
Comparative genomic hybridization of precipitated DNA · Comparative genomic hybridization of...
20
1 Supplementary methods and information Comparative genomic hybridization of precipitated DNA The karyotypes of two normal leukocytes and two MCF-7 breast cancer cells, the DNA of which had been precipitated are compiled in Supplementary Figure 1. While the profiles of the normal cells showed no significant deviation from the midline, the profiles of the MCF-7 cells exhibit multiple yet almost identical genomic aberrations (e.g., on chromosome 8) that are congruent with previously published data. Therefore, the experiments with single cells of a defined karyotype – i.e. single leukocytes and MCF-7 breast cancer cells show that cellular DNA of a single cell can be completely precipitated. Supplementary Figure 1. CGH profiles of two normal leukocytes (red) and two MCF-7 breast cancer cells (blue). Genomic DNA was isolated from the supernatant after mRNA isolation.
Transcript of Comparative genomic hybridization of precipitated DNA · Comparative genomic hybridization of...

1
Supplementary methods and information
Comparative genomic hybridization of precipitated DNA
The karyotypes of two normal leukocytes and two MCF-7 breast cancer cells, the DNA of
which had been precipitated are compiled in Supplementary Figure 1. While the profiles of
the normal cells showed no significant deviation from the midline, the profiles of the MCF-7
cells exhibit multiple yet almost identical genomic aberrations (e.g., on chromosome 8) that
are congruent with previously published data. Therefore, the experiments with single cells of
a defined karyotype – i.e. single leukocytes and MCF-7 breast cancer cells show that cellular
DNA of a single cell can be completely precipitated.
Supplementary Figure 1. CGH profiles of two normal leukocytes (red) and two MCF-7 breast
cancer cells (blue). Genomic DNA was isolated from the supernatant after mRNA isolation.

2
The ratio profiles of normal cells lie within the dashed lines that indicate the threshold of
significance. The profiles of both cancer cells show multiple and similar chromosomal
deletions and amplifications.

3
Generation and global amplification of single cell cDNA.
The following conditions were essential to obtain sufficient sensitivity and reliability. Firstly,
tRNA and rRNA had to be removed to improve PCR amplification. Secondly, we found that
random priming requires 2000-8000-times higher concentrations of primers for cDNA
synthesis than required with previously described techniques using oligo-dT primers at 10 nM
concentrations1-3. Supplementary Fig. 2A shows that higher concentrations of random primers
for cDNA synthesis lead to increased detection rates of specific transcripts (e.g. KRAS).
Surplus primer, being an effective competitor of the subsequent tailing and amplification
reaction, had to be removed before both steps. Equally, high dNTP concentrations improved
cDNA synthesis but interfered with the subsequent tailing reaction and also needed to be
removed. Capturing of mRNA on oligo-dT coated magnetic beads provided for simple
handling during mRNA isolation and buffer exchange steps. The second primer binding site
was added by homopolymer tailing. Here, poly-dC primers binding to poly-dG tails were
found to give at least a 100-fold higher sensitivity than poly-dT primers on poly-dA tails
(Supplementary Fig. 2B, compare lanes 3, 4 to 5, 6). Tailing efficiency (in Supplementary
Fig. 2B) as well as the sensitivity of the subsequent PCR of poly-dA- and poly-dG-tailed
sequences was assessed by adding a homopolymer tail of either poly-dA or poly-dG to a
defined PCR fragment. The poly-dA- and poly-dG-tailed fragments were diluted and then
amplified by PCR with poly-dT and poly-dC primers, respectively.
The most dramatic improvement was obtained when only one primer (Supplementary
Fig. 2D) was used for global PCR instead of two (Supplementary Fig. 2C). To prove this
point we isolated, diluted, reverse-transcribed and dG-tailed mRNA of HT29 cell line cells.
The cDNA synthesis primer consisted of a 3’ random hexamer (N6) and a 5’ flanking region

4
of either a poly-dC stretch (CFL5c-N6) or a sequence of all four bases (FL4-N6), introducing
either the FL4 or poly-dC sequence at the 5’ end of the generated cDNAs. Following dG-
tailing,the cDNAs were amplified with the respective primers. Use of the FL4 primer in
combination with the poly-dC binding primers (CP2, CP3) prevented amplification,
regardless of the annealing temperature chosen (Supplementary Fig. 2C, lanes 1, 2 and 4, 5).
PCR with the CP2 primer alone resulted in amplification of a wide size range of cDNA
molecules (0.2 – 3kb; Supplementary Fig. 2D, lanes 3, 4). Even highly diluted cDNA (1:200)
was still sufficient for global amplification (Supplementary Fig. 2D, lanes 1, 2). Random
hexamers (N6), octamers (N8), alone or combined with oligo-dT (dT)15 , sharing the same
poly-dC flanking region were compared. All worked well and reliably. The best results with
single cells were obtained with a combination of poly-dC-N8 (CFL5c8) and poly-dC-(dT)15
(CFL5cT).

5
Supplementary Figure 2. Experimental conditions determining amplification success. (A)
Twenty HT29 colon carcinoma cells (lanes 1-20) were individually isolated and processed.
After cell lysis in cDNA synthesis buffer containing the detergent Igepal, groups of five single
M
80 µM 8 µM 0.8 µM 0.08 µM A
A-tail
G-tail
10-5 10-3 - B
1 2 3 4 5 6 1 2 3 4 5 CP3-FL4 CP2-FL4 1:200 pure
C D
- + 1 2 3 4 5 11 6 7 8 9 10 12 13 14 15 16 17 19 18 20
3 4 5 621

6
cells were formed and reverse transcribed with four different concentrations of random cDNA
synthesis primers. Efficacy of cDNA synthesis was tested with KRAS specific PCR for each
concentration. (lanes 1-5, 80 µM; lanes 6-10, 8µM; lanes 11-15, 0.8 µM; lanes 16-20, 0.08
µM). (B) Effect of homopolymer tails on sensitivity. A 350 bp TGFA cDNA fragment was
isolated, diluted and either dA or dG tailed. Serial dilutions were tested by PCR using poly-dT
or poly-dC containing primers, respectively, and an additional primer from within the TGFA
sequence. The informative dilutions are shown in duplicates. (lanes 1+2, negative control;
lanes 3+4, 10-3 dilution; lanes 5+6, 10-5 dilution). (C) FL4-N6 primed and reverse transcribed
mRNA was dG-tailed and amplified using the CP3 + FL4 primers (lanes 1-3) or CP2 + FL4
primers at different annealing temperatures (lane 1+4, 68°C, lane 2+5 65°C, lane 3+6,
negative control). (D) An identical amount of mRNA as in C) was reverse transcribed using
the CFL5cN6 primer, and amplified with the CP2 primer (lane 3+4), which resulted in
amplification of a wide range of cDNA fragments. Even a mRNA dilution of 1:200 (lane 1+2)
resulted in a similar smear regardless of annealing temperatures (lane 1+3, 68°C; lane 2+4,
65°C; lane 5, negative control).
Primers used and mentioned in Supplementary Fig.2C and D were:
Cfl5cN6 5’-(CCC)5 GTC TAG ANN (N)6-3’
FL4N6 5’-TTT CTC CTT AAT GTC ACA GAT CTC GAG GAT TTC (N)6-3’)
CP3 5’- GCT GAA GTG GCG AAT TCC GAT GCC (C)12-3’
FL4 5’-CTC CTT AAT GTC ACA GAT CTC GAG GAT TTC-3’.

7
Comparison of single cells of different histogenetic origin
We tested the PCR amplificates from single cells for suitability of cDNA array
analysis. The specificity of the hybridisation of digoxigenin-labelled probes is depicted in
Supplementary Table 1, where the expression patterns of genes from single cells of different
histogenetic origin are shown. Only the MCF-7 and A431 cell expressed the cytokeratin
genes, markers for their epithelial origin, whereas the erythroleukemia K562 cell and EBV-
transformed B cell JY expressed genes of a hematopoetic origin, including CD33, CD37,
CD38, and kappa light chain (IGKC) in the B cell. In addition, the testis- and tumour-specific
MAGE genes were highly expressed in all cancer cells but not the virally transformed B cell.
These data show that the amplificates obtained from single cell PCR are suitable for cDNA
array analysis and produce cell type-specific gene expression patterns of single cells.
Supplementary Table 1. Expression of histogenetically informative genes by single cells
derived from different tissues.
MCF-7 A431 K562 JY ACTB + + + + EEF1A1 + + + + KRT7 + + - - KRT10 - + - - KRT13 - + - - KRT18 + + - - KRT19 + + - - EpCAM + + - - CD33 - - + + CD37 - - + + CD38 - - + - IGKC - - - + VIM - + + -

8
ITGA6 + - - - ITGB1 + - - - ITGB2 - - - + ITGB4 - - + - ITGB7 - - - + PTK2 + - - - MageA1 + - + - MageA2 + + + - MageA3 + - + - MageA6 + - + - MageA12 + + + -
Array analysis of MCF-7 breast cancer cells
Expression patterns of four MCF-7 cells were compared using a cDNA array with 109
different genes (Supplementary Table 2). 46 genes (42%) were expressed in at least one cell,
and 63 (58%) were negative for all four cells. Eighteen of the 46 (39%) expressed genes were
detected in all four cells whereas the remaining 29 (61%) were found to be heterogeneously
expressed. All housekeeping genes (EEF1A1, GHPDH, ACTB) belonged to the genes
detected in all four cells but also structural genes like the cytokeratins (KRT) 7, 8, 18, 20 and
adhesion molecules like Integrin α6 and β1 (ITGA6, ITGB1). Among the genes that were
found only in one of the cells many are regulated within the cell cycle such as CDKN2A,
CDKN1A, CDKN1B, ING1, MKI67, TK1, but also cytokeratin family members that are
usually not expressed in breast tissue (KRT10, KRT13).
Supplementary Table 2. Commonly and differentially detected transcripts of four single
MCF-7 cells.
4/4* 3/4 2/4 1/4 EEF1A1 GHPDH ACTB
KRT19 TIMP1 CTSB
ITGB4 ITGB5 TP53
KRT10 KRT13 ADAM9

9
KRT7 KRT8 KRT18 KRT20 ITGA6 ITGB1 PTK2 BSG PLAUR MMP7 CCND1 EPHA2 M4S1 (EpCAM) ABCC1 PHLDH1
CTSD CTSL ADAM10 MYC
CKM
ADAM15 ADAM17 CDKN2A CDKN1A CDKN1B ING1 MKI67 TK1 CDH1 IGF1R IGF2R TGFB VEGF DSP
* Listed are the transcripts that were detected in all four single cells (4/4), three of four (3/4),
two of four (2/4), and one of four (1/4).

10
Primer information of the gene specific PCRs
Supplementary Table 3: Sequences of oligonucleotide primers for the RT-PCR amplification
of the different genes.
MAGE 2 sense antisense
CAT TGA AGG AGA AGA TCT GCC T CAG GCT TGC AGT GCT GAC TC
MAGE 3/6 sense antisense
GGC TCG GTG AGG AGG CAA G GAT GAC TCT GGT CAG GGC AA
MAGE 4 sense antisense
CAC CAA GGA GAA GAT CTG CCT CAG GCT TGC AGT GCT GAC TCT
β-Actin sense antisense
GTG GGG CGC CCC AGG CAC CA CTC CTT AAT GTC ACG CAC GAT TTC
EF-1α sense antisense
TGC CCC AGG ACA CAG AGA CT CTG TGT CGG GGT TGT AGC CA
TNF-α sense antisense
CAG AGG GAA GAG TTC CCC AG CCT TGG TCT GGT AGG AGA CG
EMMPRIN sense antisense
CAT GCT GGT CTG CAA GTC A GTT CTC AAT GTG TAG CTC TG
EpCAM sense antisense
GCG AGT GAG AA CTA CTG CAC ATC AGC TAT GTC CAC ATC
Ki-ras sense antisense
TAT AAA CTT GTG GTA GTT GGA GC CTC TTG ACC TGC TGT GTC GA

11
Sequence-independent amplification
In order to assess the sensitivity, the potential systematic failure to amplify “difficult”
sequences and the influence of other technical factors on transcript detection we tested the
differences between cDNA preparations of single A431 cells that were split into two halves
before global amplification. In the first step, we performed gene-specific PCRs with the
amplified cDNAs of the halved cells (Supplementary Fig. 3). For comparison, we diluted
unamplified cDNA isolated from a pool of 500,000 A431 cells to such an extent that the
intensity of the β-actin band was similar to that obtained with 50% of cDNA prepared from a
single cell. After 32 cycles and with a cDNA amount corresponding to about 10,000 cells,
both β-actin signals that of the pooled control DNA as well as that of 50% of the single cell
cDNA reached the plateau phase of amplification. As shown in Supplementary Figure 3, the
variation between two cDNA halves of the same cell was very low. In two independent
experiments, each half (a+b) from six single A431 cells yielded β-actin bands of similar
intensity.
Because PCR amplification is known to be sequence-dependent, we also tested the
amplification of MAGE transcripts, very demanding with regard to primer design. Kufer et al.
had to test almost 800 primer combinations to achieve successful amplification of MAGE
genes from less than five cells demonstrating the magnitude of the problem5. MAGE genes
belong to a family of genes expressed in spermatogonal cells and exclusively in various
cancer cells, hence the designation cancer-testis genes. MAGE transcripts could be detected in
all single cells. After the same number of cycles in the specific PCR as for β-actin, the level
of MAGE expression was consistently found to be lower than that of β-actin, and the
concentration of MAGE transcripts in split single cell samples after global PCR amplification
(Supplementary Fig. 3, lanes 2-4 and 6-8) compared well to that of the control sample from

12
pooled cells (Supplementary Fig. 3, +). Although the MAGE transcripts were only weakly
expressed in the pooled cDNA from 10,000 cells (Supplementary Fig.3, +), the data obtained
from single cells were identical in 4 out of 6 paired halves of the cDNA. The failure to detect
any MAGE transcript in cell half 7a and 8b (Supplementary Fig.3) is most likely due to an
unequal distribution of the beads prior to amplification between the two halves indicating thus
the detection limit of the method. The semi-quantitative character of our amplification method
is strongly suggested by the reproducibly similar signal intensity of all β-actin and MAGE
transcripts: No artificial increase in intensity is observed in any divided cell and the intensities
of the β-actin and MAGE band closely resemble the positive control. As the yield (y) of a
PCR amplification follows the equation y = ((1+R)n -2n)x, with n being the number of cycles,
x the copy number of the original template and (1+R) the amplification efficiency of a any
sequence (R ≤ 1), Supplementary Figure 3 suggests that R is the same for all sequences under
the applied conditions. Consequently, although an exponential amplification takes place, the
ratios of the different sequences remain very similar.
Supplementary Figure 3. Gene specific PCR for β-actin and various MAGE transcripts using
unamplified pooled cDNA of A431 cells as positive control (+) and amplificates of single
76 5321 4 8 M
Actin
Mage
Mage
Mage
M - + a b a b a b a b a b a b a b a b

13
A431 cells (lane 2-4 and 6-8) that were divided into two halves (a+b) before global PCR. Two
independent experiments were performed (lane 1-4 and 5-8) with lane 1 and lane 5 being the
negative controls for the global PCR.

14
Evaluation of array hybridization of split single cells
The high sensitivity of the procedure and the diversity of the isolated and amplified
cDNA sequences was also seen with the cDNA of four halves labeled and hybridized to a
cDNA array representing 193 different gene sequences. Most transcripts could be detected in
both halves of the single cell PCR products (Supplementary Fig.4). Of the total of 148
observed signals, 95 (64%) were found in the corresponding halves, whereas 53 (36%) were
found in only one half. Figure 4 shows the distribution of the signals of the two halves from
one experiment. Hybridization intensity of the two halves for the spotted cDNAs was
classified from 0 (no signal), 1 (weak) over 2 (middle) to 3 (strong). It can be seen that for
strong signals both halves correlated well (correlation coefficient r= 0.81). Also the signals
with intermediate strength were in most cases found in both halves. Weak signals gave the
least concordant results between the two halves of a cell. This can be explained by the
stochastic nature of the cDNA priming events, an uneven distribution of low copy number
transcripts before the PCR is performed and the undefined but real detection-limit of the
method. As many transcripts are estimated to be present in a cell in copy numbers of about 5-
15 (ref. 4), it is very likely that such transcripts are particularly subjected to effects leading to
discordant results when cDNA of a single cell is divided.

15
Supplementary Figure 4. Hybridization results of two independently amplified cDNA halves
of a single cell. Each spot on the array was classified as strong (3), middle (2), weak (1) and
no signal (0). The signal intensities for each spot were compared between the two halves. X-
and y-axis give the signal intensities for each half of the cDNA of a single cell and the z-axis
the number of cDNAs. The column at position 3/3 for example means that 10 transcripts were
expressed equally strong in both halves. Ideally, the columns would form a diagonal from 3/3
over 2/2 and 1/1 to 0. However, it can be seen that the light blue columns are of similar height
at position 0/1, 1/1, 1/0, indicating that no bipartition was achieved for weak signals as was
for the strong signals. (Red columns, at least one half produced a strong signal; yellow, at
least one half produced an intermediate signal; light blue, at least one half resulted in a weak
signal; dark blue, no signal in both halves).

16
Hybridization of exemplary micrometastatic cells to Clontech Cancer1.2 and own array
From the three micrometastatic cells C, B, L mRNA was isolated and prepared as
described. As control, the procedure was performed without the addition of a cell. Seven
genes on the Clontech arrays had to be excluded from analysis because a signal was obtained
in at least one of the negative controls. These genes were coding for VBP1, CASP10, TGFB1,
HBA1, ROS1, TDGF1, and TNFR2 . The number of positive signals ranged from 5.3 %
(70/1313), 7.0 % (92/1313) to 11.8 % (155/1313) for cells from patients B, C, and L
respectively. Although, these numbers are lower than those obtained by conventional
transcriptome analysis with millions of cells, one should not forget that the number of
attributes of an individual cell is necessarily lower than that of a population. The hybridization
results are shown in Supplementary Figure 5.
Hugo B C L Hugo B C L Hugo B C L
ABCC1 HBA1 PDGFAP
ACTB hBAP PDPK1
ACTB HDAC3 PGF
ACTB HDGF PHB
ADAM15 HINT PHKB
ADAM17 HLA PHLDA1
ADAM8 HLAC PIM1
ADPRT HLADPB1 PIN1
ADSL HLADQ1B PKUA
ADSS HLADRB1 PLA2
AHR HMG2 PLAU
AK1 HMGIY PLAUR
ANT2 HNRPK PM5
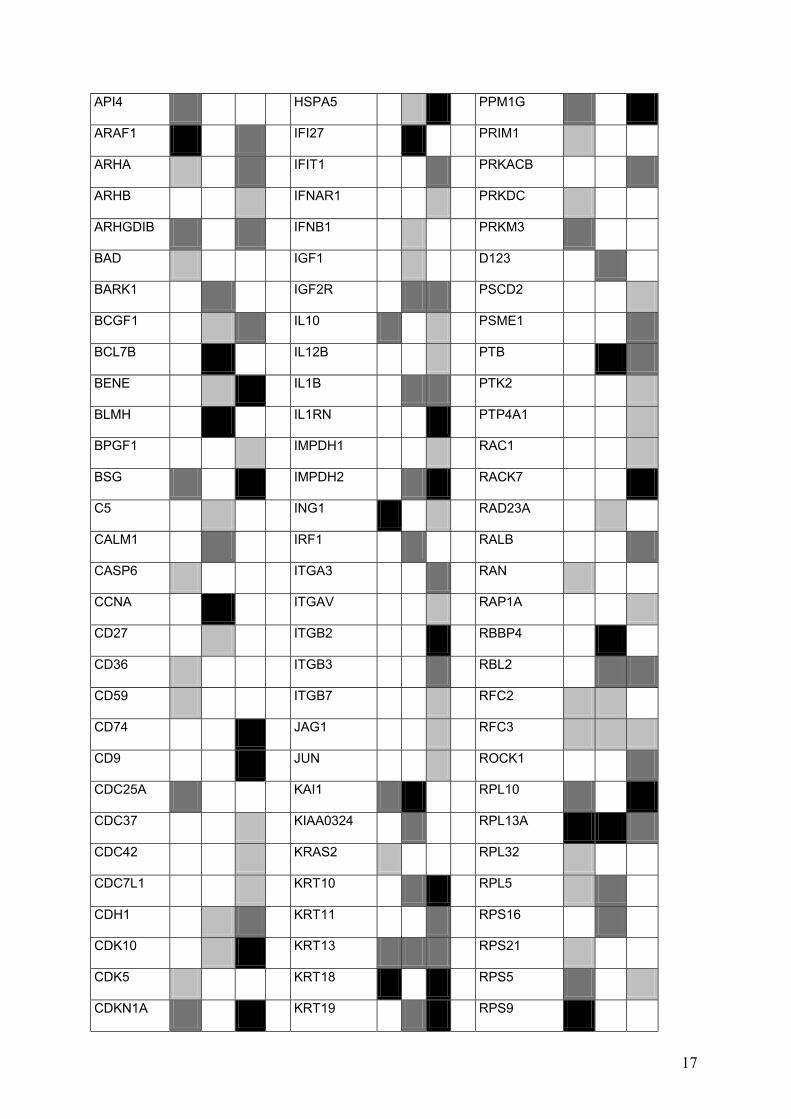
17
API4 HSPA5 PPM1G
ARAF1 IFI27 PRIM1
ARHA IFIT1 PRKACB
ARHB IFNAR1 PRKDC
ARHGDIB IFNB1 PRKM3
BAD IGF1 D123
BARK1 IGF2R PSCD2
BCGF1 IL10 PSME1
BCL7B IL12B PTB
BENE IL1B PTK2
BLMH IL1RN PTP4A1
BPGF1 IMPDH1 RAC1
BSG IMPDH2 RACK7
C5 ING1 RAD23A
CALM1 IRF1 RALB
CASP6 ITGA3 RAN
CCNA ITGAV RAP1A
CD27 ITGB2 RBBP4
CD36 ITGB3 RBL2
CD59 ITGB7 RFC2
CD74 JAG1 RFC3
CD9 JUN ROCK1
CDC25A KAI1 RPL10
CDC37 KIAA0324 RPL13A
CDC42 KRAS2 RPL32
CDC7L1 KRT10 RPL5
CDH1 KRT11 RPS16
CDK10 KRT13 RPS21
CDK5 KRT18 RPS5
CDKN1A KRT19 RPS9
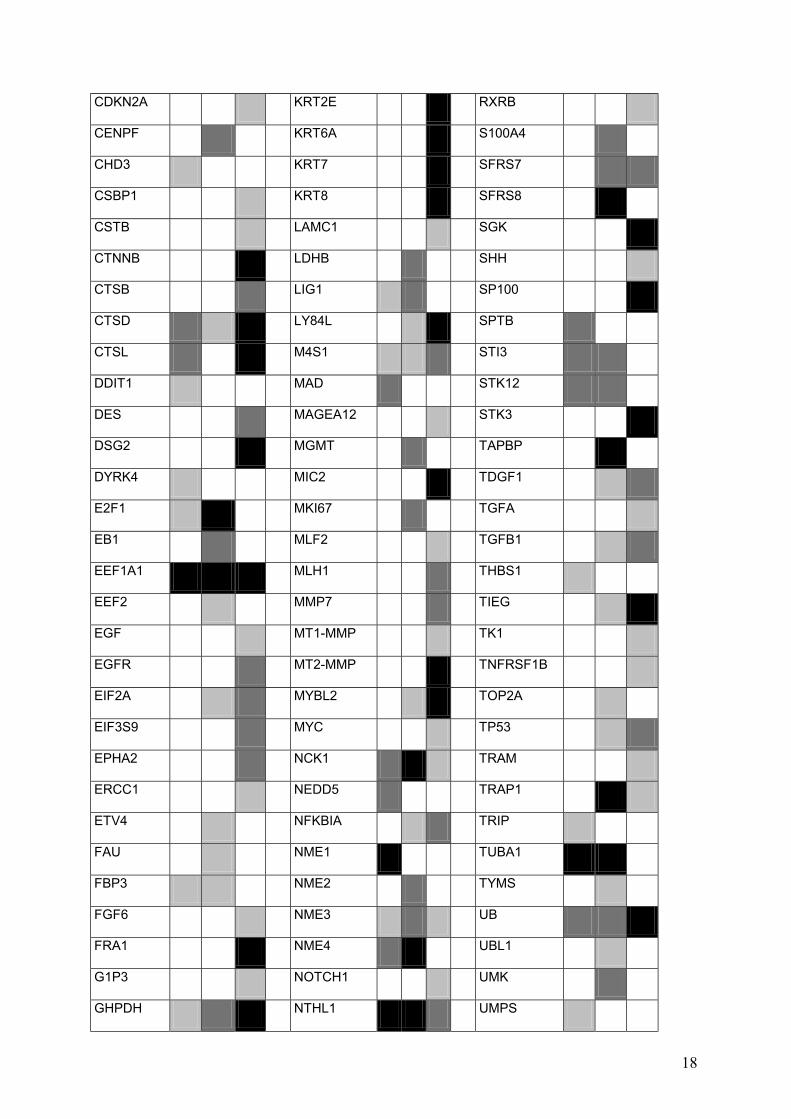
18
CDKN2A KRT2E RXRB
CENPF KRT6A S100A4
CHD3 KRT7 SFRS7
CSBP1 KRT8 SFRS8
CSTB LAMC1 SGK
CTNNB LDHB SHH
CTSB LIG1 SP100
CTSD LY84L SPTB
CTSL M4S1 STI3
DDIT1 MAD STK12
DES MAGEA12 STK3
DSG2 MGMT TAPBP
DYRK4 MIC2 TDGF1
E2F1 MKI67 TGFA
EB1 MLF2 TGFB1
EEF1A1 MLH1 THBS1
EEF2 MMP7 TIEG
EGF MT1-MMP TK1
EGFR MT2-MMP TNFRSF1B
EIF2A MYBL2 TOP2A
EIF3S9 MYC TP53
EPHA2 NCK1 TRAM
ERCC1 NEDD5 TRAP1
ETV4 NFKBIA TRIP
FAU NME1 TUBA1
FBP3 NME2 TYMS
FGF6 NME3 UB
FRA1 NME4 UBL1
G1P3 NOTCH1 UMK
GHPDH NTHL1 UMPS

19
GLG1 OSMR VEGF
GMFB PA2G4 VIM
GRP58 PAK2 VRK2
GST PCTK1
GUK1 PCTK2
HAI2 PCTK3
PDGFA
Supplementary Figure 5. Hybridization results of cell B, C, and L. Three categories of signal intensity were formed: Strong (black), middle (middle gray) and weak (light gray).

20
References
1. Brady, G. & Iscove, N.N. Construction of cDNA libraries from single cells. Methods
Enzymol 225, 611-623 (1993).
2. Brail, L.H. et al. Gene expression in individual cells: analysis using global single cell
reverse transcription polymerase chain reaction (GSC RT-PCR). Mutat-Res 406, 45-54
(1999).
3. Trumper, L.H. et al. Single-cell analysis of Hodgkin and Reed-Sternberg cells:
molecular heterogeneity of gene expression and p53 mutations. Blood 81, 3097-3115
(1993).
4. Alberts, B. Molecular biology of the cell. (Garland Publishing, New York & London;
1994).
5. Kufer, P. et al. Heterogeneous Expression of MAGE-A Genes in Occult Disseminated
Tumor Cells: A Novel Multimarker Reverse Transcription-Polymerase Chain Reaction
for Diagnosis of Micrometastatic Disease. Cancer Res 62, 251-261. (2002).


















