
Computational Spectroscopy II. ab initio Methods from part (d) Electronic Spectra Chemistry 713...
24
Computational Spectroscopy II. ab initio Methods from part (d) Electronic Spectra Chemistry 713 Updated: February 20, 2008
-
date post
19-Dec-2015 -
Category
Documents
-
view
220 -
download
1
Transcript of Computational Spectroscopy II. ab initio Methods from part (d) Electronic Spectra Chemistry 713...

Computational SpectroscopyII. ab initio Methods
from part (d) Electronic Spectra
Chemistry 713
Updated: February 20, 2008

The Born-Oppenheimer Approximation For a given molecular geometry (i.e., fixed nuclear
coordinates, R), solve the electronic Schroedinger equation:
where He is the whole molecular Hamiltonian except the nuclear kinetic energy and r represents the coordinates for all of the electrons, and e is the electronic wave function. Repeat for a range of molecular geometries R of interest to construct a potential energy surface.
The electronic energy En(R) is the potential energy in which the nuclei move.
Up to now we have just been concerned about the lowest energy electronic state, n=0.
To deal with electronic (UV/vis) spectroscopy, we also need to know some of the higher electronic surfaces (n=1, 2, …) as well.
The nuclear motion on each surface can then be solved as a separate step.
€
Heψ e,n r;R( ) = En R( )ψ e,n r;R( )
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
F.F. Crim, Spectroscopic probes and vibrational state control of chemical reaction dynamics in gases and liquids.Talk WA04, International Symposium on Molecular Spectroscopy, Columbus OH, 2006.http://molspect.chemistry.ohio-state.edu/symposium_61/symposium/Program/WA.html#WA04
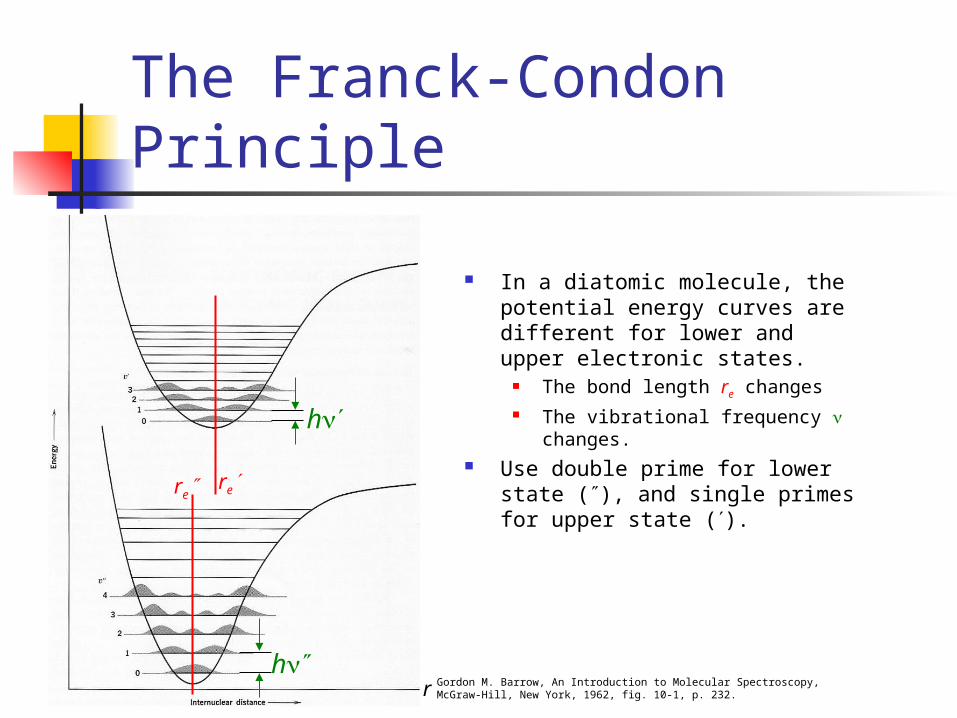
The Franck-Condon Principle
In a diatomic molecule, the potential energy curves are different for lower and upper electronic states.
The bond length re changes The vibrational frequency changes.
Use double prime for lower state (), and single primes for upper state ().
Gordon M. Barrow, An Introduction to Molecular Spectroscopy,McGraw-Hill, New York, 1962, fig. 10-1, p. 232.r
rere
h
h
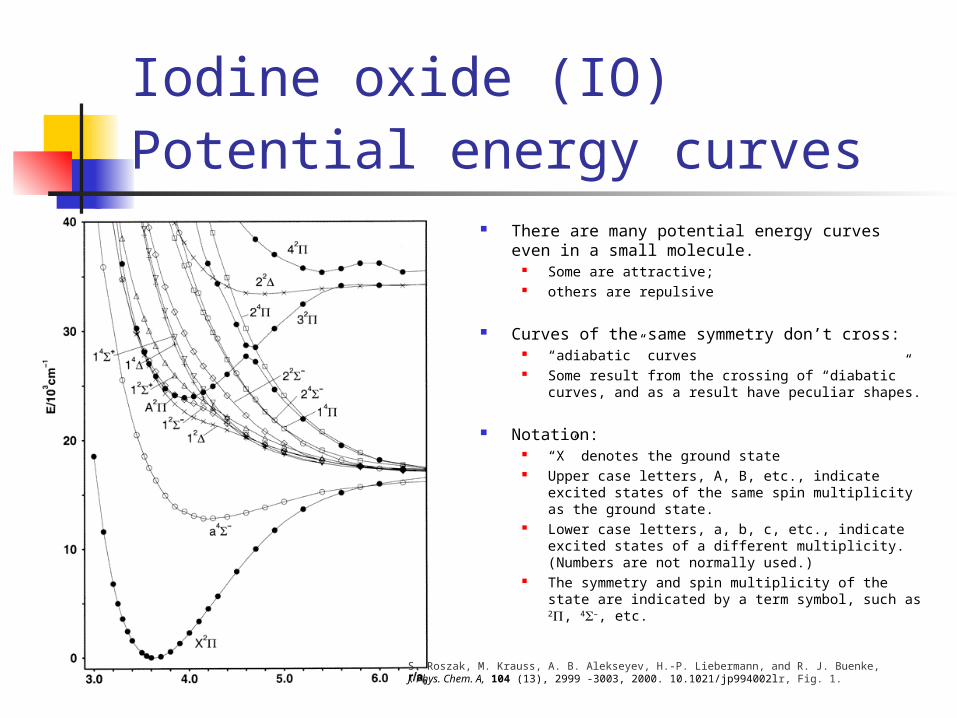
Iodine oxide (IO)
Potential energy curves There are many potential energy curves even in a
small molecule. Some are attractive; others are repulsive
Curves of the same symmetry don’t cross: “adiabatic” curves Some result from the crossing of “diabatic” curves, and
as a result have peculiar shapes.
Notation: “X” denotes the ground state Upper case letters, A, B, etc., indicate excited states of
the same spin multiplicity as the ground state. Lower case letters, a, b, c, etc., indicate excited states of a
different multiplicity. (Numbers are not normally used.) The symmetry and spin multiplicity of the state are
indicated by a term symbol, such as 2, 4–, etc.
S. Roszak, M. Krauss, A. B. Alekseyev, H.-P. Liebermann, and R. J. Buenke, J. Phys. Chem. A, 104 (13), 2999 -3003, 2000. 10.1021/jp994002lr, Fig. 1.

The Franck-Condon Principle
Electronic transitions are “vertical”, that is the nuclei don’t move while the electron(s) are being excited.
Because the upper state wavefunctions are shifted from those in the ground state and because the vibrational frequencies are different, changes in the vibrational quantum number accompany the electronic excitation.
The relative intensities of the vibrational subbands vv are given by the squares of overlap integrals, called Franck-Condon factors:
If neither the bond length, nor the vibrational frequency change, then the selection rules are v=0.
In polyatomic molecules, vibrational progressions occur in vibrational modes for which either the equilibrium position is changes or the frequency is changed.
€
v ψ ′ ′ v
2

In polyatomic molecules, vibrational progressions occur in vibrational modes for which either the equilibrium position is changes or the frequency is changed.
Therefore, a typical electronic band has a lot of vibrational structure, which extends over a few thousand cm-1.
The band origin is the frequency of the v=00 band. The band origins of electronic transitions are what we can most easily calculate with ab initio methods.
For large molecules or in the condensed phase, the vibrational structure is heavily overlapped and merges together into a wide unstructured blob (the Franck-Condon envelope).
The Franck-Condon Principle:Polyatomic molecules
A spectrum with vibrational progressions
45,000 37,000Wavenumber / cm-1
BandOrigin
Franck-Condonenvelope
Benzene
J. M. Hollas, High resolution Spectroscopy, Butterworths, London, 1982, p 393.

Selection rules for electronic spectroscopy
Spin multiplicity is conserved. Changes in vibrational motion follow the Franck-
Condon Principle Rotational transitions (J=0,1, K=0,1)
accompany each electronic+vibrational (vibronic) transition.
For molecules with a center of symmetry, the g/u symmetry changes.
Nuclear spin states are conserved. Additional rules apply in particular cases.

The fate of electronically excited molecules
1. Fluorescence: a visible or UV photon is emitted to return the molecule to its ground state.
2. Intersystem crossing: radiationless conversion of the energy back to a state of different spin multiplicity. (e.g., singlet to triplet).
- Occasionally followed by phosfluorescence: emission of a photon with a change in the spin multiplicity. (VERY weak; a long radiative lifetime.)
3. Internal conversion: radiationless conversion of the energy back to the ground state (or other state of the same spin multiplicity).
4. A Photochemical reaction- photodissociation, isomerization
5. Energy transfer to a nearby molecule
Jablonski diagram
J. I. Steinfeld, Molecules and Radiation, MIT Press, Cambridge, MA, 2nd ed, 1985, p 287.

Conical intersections
Two electronic surfaces can met like two cones touching tip to tip.
Widespread throughout electronic spectroscopy.
Act like a sink-hole that allows the system to drop through onto a lower surface.
Action spectra can be recorded by detecting photofragments.
Note that only two of the six vibrational coordinates are represented in this diagram!
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Conical intersection
h
F.F. Crim, Spectroscopic probes and vibrational state control of chemical reaction dynamics in gases and liquids. Talk WA04, International Symposium on Molecular Spectroscopy, Columbus OH, 2006.

Electronic excitationsin the orbital approximation
For electronically excited states, one or more electrons is in an orbital with higher than the lowest possible energy allowed by the Pauli principle.
Given M doubly occupied molecular orbitals, and N unoccupied orbitals (N), there are an enormous number of possible excited electronic states.
Consider cases where the ground state is closed shell, and can be represented by a single Slater determinant:
An electron in the highest occupied molecular orbital (HOMO) N/2 is excited
to a higher orbital, a:
A singly occupied orbital with no bar is spin up; one with a bar is spin down. The excited singlet state (S=0) is linear combination of two Slater
determinants:
The corresponding triplet state (S=1) has three components and is somewhat lower in energy:
€
1ψ 0 = ψ12ψ 2
2ψ 32...ψ N / 2
2
€
N / 2a = ψ1
2ψ 22ψ 3
2 ...ψ N / 2ψ a
€
1ψ N / 2a = 1
2ψ1
2ψ 22ψ 3
2...ψ N / 2ψ a − ψ12ψ 2
2ψ 32...ψ N / 2ψ a( )
€
3ψ N / 2;Ms =−1a = ψ1
2ψ 22ψ 3
2...ψ N / 2ψ a3ψ N / 2;Ms =0
a = 12
ψ12ψ 2
2ψ 32...ψ N / 2ψ a + ψ1
2ψ 22ψ 3
2...ψ N / 2ψ a( )3ψ N / 2;Ms =1
a = ψ12ψ 2
2ψ 32...ψ N / 2ψ a
a
N/2

Quick review of
Slater determinants
The Pauli principle requires that the overall wavefunction be antisymmetric with respect to the interchange of ANY two electrons.
Since determinants change sign upon the interchange of any two rows or columns, we will set-up our multi-electron wavefunctions as determinants.
Example: the ground state of lithium. The term symbol for Li is 2S.“S” means orbital angular momentum L=0; “2” indicates a doublet state, that is the spin orbital angular momentum, and
One component of the doublet is
The other component of the doublet is
€
S = 12
€
MS = − 12 ,+ 1
2
€
2ψ 0 = ψ1s2ψ 2s
=1
3!
1s 1( ) 1s 1( ) 2s 1( )
1s 2( ) 1s 2( ) 2s 2( )
1s 3( ) 1s 3( ) 2s 3( )
= 16{1s 1( )1s 2( )2s 3( ) −1s 1( )2s 2( )1s 3( ) +1s 1( )1s 2( )2s 3( )
−1s 1( )2s 2( )1s 3( ) + 2s 1( )1s 2( )1s 3( ) − 2s 1( )1s 2( )1s 3( )}
€
2ψ 0 = ψ1s2ψ 2s
=1
3!
1s 1( ) 1s 1( ) 2s 1( )
1s 2( ) 1s 2( ) 2s 2( )
1s 3( ) 1s 3( ) 2s 3( )
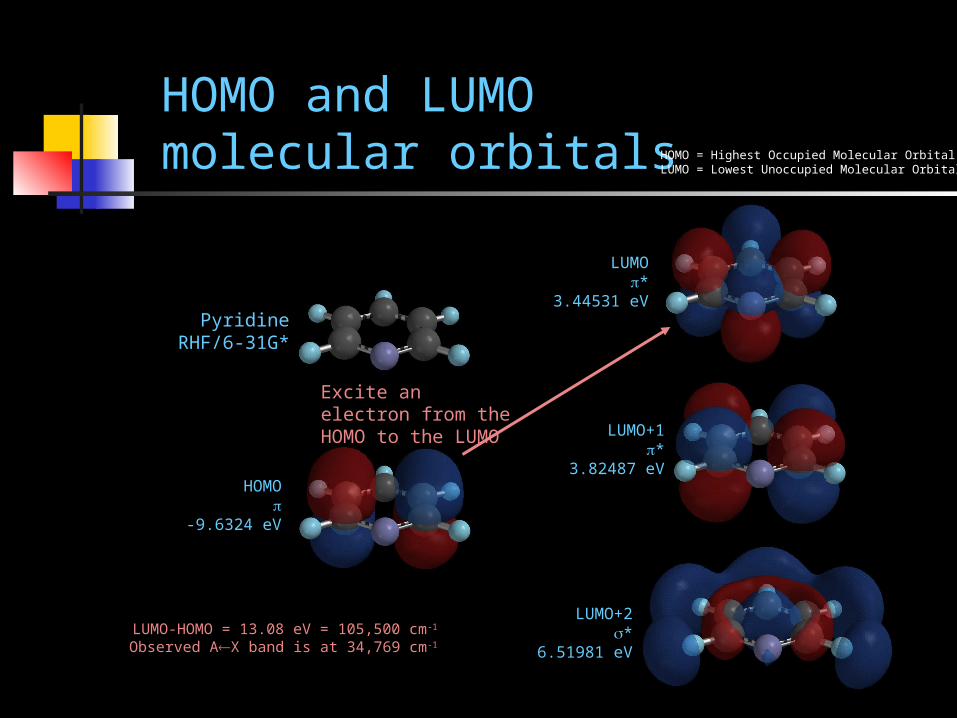
HOMO and LUMO molecular orbitals
PyridineRHF/6-31G*
HOMO
-9.6324 eV
LUMO*
3.44531 eV
LUMO+1*
3.82487 eV
LUMO+2*
6.51981 eV
HOMO = Highest Occupied Molecular OrbitalLUMO = Lowest Unoccupied Molecular Orbital
LUMO-HOMO = 13.08 eV = 105,500 cm-1
Observed AX band is at 34,769 cm-1
Excite an electron from the HOMO to the LUMO

Difficulties with the Simplest Orbital Picture
The qualitative picture on the previous slide is very appealing.
Gives our band descriptions as *, etc. Calculated Energies of excited states and transition
frequencies are much too large in the orbital approximation.
We must realize that the other electrons readjust their motions to accommodate the excited electron, thereby minimizing the total energy of the excited state.
If we use the variation method to re-optimize the excited state, then our calculation will often collapse back to the ground state.
The excited state must be kept orthogonal to the ground state.
Realize that excited states are more sensitive to basis set limitations.
Sometimes a change of symmetry or spin upon excitation will prevent the variational collapse of the excited state.
In favorable circumstances, the HF or DFT levels can be applied.
QuickTime™ and a decompressor
are needed to see this picture.
Phenyl nitrene
Cramer, p 495.

CI Singles (CIS) for Excited States
Based on the Hartree-Fock (HF) ground state and configuration interaction with single electron excitations.
With M occupied orbitals from which an electron could be excited and N possible excited orbitals that it could be promoted to gives MN interacting determinants.
Resulting wavefunctions are of approximately HF quality (meaning not really as good as we would like).
Can optimize excited state geometries and find excited state vibrational frequencies.
CIS and CIS(D) are available in Spartan and in Gaussian.

CIS excited state calculations with Spartan (04 or 06)
1. Optimize the ground state geometry at a suitable level (e.g., RHF/6-31G*)
2. At that geometry, run a single point excited state calculation (CIS or CIS(D)) to get the “vertical” UV spectrum (figure at right).
3. To get the excited state geometry and properties, optimize at the CIS level.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
acrolein

QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Acrolein UV/Vis by CIS
Spartan06 2min 21 sec for excited state calculation
Excited state optimized geometry: CIS/6-31G*C=C 1.510 AC-C 1.432 AC=O 1.209 A
Dipole moment: 0.65 Debeye
Ground state: RHF/6-31G*C=C 1.321 AC-C 1.478 A
C=O 1. 190 ADipole moment: 3.5 Debeye
/ nm
single bond!
“vertical”spectrum

Acrolein excited state vibrations by CIS
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Excited State Vibrations(IR spectrum not easily accessible by experiment)
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Ground State Vibrations
Note that the calculated ground state frequencies at the RHF/6-31G* level are systematically TOO HIGH.
What we would really like is not an excited state IR spectrum but the Franck-Condon frequencies and intensities for the UV/Vis spectrum.

Acrolein excited state vibrations by CIS
Two of the low frequency out-of-plane modes are imaginary. Implies that the excited state structure is non-planar, even though the ground state is planar. The planar (CS) structure that we found is a saddle point between two equivalent non-planar
minima. Therefore calculation of the vibrational frequencies is not valid. Spartan calculates vibrational frequencies in the excited state, but does not calculate the Franck-
Condon intensities. Repeating the calculation with the “symmetry” box unchecked did not help, so we need to start with a
non-planar initial structure.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
SPARTAN

QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
To get a starting geometry with non-planar C=CH2, I had to redraw the structure from scratch.
Convergence of the excited state geometry at the CIS/6-31G* level took much longer: 18 steps and 10 minutes.
Spartan did not recognize the ground state as acrolein and did not calculate a correct excited state spectrum.
The non-planar structure gives all real vibrational frequencies.
Both planar and non-planar excited states predict a reduction of the C=O frequency and a slight increase in the aldehyde hydrogen, which is the strongest CH stretch.
Acrolein excited state vibrations by CIS
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Excited State VibrationsPLANAR
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Ground State Vibrations
NON-PLANAR Excited state
single bond

Time-dependent DFT (TDDFT)
Based on calculating the polarization of the ground state molecule produced by an oscillating light field.
Excited state wavefunctions, geometries and frequencies are not explicitly determined.
Good for calculating UV/visible spectra, especially for low-lying excited states.
Difficulty with high lying states and for charge-transfer states.
Available in Gaussian and Spartan.

TDDFT excited state calculation with Spartan
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
EnergyDo a single point energy calculation …
Do a single point energy calculation …
… with a geometry that you previously
optimized at the same DFT level
for the ground state.
… with a geometry that you previously
optimized at the same DFT level
for the ground state.

Acrolein UV/Vis by TDDFT UV/Vis spectrum is much better than with CIS. As you would expect, the ground state IR spectrum is also much better with DFT than in the HF
calculation that we used as a starting point for the CIS calculation. But we did not get the excited state geometry, dipole moment, or vibrational frequencies. Spartan claims that it will not optimize the excited state geometry, but it might if you are willing to
let it compute for days.
QuickTime™ and a decompressor
are needed to see this picture.
QuickTime™ and a decompressor
are needed to see this picture.
Spartan06: 2 min, 7 seconds plus 3 min 24 sec for B3LYP/6-31G* geometry optimization
TDDFT B3LYP/6-31G*

Comparison of Spartan excited state calculations
CIS: UV/Vis spectra with 6-31G* basis not very accurate. The CS Excited state geometry was reasonable. Does not find excited states with lower symmetry than the ground state. Calculation produced widely varying excited state dipole moments results for the
same final structure. Spartan does not represent the excited stated orbitals in intelligible form. CIS(D) is also available and might give a more accurate UV/Vis spectrum.
TDDFT Reasonably accurate UV/Vis spectrum Other excited state properties not calculated
ZINDO A semi-empirical of calculating electronic spectra. Available in G03, but not in Spartan.

Higher level methodsfor excited states
MCSCF - multi configuration self-consistent field CASPT2 - Complete active space with electron
correlation treated perturbatively. MRCI (including MRCISD) multi reference
configuration interaction (with single and double excitations).
All of these are multi-reference methods.QuickTime™ and a
decompressorare needed to see this picture.
F.F. Crim, Spectroscopic probes and vibrational state control of chemical reaction dynamics in gases and liquids. Talk WA04, International Symposium on Molecular Spectroscopy, Columbus OH, 2006.
QuickTime™ and a decompressor
are needed to see this picture.
Cramer p 459


















