
Complement deficiency states and meningococcal disease
17
Immunol Res 1993;12:295-311 Julio Figueroa John Andreoni Peter Densen Departmentof InternalMedicine, University of IowaCollege of Medicine,IowaCity, Iowa,USA ComplementDeficiencyStates and MeningococcalDisease e ~ 1 7 6 1 7 6 1 7 6 1 7 4 Key Words Complement deficiency Neisserial infections Antibody response Host defense eel* oe*ooo,oo o o e e e e * o e e e e l e e * o o * , e * | 1 7 6 1 7 6 o , a * * . o , e o l ~ 1 7 6 1 7 4 1 7 6 Abstract Analysis of complement deficiency states has supported the role of complement in host defense and elucidated diseases associated with defective complement function. Although neisserial infection plays a prominent role in these deficiency states, examination of individuals with late complement com- ponent deficiency (LCCD) reveals a particular propensity for recurrent meningococcal disease and provides important clues to the role of complement in neisserial infections. In response to meningococcal disease, LCCD individuals produce signifi- cantly greater amounts of antilipooligosaccharide (LOS) anti- body which can kill group B meningococcus in a complement- sufficient in vitro system. Further studies of antibody cross- reactivity to other meningococci has led to a clearer under- standing of its epitopic specificity. Nevertheless, epidemiolog- ic evidence is consistent with the relative absence of protective immunity in LCCD persons following an episode of infection and supported by quantitation of antibody to capsular poly- saccharide. However, compared to anti-LOS antibodies, anti- capsular antibodies can offer immune protection to LCCD individuals via complement-dependent opsonophagocytosis - the only form of complement-mediated killing available to these persons. Thus vaccination of LCCD persons with capsu- lar antigens is considered an important means of protecting these high-risk individuals against meningococcal disease. Peter Densen, MD 9 1993 Department of Internal Medicine S. Ka~er AG, Basel University of Iowa 0257-277X/93/ 200 Hawkins Drive 0123-029552,75/0 Iowa City, IA 52242-1009 (USA)
-
Upload
julio-figueroa -
Category
Documents
-
view
212 -
download
0
Transcript of Complement deficiency states and meningococcal disease

I m m u n o l R e s 1 9 9 3 ; 1 2 : 2 9 5 - 3 1 1
Julio Figueroa John Andreoni Peter Densen
Department of Internal Medicine, University of Iowa College of Medicine, Iowa City, Iowa, USA
Complement Deficiency States and Meningococcal Disease
e ~ 1 7 6 1 7 6 1 7 6 1 7 4
Key Words Complement deficiency Neisserial infections Antibody response Host defense
e e l * o e * o o o , o o o o e e e e * o e e e e l e e * o o * , e * | 1 7 6 1 7 6 o , a * * . o , e o l ~ 1 7 6 1 7 4 1 7 6
Abstract Analysis of complement deficiency states has supported the role of complement in host defense and elucidated diseases associated with defective complement function. Although neisserial infection plays a prominent role in these deficiency states, examination of individuals with late complement com- ponent deficiency (LCCD) reveals a particular propensity for recurrent meningococcal disease and provides important clues to the role of complement in neisserial infections. In response to meningococcal disease, LCCD individuals produce signifi- cantly greater amounts of antilipooligosaccharide (LOS) anti- body which can kill group B meningococcus in a complement- sufficient in vitro system. Further studies of antibody cross- reactivity to other meningococci has led to a clearer under- standing of its epitopic specificity. Nevertheless, epidemiolog- ic evidence is consistent with the relative absence of protective immunity in LCCD persons following an episode of infection and supported by quantitation of antibody to capsular poly- saccharide. However, compared to anti-LOS antibodies, anti- capsular antibodies can offer immune protection to LCCD individuals via complement-dependent opsonophagocytosis - the only form of complement-mediated killing available to these persons. Thus vaccination of LCCD persons with capsu- lar antigens is considered an important means of protecting these high-risk individuals against meningococcal disease.
Peter Densen, MD �9 1993 Department of Internal Medicine S. Ka~er AG, Basel University of Iowa 0257-277X/93/ 200 Hawkins Drive 0123-029552,75/0 Iowa City, IA 52242-1009 (USA)

Introduction
Functional activity assigned to the comple- ment system was first described near the turn of the century [ 1 ]. The primary focus at that time was on its bactericidal and hemolytic actions. With the isolation of the complement proteins, investigators demonstrated the pres- ence of two activation pathways, which gener- ate a number ofeffector mechanisms in addi- tion to the assembly of the membrane attack complex that mediates bactericidal activity. During the second half of this century, inves- tigators explore the role of the cascade in che- motaxis, immune complex clearance, opsono- phagocytosis and host tissue injury. The re- cent characterization of complement-regula- tory proteins has provided an understanding of host protection against the indiscriminate effects of complement activation. At present, the complement system consists of 19 plasma and at least 9 membrane proteins.
Whereas antibody-antigen interactions ini- tiate the complement cascade through the classical pathway, the alternative pathway has no such requirement. Both pathways lead to the cleavage of the third component (C3). C3 plays a pivotal functional role through forma- tion of the alternative pathway C3 convertase (C3bBb), microbial opsonization (C3b and iC3b) and participation in the generation of the terminal pathway (C3b). Cleavage of the fifth component (C5) by its convertase gener- ates two fragments: C5a, which acts as a che- motactic factor and serves as a mediator of inflammation, and C5b. The latter fragment functions as the anchor for the sequential addition of the terminal components (C6-C9) to complete the membrane attack complex. Insertion of this complex leads to altered cel- lular metabolism and cell death.
Detailed studies of individuals with com- plement deficiency states have supported the importance of this system to the normal host
and have elucidated diseases associated with a deficiency in complement function [2-4]. Clinically, these patients exhibit both autoim- mune disease (especially systemic lupus ery- thematosus) and increased susceptibility to systemic infections caused by encapsulated organisms (most commonly Neisseria spe- cies). These natural experiments have en- hanced our understanding of the pathogenesis of autoimmune disease, tissue injury and host defense against microbes. This review will focus primarily on those deficiencies of plas- ma complement proteins which have been associated with an increased risk of infection.
Because complement deficiencies are un- common, much of the information about them has been derived through careful analy- sis of accumulated case reports. This ap- proach introduces numerous opportunities for bias. For example, the ethnic background of the study population is a major determi- nant of both the prevalence of complement deficiency states as well as their associated diseases. Specifically, C2 deficiency occurs predominantly in Caucasian populations; in the US its estimated frequency is 0.01%, whereas it has not been reported in the Japa- nese population [3, 5]. In contrast, C9 defi- ciency is uncommon in Caucasian popula- tions but occurs in 0.045-0.104% of normal Japanese blood donors [3, 6]. Similarly, the yearly incidence ofmeningococcal disease dif- fers among various ethnic populations [3] (fig. 1).
Analysis is also complicated by ascertain- ment bias. Traditionally, investigators have associated classical component deficiencies with rheumatologic conditions. For example, a survey of 545 patients with rheumatologic disorders found one person (0.2%) with ho- mozygous C2 deficiency and 19 individuals with definite, probable or possible heterozy- gous C2 deficiency [1 i]. In contrast, only 6 possible heterozygous C2 individuals were
296 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

Fig. 1. Relationship between the prevalence of complement defi- ciency and the incidence of menin- gococcal disease. The data were ob- tained from the literature or by per- sonal communication as described [3] and modified by recent reports from the Faroe Islands [7, 8], Nor- way [9] and Moscow [10].
c
5 0 -
4 0 -
3 0 -
20
10
o New York
Colorado
~ Israel
Iowa �9 ~ M o s c o w
S. Africa o'~nDenmONkrway Faroe Islands
9
i ! i | 1 10 100 1,000
6 Incidence, cases/lO population
identified among 509 individuals without rheumatologic disorders. This report prompted other studies confirming this asso- ciation and leading to the notion that rheuma- tologic disorders, particularly systemic lupus erythematosus, were the major clinical mani- festation of these deficiencies. In contrast, a recent prospective study of 675 individuals with invasive infections caused by encapsu- lated organisms identified two unrelated C2- deficient individuals, i.e. a frequency of 0.3 % [ 12]. Thus, the frequency of C2 deficiency in patients with these selected infections is simi- lar to that in patients with selected rheumato- logic disorders (0.3 vs. 0.2%) demonstrating that individuals with classical component de- ficiencies appear to have an increased but similar risk for both rheumatologic and infec- tious diseases. These findings underscore the potential bias inherent in disease-based popu- lation studies.
Clinical Features of Complement Deficiency States
Although the study of these patients can introduce bias associated with a retrospective literature review, such an analysis can provide important clues to the contribution of indi- vidual complement components to overall complement functions and host defense as well as to the pathogenesis of neisserial infec- tions. Excluding C1 inhibitor deficiencies, these reviews provide support for the occur- rence of four major clinical patterns: classical pathway deficiencies (C1, C4, C2), C3 defi- ciency (C3, factors H and I), alternative path- way deficiencies (properdin, factor D) and late complement component deficiencies (LCCD; C5-C9; table 1). These groups differ in their functional defects as well as in their spectrum and prevalence of associated dis- eases. To date, individuals have been de-
297

Table 1. Clinical patterns of disease in complement deficiency states
Clinical CP AP C3 LCCD variable (C1, C4, C2) (P, D) (C3, H, I) (C5-C8, C9)
Functional defect $ C' act. $ C' act. $ IC metab. ($ CTX) $IC metab. $ ops. phag. $ SBA $ imm. resp. $ imm. resp.
$ PMN $ CTX $ SBA
12 26 0 29 66 5 79 5 18 57 78 64 l 2 14 2 17
S. pneumoniae 5L meningitidis N. meningitidis ~ meningitidis N. meningitidis S. pneumoniae H. influenzae H. influenzae
Healthy, % CVD, % Systemic infection, % Median age at first infection, years Organisms 2
Infected patients with infection due to these organisms, % 40
Recurrent infection with same organism, % 6.3
91 54 99
3.5 56 50 t
CP = Classical pathway; AP =altemative pathway; IC metab. = immune complex metabolism; C'act. = com- plement activation; imm. resp. = immune response; ops. phag. = opsonophagocytosis; CTX = chemotaxis; SBA = serum bactericidal activity; CVD = collagen vascular disease; PMN = polymorphonuclear neutrophils. Adapted from Figueroa and Densen [3]. l Percentage differs for C9-deficient individuals, see table 2. 2 Listed in order of decreasing frequency.
scribed with a complete deficiency of each plasma complement component except factor B [3].
Classical Pathway Component Deficiencies Activation of the classical pathway pro-
motes opsonophagocytic killing, serum bacte- ricidal activity and removal of immune com- plexes. As discussed above, the frequency of homozygous C2 deficiency in patients with rheumatologic disease is about 0.2% or 20- fold more common than that in the general US population [11 ]. These individuals most frequently exhibit a syndrome consistent with systemic lupus erythematosus. Although ho-
mozygous C1 and C4 deficiencies are quite rare, reported individuals with these deficien- cies also suffer principally from rheumato- logic diseases [2, 3]. A review of all reported persons with classical component deficiency reveals that about 66 % have manifestations of a collagen vascular disease (table 1).
These individuals also exhibit increased susceptibility to infection. Recurrent sinopul- monary infections and meningitis caused by encapsulated organisms, especially Strepto- coccus pneumoniae, Neisseria meningitidis and Haemophilus influenzae, predominate. In individuals with invasive infections caused by these encapsulated organisms in Iowa, the reported frequency of C2 deficiency is 0.3%,
298 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

reflecting a 30-fold increase in prevalence compared with the general population [12]. The median age of first infection is 2 years. Overall, 19% of individuals with classical component deficiencies have experienced at least one documented infection with encapsu- lated organisms (table 1); 4% of individuals have had both connective tissue disease and infection with encapsulated organisms [2, 3]. Since most reported patients come from sur- veys of rheumatologic disease, these frequen- cies of infection are subject to ascertainment bias and probably represent an underestimate of the true frequency of infection in this popu- lation. The disease spectrum and onset are reminiscent of those seen in immunoglobulin- deficient individuals. Indeed, several investi- gators have noted impaired antibody re- sponses in guinea pigs and some humans with these deficiencies [13-15]. Thus, the clinical similarity to syndromes of defective humoral immunity may reflect the in vivo importance of the experimental phenomenon.
C3/Factor H and I Deficiencies As the focal point in the complement sys-
tem, C3 participates in numerous effector mechanisms responsible for inflammation and host defense. Consequently it is not sur- prising that C3 deficiency leads to multiple. severe derangements including immune com- plex disease, impaired immune responses, im- paired chemotaxis, reduced opsonophagocy- tosis and abnormal serum bactericidal activi- ty. As with classical component deficiencies, these individuals are at increased risk for rheumatologic disease, 79% demonstrating a lupus-like syndrome or systemic vasculitis (ta- ble 1) [2, 3]. These individuals have a striking disposition to severe and recurrent infections caused primarily by encapsulated organisms, which first manifests itself at an early age. Of the 19 patients reported with this deficiency, 15 have had at least one episode of bacterial
infection - particularly sinopulmonary infec- tion, bacteremia or meningitis [2, 3].
As with classical component deficiencies, the clinical similarity to immunoglobulin de- ficiencies is striking. In experimental models C3 has been shown to modulate the humoral immune response. Specifically, C3-deficient animals have low antibody responses to pri- mary immunization with bacteriophage q0X 174 (a T-cell-dependent antigen) as well as to T-independent antigens [13-16]. More- over, C3 participates in the cellular response to microbes. C3 fragments promote opsono- phagocytosis of organisms and trigger micro- bicidal mechanisms in inflammatory cells [ 17]. Overall, individuals lacking this critical component have multiple defects which are reflected in the spectrum and severity of the associated diseases.
Factors H and I are primarily responsible for the downregulation of the fluid phase, alternative pathway C3 convertase. In the ab- sence of either of these proteins the sponta- neous formation of the C3 convertase goes unchecked, leading to C3 consumption and depletion. Six of the 13 persons with factor H deficiency demonstrated evidence of glomer- ulonephritis; 2 had disease compatible with systemic lupus erythematosus [2, 3]. In factor I deficiency, the association with rheumato- logic disease is less strong; 2 of 14 reported patients had associated arthritis or vasculitis [2, 3]. Although C3 consumption predisposes to immune complex syndromes, the small amount of residual C3 in the serum of persons lacking these factors seems to lessen their risk of these disorders. These individuals demon- strate the same propensity for and spectrum of infections seen in primary C3 deficiency [2, 3]. Eight of 13 individuals with factor H and 12 of 14 persons with factor I deficiency have experienced infection caused mostly by en- capsulated organisms. Recurrent infection is also common.
299

Alternative Pathway Deficiencies Through a noncovalent association, pro-
perdin stabilizes the alternative pathway C3 convertase by a factor of 5-10 [ 18-2 I]. Al- though this effect is relatively modest in vitro, individuals with properdin deficiency exhibit a marked propensity for infections with en- capsulated organisms, especially N. meningi- tidis. Of the 70 patients described in the litera- ture with probable or confirmed properdin deficiency, 50-60% experienced at least one episode of meningococcal disease (table 1) [2, 3]. Compared with the frequency of meningo- coccal disease in the general US population, this represents a 7,000-fold increase in the risk of infection. In contrast to classical com- ponent deficiencies, patients with properdin deficiency have a later onset of infection and exhibit few recurrences (table 1). The low rate of recurrence probably reflects the high mor- tality associated with this infection in this deficiency as well as the development of spe- cific antibodies which activate the classical pathway and help prevent subsequent infec- tion. This observation supports the impor- tance of acquired immunity to Neisseria in conjunction with the complement system for protection in the normal host.
Patients with properdin deficiency do not appear to have the same propensity for rheu- matologic diseases as do individuals with either classical component or C3 deficiency states. Of the 70 persons reported, 4 individu- als from the same family had discoid lupus, and 1 was found to have progressive systemic sclerosis [3]. This difference emphasizes the importance of the classical pathway in im- mune complex clearance.
Factor D cleaves factor B to generate the alternative pathway C3 convertase, C3bBb. The 3 patients reported with this deficiency experienced infections caused by Neisseria as well as other pathogens. In contrast to proper- din deficiency, these individuals experienced
recurrent disease [3]. No data are available about the immune response of these patients, and the basis for this difference is not under- stood. However, this observation suggests that the alternative pathway convertase may be particularly important in preventing infec- tion. In theory, individuals with properdin deficiency can generate a functionally active but less stable C3 convertase able to assist the classical pathway by amplifying C3 cleavage. In contrast, in factor D deficiency the alterna- tive C3 convertase cannot be generated at all.
Late Complement Component Deficiency As a major effector mechanism of the com-
plement cascade, the membrane attack com- plex is responsible for direct complement- dependent serum bactericidal activity. Re- cent evidence suggests that it may also partici- pate in tissue injury in a wide range of dis- eases [22-26]. Perhaps for this reason, a small percentage (--5%) of reported individuals with LCCD have evidence of immune com- plex or rheumatologic disease (table I) [2, 3].
There is a marked association between LCCD and neisserial infections. This associa- tion is strongest among individuals with a deficiency of C5, C6, C7 or C8, 58% of whom have had at least one episode of neisserial infection (table 1) [2, 3]. Compared with the frequency in the general US population, LCCD increases the risk of meningococcal disease 8,000-fold. As with properdin defi- ciency, the median age at the first neisserial infection is in the second decade of life - a significant departure from that in the general population and in persons with classical com- ponent deficiencies.
Although cleavage of C5 leads to the gener- ation of potent chemotactic activity, as well as the formation of the membrane attack com- plex, the clinical features of C5 deficiency do not differ markedly from those of other termi- nal component deficiencies, suggesting that
300 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

Table 2. Comparison of meningococcal disease in LCCD states
Deficiency
C5 C6 C7 C8 C9
Number of individuals 27 77 73 73 165 With meningococcal disease 19 (70) 42 (54) 45 (62) 40 (55) 15 (9.1) Recurrent disease 8 (42) 23 (55) 14 (31) 20 (50) 0 Episodes of meningococcal disease 30 75 71 69 15
CSF + blood 23 (77) 69 (92) 56 (79) 55 (80) 15 (100) Blood 1 (3.3) 5 (6.6) 5 (7.0) 3 (4.4) 0 Unspecified 6 (20) 1 (1.3) I0 (14) 11 (16) 0
Ratio of CSF:blood 23:1 14:1 11:1 18:1 15:0
Figures in parentheses indicate percentages.
the absence of C5a does not contribute signifi- cantly to the clinical picture in these individu- als (table 2).
The association between C9 deficiency and neisserial infection is not as strong as in C5- C8 deficiency. Individuals with total C9 defi- ciency exhibit delayed but present serum bac- tericidal and hemolytic activities in vitro [27]. Initially, the few reported persons with this deficiency did not seem to have an increased risk of infection. However, Nagata et al. [6] demonstrated a 1,400-fold increased risk of meningococcal disease in C9-deficient pa- tients and confirmed a 10,000-fold increased risk of this disease in C7-deficient patients (compared with complement-sufficient con- trols). These data reinforce the association between LCCD and neisserial disease and suggest that the risk of infection in C9 defi- ciency is 5- to 10-fold less than that seen in other terminal component deficiencies (ta- ble 2). Additionally, these observations sup- port the utility of the in vitro bactericidal assay in understanding the in vivo risk of infection.
Meningococcal Disease in Complement Deficiency
Meningococcal disease is a prominent manifestation in a significant fraction of re- ported cases in all four clinical patterns of complement deficiency. In early component deficiencies, the meningococcal shares this role with the pneumococcus and H. influen- zae. In contrast, it is virtually the sole clinical manifestation in properdin deficiency and LCCD. Given this singular predisposition, studies of these deficiencies provide impor- tant clues to the role of complement in the pathogenesis of meningococcal disease.
Meningococcal disease in individuals with LCCD demonstrates several important differ- ences from that in the general population (ta- ble 3) [3]. First, the frequency of meningococ- cal disease in the US population is 0.0072%, whereas it is 9-58% in C9- and C5- to C8- deficient individuals, respectively, 1,000- to 10,000-fold increased risk of infection. After stratification for serogroup, meningococci iso- lated from LCCD individuals do not differ in their sensitivity to complement-mediated kill- ing from those isolated from complement-suf-
301

Table 3. Comparison of menin- gococcal disease in normal, LCCD and properdin-deficient individu- als
Parameter Normal C5-C8 C9 Properdin- deficient
Homozygotes - 250 165 54-70 With meningococcal disease - 146 15 25-37 Frequencyofinfection,% 0.0072 58 9.1 46-53 Median age at first episode, years 3 17 16 1 4-11.5 Recurrence rate, % 0.34 44 0 2-1.4 Relapse rate, % 0.6 7.9 0 0 Mortality/100 episodes, % 19 1.5 0 12-51.4 Infecting serogroup
Isolates 3,184 67 2 16 B, % 50 19.4 50 18.7 Y, % 4.4 32.8 0 37.5
Adapted from Figueroa and Densen [3]. Where a range is given, the first figure refers to documented cases, and the second figure refers to documented plus probable and possible cases.
ficient persons [28]. Therefore, the increased frequency of infection in LCCD is not due to an increase in infection caused by less pathogenic strains. Together, these data emphasize the importance of the membrane attack complex in the prevention of meningococcal disease.
Second, individuals with LCCD are in- fected with uncommon meningococcal sero- groups more often than normal persons [2, 3]. In their review of meningococcal infections, Fijen et al. [29] confirmed the greater fre- quency of complement deficiency among in- dividuals with disease caused by serogroups Y, W135 and probably X. This finding may be due in part to the propensity of these sero- groups to cause disease in older individuals [30]. Moreover, although group Y organisms are more susceptible to serum bactericidal action, they exhibit greater requirements for effective opsonophagocytic killing than do group B isolates [31]. In conjunction with the bactericidal defect, these findings translate into an additional degree of difficulty for the LCCD person in the elimination of these
strains compared with those that typically cause disease in the general population.
Third, individuals with LCCD deficiency experience their initial meningococcal infec- tion at an older median age than do normal persons (17 vs. 3 years) [2, 3]. Thus, the ma- jority of deficient persons pass through the period of highest risk of infection for normal individuals only to develop infection in the second decade (fig. 2). This paradox is only partially explained by the fact that deficient individuals are susceptible for life while nor- mal individuals are generally at risk only early in life. Ascertainment bias is unlikely to ex- plain this finding because this relationship is also apparent in families where ascertainment is complete. In addition, in a prospective study of the frequency of these disorders in patients with meningococcal disease, no com- plement-deficient individuals identified were younger than 15 years of age [12]. These observations suggest that unidentified factors contribute to the susceptibility of deficient individuals later in life.
302 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

120
100
80
E 6O
o_ 40 Fig. 2. The age o f f irst meningo-
coccal infection in normal and 20 complement-deficient individuals. 0 Data for normal individuals ( e )
120 were provided by Dr. J. Wenger from the Centers for Disease Con- ~, 100 o~
trol, Atlanta, Ga., USA, as pub- 80 lished [32]. Data for LCCD indi- ~.
60 viduals (11) and properdin-defi- .~ cient patients (A) are from the lit- -~ 40 erature [2, 3]. Results are expressed ~ 20 as the number of infected patients o (a) or as the cumulative percentage 0 of infections (b) by age of infection. Hatches in the horizontal axis indi- cate discontinuity in the age scale.
_ ~ ~ . . T - : " - f - ; " , ~ . : - T T
b
0 1 2 3-4 5-9 10-19 20-29 30-59 >60 Age, years
Fourth, mortality associated with menin- gococcal disease (both meningitis and menin- gococcemia) ranges between 10 and 19% in the general US population but is only 1.5- 2.4% in LCCD persons (table 3) [3]. Meningo- coccal disease is a paradigm for gram-negative sepsis. Initially, meningococcal endotoxin (li- pooligosaccharide or LOS) stimulates numer- ous host responses including complement ac- tivation, cytokine release and infiltration of inflammatory cells [33-38]. As the disease progresses, the prominent features become those of endothelial damage and consumptive coagulopathy. Several, as yet unsubstantiated, factors may contribute to the lower mortality in deficient patients: (I) lower organism inoc- ulum required to cause disease; (2) lower plas- ma endotoxin concentrations; (3) milder dis- ease, and (4) less tissue injury.
Although quantitative data are lacking, the notion that defective bactericidal activity in
LCCD might lower the infectious dose neces- sary to initiate systemic disease provides an attractive explanation for both the increased frequency of infection and improved clinical outcome. Lower infectious inocula probably correspond to lower plasma concentrations of LOS and thus less morbidity and mortality. However, because of the bactericidal defect, an initially small inoculum could proliferate unchecked, ultimately resulting in a greater overall organism burden. In this case defec- tive bacteriolysis consequent to the LCCD might result in less LOS release from the organism and milder disease. Unfortunately no data exist to compare the serum LOS con- centration in complement-deficient and -suf- ficient individuals.
Due to the highly variable nature of the reported clinical information, the issue of milder disease in LCCD persons is difficult to assess from a review of reported cases. Evi-
303

dence for this hypothesis comes from the studies of Platonov and Beloborodov [39; un- publ. data], who utilized a clinical grading sys- tem to assess the severity of meningococcal disease in 30 LCCD and 100 age-matched complement-sufficient patients. Compared with normal persons, LCCD patients experi- enced significantly fewer cases of severe or lethal disease (70 vs. 39~ of shock (14 vs. 3%) and of brain edema (25 vs. 9%). This study also confirmed the finding of Brandt- zaeg et al. [34] that the extent of complement consumption correlated with a fatal outcome in normal patients. In contrast, LCCD indi- viduals showed no differences in C 1, C4 and C2 activity during active meningococcal dis- ease compared with healthy normal or LCCD individuals.
These data support the notion that the lack of complement activation decreases tissue in- jury and mortality in deficient individuals. Brown and Lachmann [40] demonstrated that, compared with C6-sufficient rabbits, C6-deficient rabbits had improved survival after endotoxin challenge; this advantage cor- related with the preservation of the platelet count and the lack of platelet activation and release of platelet-activating factor. A possible explanation for reduced platelet activation in LCCD comes from recent experimental work showing the importance of the membrane at- tack complex in activation of many host cells. Although not generally lethal, insertion of the membrane attack complex activates leuko- cytes to release potentially toxic mediators and promotes a procoagulant state in endo- thelial cells and platelets [41-44]. In vivo, exuberant complement activation may lead to the insertion of the membrane attack complex in bystander host cells which in turn become activated to produce toxic products and a pro- coagulant state, that is, the beginning of tissue injury and the hallmark of septic shock. Since LCCD individuals are unable to generate the
membrane attack complex, these pathologic processes would not occur, allowing the defi- cient person to better tolerate a given organ- ism and endotoxin load during sepsis.
Fifth, recurrent meningococcal disease is rare in normal and properdin-deficient indi- viduals (table 3) [2, 3]. This finding suggests that prior infection provides specific and cross-reactive antibody to Neisseria in these persons. These antibodies can activate the classical pathway to prevent recurrence through either C3b-directed opsonophagocy- tosis or serum bactericidal activity. In con- trast, recurrent disease is common (44%) in persons with C5-C8 deficiency suggesting that immunity does not follow initial infec- tion in these individuals despite intact C3b- mediated phagocytosis. Support for this sug- gestion comes from scrutiny of the number of episodes of recurrent meningococcal disease in LCCD individuals [3]. This analysis indi- cates that each episode of infection is an inde- pendent event; that is prior infection does not alter the risk of infection in these persons.
Although it may not prevent disease, ac- quired immunity might ameliorate the severi- ty of subsequent infection. If so, this might help explain the reduced mortality in LCCD individuals. However, in their analysis of dis- ease severity, Platonov and Beloborodov [39; unpubl, data] observed that prior infection had no effect on the severity of subsequent infections in LCCD individuals. Moreover, the few deaths that have occurred in LCCD individuals with meningococcal diseases have not been confined to the initial infection. Thus, the occurrence and severity of menin- gococcal disease in LCCD seem to be com- pletely independent of prior disease.
304 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

3/68 3/87 4/87 5/87 I / / I I I
Healthy Birth
Y meningitis Tetravalent After Dx C7 def. vacc ine vaccine
Capsular antibody,
I.tg/ml
10/87 3/89 10/4/89 10/18/89 11/89 12/89 / / I / / I / / I I I I
C bacteremia d/c Rx Tetravalent After meningitis vaccine vaccine
A 5.0 122 31 70 33 64 48 67
C 0.4 11 26 0.7 0.4 53 33 41
Subcapsular 4+ 4+ 4+ 4+ antibody
Fig. 3. Longitudinal history ef a LCCD patient (C7 deficiency). Arrows indicate the occurrence of each meningococcal episode. Capsular antibody concentrations to serogroup A and C were quantitated by standardized ELISA. Subcapsular antibody concentrations were estimated quantitatively (0-4 + scale) by the degree of IgG immunoreactivity with separated meningococcal outer membrane antigens on immunoblots. Dx = Diagnosis; d/c Rx = discontinued therapy.
I m m u n i t y t o IV. meningitidis
Immunity to N. meningitidis is dependent upon antibody and complement. Studies by Goldschneider et al. [45, 46] established that antibody to subcapsular antigens is bacteri- cidal and important in the development of natural protective immunity against menin- gococcal disease in normal persons. This anti- body, which is acquired by the majority of the population by early adulthood, is thought to be due to the development of antibody to cross-reactive antigens on nonpathogenic Neisseria. These subcapsular antigens include surface-exposed outer membrane proteins and LOS. Antibody to these antigens is pro- duced, to a greater degree, after an episode of meningococcal disease and confers protective
immunity in a normal host. Protective immu- nity also follows vaccination with meningo- coccal capsular polysaccharides. These anti- bodies are bactericidal [46] and promote op- sonophagocytic killing of meningococci [47]. However, meningococcal infection seems to be a relatively poor inducer of capsular anti- body, and thus protective immunity in nor- mal persons seems to be due largely to anti- body to subcapsular antigens.
The typical clinical course of LCCD per- sons is represented in chronological fashion in figure 3. This person was diagnosed as having C7 deficiency at the time of his initial presen- tation in March 1987 with group Y meningo- coccal meningitis. Following infection, his se- rum contained 5- to 10-fold more antibody to subcapsular meningococcal antigens, particu-
305
Silver kD stain
4 4 . 0 - ~
29.0- ~;"2~,
18.4 -
14.3- ~
6.2- ~
3.0- ~
Convalescent J
PHS Normal LCCD
. . . . ~
m h
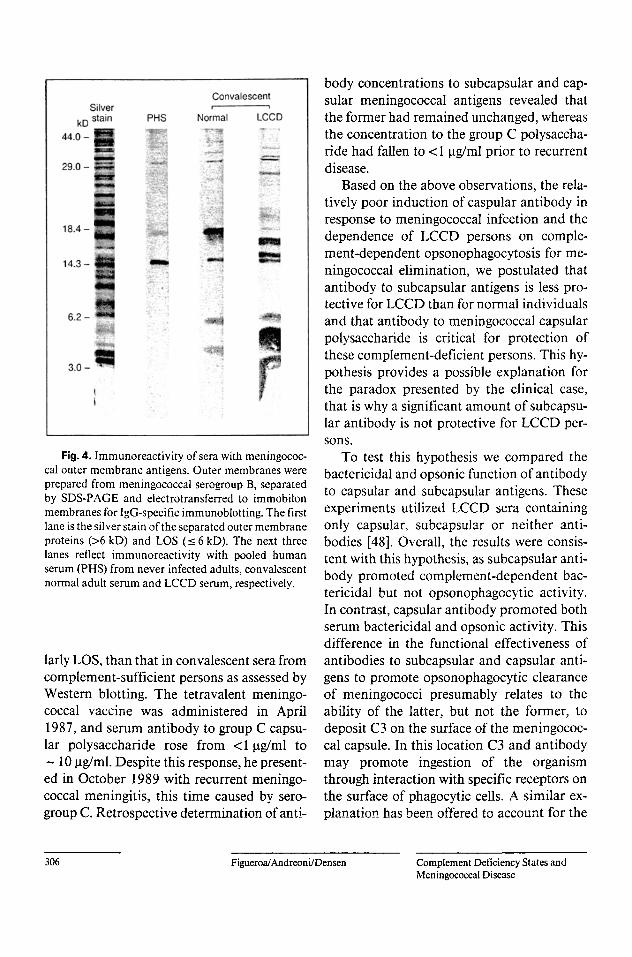
r Fig. 4. lmmunoreactivity of sera with meningococ-
cal outer membrane antigens. Outer membranes were prepared from meningococcal serogroup B, separated by SDS-PAGE and electrotransferred to immobilon membranes for IgG-specific immunoblotting. The first lane is the silver stain of the separated outer membrane proteins (>6 kD) and LOS (_< 6 kD). The next three lanes reflect immunoreactivity with pooled human serum (PHS) from never infected adults, convalescent normal adult serum and LCCD serum, respectively.
lady LOS, than that in convalescent sera from complement-sufficient persons as assessed by Western blotting. The tetravalent meningo- coccal vaccine was administered in April 1987, and serum antibody to group C capsu- lar polysaccharide rose from <1 ~tg/ml to
- 10 p.g/ml. Despite this response, he present- ed in October 1989 with recurrent meningo- coccal meningitis, this time caused by sero- group C. Retrospective determination of anti-
body concentrations to subcapsular and cap- sular meningococcal antigens revealed that the former had remained unchanged, whereas the concentration to the group C polysaccha- ride had fallen to < 1 ~tg/ml prior to recurrent disease.
Based on the above observations, the rela- tively poor induction of caspular antibody in response to meningococcal infection and the dependence of LCCD persons on comple- ment-dependent opsonophagocytosis for me- ningococcal elimination, we postulated that antibody to subcapsular antigens is less pro- tective for LCCD than for normal individuals and that antibody to meningococcal capsular polysaccharide is critical for protection of these complement-deficient persons. This hy- pothesis provides a possible explanation for the paradox presented by the clinical case, that is why a significant amount of subcapsu- lar antibody is not protective for LCCD per- sons.
To test this hypothesis we compared the bactericidal and opsonic function of antibody to capsular and subcapsular antigens. These experiments utilized LCCD sera containing only capsular, subcapsular or neither anti- bodies [48]. Overall, the results were consis- tent with this hypothesis, as subcapsular anti- body promoted complement-dependent bac- tericidal but not opsonophagocytic activity. In contrast, capsular antibody promoted both serum bactericidal and opsonic activity. This difference in the functional effectiveness of antibodies to subcapsular and capsular anti- gens to promote opsonophagocytic clearance of meningococci presumably relates to the ability of the latter, but not the former, to deposit C3 on the surface of the meningococ- cal capsule. In this location C3 and antibody may promote ingestion of the organism through interaction with specific receptors on the surface of phagocytic cells. A similar ex- planation has been offered to account for the
306 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

differential opsonophagocytic potential of an- tibodies to pneumococcal [49] and staphylo- coccal [50] cell wall and capsular polysaccha- ride. Meningococci differ from these gram- positive bacteria in that the former, but not the latter, are susceptible to complement-me- diated bactericidal activity and therefore anti- body to meningococcal subcapsular antigens may have a more clearly defined functional role in the complement-sufficient individual.
We have also been interested in why signif- icantly more antibody to outer membrane meningococcal antigens is present in conva- lescent sera from LCCD than complement- sufficient persons. The reactivity of pooled nonimmune normal serum and convalescent serum from normal or LCCD persons with SDS-PAGE separated meningococcal outer membrane proteins (>6kD) and LOS (_< 6 kD) is shown in figure 4. Clearly, conva- lescent LCCD serum contains considerably more antibody against both outer membrane proteins and LOS, although the increase is most significant for the latter. Higher concen- trations of antibodies to class I and serotype 2 outer membrane proteins in sera from C6- deficient persons experiencing meningococcal disease have been described [5 l, 52] but fur- ther studies of the immune responses to me- ningococci in LCCD individuals are lacking.
This antibody response to LOS is IgG, bac- tericidal for serogroup B meningococci in a complement-dependent bactericidal assay and cross-reactive to LOS of other meningo- coccal serogroups, gonococci and H. influen- zae. Due to its potential importance in protec- tion against group B meninogococcal disease, we have examined the epitopic specificity of this cross-reactive, bactericidal antibody. Ab- sorption of convalescent LCCD sera with the Salmonella Re595 mutant, the lipopolysac- charide of which contains only ketodeoxyoc- tonate linked to lipid A, depleted immunore- activity with Re595 but not meningcoccal or
gonococcal LOS. ELISA assays utilizing lipid A and Salmonella Re LOS as antigens confirmed the presence of minimal antibody to lipid A in LCCD sera and that antibody to Salmonella Re LOS was similar in sera from never infected and infected LCCD or normal individuals. These data indicate that this antibody is not directed against ketodeoxyoctonate or lipid A, which are present in all LOS structures. In con- trast, absorption with group B meningococci depleted all reactivity except that for the Sal- monella Re mutant. Recent experiments place the epitopic specificity of this antibody in the oligosaccharide chain of the LOS molecule.
The immunologic basis for the quantita- tive difference in the specific antibody re- sponse to meningococcal LOS of LCCD com- pared with normal individuals is unknown. Possible factors contributing to this response include a greater organism load, intracellular survival within phagocytic cells, the absence of meningococcal bacteriolysis in LCCD plas- ma and altered antigen presentation conse- quent to the maintenance of outer membrane integrity.
Vaccination
Our data strongly suggest that antibody to meningococcal capsular polysaccharide is critical for the protection of LCCD persons from meningococcal disease. Consequently we assessed the LCCD individuals' response to the tetravalent meningococcal polysaccha- ride vaccine to determine if vaccination is effective in conferring immunity. The total antibody response to meningococcal sero- group A and C capsular polysaccharides was compared among individuals with LCCD, their family members and unrelated comple- ment-sufficient persons 3-4 weeks following vaccination. The median age in all three groups was similar, and there were no individ-
307

1,000
100
of CO {3_
lo
o
o
- - - - g
Group A Group C
o
o
8 0 o
I ~ ' - ._e_ o
o
o
n=9
o
-8- o o
n = 1 0
LCCD CS LCCD CS
Fig.& Maintenance of antibody response to the meningococcal capsular polysaccharide vaccine in LCCD and complement-sufficient (CS) individuals. Antibody concentrations to mcningococcal poly- saccharide (PSS) were determined 2-2.5 years following initial vaccination and expressed as a percentage of the concentration of these antibodies in individual sera obtained 3-4 weeks following vaccination. The horizontal bar represents the mean percent change in antibody concentration. The difference between the means for the two groups was not significant for either type of polysaccharide although it approached significance for group C (p = 0.065). The number of individuals who were unable to maintain 50 % of their initial antibody response (dashed line) was not statistically different for group A. Significantly fewer LCCD than CS individuals maintained a 50% response to group C polysaccharide (p < 0.02).
uals less than 10 years of age in any of the groups. The difference in mean antibody con- centrations among the various groups was not significant. For example, for meningococcal C polysaccharide the median values for unre- lated completed-sufficient, complement-suf- ficient family members, heterozygous and homozygous complement-deficient family members were 38.9, 107, 19.2 and 37.6 ~tg/ ml, respectively. These data support the premise that the initial response of LCCD
persons to polysaccharide vaccines is normal. However, longitudinal studies suggest that LCCD persons may have a more rapid decline than complement-sufficient individuals in certain serogroup-specific polysaccharide an- tibodies. As the clinical case demonstrates, maintenance of adequate capsular antibody concentrations over time appears to be a cru- cial aspect of host defense against meningo- coccal disease of LCCD individuals. For sero- group A polysaccharide, 4 of 7 versus 7 of 10
308 Figueroa/Andreoni/Densen Complement Deficiency States and

in the LCCD and heterozygous/normal groups, respectively, maintained 50% or greater of their initial response to the vaccine (p = 0.50). In contrast, only 2 of 7 LCCD com- pared with 9 of 10 heterozygous deficient or normal persons maintained 50% or more of their initial serogroup C capsular antibody concentration (p < 0.02) 2-2.5 years following vaccination (fig. 5). Although the initial vacci- nation responses of these individuals to the Y and W135 components of the meningococcal vaccine were not available, deficient individ- uals demonstrated a lower mean antibody concentration to group Y capsular polysac- charide 2-2.5 years following vaccination compared with complement-sufficient indi- viduals (18.3 vs. 122 units/ml, respectively). The mean concentration to W135 capsular polysaccharide did not differ between the two groups (64.8 vs. 79.8 units/ml, respectively).
Treatment
Our data indicate that antibody to menin- gococcal capsular polysaccharides is critical for protection of LCCD individuals from me- ningococcal disease. Thus, vaccination with the tetravalent meningococcal vaccine to pro- mote protective immunity in these persons is strongly indicated. The longitudinal data sug- gest that LCCD persons may have a more rap- id decline in the concentration of certain sero- group-specific polysaccharide antibodies and thus may become susceptible to meningococ- cal disease when this concentration falls be- low a critical threshold. Based on the opsono- phagocytic assays and the aforementioned clinical case this antibody threshold appears to be about at 1-2 ~tg/ml. Thus it seems rea- sonable to quantitate capsular antibody ap- proximately 2 years after vaccination and to revaccinate if the concentration of these anti- bodies is less than this amount.
The hypothesis presented above and the availability of the tetravalent meningococcal capsular polysaccharide vaccine provide a ra- tionale to prevent meningococcal disease, not only in individuals with LCCD but also in persons with defects affecting the classical and alternative pathways. Antibody to capsu- lar polysaccharide in individuals with a classi- cal pathway defect should facilitate the use of the alternative pathway thereby enhancing both opsonophagocytosis and direct bacteri- cidal activity. However, 4- to 8-fold greater amounts of capsular antibody are required to produce a degree of meningococcal killing in C2-deficient serum comparable to that in nor- mal serum. High levels of antibody may also facilitate the use of the classical pathway in C2-deficient serum in an alternative-path- way-dependent manner [53].
In vitro studies similar to those described here have been performed in sera from pro- perdin-deficient individuals. Response to the meningococcal vaccine in these individuals is associated with efficient utilization of the classical pathway and enhancement of both opsonophagocytosis and direct bactericidal activity. The high mortality from meningo- coccal disease in these patients makes vacci- nation mandatory [3, 54-56]. The efficacy of penicillin prophylaxis has been assessed by Potter et al. [56] and should be used as an adjunct to vaccination when group B menin- gococcal disease is epidemic or when recur- rent episodes of disease occur over a brief period of time [57].
309

�9 �9 i o Q �9 �9 �9 o o �9 �9 �9 �9 �9 � 9 1 4 9 1 4 9 �9 �9 � 9 1 4 9 �9 �9 �9 �9 � 9 �9 �9 �9 �9 o � 9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 | �9 �9 �9 �9 �9 �9 �9 �9 �9 � 9 1 4 9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 � 9 1 4 9 �9 �9 * �9 �9 �9 � 9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 �9 � 9 1 4 9 �9 �9
References
1 Ross GD: Introduction and history of complement research; in Ross GD (ed): Immunobiology of the Complement System. Orlando, Aca- demic Press, 1986, pp 1-20.
2 Ross SC, Densen P: Complement deficiency states and infection: Epi- demiology path�9 and conse- quences of Neisseria and other in- fections in an immune deficiency. Medicine (Baltimore) 1984;63:243- 273.
3 Figueroa JE, Densen P: Infectious diseases associated with comple- ment deficiencies. Clin Microbiol Rev 1991;4:359-395.
4 Sj6holal AG: Inherited complement deficiency states: Implications for immunity and immunological dis- ease. Acta Pathol Microbiol lmmu- nol Scand (C) 1990;98:861-874.
5 Alper CA, Awdeh Z, Yunis E J: Fre- quency of the C2 deficiency gene among normal Caucasians (ab- stract). Complement 1987;4: L25.
6 Nagata M, Hara T, Aoki T, Mizuno Y, Akeda H, Inaba S, Tsumoto K, Ueda K: Inherited deficiency of ninth component of complement: An increased risk of meningococcal meningitis. J Pediatr 1989; 114:260- 264.
7 Rasmussen JM, Tcisner B, Weihe P, Mathiassen B, Petersen T, Isager H: Screening for complement deficien- cies in patients surviving from epi- demic meningococcal disease. J Clin Lab Immunol 1988;25:161-165.
8 Weihe P, Mathiassen B, Rasmussen JM, Pctersen T, Isager H: An epi- demic outbreak of group B menin- gococcal disease on the Faroe Is- lands. Scand J Infect Dis 1988;20: 291-296.
9 Rogde S, Ho'iby EA, Teisberg P, Olaisen B: Genetic aspects of com- plement component C8 in Norwe- gian meningococcal disease pa- tients. Scand J Infect Dis 1990;22: 673-679.
10 Belodorodov VB, Plat�9 AE: Me- ningococcal disease in the USSR in patients with deficiencies in rate complement components; in Acht- man M, Kohl P, Marchal C, MoreUi G, Seiler A, Thiesen B (eds): Neis- seriae 1990. Berlin, de Gruyter & Co, 1991, pp 659-663.
11 Glass D, Raum D, Gibson D, Still- man JS, Schur PH: Inherited defi- ciency of the second component of complement. Rheumatic disease as- sociation. J Clin Invest 1976;58: 853.
12 Densen P, Sanford M, Burke T, Densen E, Wintermeyer L: Prospec- tive study of the prevalence of com- plement deficiency in meningitis. (abstract). Program Abstr 30th In- tersci Congr Antimicrob Agents, Am Soc Microbiol, Washington, D.C., 1990, p 140.
13 Brttger EC, Bitter-Suermann D: Complement and the regulation of humoral immune responses. Immu- nol Today 1987;8:261-264.
14 Ochs HD, Wedgwood R J, Frank MM, Heller SR, Hosea SW: The role of complement in the induction of antibody responses. Clin Exp lm- muno11983;53:208-216.
15 Ochs HD, Wedgwood ILl', Heller SR, Beatty PG: Complement, mem- brane glycoproteins, and comple- ment receptors: Their role in regula- tion of the immune response. Clin Immunol lmmunopathol 1986;40: 94-104.
16 O'Neil KM, Ochs HD, Holler SR, Cork LC, Morris JM, Winkelstein JA: Role of C3 in humoral immuni- ty: Defective antibody production in C3-deficient dogs. J Immunol 1988; 140:1939-1945.
17 Densen P, Mandell GL: Granulocyt- ic phagocytes; in Mandell GL, Dou- glas RG Jr, Bennett JE (eds): Princi- ples and Practice of Infectious Dis- eases. New York, Churchill Living- stone, 1990, pp 81-101.
18 Fearon DT, Austen KF: Properdin: Initiation of the alternative comple- ment pathway. Proc Natl Acad Sci USA 1975;72:3220-3224.
19 Fearon DT, Austen KF: Properdin: Binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med 1975;142:856-863.
20 Fearon DT, Austen KF: Current concepts in immunology: The alter- native pathway of complement - A system for host resistance to micro- bial infection. N Engl J Med 1980; 303:259-263.
21 Pangburn MK: The alternative pathway; in Ross GD (ed): Immuno- biology of the complement system. Orlando, Academic Press, 1986, pp 45-62.
22 Parra G, Takekoshi Y, Striegel J, Vernier RL, Michael AF: Acute se- rum sickness in normal and C6 defi- cient rabbits: Role of membrane at- tack complex. Int J Exp Pathol 1992;73:299-312.
23 Cybulsky AV, Rennke HG, Feint- zeig ID, Salant D J: Complement- induced glomerular epithelial cell injury: Role of the membrane attack complex in rat membranous ne- phropathy. J Clin Invest 1986;77: 1096-1107.
24 Halstensen TS, Mollnes TE, Fausa O, Brandtzaeg P: Deposits of termi- nal complement complex (TCC) in muscularis mucosae and submuco- sal vessels in ulcerative colitis and Crohn's disease of the colon. Gut 1989;30:361-366.
25 Rus HG, Niculescu F, Vlaicu R: Presence of C5b-9 complement complex and S-protein in human myocardial areas with necrosis and sclerosis, lmmunol Lett 1987;16: 15-20.
26 Rus HG, Niculescu F, Constantines- cu E, Cristca A, Vlaicu R: Immu- noelectron-microscopic localization of the terminal C5b-9 complement complex in human atherosclerotic fibrous plaque. Atherosclerosis 1993;61:35-42.
27 Harriman GR, Esser AF, Podack ER, Wunderlich AC, Braude AI, Lint TF, Curd JG: The role of C9 in complement-mediated killing of Neisseria. J Immunol 1981;127: 2386-2390.
28 Ross SC, Berberich HM, Densen P: Natural serum bactericidal activity against Neisseria meningitidis iso- lates from disseminated infections in normal and complement-defi- cient hosts. J Infect Dis 1985;152: 1332-1335.
29 Fijen CA, Kuijper EJ, Hannema AJ, Sj6holm AG, van Putten JP: Com- plement deficiencies in patients over ten years old with meningococ- cal disease due to uncommon set�9 groups. Lancet 1989;ii:585-588.
310 Figueroa/Andreoni/Densen Complement Deficiency States and Meningococcal Disease

30 Anonymous: Analysis of endemic meningocoocal disease by serogroup and evaluation of chemoprophylax- is. J Infect Dis 1976;134:201-204.
31 Ross SC, Rosenthal J, Berberich HM, Densen P: Killing of Neisseria meningitidis by human neutrophils: Implications for normal and com- plement-deficient individuals. J In- fect Dis 1987;155:1266-1275.
32 Wenger JD, Hightower AW, Fack- lam RR, Gaventa S, Broome CV: Bacterial meningitis in the United States, 1986: Report of a multistate surveillance study. J Infect Dis 1990;162:1316-1323.
33 Brandtzaeg P, Joo GB, Brusletto B, Kierulf P: Plasminogen activator in- hibitor 1 and 2, alpha-2-antiplasmin, plasminogen, and endotoxin levels in systemic meningococcaldisease. Thromb Res 1990;57:271-278.
34 Brandtzaeg P, Kierulf P, Gaustad P, Skulberg A, Bruun JN, Halvorsen S, Srrensen E: Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococ- cal disease. J Infect Dis 1989;159: 195-204.
35 Brandtzaeg P, Mollnes TE, Kierulf P: Complement activation and en- dotoxin levels in systemic meningo- coocal disease. J Infect Dis 1989; 160:58-65.
36 Brandtzaeg P, Sandset PM, Joo GB, Ovstebo R, Abildgaard U, Kierulf P: The quantitative association of plas- ma endotoxin, antithrombin, pro- tein C, extrinsic pathway inhibitor and fibrinopeptide A in systemic meningoooccal disease. Thromb Res 1989;55:459-470.
37 Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T: The com- plex pattern of cytokines in serum from patients with meningocoocal septic shock: Association between interleukin 6, interleukin 1, and fa- tal outcome. J Exp Med 1989;169: 333-338.
38 Zwahlen A, Waldvogel FA: Magni- tude of bacteremia and complement activation during Neisseria meningi- tidis infection: Study of two co-pri- mary cases with different clinical presentations. Eur J Clin Microbiol 1984;3:439--441.
39 Platonov AE, Beloborodov VB: Late complement component deficiency (LCCD) in the USSR: The situation in 1991 (abstract). Complement In- flamm 1991;8:211.
40 Brown DL, Lachmann P J: The be- haviour of complement and plate- lets in lethal endotoxin shook in rab- bits. Int Arch Allergy 1973;45:193- 205.
41 H~insch GM, Seitz M, Martinotti G, Betz M, Rauterberg EW, Gemsa D: Macrophages release arachidonic acid, prostaglandins E2, and throm- boxane in response to late comple- ment components. J Immuno11984; 133:2145-2150.
42 lmagawa DK, Osifchin NE, Pazne- kas WA, Shin ML, Mayer MM: Consequences of ceil membrane at- tack by complement: Release of ara- chidonate and formation of inflam- matory derivatives. Proc Natl Acad Sci USA 1983;80:6647-6651.
43 Hamilton KK, Hattori R, Esmon CT, Sims P J: Complement proteins C5b-9 induce vesiculation of the en- dothelial plasma membrane and ex- pose catalytic surface for assembly of the prothrombinase enzyme com- plex. J Biol Chem 1990;265:3809- 3814.
44 Sims P J, Wiedmer T: The response of human platelets to activated com- ponents of the complement system. lmmunol Today 1991;12:338-342.
45 Goldschneider I, Gotschlich EC, Ar- tenstein MS: Human immunity to the meningoooocus. I. The role of humoral antibodies. J Exp Med 1969;129:1307-I 326.
46 Goldschneider I, Gotschlich EC, Ar- tenstein MS: Human immunity to the meningococcus. 11. Develop- ment of natural immunity. J Exp Med 1969;129:1327-1348.
47 Roberts RB: The relationship be- tween group A and group C menin- gocoocal polysaccharides and serum opsonins in man. J Exp Med 1970; 131:499-513.
48 Andreoni J, K~iyhty H, Densen P: Vaccination and the role of capsular polysaccharide antibody in pre- vention of recurrent meningocoocal disease in late complement compo- nent deficient individuals. J Infect Dis 1993;168:227-231.
49 Brown EJ, Joiner K.A, Cole RM, Berger M: Localization of comple- ment component 3 on Streptococcus pneumoniae: Anti-capsular anti- body causes complement deposition on the pneumoooocal capsule. Infect Immun 1983;39:403-409.
50 Wilkinson B J, Sisson SP, Kim Y, Peterson PK: Localization of the third component of complement on the cell wall of encapsulated Staphy- lococcus aureus M: Implications for the mechanism of resistance to phagooytosis. Infect Immun 1979; 26:1159-1163.
51 Orren A, Warren RE, Potter PC, Jones AM, Lachmann P J, Poolman JT: Antibodies to meningocoocal class 1 outer membrane proteins in South African complement-defi- cient and complement-sufficient subjects. Infect Immun 1992;60: 4510-4516.
52 Orren A, Potter PC, Cooper RC, du Toit E: Deficiency of the sixth com- ponent of complement and suscepti- bility to Neisseria meningitMis in- fections: Studies in 10 families and five isolated cases. Immunology 1987;62:249-253.
53 Steuer KLK, Sloan LB, Oglesby TJ, Farries TC, Nickells MW, Densen P, Harley JB, Atkinson JP: Lysis of sensitized sheep erythrocytes in hu- man sera deficient in the second component of complement. J Im- munol 1989;143:2256-2261.
54 Densen P, Weiler JM, Griffiss JM, Hoffman LG: Familial properdin deficiency and fatal meningoooc- cemia: Correction of the bactericidal defect by vaccination. N Engl J Med 1987;316:922-926.
55 Stiderstrom C, Braconier JH, Dani- elsson D, Sj6holm AG, Thuresson B: Immune response to tetravalent meningococcal vaccine: Opsonic and bactericidal functions of normal and properdin deficient sera. Eur J Clin Microbiol Infect Dis 1989;8: 220-224.
56 Potter PC, Frasch CE, van der Sande W J, Cooper RC, Patel Y, Or- ren A: Prophylaxis against Neisseria meningitidis infections and anti- body responses in patients with defi- ciency of the sixth component of complement. J Infect Dis 1990;161: 932-937.
57 Densen P, Brown EJ, O'Neill GJ, Tedesco F, Clark RA, Frank MM, Webb D, Myers J: Inherited defi- ciency of C8 in a patient with recur- rent meningoooocal infections: Fur- ther evidence for a dysfunctional C8 molecule and nonlinkage to the HLA system. J Clin Immunol 1983; 3:90-99.
311











![[Type text] Meningococcal vaccinesncirs.org.au/sites/default/files/2020-02/Meningococcal... · 2020-02-10 · Meningococcal vaccines . f. or Australians | NCIRS Fact sheet: April](https://static.fdocuments.net/doc/165x107/5f3b89378aca2557ce785a5e/type-text-meningococcal-2020-02-10-meningococcal-vaccines-f-or-australians.jpg)






