
Combinatorial Chemistry Advanced Medicinal Chemistry (Pharm 5219): Section A Ref.: An Introduction...
60
Combinatorial Chemistry Advanced Medicinal Chemistry (Pharm 5219): Section A Ref.: An Introduction to Medicinal Chemistry, 3 rd ed. 2005, G.L.Patrick, Oxford University press Md. Saifuzzaman Assoc. Professor [email protected]
-
Upload
liliana-reeves -
Category
Documents
-
view
240 -
download
6
Transcript of Combinatorial Chemistry Advanced Medicinal Chemistry (Pharm 5219): Section A Ref.: An Introduction...

Combinatorial Chemistry
Advanced Medicinal Chemistry (Pharm 5219): Section A
Ref.: An Introduction to Medicinal Chemistry, 3rd ed. 2005, G.L.Patrick, Oxford University press
Md. SaifuzzamanAssoc. [email protected]

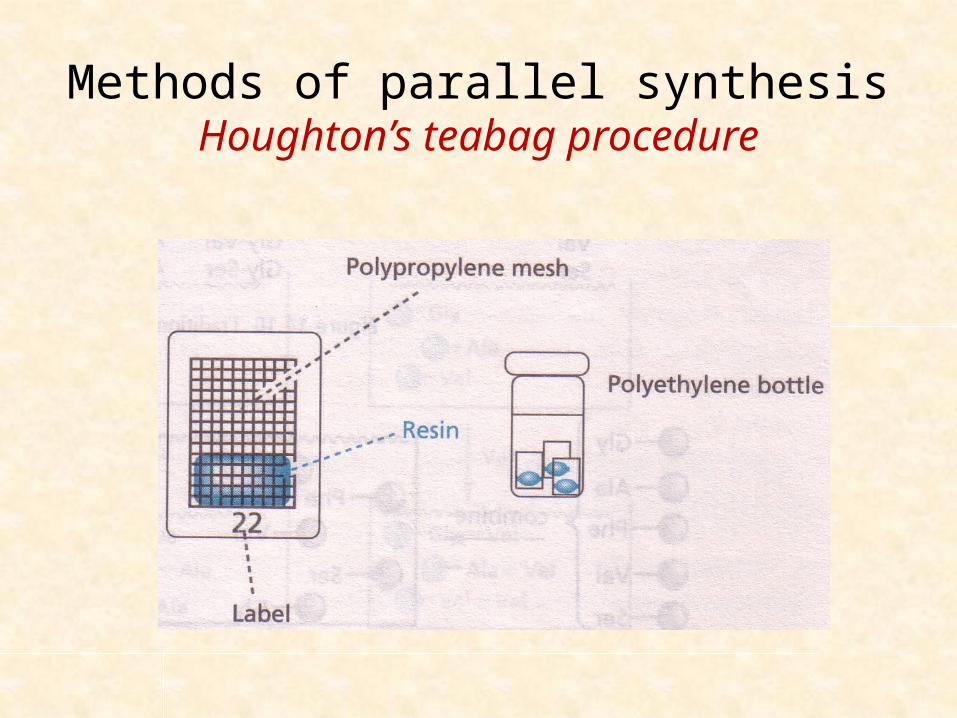
Methods of parallel synthesisHoughton’s teabag procedure
A manual approach to parallel synthesis
More than 150 peptides at a time
Polymeric support resin (100mg) – sealed in polypropylene meshed containers (3x4cm) – known as teabag
Each teabag is labelled
Methods of parallel synthesisHoughton’s teabag procedure

Methods of parallel synthesis
Houghton’s teabag procedure
Teabags – placed in PE bottles (reaction vessels)
First a.a. added to resin ( different a.a. in different bottle)
All teabags in a specific bottle – have same a.a.
Teabags from every bottle – combined in 1 vessel for deprotection and washing all at a time.

Methods of parallel synthesis
Houghton’s teabag procedure
Teabags – redistributed in bottles for addition of second a.a., recombined for deprotection & washing, redistributed for addition of next a.a. and so on.
Advantages – cheap, no need of expensive equipment
Disadvantages – manual, so limited quantity & speed.

Methods of parallel synthesis
automated parallel synthesis
Synthesis of 6, 12, 42, 96, or 144 structures depending on instrument and size of reaction tubes
Solvents, starting materials & reagents – added automatically using syringes
Removal of solvent, washing & liquid-liquid separations – also automatic
Reaction – can be stirred & carried out under inert atmos.
Reaction – can be heated & cooled as required.

Methods of mixed combinatorial synthesisGeneral principles
Designed to produce a mixture of products in each vessel from wide range of starting materials & reagents
Doesn’t mean that all starting materials should be put in one flask
Planning has to go to design a reaction to minimize efforts & to maximize outputs

Methods of mixed combinatorial synthesisGeneral principles
For example, if we plan to synthesize all dipeptides of 5 different a.a.,
Using orthodox chemistry, we would synthesize one at a time
25 possible dipeptides, so 25 separate experiments

Methods of mixed combinatorial synthesisGeneral principles
Using combinatorial synthesis, same products with far less effort
All 5 a.a. sperately bound to resin beads, mixed together & treated with second a.a. to produce all possible dipeptides in 5 experiments

Methods of mixed combinatorial synthesisGeneral principles
Mixtures – tested for activity; if positive, emphasis on identifying active dipeptides & if negative, mixtures – ignored & stored.
Large numbers of mixtures –can be generated; many are inactive
But they are not discarded (though no lead compound for the target but may contain lead for a different target)

Methods of mixed combinatorial synthesisGeneral principles
All the mixtures – stored & referred to combinatorial or compound libraries.
Combinatorial library acts as a source of potential new leads.

Methods of mixed combinatorial synthesisGeneral principles
Thousands or millions of different structures can be produced
As quantity is extremely small, huge no. of compounds – can be stored & used for further study
Though exact structure is not known, a general idea of type of structure based on type of synthesis and reagents used

The mix and split method
If huge quantities of different compounds, important to minimize the efforts involved
An example illustrating the mix and split method:
To make all possible tripeptides of 3
different a. a. (Gly, Val & Ala)

The mix and split method
Stage 1: Link each amino acid to a solid support

The mix and split method
Stage 2: Mix the beads together and separate into 3 equal portions

The mix and split method
Stage 3: React each portion with a different a.a
All 9 possible dipeptides – synthesized in 3 separate experiments
Samples of each portion – retained for recursive deconvolution.

The mix and split method
Stage 4: isolate all the beads, mix them together and split into 3 equal portions. Each portion will now have all nine possible dipeptides

The mix and split methodStage 5: react each portion with one of 3 a.a.
All 27 possible tripeptides – synthesized in 3 experiments

Isolating the active component in a mixture: deconvolution
Isolating & identifying the most active compound in a mixture – deconvolution
1. Micromanipulation
2. Recursive deconvolution
3. Sequential release

Micromanipulation
Each bead contains only one structural product
Individual beads – separate & product – cleaved & tested
Aided by colorimetric analysis (test activity when still bound)
Active beads – distinguished by colour reaction and can be picked out.
Disadvantage: tedious process & problematic with large quantities of beads.

Recursive deconvolution
Useful in cutting down amount of work involved
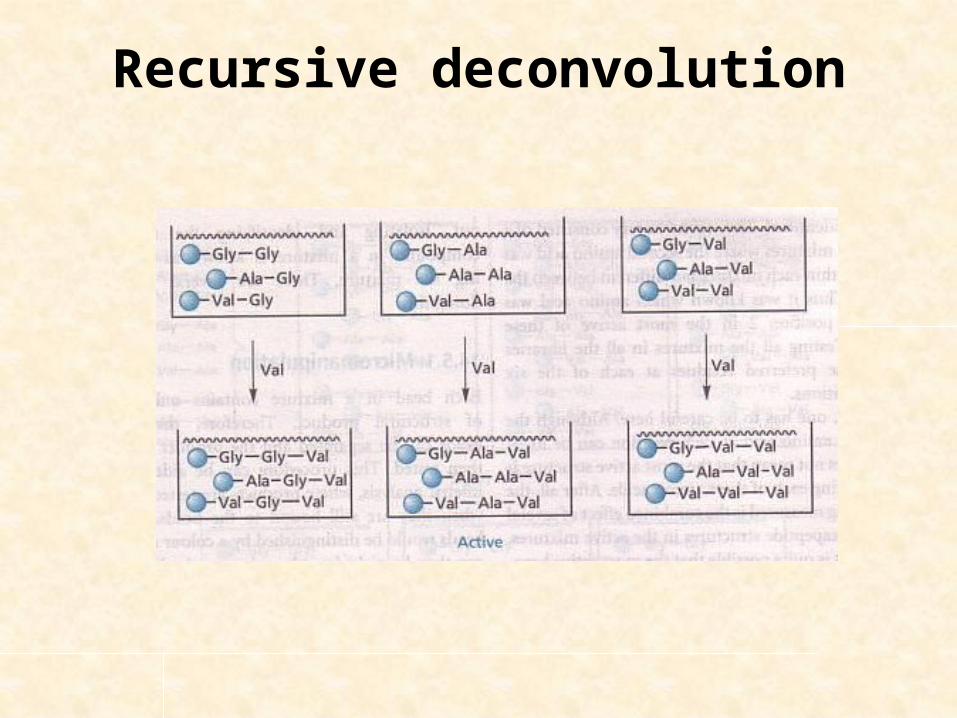
Let us consider the libraries of tripeptides (already discussed)
3 final mixtures – suppose 1 mixture shows activity
Could you synthesize all nine possible tripeptides separately?
No, you have samples of the dimer mixtures produced in synthesis.

Recursive deconvolution
Suppose third tripeptide mixture showed activity, that means the active tripeptide has Val in N-terminus
Take 3 dipeptide mixtures (retained previously) & link Val to each mixture.
This gives 9 tripeptides in 3 mixtures where 2nd & 3rd a.a. are same in each mixture.
Recursive deconvolution

Recursive deconvolution
Test 3 mixtures, if 1 is active we can identify 2nd & 3rd a.a.
Suppose mixture containing Ala (2nd) & Val (3rd) is the active mixture.
Finally 3 component tripeptides in the active mixture – individually synthesized & tested.

Sequential release
Linkers – allow release of certain percentage of product from bead
Process – repeated releasing product sequentially
Mixture of beads – treated to release some of bound product for testing.

Sequential release
If the mixture is active, beads – split into smaller mixture, further product – released & tested
Whole process – repeat several times until active bead is identified.

Structure determination of active compounds
Direct structure determination of components – much difficult
But huge advancements in mass, NMR, Raman, infrared and ultraviolet spectrophotomentry
Peptides – sequenced while attached to bead.
Tagging procedure – can be used

Tagging
Two molecules – built up on same bead
One is intended structure; other is a molecular tag (peptide or oligonucleotide) as a code for each step of synthesis
Bead – has multiple linker linking both target structures & molecular tags
Starting material is added to 1 part & encoding a.a. or necleotide to another part.

TaggingAfter each stage, an a.a. or nucleotide is added to growing tag
to indicate what reagent was used
Example of multiple linker – Safety Catch Linker (SCAL) which has lys & try, both having a free amino group.
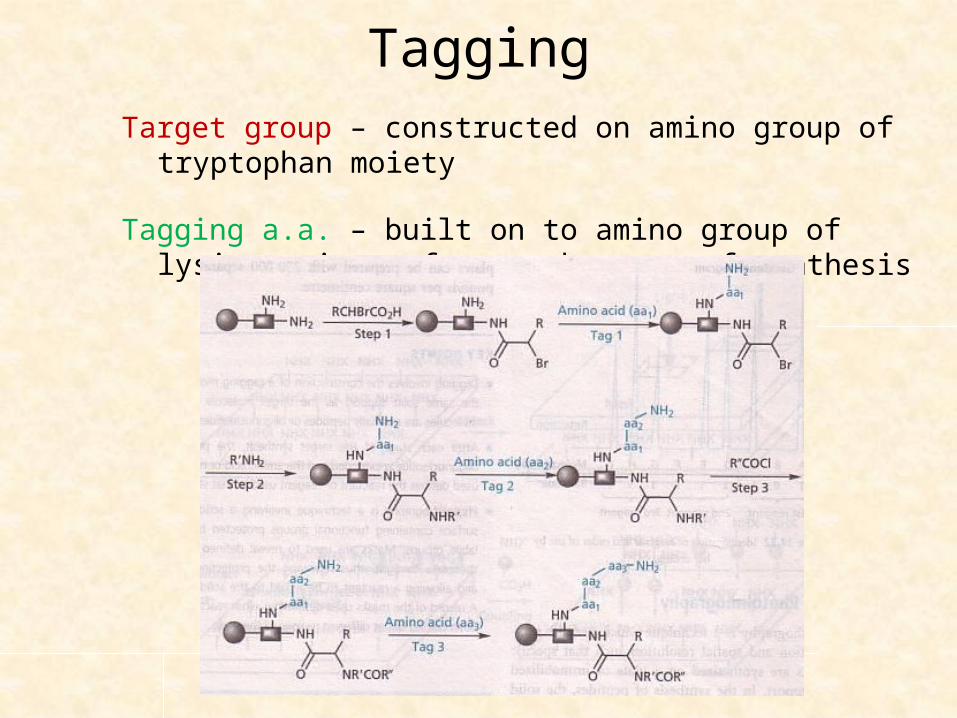
TaggingTarget group – constructed on amino group of tryptophan moiety
Tagging a.a. – built on to amino group of lysine moiety after each stage of synthesis

TaggingBy end of the process, there is a tripeptide tag where each a.a
defines the identity of variable groups R1, R2 & R3 in target structure
Target group – cleaved by reducing 2 sulfoxide groups in SCAL, treat with acid
Tripeptide sequence – still attached to bead, sequenced to identify structure of released compound
Same strategy – with oligonucleotide as tag,
Additionally oligonucleotide – amplified by replication and read by DNA sequencing

Tagging
Drawbacks:
Time consuming
Require elaborate instrumentation
Coding structure adds extra restraints on protection & imposes limitations on reaction
For oligonucleotides, inherent instability
Possibility of unexpected reaction resulting in unwanted structure

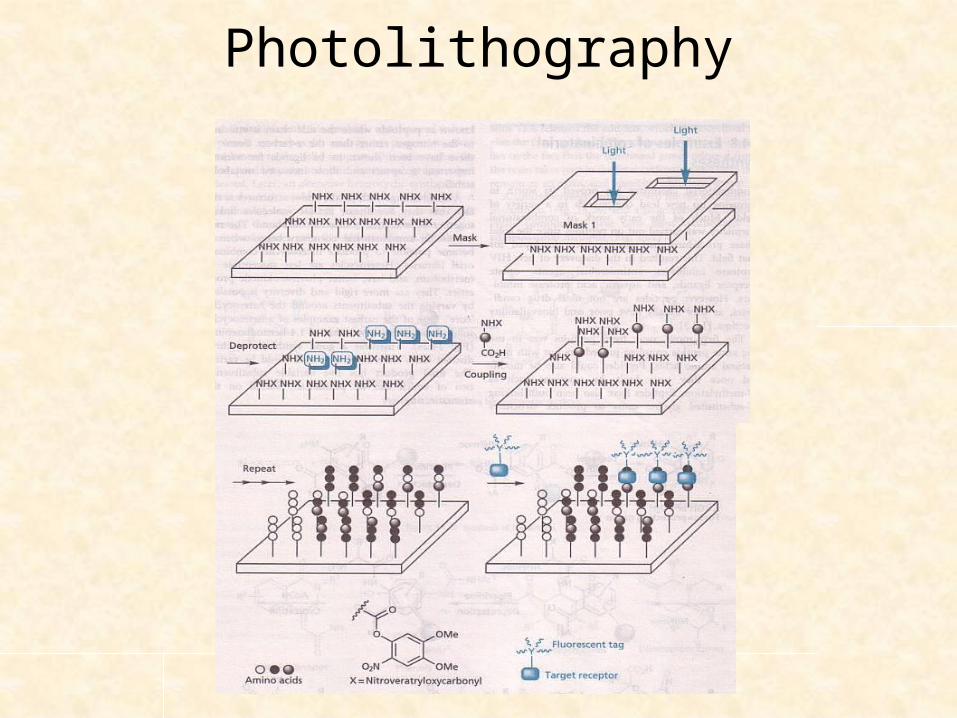
Photolithography
A technique that permits miniaturization and spatial resolution such that specific products are synthesized on a plate of immobilized solid support.
For synthesis of peptides, solid support surface contains an a.a. protected by photolabile group, nitroveratryloxycarbonyl (NVOC)

Photolithography
With mask part of surface – exposed to light – deprotection
Plate is treated with protected a.a; coupling reaction only on deprotected region of the plate
Plate – wash to remove excess a.a.

Photolithography
The process – can be repeated on a different region using a different mask, so different peptide chains can be built on different parts of the plate
Sequences can be known from record of masks used.
Photolithography

Photolithography
Incubation of the plate with a protein receptor – to detect active compounds that bind to receptor
More convenient method – using a fluorescently tagged receptor
Only regions of plate containing active compounds bind to receptor and fluoresce.
Fluorescence intensity – measured using fluorescene microscopy and a measure of affinity of the compound to receptor.
Also detection by radioactivity or chemiluminescence.

Photolithography
Photodeprotection can be achieved in high resolution
At 20-µm resolution, plates can be made with 250,000 compounds /cm2.

Limitations of combinatorial synthesis
Total natural a.a = 20
Total possible decapeptides = 10,240 billion
For statistical reason, no. of beads should exceed no. of target molecules by a factor of 10
e.g., if 5 beads for each of 3.2 million components of a pentapeptide library and 1/5 is taken as sample; probability of getting all peptides is only 63%

Limitations of combinatorial synthesis
If you use required excess of beads,
beads for complete dipeptide library = 8.4 mg
beads for complete tetrapeptide library = 3.4 gm (still good!!)
beads for complete decapeptide library = 215.3 tonnes!!!!!!!!

Dynamic combinatorial chemistry
An exciting development in new lead discovery as an alternative to classic mix and split combinatorial synthesis
In classic method, stable products are synthesized with particular route & building blocks. Then products are screened to find the most active compound.
In dynamic combinatorial chemistry, synthesize all different compounds in 1 flask at same time, screen them in situ as they are being formed; thus identify the most active compound in a much shorter period of time.

Dynamic combinatorial chemistryBest way of screening is to have the desired target in reaction flask
along with building blocks. Active compounds bind to target as soon as they are formed.
Reactions should be reversible. Products are constantly being formed and then breaking back down to building blocks. Advantage is amplification. Active compounds become bound to target and removed from equilibrium mixture. Equilibrium is shifted such that more active product is formed. Thus target serves both to screen and to amplify active compounds.
Necessary to freeze equilibrium reaction to identify active compounds. A further reaction can be carried out which converts all equilibrium products into stable products that cannot revert back to starting materials

Dynamic combinatorial chemistry
Limitations:
Condition such that target does not react with any building block or unstable under reaction conditions
Target is normally in aqueous environment, so reactions have to be in aqueous solution.
Reactions should undergo fast equilibrium rates to allow amplification
Avoid using some building blocks which are more reactive than others, as this would bias the equilibrium and confuse the identification.

Planning & designing a combinatorial synthesis
‘Spider like’ scaffolds’
To find a new lead compound, we need a large no. of diverse structures.
Best to synthesize ‘spider-like’ molecules consisting of a central body (centroid/scaffold) & various arms (substituents)

Planning & designing a combinatorial synthesis
‘Spider like’ scaffolds’
Arms contain different functional groups to probe a binding site
Chance of success is greater if arms are spreaded around scaffold
Allows more theoretical explanation of 3D space or conformational space around the molecule

Planning & designing a combinatorial chemistry
‘Spider like’ scaffolds’
Plan in advance such that synthesized molecules contain different functional groups on arms & different distances from scaffold
In general, this approach increases the chances of finding a lead compound that interacts with a target binding site.

Planning & designing a combinatorial chemistry
Designing ‘drug-like’ molecules
Compounds with good binding interactions do not necessarily make good medicines.
Pharmacokinetic issues also to be taken into account.
Certain restrictions to type of molecules in order to increase chance of getting orally active lead compounds

Planning & designing a combinatorial chemistry
Designing ‘drug-like’ molecules
Chances of oral activity is increased if structure obeys Lipinski’s rule of five:
M.W < 500
Log P < +5
H-bond donating groups ≤ 5
H-bond accepting groups ≤ 10

Planning & designing a combinatorial chemistry
Designing ‘drug-like’ molecules
Groups should be avoided:
Esters (liable to easy metabolism)
Alkylating groups (toxic)
Aromatic amino groups (toxic)

Planning & designing a combinatorial chemistry
Scaffolds
Synthesized by synthetic route used for combinatorial synthesis
Synthesis determines no. & variety of substituents
Ideal scaffold is small & allows a wide variety of substituents

Planning & designing a combinatorial chemistry
Scaffolds
Its substituents are dispersed widely around its structure (spider-like), not restricted to part of structure (tadpole-like)
Synthesis allows substituents to be varied independently of each other.

Planning & designing a combinatorial chemistry
Scaffolds
Can be flexible (peptide backbone) or rigid (a cyclic system)
May contain groups useful for binding interactions
Some scaffolds are common called ‘privileged scaffolds’.
e.g., benzodiazepine, hydantoin, tetrahydroisoquinoline, benzenesulfonamide, etc.

Planning & designing a combinatorial chemistry
Substituent variation
Choice of substituents depends on availability and diversity required
Consider followings:
Structure, size, shape, lipophilicity, dipole moment, electrostatic charge and functional groups present.

Planning & designing a combinatorial chemistry
Designing compound libraries for lead optimization
To optimize a lead, consider following factors for planning of variations:
biological & physical properties of compound
binding interactions, and
potential problems of particular substituent

Planning & designing a combinatorial chemistry
Computer-designed libraries
Computer software programs to design more focused combinatorial compound libraries.
Descriptors: log P, molecular weight, no. of H-bond donors, no. of H-bond acceptors, no. of rotatable bonds, aromatic density, degree of branching in structure, and presence or absence of specific functional groups.

Testing for activity
High-throughput screening (HTS)
A process of biological testing of a large quantity of structures quickly and automatically
HTS was developed before combinatorial chemistry and acted as driving force for synthesizing huge quantity of structures to meet rapid & efficient biological testing process (HTS)

Testing for activity
High-throughput screening (HTS)
Compounds tested on 96 well plate, capacity of each well being 100 μl
Currently same size plate of 1536 wells containing 1-10 μl
Fluorescence, chemiluminescence developed for simultaneous identification of active wells.

Testing for activity
High-throughput screening (HTS)
Next major advancement is microfluidics that involves manipulation of tiny volumes of liquids in confined space.
Microfluidic circuits to control fluid electronically, separation using capillary electrophoresis
New machines for both ultra-small-scale synthesis & miniaturized analysis
10x10 cm silicon wafer for 105 synthesis/bioassay on nl scale.

Testing for activity
Screening ‘on bead’ vs ‘off bead’
Structures can be tested when still attached to solid phase
It involves interactions with targets tagged to enzyme, fluorescent probe, radionuclide or chromophore.
Rapid & 108 beads can be screened
Active beads picked out by micromanipulation & structure of active compound determined.

Testing for activity
Screening ‘on bead’ vs ‘off bead’
False negative may be obtained if solid phase sterically interferes assay
In such case, better to release drug from solid phase and test to avoid false (-)ve
However, some compounds are insoluble in test assay and give false (-)ve result in solution but (+)ve result when attached to bead.


















