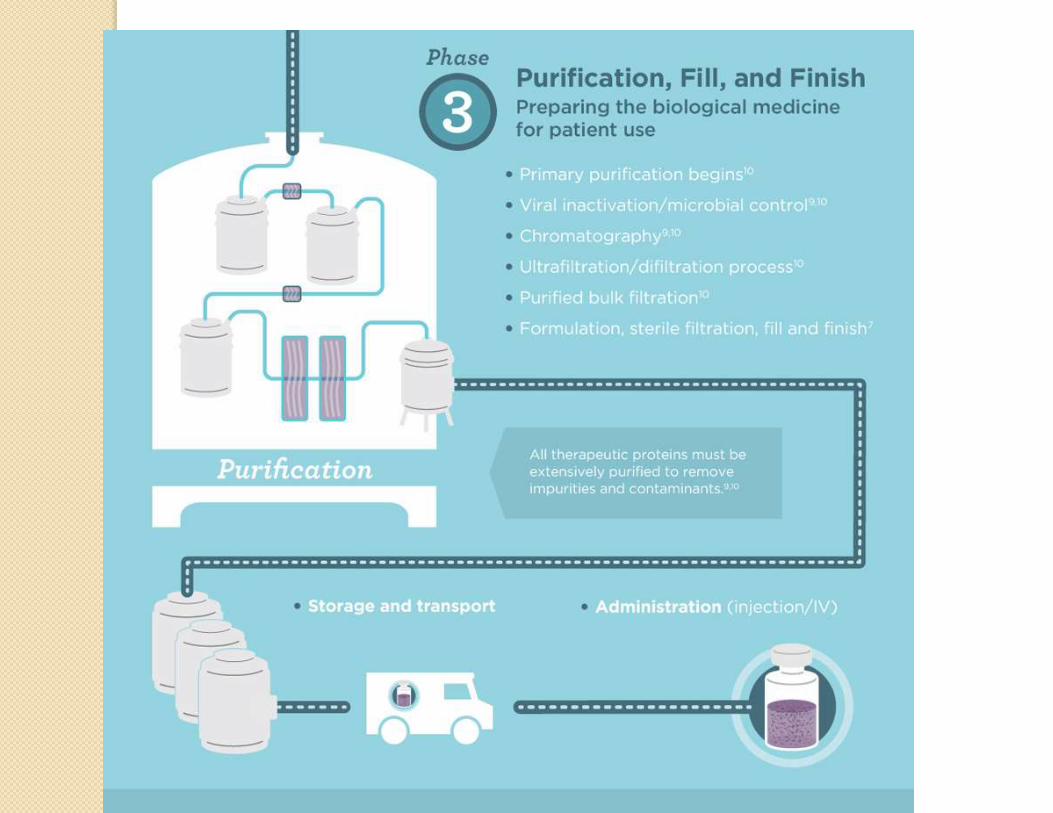
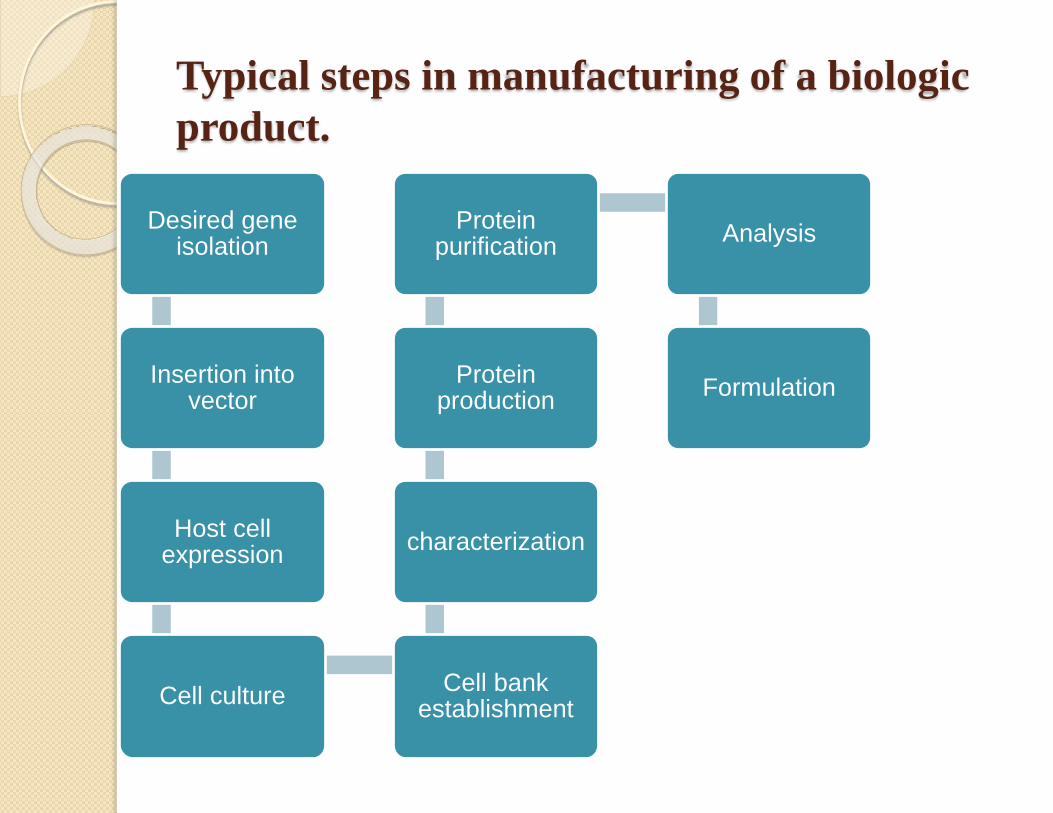
Desired gene isolation
Insertion into vector
Host cell expression
Cell cultureCell bank
establishment
characterization
Protein production
Protein purification
Analysis
Formulation
Typical steps in manufacturing of a biologic
product.

Regulation
• The regulatory pathway for approval of Biosimilars is
more complex than for the generic innovator product
because the design of a scientifically valid study to
demonstrate the similarity of a highly process-
dependent product is not easy.
• Modest differences may have clinical implications and
pose a significant risk to patient safety.
• Therefore, it is considered necessary that Biosimilars
must be assessed for clinical efficacy and safety by
valid preclinical and clinical studies before marketing
approval.

Regulation:
European Union• The European Union became the first region globally
to introduce a particular regulatory framework for
Biosimilars developed by EMEA’s Committee for
Medicinal Products for Human Use (CHMP).
• It consists of an overarching guideline , a guideline
on quality issues, a guideline on non-clinical and
clinical issues , as well as class-specific guideline
annexes describing the non-clinical and clinical
requirements for specific classes of new products. In
addition to these guidelines, product-class-specific
guidelines have been issued for the development of
Biosimilars based on recombinant erythropoietin,
somatotropin, human granulocyte colony-stimulating
factor, human insulin, recombinant IFN-a, and low
molecular weight heparins.

Regulation:
United States• In the US, after the approval of Biosimilars
Omnitropein 2006, the FDA stated that no other
Biosimilars will be approved until a specific
regulation has been issued.
• The Pathway for Biosimilars Act of 2009 and the
Patient Protection and Affordable Care Act of 2010
have provided greater clarity, and a reasonably clear
mandate from the US.

Regulation:
India• The “Guidelines on Similar Biologics” prepared by
Central Drugs Standard Control Organization
(CDSCO) and the Department of Biotechnology
(DBT) lay down the regulatory pathway for a similar
biologic claiming to be similar to an already
authorized reference biologic.
• The guidelines address the regulatory pathway
regarding manufacturing process and quality aspects
for similar biologics.
• These guidelines also address the pre-market
regulatory requirements including comparability
exercise for quality, preclinical and clinical studies
and post market regulatory requirements for similar
biologics. This was decided in the year 2012.
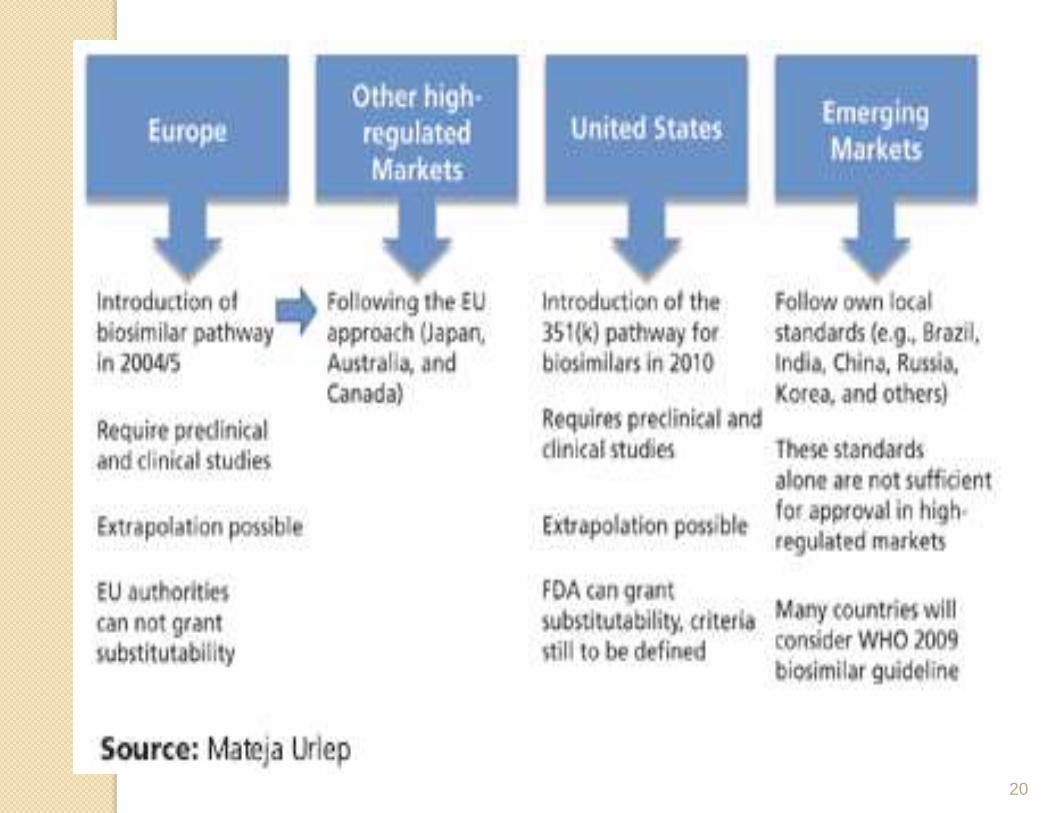
20

Approval pathway for biosimilars
In the US, the Biologics Price Competition and Innovation Act (BPCI Act, 2009) provided the pathway to create an abbreviated licensure procedure for biologic products that are demonstrated to be biosimilar to or interchangeable with a Food and Drug Administration (FDA) licensed biologic product.
In February 2012, the FDA issued three guidelines that list the requirements for biosimilar registration. The topics covered include scientific and quality considerations to demonstrate biosimilarity to a reference product and a guidance that clarifies the BPCI Act implementation.
21

Approval pathway for biosimilars
According to these guidelines, the FDA will consider
different aspects when evaluating biosimilarity, such
as product formulation, complexity, and stability
which will have a risk-based approach and will
depend on the degree of knowledge of the product
characteristics, as well as clinical experience with the
reference one.
The FDA intends to use a risk-based and facts-
focused approach for review of applications of
biosimilars, although it faces several challenges.
Once a biologic medicine has been demonstrated to
be biosimilar to the reference product, an abridged
development program for the biosimilar medicine
can be carried out. 22

Approval pathway in IndiaIn India, similar biologics are regulated by the:
1. Drugs and Cosmetics Act, 1940 (Drugs and Cosmetics Act);
2. Drugs and Cosmetics Rules, 1945 (as amended from time to
time);
3. Rules for the Manufacture, Use, Import, Export and Storage
of Hazardous Microorganisms and Genetically Engineered
Organisms or Cells, 1989 (Rules 1989) notified under the
Environment (Protection) Act, 1986;
4. Recombinant DNA Safety Guidelines, 1990;
5. Guidelines for Generating Preclinical and Clinical Data for
rDNA Vaccines, Diagnostics and other Biologicals, 1999;
6. The Central Drugs Standard Control Organization
(CDSCO) Guidance for Industry, 2008 {including: (a)
Submission of Clinical Trial Application for Evaluating Safety
and Efficacy
23

Approval pathway in India
; (b) Requirements for Permission of New Drugs Approval; (c)
Post Approval Changes in Biological Products: Quality,
Safety and Efficacy Documents; and (d) Preparation of the
Quality Information for Drug Submission for New Drug
Approval: Biotechnological/Biological Products}; and
7. Guidelines and Handbook for Institutional Biosafety
Committees (IBSCs), 2011.
In 2012, CDSCO, in collaboration with the DBT, issued
the Guidelines on Similar Biologics: Regulatory Requirements
for Marketing Authorization in India (Guidelines). The
Guidelines detail the regulatory requirements, such as data
requirements for the manufacturing, characterization,
preclinical studies and clinical trials, for receiving marketing
authorization of similar biologics. The Guidelines are
applicable for similar biologics developed in or imported into
India.24

Approval pathway in India According to the Guidelines, similar biologics are developed
through a sequential process designed to demonstrate the
similarity, by extensive characterization studies, of the
molecular and quality attributes of the similar biologic with a
reference biologic.
It is essential that the testing of the similar biologic be
sufficient to ensure that the product meet acceptable levels of
safety, efficacy and quality to ensure public health.
Generally, a reduction in data requirements is possible for
preclinical and or clinical components of the development
program by demonstrating comparability of the product (to
the reference biologic) and consistency in the production
process.
If any significant differences in safety, efficacy and quality
between the similar biologic and the reference biologic are
identified, more extensive preclinical and clinical evaluation
will be necessary. It is quite likely in this instance that the
product may not qualify as a similar biologic. 25

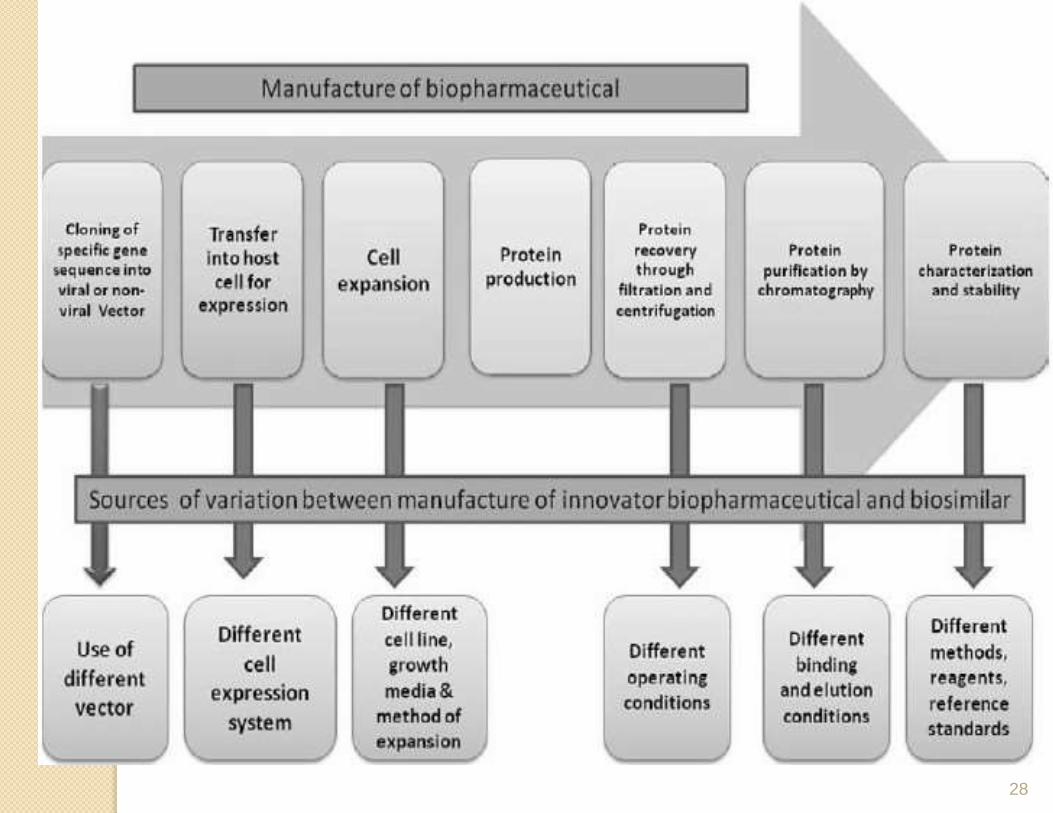
Quality, efficacy and safety
• The quality, safety, and efficacy of a Biosimilars
product must be approved by the relevant regulatory
body before marketing approval can be gained.
• . The quality comparison between the Biosimilars
and the innovator product is crucial, because the
quality of a protein product affects its safety and
efficacy.
• Towards the particular manufacturing process used,
biopharmaceuticals exhibited great sensitivity, and
variation in product quality was commonly observed,
even when the exact same process of manufacturing
was used.
• Variability of source material has also been known to
affect product quality. Thus the product is affected
both by the host cell and the processing steps that
follow.

Quality, efficacy and safety
• The recent guidelines of the International Conference
on HarmonizationQ8 on pharmaceutical
development,47 and the roll-out of the Quality by
Design48 and Process Analytical Technology
initiatives from the FDA have improved
understanding of the impact of manufacturing
processes and their starting materials, on product
quality.
28

Methods for QSE assessment of Biosimilars
Attributes Methods
Primary sequence (peptide map
and amino acid
sequence analysis),
immunogenicity (immunoassay)
other identity indicators
IE, HPLC, gel electrophoresis
Potency Cell-based bioassay, gene
expression bioassay, ADCC, CDC
Conformation Near/far UV circular dichroism
spectroscopy, Fourier transform
infrared
spectroscopy, X ray
crystallography and differential
scanning calorimetry

Methods for QSE assessment of Biosimilars
Attributes Methods
Host cell proteins ELISA, DNA,
endotoxin (Limulus
amebocyte lysate
assay)
Binding Cell assays,
spectroscopy, ELISA
Biological activity Cell assays, animal
models

Pharmacovigilance
• Pharmacovigilance is particularly concerned with adverse
drug reactions.
• The most critical safety concern relating to
biopharmaceuticals (including Biosimilars) is
immunogenicity.
• Minimization of immunogenicity has to begin at the
molecule design stage by reducing or eliminating antigenic
epitopes and building in favorable physical and chemical
properties.
• Pharmacovigilance is important in the Biosimilars market
because of the limited ability to predict clinical
consequences of seemingly innocuous changes in the
manufacturing process and the scientific information gap.

Pharmacovigilance• Pharmacovigilance systems should differentiate
between innovator product and Biosimilars products, so
that effects of Biosimilars are not lost in the back–
ground of reports on innovator products.
• Further, the risk management plan for Biosimilars
should focus on increasing pharmacovigilance
measures, identify immunogenicity risk, and implement
special post-marketing surveillance.
• Although International Nonproprietary Names (INNs)
served as a useful tool in worldwide
pharmacovigilance, for biological products, they should
not be relied upon as the only means of product
identification.
• In addition, biological products should always be
commercialized with a brand name or the INN plus the
manufacturer’s name.

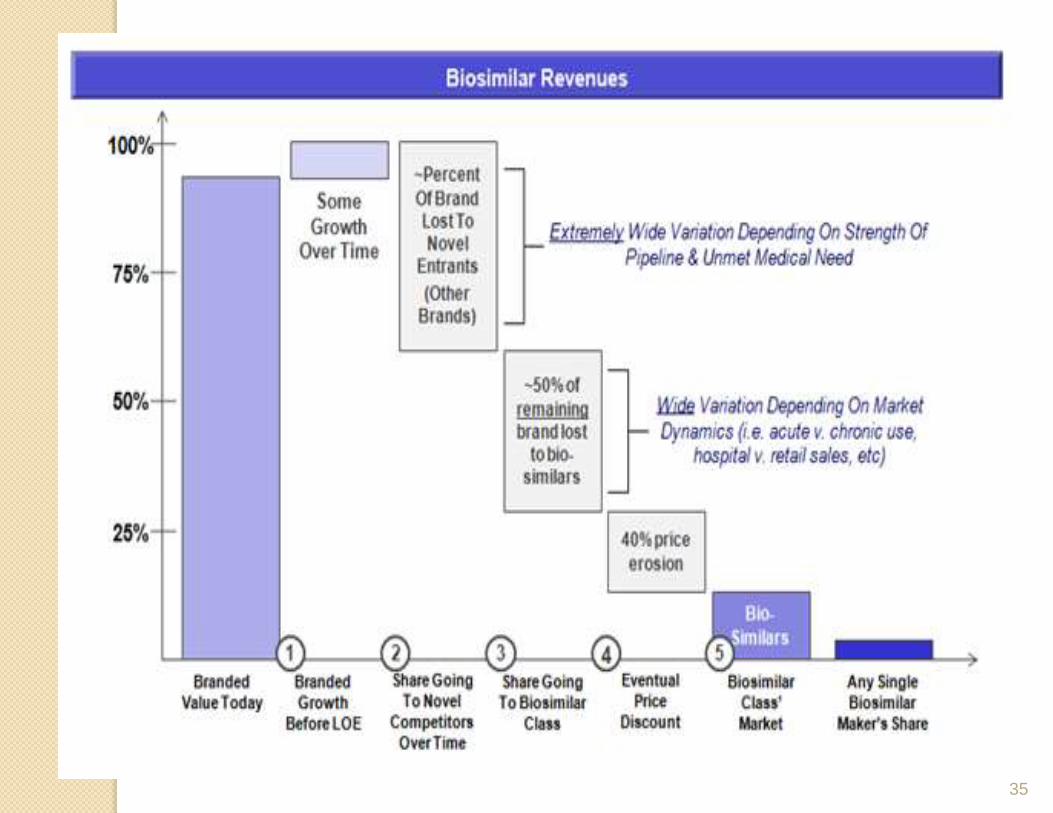
Commercial opportunities
• Biosimilars development is a landmine of
complexities with respect to regulatory,
manufacturing, and marketing aspects, making it one
of the most expensive development propositions in
the pharmaceutical industry.
• Like generic pharmaceuticals, Biosimilars enter the
market with the aim of reducing healthcare cost, but
entry to the Biosimilars market carries higher costs,
greater risks, and more time and expertise in relation
to the clinical development of these products.
• The considerable costs to obtain FDA approval, and
the substantial costs to develop manufacturing
capacity, will limit the number of Biosimilars
competitors.

Commercial opportunities
• The type and amount of resources required for biosimilar
development can create high barriers of entry, not just for small to
mid-sized companies, but even for the larger, well-established
generics players and global biopharmaceutical companies.
• Gaining market share for a biosimilar could be challenging when
there is no added benefit over the innovator and insignificant cost
savings. The price decrease can be achieved when multiple
biosimilars are introduced to the market.
• On the other hand, if a substantial price decrease is not viable for a
biosimilar, a better strategy seems to be to develop a biosimilar as a
new product.
• It would benefit the sponsor to use a scientific rationale and its own
nonclinical and clinical testing, most of which will be required
anyway, to develop its product as a unique innovator product, and
gain the benefit of extended market exclusivity.
35

Development of next-generation products
• Competition in biopharmaceuticals is dynamic and
many biologicals have next generation products in
development
• Roche is developing subcutaneous injection
presentations for its Rituxan and Herceptin products
• Biogen is developing a PEGylated version of its
interferon beta 1-a product for multiple sclerosis
• Next generation products may be in the planning
stages for Avastin and Remicade
37

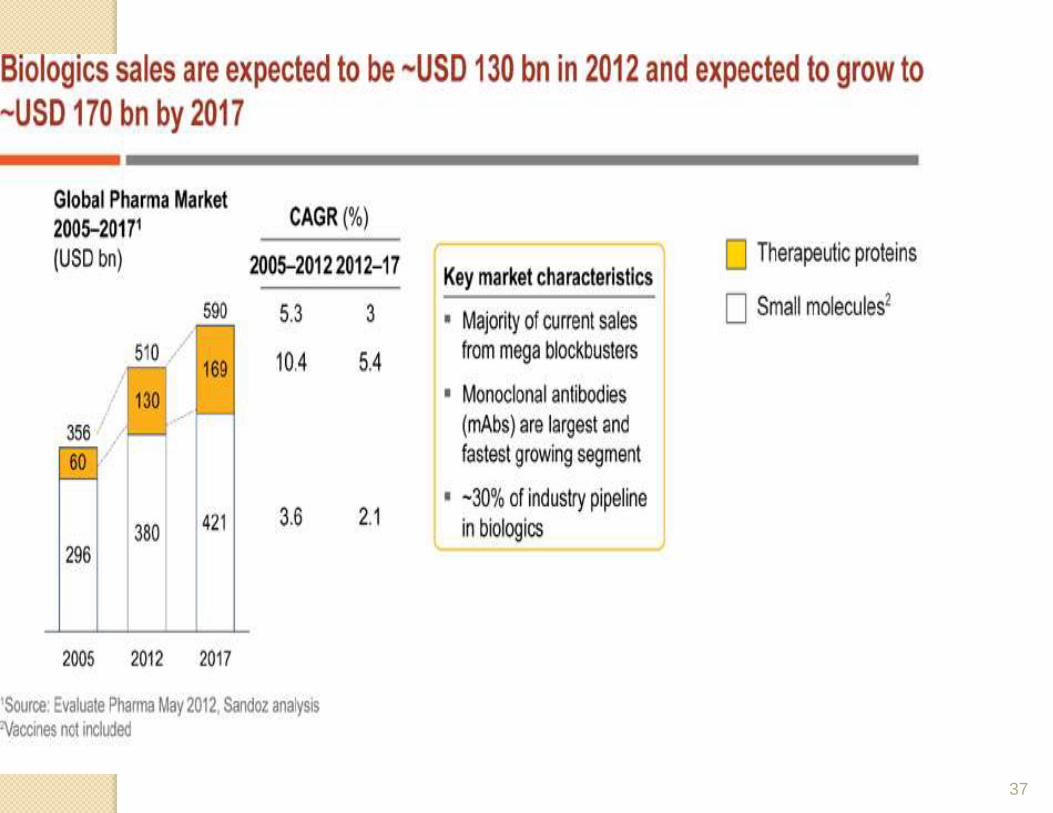
Market advantages Biopharmaceutical drugs have outperformed the
pharmaceutical market as a whole largely due to two factors:
1)They address areas of clinical need that are unmanageable with conventional therapeutics (including many cancers and genetic diseases)
2) They are able to command a premium price.
Datamonitor, for example, forecast growth in biopharmaceuticals of 11% a year between 2004 and 2010 compared to 3.4% annually for the total market. Currently, the USA accounts for 55% of the biopharmaceuticals market.
38

Market advantages
Usually, the imminent expiry of a drug’s
patent leads to companies developing cheaper,
bioequivalent versions of the original brand
(generics), followed by intense price
competition. This approach to the
biopharmaceuticals market can yield
significant reward.
Biopharmaceuticals’ commercial value derives
from their ability to address otherwise unmet
need.
39

Market advantages Genzyme’s Cerezyme (imiglucerase) offers a case in
point. Cerezyme treats Gaucher’s Disease, which
occurs because of an inherited deficiency in an
enzyme called glucocerebrosidase. As a result, levels
of a fat called glucosylceramide rise excessively,
which grossly enlarges the liver, spleen, bone
marrow and other organs leading to numerous
potentially fatal complications and considerable
morbidity among those who survive. Before
imiglucerase, there were no effective treatments.
Cerezyme markedly improves the prognosis of
people affected by Gaucher’s Disease. Cerezyme,
which addresses this previously unmet need, is priced
at around $200 000 per patient per year.40

Market challenges
Many commercially important biopharmaceuticals, including monoclonal antibodies (MAbs) such as Herceptin (trastuzumab), Rituxan (rituximab) and Humira (adalimumab), were launched fairly recently and will not be open to generic competition for many years.
They are protected by a complex series of patents that even the biggest, most experienced generics companies find impenetrable.
41

Market challenges
The commercial and scientific hurdles facing biopharmaceuticals hinder the entry of generic biopharmaceuticals and mean that companies that want to develop biosimilars will need to rethink some fundamental assumptions about the generics market and work according to new business models.
The innate variation and the lack of established methods to determine bioequivalence mean that regulators are likely to be much stricter when considering an application for marketing approval of biosimilars than they are with conventional generics.
42

Market challenges
As a result, regulators will require more extensive clinical testing for biosimilars than for conventional generics.
The cost of manufacturing a biopharmaceutical is much higher than that of a conventional generic. The estimated cost to develop a biosimilar is estimated to be in the range $10-40 million, largely because of the need for extensive safety and efficacy testing. This compares with $1-2 million for a traditional generic.
43

Market challenges
Physicians will be cautious about the
relative safety and efficacy of biosimilars
in the short term at least. Therefore, the
market may develop slowly, which is one
reason why the commercial rewards are
likely to be limited in the short term.
44

The role of hospital pharmacists• It is of utmost importance that the hospital pharmacist is aware that
the innovator products and biosimilars are not interchangeable,
because patients must be carefully monitored if their treatment is
changed between products.
• Moreover, patient welfare is foremost and for pharmacists, the
knowledge that biosimilars are not generics, and the possible
implications for clinical outcomes when products are switched, will
help ensure patient safety.
• Systematic checklists have been proposed for the evaluation of
biopharmaceuticals coming on to the market, which have provided
additional reassurance for the pharmacist.
• For example, the Pharmacy Checklist for Retacrit (epoetin zeta)
provides information on manufacturing, protein and product
formulation, batch consistency, supply reliability, good handling
practice, clinical efficacy, and clinical safety and tolerability.

Summary and conclusions
• Biologics represent a major structural change in terms of
innovation, new indications, costs, and competition
• Biosimilars have large potential commercial
opportunities but they also face high regulatory and
other hurdles compared to generic drugs for chemically
derived drugs
• Biosimilar cost savings are expected to be modest, but
scientific advances eventually could lead to easier entry
and more robust price competition.

References
1. Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and
pharmacological classification. Nat Reviews Drug Discovery.
2. Genentech Inc. Corporate Chronology. 1982. http://www.gene.com/
gene/about/corporate/history/timeline.html.
3. Global Biopharmaceutical Market Report (2010–2015) IMARC
October 29, 2010:234 Pages. Pub ID: IMRC2849563.
4. http://www.icis.com/Articles/2010/02/15/9333235/follobw-on-
biologicspresent-opportunity-to-big-pharma.html.
5. Roger SD, Goldsmith D. Biosimilars: it’s not as simple as cost
alone.J Clin Pharm Ther. 2008;33:459–464.
6. Avidor Y, Mabjeesh NJ, Matzkin H. Biotechnology and drug
discovery: from bench to bedside. South Med J. 2003;96:1174–1186.

References7. IMS Health. IMS Webinar: Biologics. 2009.
http://www.imshealth.com/portal/site/imshealth/menuitem.a67578132
5ce246f7cf6bc429418c22a
/?vgnextoid=a0c22e9b65802210VgnVCM100000ed152ca2RCRD&v
gnextfmt=default.
8. BIO. Biotechnology Industry Facts. 2009.
http://bio.org/speeches/pubs/er/statistics.asp.
9. Hincal F. An introduction to safety issues in biosimilars/follow-on
biopharmaceuticals. J Med CBR Def. 2009;7:1–18.
10. Ledford H. Biosimilar drugs poised to penetrate market. Nature.
2010
11. Shaldon S. Biosimilars and biopharmaceuticals: what the
nephrologist needs to know – a position paper by the ERA-EDTA
Council. Nephrol Dial Transplant. 2009

References
13. De Groot AS, Scott DW. Immunogenicity of protein therapeutics.
Trends Immunol. 2007;28:482–490.
14. Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR. Rational
design and engineering of therapeutic proteins. Drug Discovery
today
15. Revers L, Furczon E. An introduction to biologics and biosimilars.
Part II: subsequent entry biologics: biosame or biodifferent? Can
Pharmacists J (CPJ/RPC). 2010;143:184–191.
16. Crommelin DJA, Storm G, Verrijk R, Leede L, Jiskoot W, Hennink
WE.Shifting paradigms: biopharmaceuticals versus low molecular
weight drugs.
17. Biosimilars: an overview, Bhupinder Singh Sekhon Vikrant
Saluja,Institute of Pharmacy, PCTE Group of Institutes, Near
Baddowal Cantt,(Ludhiana), India

References
18. Biosimilars – Science, status, and strategic
perspective, Georg-Burkhard Kresse ,European
Journal of Pharmaceutics and
Biopharmaceutics,2009.
19. Biosimilars by Sandoz: Capturing the future
opportunity,Ameet Mallik,Global Head, Sandoz
Biopharmaceuticals and Oncology Injectables.
20.Biosimilar Biological Products, Rachel E. Sherman,
MD, MPH,Associate Director for Medical Policy
Center for Drug Evaluation and Research.
21. www.fda.gov
22.WHO. Guidelines on Evaluation of Similar
Biotherapeutic Products (SBPs) WHO; Geneva,
Switzerland: 2009.

51


















