
Combined inhibition of 20-hydroxyeicosatetraenoic acid ...
19
Clinical Science (2010) 118, 617–632 (Printed in Great Britain) doi:10.1042/CS20090459 617 Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and of epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension- induced end-organ damage in Ren-2 transgenic rats Vˇ era ˇ CERT ´ IKOV ´ A CH ´ ABOV ´ A ∗ † 1 , Agnieszka WALKOWSKA‡ 1 , Elzbieta KOMPANOWSKA-JEZIERSKA‡, Janusz SADOWSKI‡, Petr KUJAL†§, Zdenka VERNEROV ´ A†§, Zdeˇ na VA ˇ NOURKOV ´ A†, Libor KOPKAN†, Herbert J. KRAMER¶, John R. FALCK ∗∗ , John D. IMIG††, Bruce D. HAMMOCK‡‡, Ivana VAN ˇ E ˇ CKOV ´ A† and Ludˇ ek ˇ CERVENKA†§§ ∗ Department of Nephrology, 1st Medical Faculty, Charles University, Prague, Czech Republic, †Department for Experimental Medicine, Institute for Clinical and Experimental Medicine, 1958/9 V´ ıdeˇ nsk´ a, CZ-140 21 Prague 4, Czech Republic, ‡Laboratory of Renal and Body Fluid Physiology, Mossakowski Medical Research Centre, Polish Academy of Science, Warsaw, Poland, §Department of Pathology, Third Faculty of Medicine, Charles University, Prague, Czech Republic, Center for Cardiovascular Research, Prague, Czech Republic, ¶Section of Nephrology, Medical Policlinic, Department of Medicine, University of Bonn, Bonn, Germany, ∗∗ Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 606-538, U.S.A., ††Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI 53226, U.S.A., ‡‡Department of Entomology, University of California at Davis Cancer Center, Davis, CA 95616, U.S.A., and §§Department of Physiology, 2nd Medical Faculty, Charles University, Prague, Czech Republic A B S T R A C T Recent studies have shown that the renal CYP450 (cytochrome P450) metabolites of AA (arachidonic acid), the vasoconstrictor 20-HETE (20-hydroxyeicosatetraenoic acid) and the vasodilator EETs (epoxyeicosatrienoic acids), play an important role in the pathophysiology of AngII (angiotensin II)-dependent forms of hypertension and the associated target organ damage. The present studies were performed in Ren-2 renin transgenic rats (TGR) to evaluate the effects of chronic selective inhibition of 20-HETE formation or elevation of the level of EETs, alone or in combination, on the course of hypertension and hypertension-associated end-organ damage. Both young (30 days of age) prehypertensive TGR and adult (190 days of age) TGR with established hypertension were examined. Normotensive HanSD (Hannover Sprague–Dawley) rats served Key words: cytochrome P450 (CYP450), end-organ damage, hypertension, renin–angiotensin system (RAS), soluble epoxide hydrolase. Abbreviations: AngII, angiotensin II; AA, arachidonic acid; CYP450, cytochrome P450; DDMS, N-methylsulfonyl-12,12- dibromododec-11-enamide; DHETE, dihydroxyeicosatrienoic acid; EET, epoxyeicosatrienoic acid; GSI, glomerulosclerosis index; HanSD, Hannover Sprague–Dawley; 20-HETE, 20-hydroxyeicosatetraenoic acid; LVW/TL, ratio of left ventricular weight to tibial length; MAP, mean arterial pressure; NE, noradrenaline (norepinephrine); NCND, N-cyclohexyl-N-dodecyl urea; RAS, renin– angiotensin system; RBF, renal blood flow; RVR, renal vascular resistance; SBP, systolic blood pressure; sEH, soluble epoxide hydrolase; TGR, Ren-2 renin transgenic rats; VSMC, vascular smooth muscle cell. 1 These authors contributed equally to the study and should be considered as joint first authors. Correspondence: Dr Ludˇ ek ˇ Cervenka (email [email protected]). C The Authors Journal compilation C 2010 Biochemical Society www.clinsci.org Clinical Science
Transcript of Combined inhibition of 20-hydroxyeicosatetraenoic acid ...

Clinical Science (2010) 118, 617–632 (Printed in Great Britain) doi:10.1042/CS20090459 617
Combined inhibition of20-hydroxyeicosatetraenoic acid formation and
of epoxyeicosatrienoic acids degradationattenuates hypertension and hypertension-
induced end-organ damage in Ren-2transgenic rats
Vera CERTIKOVA CHABOVA∗†1, Agnieszka WALKOWSKA‡1, ElzbietaKOMPANOWSKA-JEZIERSKA‡, Janusz SADOWSKI‡, Petr KUJAL†§,Zdenka VERNEROVA†§, Zdena VANOURKOVA†‖, Libor KOPKAN†‖,Herbert J. KRAMER¶, John R. FALCK∗∗, John D. IMIG††, Bruce D. HAMMOCK‡‡,Ivana VANECKOVA†‖ and Ludek CERVENKA†§§∗Department of Nephrology, 1st Medical Faculty, Charles University, Prague, Czech Republic, †Department for ExperimentalMedicine, Institute for Clinical and Experimental Medicine, 1958/9 Vıdenska, CZ-140 21 Prague 4, Czech Republic,‡Laboratory of Renal and Body Fluid Physiology, Mossakowski Medical Research Centre, Polish Academy of Science, Warsaw,Poland, §Department of Pathology, Third Faculty of Medicine, Charles University, Prague, Czech Republic, ‖Center forCardiovascular Research, Prague, Czech Republic, ¶Section of Nephrology, Medical Policlinic, Department of Medicine,University of Bonn, Bonn, Germany, ∗∗Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas,TX 606-538, U.S.A., ††Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI 53226,U.S.A., ‡‡Department of Entomology, University of California at Davis Cancer Center, Davis, CA 95616, U.S.A., and§§Department of Physiology, 2nd Medical Faculty, Charles University, Prague, Czech Republic
A B S T R A C T
Recent studies have shown that the renal CYP450 (cytochrome P450) metabolites of AA(arachidonic acid), the vasoconstrictor 20-HETE (20-hydroxyeicosatetraenoic acid) and thevasodilator EETs (epoxyeicosatrienoic acids), play an important role in the pathophysiology ofAngII (angiotensin II)-dependent forms of hypertension and the associated target organ damage.The present studies were performed in Ren-2 renin transgenic rats (TGR) to evaluate the effectsof chronic selective inhibition of 20-HETE formation or elevation of the level of EETs, alone or incombination, on the course of hypertension and hypertension-associated end-organ damage. Bothyoung (30 days of age) prehypertensive TGR and adult (190 days of age) TGR with establishedhypertension were examined. Normotensive HanSD (Hannover Sprague–Dawley) rats served
Key words: cytochrome P450 (CYP450), end-organ damage, hypertension, renin–angiotensin system (RAS), soluble epoxidehydrolase.Abbreviations: AngII, angiotensin II; AA, arachidonic acid; CYP450, cytochrome P450; DDMS, N-methylsulfonyl-12,12-dibromododec-11-enamide; DHETE, dihydroxyeicosatrienoic acid; EET, epoxyeicosatrienoic acid; GSI, glomerulosclerosis index;HanSD, Hannover Sprague–Dawley; 20-HETE, 20-hydroxyeicosatetraenoic acid; LVW/TL, ratio of left ventricular weight to tibiallength; MAP, mean arterial pressure; NE, noradrenaline (norepinephrine); NCND, N-cyclohexyl-N-dodecyl urea; RAS, renin–angiotensin system; RBF, renal blood flow; RVR, renal vascular resistance; SBP, systolic blood pressure; sEH, soluble epoxidehydrolase; TGR, Ren-2 renin transgenic rats; VSMC, vascular smooth muscle cell.1 These authors contributed equally to the study and should be considered as joint first authors.Correspondence: Dr Ludek Cervenka (email [email protected]).
C© The Authors Journal compilation C© 2010 Biochemical Society
www.clinsci.org
Clin
ical
Sci
ence

618 V. Certıkova Chabova and others
as controls. The rats were treated with N-methylsulfonyl-12,12-dibromododec-11-enamide toinhibit 20-HETE formation and/or with N-cyclohexyl-N-dodecyl urea to inhibit soluble epoxidehydrolase and prevent degradation of EETs. Inhibition in TGR of 20-HETE formation combined withenhanced bioavailability of EETs attenuated the development of hypertension, cardiac hypertrophy,proteinuria, glomerular hypertrophy and sclerosis as well as renal tubulointerstitial injury. Thiswas also associated with attenuation of the responsiveness of the systemic and renal vascularbeds to AngII without modifying their responses to noradrenaline (norepinephrine). Our findingssuggest that altered production and/or action of 20-HETE and EETs plays a permissive role inthe development of hypertension and hypertension-associated end-organ damage in this model ofAngII-dependent hypertension. This information provides a basis for a search for new therapeuticapproaches for the treatment of hypertension.
INTRODUCTION
Chronic kidney disease and end-stage renal diseasepresent severe medical problems; their incidence hasbeen increasing steadily, especially in industrializedcountries [1]. For both conditions, hypertension remainsone of the most important risk factors even thoughantihypertensive treatment has significantly advancedover the recent decades [2]. It is generally accepted thatinappropriate activation of the RAS (renin–angiotensinsystem) is a major factor in the development of AngII(angiotensin II)-dependent forms of hypertension [3].However, the role of the RAS in the pathophysiologyof hypertensive target organ damage is still poorlyunderstood.
It is well recognized that CYP450 (cytochromeP450)-dependent metabolites of AA (arachidonic acid),20-HETE (20-hydroxyeicosatetraenoic acid) and EETs(epoxyeicosatrienoic acids), play an important role in theregulation of renal tubular ion transport and renal andsystemic vascular tone [4,5]. Moreover, recent studiesstrongly suggest that altered production and/or actionof CYP450-dependent metabolites contribute to thedevelopment of AngII-dependent forms of hypertension[6–11].
The hypertensive rat transgenic for the mouse Ren-2renin gene [TGR; strain name TGR(mRen2)27] repres-ents a unique AngII-dependent animal model in whichthe development of hypertension is attributable to asingle gene alteration [12]. We have found recentlythat TGR exhibit increased intrarenal levels of 20-HETE and, simultaneously, an intrarenal deficiencyof EETs [13]. 20-HETE is a vasoconstrictor and iscommonly regarded as a natriuretic agent, a combinationof properties forming the background for both its pro-and anti-hypertensive potential [4,6,7]. On the otherhand, we have recently provided evidence that 20-HETEmay induce antinatriuresis in TGR [13]. EETs exhibitantihypertensive properties related to their vasodilatorand natriuretic potency [4–6].
In the present study, we tested the hypothesisthat in TGR, a monogenic model of AngII-dependent
hypertension, increased intrarenal 20-HETE combinedwith intrarenal deficiency of EETs may at least partiallyaccount for the enhanced renal and systemic vascularresponsiveness to AngII and thereby contribute tothe development and/or maintenance of hypertension.To examine this possibility, we evaluated the possibleantihypertensive effects of chronic selective inhibitionof 20-HETE formation and of an elevation of EETlevels in young prehypertensive heterozygous TGRrats; the effect of the treatment on the associatedhypertension-induced end-organ damage was alsoexamined. In addition, to make the study more relevantto the clinical condition of hypertensive patients, weassessed the effects of chronic selective inhibition of20-HETE production and of enhanced bioavailabilityof EETs on blood pressure and target organs in adultTGR with established hypertension.
Finally, to gain a more detailed insight intothe mechanism(s) underlying the potential beneficial(antihypertensive) effects of selective pharmacologicalinterventions into the CYP450-dependent metabolism ofAA, experiments were performed in which systemic andrenal vascular responses to AngII and NE [noradrenaline(norepinephrine)] were evaluated in TGR and HanSD(Hannover Sprague–Dawley) rats. TGR and HanSD ratswere either untreated or exposed to chronic reductionof 20-HETE production, or to an increase in thelevel of EETs, or they were subjected to both (combined)treatments at the same time.
MATERIALS AND METHODS
Ethical approval and animalsThe studies were performed in accordance with guidelinesand practices established by the Animal Care and UseCommittee of the Institute for Clinical and ExperimentalMedicine. All animals used in the present study werebred at the Department of Experimental Medicine of theInstitute for Clinical and Experimental Medicine, Prague,from stock animals supplied by the Max DelbruckCenter for Molecular Medicine, Berlin, Germany, which
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 619
is accredited by the Czech Association for Accreditationof Laboratory Animal Care. The animals were kept on a12-h/12-h light/dark cycle. Throughout the experiments,rats were fed a normal salt, normal protein diet (0.45 %NaCl, 19–21 % protein) produced by SEMED and hadfree access to tap water.
ChemicalsDDMS (N-methylsulfonyl-12,12-dibromododec-11-enamide), a selective inhibitor of 20-HETE formation[14], was administered using osmotic minipumpsimplanted at the dorsal neck; the concentration usedallowed the delivery of 2 mg/day. As tested in ourpreliminary experiments, this dose blocks 20-HETEformation selectively and persistently [13].
NCND (N-cyclohexyl-N-dodecyl urea) selectivelyinhibits sEH (soluble epoxide hydrolase), the enzymeresponsible for the conversion of EETs into DHETEs(dihydroxyeicosatrienoic acids), the biologically inactivemetabolites of EETs. NCND was administered i.p.(intraperitoneally) at a dose of 10 mg · kg−1 of bodyweight · day−1, as described previously [15]. It has beendemonstrated that this dose selectively blocks sEHactivity and increases the bioavailability of EETs [16].
Experimental protocols
Series 1: inhibition of 20-HETE formation and of sEHactivity starting in young prehypertensive rats (earlytreatment protocol)Male heterozygous TGR rats, aged 28 days, and age-matched male HanSD rats from several litters wererandomly assigned to experimental groups, to makesure that the animals from a single litter did notprevail in any of the groups. Beginning from 30 days ofage, SBP (systolic blood pressure) was measured everysecond day in appropriately trained conscious animalsby tail-plethysmography, using a tail-cuff apparatus(MC 4000; Hatteras Instruments); in all cases, a meanSBP of four measurements was taken. This approachis regularly employed in our laboratory, and a closecorrelation between tail-plethysmography and directblood pressure measurements with an indwelling catheterwas established [11,17,18]. At 80 and 140 days of age, theanimals were placed in individual metabolic cages, andafter appropriate habituation training, their 24-h urinewas collected for sodium and water excretion and proteindetermination. At the end of the experiment (150 daysof age), animals were again placed in metabolic cages,food was withheld to prevent contamination of urinesamples and 12-h urine samples were collected on dry ice.The samples were stored at − 80 ◦C until assayed. Theurinary 20-HETE, EETs and DHETEs concentrationswere measured by ELISA using commercially availablekits, according to the manufacturer’s instructions (Detroit
R&D Inc.). Thereafter, rats were killed by decapitation,and AngII levels in plasma, whole right kidney andleft ventricular heart tissue were measured by RIA asdescribed in detail in our previous studies [19–21]. Weused the LVW/TL (ratio of left ventricular weight to tibiallength) to evaluate the degree of cardiac hypertrophy,since we have demonstrated previously that this is themost suitable index to assess cardiac hypertrophy [21].
To assess renal glomerular damage, the left kidney wasquickly removed, fixed in 4 % formaldehyde, dehydratedand embedded in paraffin. The sections stainedwith haematoxylin–eosin and PAS (periodic acid-Schiffreaction) were examined and evaluated in a blind-testfashion. Fifty glomeruli in each kidney were examinedon a semiquantitative scale as described previously[22]: grade 0, entire glomerulus normal; grade 1,sclerotic area up to 25 % (minimal sclerosis);grade 2, sclerotic area 25–50 % (moderate sclerosis);grade 3, sclerotic area 50–75 % (moderate-to-severesclerosis); grade 4, sclerotic area 75–100 % (severesclerosis). The GSI (glomerulosclerosis index) wascalculated using the following formula: GSI = [(1 × n1)+(2 × n2)+(3 × n3)+(4 × n4)]/(n0+n1+n2+n3+n4), wherenx is the number of glomeruli in each grade ofglomerulosclerosis.
Cortical tubulointerstitial injury was evaluated asdescribed by Nakano et al. [23] for inflammatorycell infiltration, tubular dilatation and/or atrophy, orinterstitial fibrosis and was graded semiquantitativelyusing the following scale: grade 0, no abnormal findings;1, mild (< 25 % of the cortex); 2, moderate (25–50 % ofthe cortex); 3, severe (> 50 % of the cortex). Lesions wereassessed for at least 30 random and non-overlapping fieldsin the renal cortex.
Morphometric evaluation of the glomerular volumewas made in the same kidney sections that were examinedfor morphological changes, using the method developedby Weibel [24] and validated by Lane et al. [25]using the Nikon NIS-Elements AR 3.1 morphometricprogram. Briefly, this method consists of determi-nation of mean glomerular profile area and calculationof mean glomerular volume from the following formula:glomerular volume = area1.5 × 1.38/1.01, where 1.38 is β,the shape coefficient for a sphere, and 1.01 is the sizedistribution coefficient assuming a 10 % coefficient ofvariation.
Since osmotic minipumps (M 2006; Alzet) have anoperating time of 42 days, they were replaced by newminipumps containing DDMS on days 68 and 108 of theexperiment. Control rats received osmotic minipumpscontaining saline vehicle and i.p. saline vehicle at the sametime as treated rats. The following experimental groupswere investigated: (i) HanSD rats+vehicle (n = 15); (ii)TGR+vehicle (n = 16); (iii) TGR+DDMS (n = 18); (iv)TGR+NCND (n = 16); and (v) TGR+DDMS+NCND(n = 18).
C© The Authors Journal compilation C© 2010 Biochemical Society

620 V. Certıkova Chabova and others
Series 2: inhibition of 20-HETE formation and of sEH activitystarting in adult hypertensive rats (late treatment protocol)In this series, animals were randomly assigned toexperimental groups and remained untreated fromweaning until 188 days of age. At the age of 189 days,the appropriate treatment(s) was started as describedabove. At 185, 260 and 300 days of age, the animals wereplaced in individual metabolic cages, and their 24-h urinewas collected for protein determination. New minipumpscontaining DDMS were implanted on days 228 and 268of the experiment. At the end (310 days of age), theanimals were again placed individually in metabolic cages,and their 12-h urine was collected for determinationof 20-HETE, EETs and DHETEs as described above.Thereafter, the rats were killed for morphologicalexamination and measurements of AngII levels asdescribed above. The following experimental groupswere investigated: (i) HanSD rats+vehicle (n = 12); (ii)TGR+vehicle (n = 15); (iii) TGR+DDMS (n = 15); (iv)TGR+NCND (n = 15); and (v) TGR+DDMS+NCND(n = 15).
Series 3: MAP (mean arterial pressure) and RBF (renal bloodflow) responses to AngII and NE during inhibition of20-HETE formation and/or of sEH in the early treatmentprotocolIn this series, TGR and HanSD rats were subjectedto the same experimental protocols as described above(series 1). At the end of the experiments (150 days ofage), the animals were fasted overnight and anaesthetizedwith sodium thiopental (50 mg/kg of body weight). Theywere placed on a thermoregulated table to maintainbody temperature at 37–37.5 ◦C. A tracheotomy wasperformed to maintain a patent airway, and the exteriorend of the tracheal cannula was placed inside a smallplastic chamber into which a humidified 95 % O2/5 %CO2 mixture was continuously passed. The right jugularvein was cannulated with PE-50 tubing for infusion ofsolutions and of additional anaesthetic as required, andfor intravenous drug administration. The right femoralartery was cannulated to allow continuous monitoringof MAP and blood sampling. MAP was monitoredwith a pressure transducer (model MLT 1050) andrecorded on the computer using a computerized data-acquisition system (PowerLab/4SP; AD Instruments).The left kidney was exposed via a flank incision, isolatedfrom the surrounding tissue and placed in a lucite cup.A tapered PE-10 catheter was inserted into the leftrenal artery via the left femoral artery, for selectiveintrarenal administration. This catheter was kept patentby a continuous infusion of heparinized isotonic salineat a rate of 4 μl · min−1 throughout the experiment.A PE-10 catheter was inserted into the left ureter.During surgery, an isotonic saline solution containingBSA (6 %) was infused at a rate of 20 μl · min−1. An
ultrasonic transit-time flow probe (1RB) connected toa Transonic flowmeter (Transonic Systems) was placedaround the left renal artery, and RBF was recordedusing a computerized data-acquisition system. After a30-min equilibration period, the experimental protocolwas started to assess the responses of MAP and RBFto systemic (intravenous) and intrarenal bolus doses ofAngII or NE. The vasoactive agents were dissolvedin 100 or 20 μl of isotonic saline for intravenous andintrarenal administration, respectively. After intravenousdrug administration, a bolus of 100 μl of isotonic salinewas injected to ensure a rapid and complete deliveryof the agent into the systemic circulation. Just beforeintrarenal administration of either agent, the intrarenalinfusion rate was increased from 4 to 40 μl/min to deliverthe entire dose rapidly into the kidney; thereafter, thebasal rate of the intrarenal infusion was restored. Weand others have demonstrated previously that a 10-minperiod between injections is sufficient for MAP and RBFrecovery [11,26,27]. MAP responses to intravenous bolusdoses of AngII (50 ng) and thereafter of NE (150 ng)were studied in the following experimental groups in eachseries: (i) HanSD rats+vehicle (n = 8); (ii) TGR+vehicle(n = 8); (iii) TGR+DDMS (n = 7); (iv) TGR+NCND(n = 7); and (v) TGR+DDMS+NCND (n = 7).
Thereafter, the RBF responses to intrarenal bolus dosesof AngII (10 ng) and of NE (30 ng) were determined inthe same experimental groups.
Series 4: MAP and RBF responses to AngII and NE duringinhibition of 20-HETE formation and/or of sEH activity inthe late treatment protocolIn this series, TGR and HanSD rats were subjected to thesame experimental protocols as described in series 2. Atthe end of the experiment (301 days of age), the animalswere prepared in a manner described above (series 3),and MAP and RBF responses to the same doses of AngIIand NE were determined in the following groups: (i)HanSD rats+vehicle (n = 6); (ii) TGR+vehicle (n = 7);(iii) TGR+DDMS (n = 8); (iv) TGR+NCND (n = 9); (v)TGR+DDMS+NCND (n = 7).
Statistical analysis and calculationsStatistical analysis of the data was performed usingGraph-Pad Prism software (Graph Pad Software).All values are expressed as means +− S.E.M. Two-way repeated-measures ANOVA was used to detectdifferences within each experimental group. One-way ANOVA was employed when appropriate. TheRVR (renal vascular resistance) was calculated asthe MAP/RBF ratio during the 30-min equilibrationperiod, using a computerized data-acquisition system(PowerLab/4SP; ADInstruments). Statistical significancewas defined as P < 0.05.
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 621
Figure 1 Changes in SBP (A), proteinuria (B), heart left ventricle weight to tibia length ratio (C) and GSI (D) in untreatedTGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats and in TGR treated either with DDMSor NCND or their combinationResults are taken from the ‘early’ treatment protocol. *P < 0.05 compared with untreated TGR; #P < 0.05 compared with the values in each of TGR groups.
RESULTS
Series 1: effects of ‘early’ inhibition of20-HETE formation or/and of sEH activityAs shown in Figure 1(A), DDMS treatment did notalter the development or the final level of hypertensionin TGR, whereas NCND did retard its developmentsignificantly. The increase in the pressure was less steepin NCND-treated rats than in untreated TGR, and atthe age of 79 days, the SBP levels were 181 +− 4 and196 +− 4 mmHg, respectively (P < 0.05). Thereafter, theprogress of hypertension was similar, and the final SBPdid not significantly differ between NCND-treated anduntreated TGR (202 +− 6 compared with 199 +− 5 mmHg,respectively). In contrast, the combined treatment withDDMS and NCND both attenuated the progressionof hypertension and lowered the final SBP to a levelthat was significantly different from that in untreatedTGR (178 +− 3 compared with 199 +− 5 mmHg, P < 0.05).
Nevertheless, the level was still distinctly higher thanthat in HanSD rats, which remained normotensivethroughout the experiment.
Figure 1(B) shows that HanSD rats exhibited onlyminor proteinuria over the entire duration of theexperiment. In TGR, a pronounced proteinuria wasobserved already during the early phase of hypertension,at the age of 80 days [26.4 +− 2.8 mg · (24 h)−1 comparedwith 6.5 +− 1.4 mg · (24 h)−1 seen in HanSD rats]. Theproteinuria further increased to 34.8 +− 2.2 mg · (24 h)−1
in the phase of established hypertension (day 140 ofage). The treatment with DDMS did not affect thedegree of proteinuria in TGR. On the other hand,NCND attenuated proteinuria during the early but notin the established phase of hypertension. In contrast,combined treatment with DDMS and NCND distinctlyameliorated the proteinuria in the early as well asin the established phase of hypertension [8.7 +− 1.5and 12.2 +− 1.1 mg · (24 h)−1, respectively, compared with
C© The Authors Journal compilation C© 2010 Biochemical Society

622 V. Certıkova Chabova and others
26.4 +− 2.8 and 34.8 +− 2.2 mg · (24 h)−1] observed inuntreated TGR (P < 0.05 for both the early and theestablished phase).
As shown in Figure 1(C), untreated TGR developeddistinct cardiac hypertrophy, expressed as the ratio ofLVW/TL, which was not attenuated by DDMS orNCND given alone. In contrast, the combined treatmentwith DDMS and NCND substantially attenuated thedegree of cardiac hypertrophy (25.9 +− 0.6 comparedwith 32.6 +− 0.7 observed in untreated TGR, P < 0.05);however, the levels observed in HanSD rats were notattained.
As shown in Figure 1(D), untreated TGR exhibiteda GSI that was substantially higher than in HanSDrats (0.64 +− 0.05 compared with 0.11 +− 0.05, P < 0.05).The treatment with DDMS or NCND given alonedid not prevent the development of glomerulosclerosis.In contrast, the combined treatment with DDMS andNCND significantly reduced the GSI, even though it didnot decrease to the level observed in HanSD rats.
As shown in Figure 2(A), untreated TGR hadmarkedly higher degree of kidney tubulointerstitialinjury, compared with HanSD rats (0.025 +− 0.005compared with 0.002 +− 0.001, P < 0.05). The treatmentwith DDMS alone did not attenuate the injury inTGR. In contrast, the treatment of TGR with NCNDalone markedly inhibited the development of kidneytubulointerstitial injury, compared with untreated TGR,and the combined treatment with DDMS and NCNDcompletely prevented the development of the injury(Figure 2A).
The glomerular volume in untreated TGR was signific-antly greater than in HanSD rats (1.43 +− 0.065 comparedwith 1.08 +− 0.025 × 106 μm3, P < 0.05) (Figure 2B). Thetreatment with DDMS alone did not change the volumein TGR compared with untreated TGR. In contrast, thetreatment of TGR with NCND alone distinctly reducedthe volume, and the combined treatment with DDMS andNCND normalized it, bringing the values down to thelevels observed in HanSD rats.
As shown in Figure 3(A), the urinary excretion of 20-HETE in untreated TGR was significantly higher thanin HanSD rats [1480 +− 165 compared with 460 +− 90 ng(12 h)−1, P < 0.05]. The treatment with NCND didnot alter the urinary excretion of 20-HETE. However,as expected, the treatment with DDMS alone, orthe combined treatment with NCND and DDMSsignificantly reduced 20-HETE excretion down to thelevels observed in HanSD rats.
In contrast, urinary excretion of EETs was significantlylower in TGR than in HanSD rats [640 +− 120 comparedwith 1295 +− 110 ng (12 h)−1, P < 0.05]. Treatment withDDMS did not alter EETs excretion, whereas thetreatment with NCND, alone or in combination withDDMS, significantly increased it to the levels observed inHanSD rats (Figure 3B).
Figure 2 Kidney tubulointerstitial injury score (A) andglomerular volume (B) in untreated TGR (heterozygousRen-2 renin transgenic rats) and HanSD (transgene-negative)rats and in TGR treated either with DDMS or NCND or theircombinationResults are taken from the ‘early’ treatment protocol. #P < 0.05 comparedwith untreated TGR; @P < 0.05 compared with the values for TGR+vehicle,TGR+DDMS and TGR+NCND groups.
Figure 3(C) shows that the urinary excretion ofDHETEs, the biologically inactive metabolites of EETs,was significantly higher in untreated TGR than in HanSDrats [920 +− 60 compared with 490 +− 60 ng (12 h)−1,P < 0.05]. The treatment with DDMS did not significantlychange DHETEs excretion, but treatment with NCNDalone or in combination with DDMS significantlydecreased it to the levels observed in HanSD rats.
Figure 3(D) summarizes the results on the intrarenalavailability of biologically active epoxygenase metabol-ites when expressed as the EETs/DHETEs ratio. Thisratio was significantly lower in untreated TGR thanin HanSD rats (0.7 +− 0.07 compared with 2.64 +− 0.76,P < 0.05). It was not significantly changed by DDMStreatment. However, treatment with NCND, alone or
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 623
Figure 3 Urinary excretion of 20-HETE (A), EETs (B), DHETEs (C) and the urinary EETs to DHETEs excretion ratio (D) inuntreated TGR (heterozygous Ren-2 renin transgenic rats) and in HanSD (transgene-negative) rats and in TGR treated withDDMS, NCND or combination of the two agentsResults are taken from the ‘early’ treatment protocol. *P < 0.05 compared with untreated TGR.
in combination with DDMS, significantly increased thisratio to the levels observed in HanSD rats.
Plasma, kidney and left heart ventricle AngII levelswere significantly higher in untreated TGR than inHanSD rats; in the former, the levels were not altered byany of the three treatment modalities. In addition, urinaryoutput and urinary sodium excretion were not signific-antly different among experimental groups at any time ofthe experiment (see Supplementary Table S1 available athttp://www.ClinSci.org/cs/118/cs1180617add.htm).
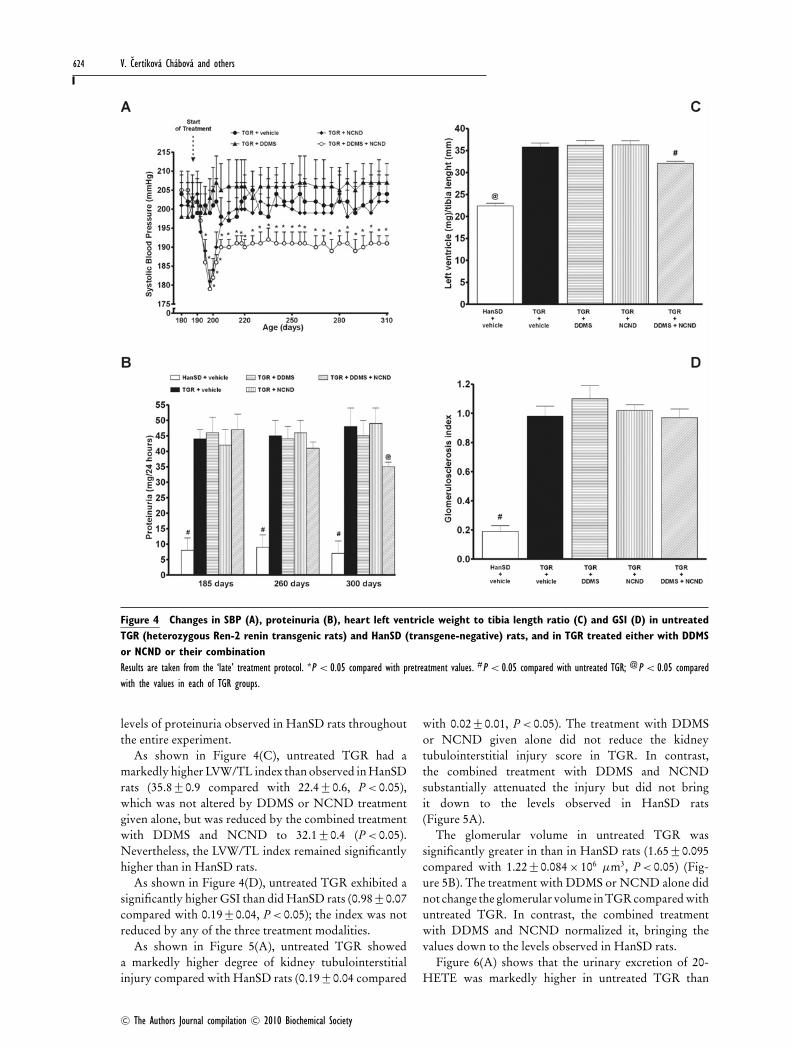
Series 2: effects of ‘late’ inhibition of20-HETE formation and/or sEH activityAs shown in Figure 4(A), untreated TGR remaineddistinctly hypertensive throughout the experiment, witha final SBP of 204 +− 5 mmHg, whereas HanSD ratswere invariably normotensive, with a final SBP of138 +− 3 mmHg. In TGR, the treatment with DDMS did
not significantly change SBP at any time point. Within thefirst 8 days of treatment, NCND significantly decreasedSBP from 204 +− 6 to 181 +− 3 mmHg (P < 0.05), but duringthe further 10 days, it returned to the levels observed inuntreated TGR, reaching a final value of 202 +− 7 mmHg.In contrast, the combined treatment with DDMS andNCND resulted in a distinct decrease in SBP from 205 +− 4to 179 +− 2 mmHg (P < 0.05) within 8 days. Thereafter,despite a gradual return of the pressure towards theoriginal hypertensive levels, SBP remained significantlylower than in untreated TGR (191 +− 2 compared with204 +− 5 mmHg, P < 0.05).
Figure 4(B) shows that on day 185, the basal values ofproteinuria were markedly higher in TGR than in HanSDrats. This pattern was not reduced by chronic treatmentwith either DDMS or NCND alone. In contrast, thecombined treatment with DDMS and NCND decreasedproteinuria from 47.4 +− 4.8 to 34.9 +− 1.1 mg · (24 h)−1
(P < 0.05). This was still substantially higher than the low
C© The Authors Journal compilation C© 2010 Biochemical Society
624 V. Certıkova Chabova and others
Figure 4 Changes in SBP (A), proteinuria (B), heart left ventricle weight to tibia length ratio (C) and GSI (D) in untreatedTGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated either with DDMSor NCND or their combinationResults are taken from the ‘late’ treatment protocol. *P < 0.05 compared with pretreatment values. #P < 0.05 compared with untreated TGR; @P < 0.05 comparedwith the values in each of TGR groups.
levels of proteinuria observed in HanSD rats throughoutthe entire experiment.
As shown in Figure 4(C), untreated TGR had amarkedly higher LVW/TL index than observed in HanSDrats (35.8 +− 0.9 compared with 22.4 +− 0.6, P < 0.05),which was not altered by DDMS or NCND treatmentgiven alone, but was reduced by the combined treatmentwith DDMS and NCND to 32.1 +− 0.4 (P < 0.05).Nevertheless, the LVW/TL index remained significantlyhigher than in HanSD rats.
As shown in Figure 4(D), untreated TGR exhibited asignificantly higher GSI than did HanSD rats (0.98 +− 0.07compared with 0.19 +− 0.04, P < 0.05); the index was notreduced by any of the three treatment modalities.
As shown in Figure 5(A), untreated TGR showeda markedly higher degree of kidney tubulointerstitialinjury compared with HanSD rats (0.19 +− 0.04 compared
with 0.02 +− 0.01, P < 0.05). The treatment with DDMSor NCND given alone did not reduce the kidneytubulointerstitial injury score in TGR. In contrast,the combined treatment with DDMS and NCNDsubstantially attenuated the injury but did not bringit down to the levels observed in HanSD rats(Figure 5A).
The glomerular volume in untreated TGR wassignificantly greater in than in HanSD rats (1.65 +− 0.095compared with 1.22 +− 0.084 × 106 μm3, P < 0.05) (Fig-ure 5B). The treatment with DDMS or NCND alone didnot change the glomerular volume in TGR compared withuntreated TGR. In contrast, the combined treatmentwith DDMS and NCND normalized it, bringing thevalues down to the levels observed in HanSD rats.
Figure 6(A) shows that the urinary excretion of 20-HETE was markedly higher in untreated TGR than
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 625
Figure 5 Kidney tubulointerstitial injury score (A) andglomerular volume (B) in untreated TGR (heterozygousRen-2 renin transgenic rats) and HanSD (transgene-negative)rats and in TGR treated with DDMS or NCND or theircombinationResults are taken from the ‘late’ treatment protocol. #P < 0.05 compared withuntreated TGR; @P < 0.05 compared with the values for the remaining fourgroups.
in HanSD rats [2142 +− 194 compared with 355 +− 105 ng(12 h)−1, P < 0.05]. The treatment with DDMS, aloneor in combination with NCND, significantly reduced20-HETE excretion to levels not significantly differentfrom those in HanSD rats. The treatment with NCNDalone did not alter 20-HETE excretion.
As shown in Figure 6(B), urinary excretion of EETswas significantly lower in TGR than in HanSD rats[340 +− 85 compared with 706 +− 102 ng (12 h)−1, P < 0.05]and was not altered by DDMS treatment. In contrast,treatment with NCND, alone or in combination withDDMS, elicited substantial increases of EETs excretion.Urinary excretion of DHETEs was significantly higher inuntreated TGR than in HanSD rats [1184 +− 144 comparedwith 535 +− 85 ng (12 h)−1, P < 0.05] and, again, was not
altered by DDMS treatment. In contrast, treatmentwith NCND, alone or in combination with DDMS,significantly reduced DHETEs excretion in TGR to thelevels observed in HanSD rats (Figure 6C).
Figure 6(D) shows that the urinary EETs/DHETEsratio was distinctly lower in untreated TGR than inHanSD rats (0.29 +− 0.05 compared with 1.32 +− 0.22). Thiswas not significantly modified by DDMS treatment,whereas treatment with NCND, alone or in combinationwith DDMS, significantly increased this ratio to the levelobserved in HanSD rats.
Plasma, kidney and left heart ventricle AngII levelswere significantly higher in untreated TGR than inHanSD rats and, this pattern was not altered by any ofthe three treatment modalities. Moreover, urinary outputand urinary sodium excretion were not significantlydifferent among experimental groups at any time ofexperiment (see Supplementary Table S2 available athttp://www.ClinSci.org/cs/118/cs1180617add.htm).
Series 3: MAP and RBF responses to AngIIand NE in the ‘early’ treatment protocolBasal values for MAP, RBF and RVR (averagevalues from the equilibration period) are sum-marized in Supplementary Table S3 (available athttp://www.ClinSci.org/cs/118/cs1180617add.htm). Asshown in Figure 7(A), the responses of MAP tointravenous bolus administration of AngII, 50 ng, weresignificantly greater in untreated TGR than in HanSDrats (+32 +− 3 compared with +14 +− 2 mmHg, P < 0.05).Treatment with either DDMS or NCND alone did notmodify the responses, whereas the combined treatmentwith DDMS and NCND did attenuate them significantly.However, the responses of MAP remained significantlygreater than in HanSD rats.
Similarly, as shown in Figure 7(B), the intrarenal ad-ministration of AngII, 10 ng, decreased RBF significantlymore in untreated TGR than in HanSD rats (−31 +− 3compared with −14 +− 2 %, P < 0.05). The treatment witheither DDMS or NCND alone did not attenuate the RBFdecreases, whereas in contrast, a distinct attenuation wasseen with the combined treatment (−22 +− 2 comparedwith −31 +− 3 % in untreated TGR, P < 0.05). As shownin Figures 7(A) and 7(B), an intravenous or intrarenaladministration of NE elicited similar increases in MAPand decreases in RBF in TGR and HanSD rats; in theformer, the responses were not altered by any of the threetreatment modalities.
Series 4: MAP and RBF responses to AngIIand NE in the ‘late’ treatment protocolBasal values for MAP, RBF and RVR (average valuesfrom the equilibration period) are summarized inSupplementary Table S3. As shown in Figure 8(A), anintravenous bolus of 50 ng of AngII increased MAP
C© The Authors Journal compilation C© 2010 Biochemical Society

626 V. Certıkova Chabova and others
Figure 6 Urinary excretion of 20-HETE (A), EETs (B), DHETEs (C) and the urinary EETs to DHETEs excretion ratio (D) inuntreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated withDDMS or NCND, alone or combinedResults are taken from the ‘late’ treatment protocol. *P < 0.05 compared with untreated TGR.
significantly more in untreated TGR than in HanSDrats (+35 +− 3 compared with +16 +− 3 mmHg, P < 0.05).The treatment with either DDMS or NCND alonedid not alter the responses, whereas the combinedtreatment did attenuate them significantly. Similarly, asshown in Figure 8(B), the intrarenal bolus administrationof AngII, 10 ng, elicited significantly larger decreasesin RBF in untreated TGR than in HanSD rats(−35 +− 4 compared with −16 +− 2 %, p < 0.05). Theresponses were not modified by treatment witheither DDMS or NCND alone but were significantlyreduced by the combined treatment with DDMS andNCND.
As shown in Figures 8(A) and 8(B), an intravenous orintrarenal administration of NE elicited similar increasesin MAP and decreases in RBF in TGR and HanSD rats;in the former, the responses were not altered by any ofthe three treatment modalities.
DISCUSSION
After initial controversies regarding the role of theRAS in the pathophysiology of hypertension in TGR[12,28,29], more recent studies have clearly demonstratedthat enhanced formation of AngII leading to elevation ofits plasma and kidney tissue levels, and an exaggerationof peripheral and renal vascular responsiveness toAngII, are the main pathogenetic factors in thedevelopment and maintenance of hypertension inTGR [19,20,26,27]. Notwithstanding this conclusion,the important role of CYP450-derived eicosanoidsin the regulation of renal function and blood pressureas well as in the pathophysiology of hypertension andthe associated hypertension-induced end-organ damagehas recently been documented for AngII-dependentforms of hypertension [6–11,16,30–32]. Moreover,disturbed intrarenal production and diminished action of
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 627
Figure 7 Maximum increases in MAP in response tointravenous bolus administration of AngII and NE (A)and maximum decreases in RBF in response to intrarenalbolus administration of AngII and NE (B) in untreatedTGR (heterozygous Ren-2 renin transgenic rats) and HanSD(transgene-negative) rats, and in TGR treated with DDMSand NCND, alone or combinedResults are taken from the ‘early’ treatment protocol. #P < 0.05 compared withuntreated TGR; @P < 0.05 compared with the values in each of TGR groups.
20-HETE, combined with intrarenal deficiency of EETs(a consequence of increased sEH-mediated conversioninto biologically inactive DHETEs), have been implicatedin the pathophysiology of hypertension in TGR [11,13].Evaluation of the proposed in vivo functional role of20-HETE and EETs is difficult. One potential approachis to apply pharmacological interventions aimed at anabolishment or substantial reduction of the agents’activity. This requires their chronic selective blockade andprolonged follow-up studies of transgenic rats subjectedto the treatment and parallel studies of appropriatecontrol animals. The present study evaluates the effectof such blockade on the development of hypertensionand of the end-organ damage in TGR.
Figure 8 Maximum increases in MAP in response tointravenous bolus administration of AngII and NE (A)and maximum decreases in RBF in response to intrarenalbolus administration of AngII and NE (B) in untreatedTGR (heterozygous Ren-2 renin transgenic rats) and HanSD(transgene-negative) rats, and in TGR treated with DDMSand NCND, alone or combinedResults are taken from the ‘late’ treatment protocol. #P < 0.05 compared withuntreated TGR; @P < 0.05 compared with the values in each of TGR groups.
20-HETE has been shown to promote vasocon-striction via mechanisms involving inhibition of large-conductance, calcium-activated potassium channels [33],activation of calcium channel conductance leading todepolarization of the VSMC (vascular smooth musclecell) [34] and activation of Rho-kinase, which causessensitization of the contractile apparatus to calcium[35]. 20-HETE is also known to augment vascularreactivity to vasoconstrictor agents [36,37] and to playa critical role as a second messenger for AngII-mediatedvasoconstriction [7,8]. Overall, it has been claimed thatenhanced formation of 20-HETE in the vasculature is thebasis of its prohypertensive action and contributes tothe development and maintenance of hypertension insome experimental models [7–9,30,38].
C© The Authors Journal compilation C© 2010 Biochemical Society

628 V. Certıkova Chabova and others
In addition to its action on the vasculature, 20-HETE inhibits sodium reabsorption in the proximaltubule [39] and in the thick ascending limb of theloop of Henle [40]. Accordingly, it has been postulatedthat increased 20-HETE levels along the renal tubuleshould promote natriuresis and thereby oppose itsconstrictor action on the vasculature and the developmentof hypertension [4,6,30]. The notion that intrarenal20-HETE also exerts an antihypertensive action isstrengthened by the demonstration that chronic blockadeof 20-HETE formation promotes the development ofsalt-sensitive hypertension in otherwise salt-resistantrats [41]. In a clear contrast with the transportinhibitory effect of 20-HETE observed in normotensiverats and in a number of experimental or genetichypertensive rat models, in a recent study, we foundthat in hypertensive TGR, increased intrarenal 20-HETE concentrations cause sodium retention withoutdecreasing renal haemodynamics, which suggests anincrease in the tubular sodium transport [13]. Thus, inhypertensive TGR, 20-HETE could be prohypertensiveboth owing to its vasoconstrictor and to the sodium-retaining activity.
EETs have been consistently shown to cause vaso-dilatation through stimulation of the large-conductance,calcium-activated potassium channels (KCa) [5]. Theyhave also been identified as an EDHF (endothelium-derived hyperpolarizing factor) that mediates nitricoxide- and prostaglandin-independent vasodilatation,and have been found to oppose the vasoconstrictoractions of AngII [42–45]. Moreover, EETs inhibit tubularsodium reabsorption in the renal proximal tubule [46]and in the cortical collecting duct [47]. Lastly, anincreasing body of evidence suggests that EETs exhibitcardiovascular and renal protective actions [48].
A major finding of the present study is thatcombined chronic inhibition of 20-HETE synthesis(DDMS) and of sEH activity (NCND), when appliedin young prehypertensive TGR, significantly attenuatedthe development of hypertension and had considerablenephroprotective and cardioprotective effects, includingreduction of proteinuria, glomerulosclerosis, kidneytubulointerstitial injury and cardiac hypertrophy. Thiswas associated with reduction of urinary excretionof 20-HETE and DHETEs, and elevation of urinaryexcretion of EETs to the levels observed in normotensiveHanSD rats. Of special interest is our finding thatnephroprotective effects of the combined treatment withDDMS and NCND were associated with restoration ofglomerular volume to values observed in normotensiveHanSD rats. Given a strong correlation between theglomerular size and the degree of glomerulosclerosis,these findings are in good agreement with thedemonstration that the nephroprotective effect ofantihypertensive therapy is also associated with thereduction of the size of the glomerulus [49,50].
Remarkably, isolated chronic 20-HETE inhibition didnot significantly affect the development of hypertensionor of the end-organ damage, even though the urinaryexcretion of 20-HETE was reduced to levels close tothose in the animals treated with the combination ofDDMS and NCND. Similarly, chronic isolated inhibitionof sEH increased the intrarenal availability of biologicallyactive epoxygenase metabolites 3-fold, but elicited onlytransitory antihypertensive effects and did not alter theextent of the final end-organ damage. Nevertheless, it isimportant to emphasize that in TGR, the treatment withNCND alone exhibited some minor nephroprotectiveactions, e.g. the degree of tubulointerstitial injury wasreduced, and glomerular hypertrophy was prevented.However, these nephroprotective effects were minimalcompared with those of the combined treatment withDDMS and NCND. On the whole, the present findingsagree well with those of our previous studies withTGR, which showed that altered production and/oraction of CYP450-derived metabolites contributes tothe development of hypertension and of the associatedend-organ damage [11,13]. However, this is the firststudy clearly showing that only the combination ofthe blockade of 20-HETE formation and of sEHinhibition exerts long-term antihypertensive and renaland cardiovascular protective actions in the monogenicmodel of AngII-dependent hypertension.
It is difficult to explain why the beneficial effects wereachieved exclusively with the combined DDMS andNCND treatment. Since in most situations 20-HETE hasboth pro- and anti-hypertensive properties, one cannoteasily predict the consequence of its inhibition on bloodpressure [4,6]. However, we have shown earlier that inTGR rats, 20-HETE causes sodium retention rather thannatriuresis [13], which means that both components,its vascular and tubular actions, should promote thedevelopment of hypertension. Therefore, we assumedthat in this AngII-dependent model, a chronic andselective inhibition of 20-HETE formation should bemore effective in attenuating blood pressure elevation,and possibly also the associated organ damage, than couldbe expected in other models of hypertension. However,we found that although DDMS treatment lowered theelevated urinary excretion of 20-HETE to the levelsobserved in normotensive HanSD rats, it did not exhibitany antihypertensive and organ-protective action.Moreover, chronic treatment with DDMS did not alterplasma and kidney AngII levels and did not attenuate theenhanced systemic and renal vascular responsiveness toAngII.
Similar to the effects of specific 20-HETE inhibition,our TGR chronic inhibition of sEH activity alone byNCND did not exhibit long-term antihypertensive andorgan-protective actions, even though the availability ofbiologically active epoxygenase products was substan-tially increased. Most probably, it is not tissue EETs or
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 629
tissue 20-HETE but the elevated AngII concentrationsand the augmented vascular responsiveness to AngII,which are the dominant factors in the developmentof hypertension and in the end-organ damage in thistransgenic AngII-dependent model. This is in contrastto the results obtained in rats rendered hypertensiveby chronic infusion of AngII in whom the endogenousRAS activity is substantially suppressed [3,16]. In theseanimals, isolated pharmacological manipulations of theCYP450 pathway of AA metabolism clearly reducedblood pressure and the end-organ damage [6,32,51].Overall, the results strengthen our earlier conclusionsthat the substantial chronic elevation of RAS activity isthe crucial step in the pathophysiology of hypertensionin the TGR model [19,20,27].
We found that the combination of suppression of 20-HETE production with an elevation of EETs can partiallycounteract the vasoconstrictor actions of chronicallyelevated circulating and tissue AngII levels in TGR andcan partly attenuate the development of hypertension andhypertension-induced end-organ damage. The reasonswhy the combined treatment was generally effective whilethe isolated treatment, e.g. with NCND, was not, may becomplex. It should be kept in mind that in TGR withestablished hypertension, the isolated inhibition of sEHdid not lower blood pressure or attenuate proteinuria,whereas the combined treatment had appreciable effects.This could be related to the very high basal sEH activity(in TGR the DHETE excretion increased with age),and also to a high rate of 20-HETE synthesis; theurinary excretion of 20-HETE was almost twice thatseen in young TGR and 7-fold higher than in oldHanSD. One might speculate that the extremely high 20-HETE synthesis diminished the amount of the substrateAA available for the synthesis of EETs and decreasedtheir synthesis rate, in accordance with the repeatedlyproposed hypothesis assuming ‘substrate shifts’ betweenthe individual pathways of AA metabolism [4,52–55].At a reduced EETs synthesis rate, an inhibition oftheir degradation with NCND alone would not increaseavailable EETs very effectively. On the other hand, asimultaneous suppression of 20-HETE synthesis (DDMStreatment) would, in addition to the specific direct effect,make the AA substrate available for the epoxygenasesubpathway of CYP450-dependent AA metabolism,and at a high EETs synthesis rate, their action wouldincrease more than with NCND treatment alone; thefinal result would be an appreciable reduction of bloodpressure and proteinuria. It is commonly recognizedthat antihypertensive therapy is more effective whenapplied early during the development rather than duringestablished hypertension [56]. Therefore, in a separateseries of experiments, we evaluated the effects of chronicblockade of 20-HETE formation and sEH inhibition onblood pressure and on proteinuria, an index of kidneydamage, in the TGR with sustained elevation of blood
pressure, i.e. a condition that more closely resemblesclinical hypertension. We found that in this setting –similar to the first series of experiments – a reductionin blood pressure, proteinuria and cardiac hypertrophywas seen only with the combined treatment with DDMSand NCND. The improvement was associated with areduction in the urinary excretion of 20-HETE andDHETEs and an increase in the urinary excretion ofEETs to levels observed in normotensive HanSD rats.Thus, the findings strengthen our conclusion based onthe results of ‘early’ treatment experiments. It shouldbe noticed, however, that the amelioration of end-organdamage was significantly smaller than that observedwhen the same treatment was started immediately afterweaning.
After the late-onset combined therapy, proteinuriaand cardiac hypertrophy were only modestly attenuated,whereas tubulointerstitial injury was markedly attenu-ated, and the glomerular hypertrophy was prevented.There was no difference between untreated TGR andTGR treated with the combination of DDMS and NCNDwith regard to the index of glomerulosclerosis – thiswas so, despite a similar blood pressure-lowering effectobserved with the early and late-treatment protocols.The failure of the combined treatment to inhibitthe progress of glomerulosclerosis could be relatedto the fact that renal tissue AngII and 20-HETEprogressively increased in TGR and were much higherin the phase of established hypertension than in theyoung prehypertensive rats. It should be noticed thatAngII, among other actions, activates Ras/mitogen-activated protein kinase by increasing the synthesis ofCYP4A-derived 20-HETE; this stimulates proliferationof the VSMCs [57–60]. One might speculate thatprolonged exposure of the glomeruli to increasingconcentrations of AngII resulted in proliferation-basedchanges that could not be reversed by our combinedtherapy. Interestingly, cardiac hypertrophy could still beattenuated by the combined treatment, possibly becausethe plasma concentration, unlike kidney tissue AngII,did not increase with the age of the TGR. Overall, inthe TGR with established hypertension, alterations of20-HETE and EETs activities have only limited effectson end-organ damage. This suggests that the CYP450-dependent active agents play a ‘permissive’ rather thancausal role in the pathophysiology of hypertension andthe associated end-organ damage in TGR.
We found that only the combined blockade of 20-HETE formation and of sEH activity significantlyattenuated MAP and RBF responses to bolus injectionsof AngII; this was similar when examined in youngprehypertensive TGR and in the rats with establishedhypertension. The responses to NE were not altered.These findings are in accordance with the previousstudies showing that TGR display an exaggeratedsystemic and renal vascular reactivity to AngII [26,27].
C© The Authors Journal compilation C© 2010 Biochemical Society

630 V. Certıkova Chabova and others
However, our present findings are the first to clearlyshow that, in contrast to rats rendered hypertensiveby chronic subpressor AngII infusion [7,8,16,28,32,51],in TGR, an augmented vascular responsiveness toAngII is not attenuated by an isolated blockade of 20-HETE formation or an isolated enhancement of EETsbioavailability. The fact that an alteration of the activitiesof the two types of CYP450 metabolites does result in anappreciable attenuation of the vasoconstriction indicatesthat an augmentation of the vascular responsivenessto AngII is one of the mechanism(s) underlying thebeneficial antihypertensive actions of combined DDMSand NCND treatment in TGR.
In summary, the results show that selective blockade of20-HETE formation and of sEH activity, when appliedchronically and in combination in young prehypertensiveTGR, suppresses the development of hypertension andprovides a substantial protection from the associatedcardiac hypertrophy, proteinuria and glomerulosclerosis.Also, in adult TGR with established hypertension, thecombined treatment with DDMS and NCND reducedblood pressure and slightly decreased proteinuria andcardiac hypertrophy. We found also that a reductionin 20-HETE combined with an increase in EETs levelsattenuated the enhanced vascular responsiveness to AngIIin TGR. Taken together, the findings strongly suggest thatalteration in the production and/or action of 20-HETEand EETs plays a permissive role in the developmentof hypertension and hypertension-associated end-organdamage in this AngII-dependent model. This informationderived from our present study should be consideredin attempts at the development of new therapeuticapproaches or tools for the treatment of early stages ofhuman hypertension.
FUNDING
L.C. was supported by the Czech Science Foundation(GACR) [grant number 305/08/J006]. A.W. was partlysupported by the Center for Cardiovascular Research[grant number 1M6798582302] and the Institute forClinical and Experimental Medicine [grant number MZO00023001]. V.C.C. was supported by the Internal GrantAgency of the Ministry of Health of the Czech Republic[grant number NS/10500-3]. Z.V. was supported bythe Internal Grant Agency of the Ministry of Healthof the Czech Republic [grant number NS/10499-3].L.K. was supported by the Grant Agency of Academyof Science of Czech Republic [grant number GAAV-IAA 511-700-701] and the Internal Grant Agency ofthe Ministry of Health of the Czech Republic [grantnumber NS/9699-4]. I.V. was supported by the GACR[grants numbers 305/07/0167, 305/07/J004]. H.J.K. wassupported by the German Research Foundation (DFG),Bonn [grant numbers Kra 436/14-2, 436 TSE 113/57/0-
1]. J.D.I. was supported the National Institutes ofHealth (NIH) [grant numbers HL59699, DK38226] andAdvancing a Healthier Wisconsin. J.R.F. was supportedby the Robert A. Welch Foundation and the NIH[grant number GM31278]. B.D.H. was supported bythe National Institute of Enviromental Health Sciences(NIEHS) [grant number R01 ES002710] and the NIEHSSuperfund Program [grant number P42 ES004699].
REFERENCES
1 US Renal Data System: USRDS 2007. (2008) Chronickidney disease. Am. J. Kidney Dis. 51, S63–S80
2 Bakris, G. L. and Ritz E., G. L. (2009) The message forWorld Kidney Day 2009: hypertension and kidney disease:a marriage that should be prevented. J. Clin. Hypertens. 11,144–147
3 Kobori, H., Nangaku, M., Navar, L. G. and Nishiyama, A.(2007) The intrarenal renin–angiotensin system: fromphysiology to the pathobiology of hypertension andkidney disease. Pharmacol. Rev. 59, 251–287
4 Roman, R. J. (2002) P-450 metabolites of arachidonic acidin the control of cardiovascular function. Physiol. Rev. 82,131–185
5 Campbell, W. B. and Falck, J. R. (2007) Arachidonic acidmetabolites as endothelium-derived hyperpolarizingfactors. Hypertension 49, 590–596
6 Capdevila, J. H, Falck, J. R. and Imig, J. D. (2007) Role ofthe cytochrome P450 arachidonic acid monooxygenases inthe control of systemic blood pressure and experimentalhypertension. Kidney. Int. 72, 683–689
7 Alonso-Galicia, M., Maier, K. G., Greene, A. S., Cowley,A. W. and Roman, R. J. (2002) Role of20-hydroxyeicosatetraenoic acid in the renal andvasoconstrictor actions of angiotensin II. Am. J. Physiol.Integr. Comp. Physiol. Physiol. 283, R60–R68
8 Joly, E., Seqqat, R., Flamion, B., Caron, N., Michal, A.,Imig, J. D. and Kramp, R. (2006) Increased renal vascularreactivity to angiotensin II after unilateral nephrectomy inthe rat involves 20-HETE. Am. J. Physiol. Integr. Comp.Physiol. 291, R977–R986
9 Muthalif, M. M., Karzoun, N. A., Gaber, L., Khandekar,Z., Benter, I. F., Saeed, A. F., Parmentier, J. H., Estes, A.and Malik, K. U. (2000) Angiotensin II-inducedhypertension: contribution of Ras GTPase/mitogen-activated protein kinase and cytochrome P-450metabolites. Hypertension 36, 604–609
10 Kaergel, E., Muller, D. N., Honeck, H., Theuer, J.,Shagdarsuren, E., Mullally, A., Luft, F. C. and Schunck,W. H. (2002) P450-dependent arachidonic acid metabolismand angiotensin II-induced renal damage. Hypertension40, 273–279
11 Certıkova Chabova, V., Kramer, H. J., Vaneckova, I.,Vernerova, Z., Eis, V., Tesar, V., Skaroupkova, P., Thumova,M., Schejbalova, S., Huskova, Z., Vanourkova, Z., Kolsky,A., Imig, J. D. and Cervenka, L. (2007) Effects of chroniccytochrome P-450 inhibition on the course ofhypertension and end-organ damage in Ren-2 transgenicrats. Vasc. Pharmacol. 47, 145–159
12 Mullins, J. J., Peters, J. and Ganten, D. (1990) Fulminanthypertension in transgenic rats harboring the mouse Ren-2gene. Nature 344, 541–544
13 Certıkova Chabova, V., Kramer H.J., Vaneckova, I.,Thumova, M., Skaroupkova, P., Tesar, V., Falck, J.R., Imig,J.D., Cervenka and L., Cervenka (2007) The roles ofintrarenal 20-hydroxyeicosatetraenoic andepoxyeicosatrienoic acids in the regulation of renalfunction in hypertensive Ren-2 transgenic rats. KidneyBlood Press. Res. 30, 335–346
C© The Authors Journal compilation C© 2010 Biochemical Society

CYP450 metabolites and end-organ damage 631
14 Wang, M. H., Brand-Schieber, E., Zand, B. A., Nguyen, X.,Falck, J. R., Balu, N. and Schwartzman, M. L. (1998)Cytochrome P-450-derived arachidonic acid metabolism inthe rat kidney: characterization of selective inhibitors.J. Pharmacol. Exp. Ther. 284, 966–973
15 Morisseau, C., Goodrow, W. H., Dowdy, D., Zheng, J.,Greene, J. F., Sanborn, J. R. and Hammock, B. D. (1999)Potent urea and carbamate inhibitors of soluble epoxidehydrolase. Proc. Nat. Acad. Sci. U.S.A. 96, 8849–8854
16 Imig, J. D., Zhao, X., Capdevila, J. H., Morisseau, C. andHammock, B. D. (2002) Soluble epoxide hydrolaseinhibition lowers arterial blood pressure in angiotensin IIhypertension. Hypertension 39, 690–694
17 Vaneckova, I., Kramer, H. J., Backer, A., Schejbalova, S.,Vernerova, Z., Eis, V., Opocensky, M., Dvorak, P. andCervenka, L. (2006) Early-onset endothelin receptorblockade in hypertensive heterozygous Ren-2 rats. Vasc.Pharmacol. 45, 163–170
18 Opocensky, M., Kramer, H. J., Backer, A., Vernerova, Z.,Eis, V., Cervenka, L. and Vaneckova, I., (2006) Late-onsetendothelin-A receptor blockade substantially improvessurvival rate in homozygous hypertensive Ren-2 rats.Hypertension 48, 965–971
19 Huskova, Z., Kramer, H. J., Vanourkova, Z. and Cervenka,L. (2006) Effects of changes in sodium balance on plasmaand kidney angiotensin II levels in anesthetized andconscious Ren-2 transgenic rats. J. Hypertens. 24, 517–527
20 Huskova, Z., Kramer, H. J., Thumova, M., Vanourkova, Z.,Burgelova, M., Teplan, V., Maly, J. and Cervenka, L. (2006)Effects of anesthesia on plasma and kidney ANG II levelsin normotensive and ANG II-dependent hypertensive rats.Kidney Blood Press. Res. 29, 74–83
21 Vanourkova, Z., Kramer, H. J., Huskova, Z., Vaneckova, I.,Opocensky, M., Certıkova Chabova, V., Tesar, V.,Skaroupkova, P., Thumova, M., Dohnalova, M., Mullins,J. J. and Cervenka, L. (2006) AT1 receptor blockade issuperior to conventional triple therapy in protectingagainst end-organ damage in Cyp1a1-Ren-2 transgenic ratswith inducible hypertension. J. Hypertens. 24, 2465–2472
22 Sandberg, M. C. and Laborde, C. (2004)Glomerulosclerosis and tubulointerstitial fibrosis areattenuated with 17-β-estradiol in the aging Dahl saltsensitive rat. J. Am. Soc. Nephrol. 15, 1546–1556
23 Nakano, Y., Hirano, T., Uehara, K., Nishibayashi, S.,Hattori, K., Aihara, M. and Yamada, Y. (2008) New ratmodel induced by anti-glomerular basement membraneantibody shows severe glomerular adhesion in early stageand quickly progresses to end-stage renal failure. Pathol.Int. 58, 361–370
24 Weibel, E. R. (1979) Stereological Methods, PracticalMethods for Biological Morphometry, pp. 44–46,Academic Press, London
25 Lane, P. H., Steffes, M. W. and Mauer, M. S. (1992)Estimation of glomerular volume: a comparison of fourmethods. Kidney Int. 41, 1085–1089
26 Jacinto, S. M., Mullins, J. J. and Mitchell, K. D. (1999)Enhanced renal vascular responsiveness to angiotensin II inhypertensive ren-2 transgenic rats. Am. J. Physiol. 276,F315–F322
27 Kopkan, L., Kramer, H. J., Huskova, Z., Vanourkova, Z,,Skaroupkova, P., Thumova, M. and Cervenka, L. (2005)The role of intrarenal angiotensin II in the development ofhypertension in Ren-2 transgenic rats. J. Hypertens. 23,1531–1539
28 Lee, M. A., Bohm, M., Paul, M., Bader, M., Ganten, U. andGanten, D. (1996) Physiological characterization of thehypertensive transgenic rat TGR(mRen2)27. Am. J.Physiol. 270, E919–E929
29 Mitchell, K. D., Jacinto, S. M. and Mullins, J. J. (1997)Proximal tubular fluid, kidney, and plasma levels ofangiotensin II in hypertensive Ren-2 transgenic rats. Am. J.Physiol. 273, F246–F253
30 Sarkis, A., Lopez, B. and Roman, R. J. (2004) Role of20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoicacids in hypertension. Curr. Opin. Nephrol. Hypertens.13, 205–214
31 Zhao, X., Pollock, D. M., Zeldin, D. C. and Imig, J. D.(2003) Salt-sensitive hypertension after exposure toangiotensin is associated with inability to upregulate renalepoxygenases. Hypertension 42, 775–780
32 Jung, O., Brandes, R. P., Kim, I-H., Schweda, F., Schmidt,R., Hammock, B. D., Busse, R. and Fleming, I. (2005)Soluble epoxide hypdrolase is a main effector ofangiotensin II-induced hypertension. Hypertension 45,759–765
33 Zou, A. P., Fleming, J. T., Falck, J. R., Jacobs, E. R.,Gebremedhin, D., Harder, D. R. and Roman, R. J. (1996)20-HETE is an endogenous inhibitor of thelarge-conductance Ca2+-activated K+ channel in renalarterioles. Am. J. Physiol. 270, R228–R237
34 Gebremedhin, D., Lange, A. R., Narayanan, J., Aebly,M. R., Jacobs, E. R. and Harder, D. R. (1998) Cat cerebralarterial smooth muscle cells express cytochrome P4504A2 enzyme and produce the vasoconstrictor 20-HETEwhich enhances L-type Ca2+ current. J. Physiol. 507,771–781
35 Randriamboavonjy, V., Busse, R. and Fleming, I. (2003)20-HETE-induced contraction of small coronary arteriesdepends on the activation of Rho-Kinase. Hypertension41, 801–806
36 Imig, J. D., Pham, B. T., LeBlanc, E. A., Reddy, K. M.,Falck, J. R. and Inscho, E. W. (2000) Cytochrome P450 andcyclooxygenase metabolites contribute to the endothelin-1afferent arteriolar vasoconstrictor and calcium responses.Hypertension 35, 307–312
37 Oyekan, A. O. and McGiff, J. C. (1998) CytochromeP-450-derived eicosanoids participates in the renalfunctional effects of ET-1 in the anaesthetized rat. Am. J.Physiol. 274, R52–R61
38 Singh, H. and Schwartzman, M. L. (2008) Renal vascularcytochrome P450-derived eicosanoids inandrogen-induced hypertension. Pharmacol. Rep. 60,29–37
39 Quigley, R., Baum, M., Reddy, K. M., Griener, J. C. andFalck, J. R. (2000) Effects of 20-HETE and 19(S)-HETEon rabbit proximal straight tubule volume transport. Am.J. Physiol. Renal Physiol. 278, F949–F953
40 Yu, M., Lopez, B., Dos Santos, E. A., Falck, J. R. andRoman, R. J. (2007) Effects of 20–HETE onNa+–K+–ATPase activity in the thick ascending loop ofHenle. Am. J. Physiol. Integr. Comp. Physiol. 292,R2400–R2405
41 Hoagland, K. M., Flasch, A. K. and Roman, R. J. (2003)Inhibitors of 20-HETE formation promote salt-sensitivehypertension in rats. Hypertension 42, 669–673
42 Imig, J. D., Zhao, X., Falck, J. R., Wei, S. and Capdevila,J. H. (2001) Enhanced renal mircovascular reactivity toangiotensin II in hypertension is ameliorated by thesulfonimide analog of 11,12-epoxyeicosatrienoic acid.J. Hypertens. 19, 983–992
43 Imig, J. D., Falck, J. R., Wei, S. and Capdevila, J. H. (2001)Epoxygenase metabolites contribute to nitricoxide-independent afferent arteriolar vasodilatation inresponse to bradykinin. J. Vasc. Res. 38, 247–255
44 Wang, D., Borrego-Conde, L. J., Falck, J. R., Sharma,K. K., Wilcox, C. S. and Umans, J. G. (2003) Contributionof nitric oxide, EDHF, and EETs toendothelium-dependent relaxation in renal afferentarterioles. Kidney Int. 63, 2187–2193
45 Kohagure, K., Endo, Y., Ito, O., Arima, S., Omata, K. andIto, S. (2000) Endogenous nitric oxide andepoxyeicosatrienoic acids modulate angiotensin II-inducedconstriction in the rabbit afferent arteriole. Acta Physiol.Scand. 168, 107–112
C© The Authors Journal compilation C© 2010 Biochemical Society

632 V. Certıkova Chabova and others
46 Madhun, Z. T., Goldthwait, D. A., McKay, D., Hopfer, U.and Douglas, J. G. (1991) An epoxygenase metabolite ofarachidonic acid mediates angiotensin II-induced rises incytosolic calcium in rabbit proximal tubule epithelial cells.J. Clin. Invest. 88, 456–461
47 Sakairi, Y., Jacobson, H. R., Noland, D. T., Capdevila,J. H., Falck, J. R. and Breyer, M. D. (1995) 5,6-EETinhibits ion transport in collecting duct by stimulatingendogenous prostaglandin synthesis. Am. J. Physiol. 268,F931–F939
48 Imig, J. D. (2005) Epoxide hydrolase and epoxygenasemetabolites as therapeutic targets for renal diseases. Am. J.Physiol. Renal Physiol. 289, F496–F503
49 Yoshida, Y., Kawamura, T., Ikoma, M., Fogo, A. andIchikawa, I. (1989) Effects of antihypertensivedrugs on glomerular morphology. Kidney Int. 36,626–635
50 Dworkin, L. D., Feiner, H. D., Parker, M. and Tolbert, E.(1991) Effects of nifedipine and enalapril on glomerularstructure and function in uninephrectomized SHR. KidneyInt. 39, 1112–1117
51 Imig, J. D. and Hammock, B. D., (2009) Soluble epoxidehydrolase as a therapeutic target for cardiovasculardiseases. Nat. Rev. Drug Discov. 8, 794–805
52 McGiff, J. C. (1991) Cytochrome P-450 metabolism ofarachidonic acid. Ann. Rev. Pharmacol. Toxicol. 31,339–169
53 Makita, K., Falck, J. R. and Capdevila, J. H. (1996)Cytochrome P450, the arachidonic acid cascade, andhypertension: new vistas for old enzyme system. FASEB J.10, 1456–1463
54 Sakuma, S., Usa, K. and Fujimoto, Y. (2006)15-hydroperoxyeicosapentaenoic acid, but noteicosapentaenoic acid, shifts arachidonic acid away fromcyclooxygenase pathway into acyl-CoA synthetasepathway in rabbit kidney medulla microsomes.Prostaglandins Leukotriene Essent. Fatty Acids 75, 69–74
55 Spector, A. A. (2009) Arachidonic acid cytochrome P450epoxygenase pathway. J. Lipid. Res. 50, S52–S60
56 Zicha, J. and Kunes, J., (1999) Ontogenetic aspects ofhypertension development: analysis in the rat. Physiol.Rev. 79, 1227–1282
57 Muthalif, M. M., Benter, L. F., Karzoun, N., Fatima, S.,Harper, J., Uddin, M. R. and Malik, K. U. (1998)20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin dependent protein kinase II-inducedmitogen-activated protein kinase activation in vascularsmooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 95,12701–12706
58 Parmentier, J-H., Muthalif, M. M., Nishimoto, A. T. andMalik, K. U. (2001) 20-Hydroxyeicosatetraenoic acidmediates angiotensin II-induced phospholipase Dactivation in vascular smooth muscle cells. Hypertension37, 623–629
59 Yaghini, F. A., Zhang, C., Parmentier, J. H., Estes, A. M.,Jafari, N., Schaefer, S. A. and Malik, K. U. (2005)Contribution of arachidonic acid metabolites derived viacytochrome P4504A to angiotensin II-induced neointimalgrowth. Hypertension 45, 1182–1187
60 Camara, N. O., Martins, J. O., Landgraf, R. G. and Jancar,S. (2009) Emerging roles for eicosanoids in renal diseases.Curr. Opin. Nephrol. Hypertens. 18, 21–27
Received 9 September 2009/17 December 2009; accepted 5 January 2010Published as Immediate Publication 5 January 2010, doi:10.1042/CS20090459
C© The Authors Journal compilation C© 2010 Biochemical Society

Clinical Science (2010) 118, 617–632 (Printed in Great Britain) doi:10.1042/CS20090459
S U P P L E M E N T A R Y O N L I N E D A T A
Combined inhibition of20-hydroxyeicosatetraenoic acid formation and
of epoxyeicosatrienoic acids degradationattenuates hypertension and hypertension-
induced end-organ damage in Ren-2transgenic rats
Vera CERTIKOVA CHABOVA∗†1, Agnieszka WALKOWSKA‡1, ElzbietaKOMPANOWSKA-JEZIERSKA‡, Janusz SADOWSKI‡, Petr KUJAL†§,Zdenka VERNEROVA†§, Zdena VANOURKOVA†‖, Libor KOPKAN†‖,Herbert J. KRAMER¶, John R. FALCK∗∗, John D. IMIG††, Bruce D. HAMMOCK‡‡,Ivana VANECKOVA†‖ and Ludek CERVENKA†§§∗Department of Nephrology, 1st Medical Faculty, Charles University, Prague, Czech Republic, †Department for ExperimentalMedicine, Institute for Clinical and Experimental Medicine, 1958/9 Vıdenska, CZ-140 21 Prague 4, Czech Republic,‡Laboratory of Renal and Body Fluid Physiology, Mossakowski Medical Research Centre, Polish Academy of Science, Warsaw,Poland, §Department of Pathology, Third Faculty of Medicine, Charles University, Prague, Czech Republic, ‖Center forCardiovascular Research, Prague, Czech Republic, ¶Section of Nephrology, Medical Policlinic, Department of Medicine,University of Bonn, Bonn, Germany, ∗∗Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas,TX 606-538, U.S.A., ††Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI 53226,U.S.A., ‡‡Department of Entomology, University of California at Davis Cancer Center, Davis, CA 95616, U.S.A., and§§Department of Physiology, 2nd Medical Faculty, Charles University, Prague, Czech Republic
1 These authors contributed equally to the study and should be considered as joint first authors.Correspondence: Dr Ludek Cervenka (email [email protected]).
C© The Authors Journal compilation C© 2010 Biochemical Society

V.CertıkovaChabova
andothers
Table S1 Kidney and left heart ventricle AngII levels and urinary sodium and water excretion in individual experimental groupsResults are taken from the ‘early’ treatment protocol. Values are means+− S.E.M. HanSD, transgene-negative normotensive rats; TGR, heterozygous Ren-2 renin transgenic hypertensive rats. *P<0.05 compared with the corresponding values in theother four groups.
Group
HanSD+vehicle TGR+vehicle TGR+DDMS TGR+NCND TGR+DDMS+NCND
Parameter Age (days) . . . 80 140 150 80 140 150 80 140 150 80 140 150 80 140 150
Plasma AngII (fmol/ml) 19 +− 3* 53 +− 4 56+− 4 51+− 4 56 +− 3Kidney AngII (fmol/g) 42 +− 2* 101+− 10 97+− 5 95+− 4 96 +− 4Heart AngII (fmol/g) 24+− 2* 32 +− 2 33+− 2 33+− 3 34 +− 3Na excretion (μmol/24 h) 654+− 48 672+− 51 694+− 65 637+− 41 688+− 56 684+− 61 634+− 55 652+− 48 614+− 73 664+− 56Water excretion (ml/24 h) 8.8+− 0.7 9.2 +− 0.6 8.9 +− 0.9 9.4 +− 1.1 8.7 +− 0.8 8.9+− 0.9 9.6 +− 0.8 8.5+− 0.9 9.1 +− 0.5 9.2 +− 0.8
Table S2 Kidney and left heart ventricle AngII levels and urinary sodium and water excretion in individual experimental groupsResults are taken from the ‘late’ treatment protocol. Values are means+− S.E.M. HanSD, transgene-negative normotensive rats; TGR, heterozygous Ren-2 renin transgenic hypertensive rats. *P<0.05 compared with the corresponding values in theother four groups.
Group
HanSD+vehicle TGR+vehicle TGR+DDMS TGR+NCND TGR+DDMS+NCND
Parameter Age (days) . . . 80 140 150 80 140 150 80 140 150 80 140 150 80 140 150
Plasma AngII (fmol/ml) 16+− 3* 46 +− 4 47+− 5 45+− 5 44 +− 5Kidney AngII (fmol/g) 38+− 4* 129+− 22 114+− 15 126+− 14 124+− 11Heart AngII (fmol/g) 20+− 2* 30 +− 2 29+− 2 30+− 3 31 +− 3Na excretion (μmol/24 h) 724+− 59 772+− 82 738+− 59 737+− 61 757+− 54 744+− 69 755+− 69 782+− 78 759+− 52 709+− 79Water excretion (ml/24 h) 9.8+− 0.6 9.9 +− 0.8 9.7 +− 0.9 9.6 +− 0.6 9.7 +− 0.7 9.1 +− 0.7 9.2+− 0.9 9.9+− 0.5 9.2 +− 0.5 9.1 +− 0.4
C ©The
AuthorsJournalcompilation
C ©2010
BiochemicalSociety

CYP450 metabolites and end-organ damage
Table S3 Baseline values of arterial blood pressure (MAP), RBF and RVR in individual experimental groupsResults are from series evaluating MAP and RBF responses to bolus injections of AngII and NE. Values are means+− S.E.M. Abbreviation are as given in Tables S1and S2. *P<0.05 compared with the corresponding values in TGR+vehicle, TGR+DDMS and TGR+DDMS groups; #P<0.05 compared with the values in the fourremaining groups.
Series Group MAP (mmHg) RBF (ml · min−1 · g−1) RVR (mmHg · ml−1 · min−1 · g−1)
‘Early’ treatment protocol HanSD+vehicle 121+− 4# 6.58+− 0.22 18.39+− 0.61#
TGR+vehicle 189+− 4 6.41+− 0.21 29.49+− 0.52TGR+DDMS 191+− 5 6.52+− 0.28 29.29+− 0.49TGR+NCND 188+− 4 6.53+− 0.29 28.79+− 0.56TGR+DDMS+NCND 166+− 4* 6.61+− 0.24 25.11+− 0.43*
‘Late’ treatment protocol HanSD+vehicle 132+− 4# 6.22+− 0.31 21.22+− 0.29#
TGR+vehicle 191+− 4 6.12+− 0.29 31.21+− 0.37TGR DDMS 192+− 3 6.09+− 0.31 31.53+− 0.41TGR+NCND 189+− 4 6.11+− 0.31 30.93+− 0.42TGR+DDMS+NCND 161+− 4* 6.36+− 0.44 25.31+− 0.52*
Received 9 September 2009/17 December 2009; accepted 5 January 2010Published as Immediate Publication 5 January 2010, doi:10.1042/CS20090459
C© The Authors Journal compilation C© 2010 Biochemical Society


















