
Colloid Stability
14
Chapter 3 Colloid Stability John Eastman 3.1 Introduction One of the important aspects of the study of colloidal dispersions is understanding their stability so that we can manipulate the state of dispersions for particular applications. Charge stabil isation is one means by which this can be achieved and we can manipu late the stability through changes in the chemical environment such as salt concentration, ion type and pH. We must understand how this works both in quiescent systems and when external fields, such as gravitational or shear fields, are present. So what do we mean by stability? This very much depends on the circumstances which are being considered. We can define stability in colloidal systems either in terms of their tendency to aggregate or in terms of their tendency to sediment under the action of gravity. In this chapter we will focus on the stability to aggregation and look at the factors which control this stability. We will study this by considering the interaction between two rep- rese ntativ e particle s in the system. By considering what happens when two particles come toget her (during a Bro wnian collision) we can pred ict the stability of the whole syste m by looking at the form of the colloidal pair potential. 3.2 The Colloidal Pair Potential The pair potential is the total potential energy of interaction between two colloidal particles as the separation or distance between them is varied. Formally it is a free energy, and we calculate it by simply summing the various components that we can identify. The calculation is normally done for two particles in isolation, i.e . at infinite dilution. In a concentrated system (a condensed phase), multi-body interactions should be accounted for, and then we would refer to the potential of mean force. However we get an adequate estima tion of the total potential in a concentrate d system simply by the summa tion of the interaction from the nearest neighbours. It is this interaction energy that gov erns the stability of colloidal dispers ions and which we effectively manipulate whenever we make a change to a formulation.
-
Upload
ceyda-oeksel -
Category
Documents
-
view
245 -
download
0
Transcript of Colloid Stability

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 1/14
Chapter 3
Colloid Stability
John Eastman
3.1 Introduction
One of the important aspects of the study of colloidal dispersions is understanding their
stability so that we can manipulate the state of dispersions for particular applications.
Charge stabilisation is one means by which this can be achieved and we can manipulate
the stability through changes in the chemical environment such as salt concentration, ion
type and pH.
We must understand how this works both in quiescent systems and when external fields,
such as gravitational or shear fields, are present.
So what do we mean by stability? This very much depends on the circumstances which
are being considered. We can define stability in colloidal systems either in terms of their
tendency to aggregate or in terms of their tendency to sediment under the action of gravity.
In this chapter we will focus on the stability to aggregation and look at the factors which
control this stability. We will study this by considering the interaction between two rep-
resentative particles in the system. By considering what happens when two particles come
together (during a Brownian collision) we can predict the stability of the whole system by
looking at the form of the colloidal pair potential.
3.2 The Colloidal Pair Potential
The pair potential is the total potential energy of interaction between two colloidal particles
as the separation or distance between them is varied. Formally it is a free energy, and we
calculate it by simply summing the various components that we can identify.
The calculation is normally done for two particles in isolation, i.e. at infinite dilution. In
a concentrated system (a condensed phase), multi-body interactions should be accounted
for, and then we would refer to the potential of mean force. However we get an adequate
estimation of the total potential in a concentrated system simply by the summation of the
interaction from the nearest neighbours.
It is this interaction energy that governs the stability of colloidal dispersions and which
we effectively manipulate whenever we make a change to a formulation.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 2/14
Colloid Stability 37
The components of the pair potential that we are most interested in for charged colloids
are those due to the attractive van der Waals forces and the repulsive force between similarly
charged particles.
3.2.1 Attractive forces
Molecules with a permanent dipole will attract similar molecules as the dipoles align. They
will also induce a dipole in an adjacent neutral atom or molecule and cause an attraction.
This is relatively easy to understand; however, the motion of the electrons in any atom cause
rapidly fluctuating dipoles. This leads to the London dispersion interaction as the oscillating
dipoles become coupled. Even neutral atoms have a fluctuating dipole due to the motion
of the electrons around the nucleus. It is energetically more favourable for adjacent atoms
to be oscillating in unison. This is the interaction which we recognise from the non-ideal
behaviour of the inert gases.This interaction is non-directional, so that when large assemblies of atoms are considered,
different dipolar orientations do not cancel each other. Colloidal particles are of course
large assemblies of atoms and hence the van der Waals forces from the London dispersion
interaction act between particles to cause attraction (see Figure 3.1).
The early calculations were due to Hamaker and de Boer [1]. The route is to sum the
interaction of one atom in a particle with each atom in the adjacent particle. That interaction
is then summed over all the atoms in the first particle.
The result is a long range interaction – much longer range than the interaction between
two isolated atoms. The range of the interaction is comparable with the radii of colloidal
particles.
The attractive potential energy is directly proportional to a particle radius (a ), a material
constant – the Hamaker [1] constant ( A) – and is inversely proportional to distance of
separation (h):
V A = −A
12
1
x (x + 2)+
1
(x + 1)2+ 2 ln
x (x + 2)
(x + 1)2
, where x =
h
2a (3.1)
When the particle separation is small (h 2a ) this reduces to a simple form of
V A = −Aa
12h(3.2)
a h
Figure 3.1 London forces between atoms in two adjacent colloidal particles.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 3/14
38 Colloid Science: Principles, Methods and Applications
Table 3.1 Hamaker constants for various materials
Particles Hamaker constant (J/10−20) Media Hamaker constant ( J/10−20)
Poly(tetrafluorethylene) 3.8 Water 3.7Poly(methyl methacrylate) 7.1 Pentane 3.8
Poly(styrene) 7.8 Ethanol 4.2
Silica (fused) 6.5 Decane 4.8
Titanium dioxide 19.5 Hexadecane 5.1
Metals (Au, Ag, Pt, etc.) ∼40 Cyclohexane 5.2
The Hamaker constant is a function of both the electronic polarisability and the density of
the material. When particles are immersed in a medium the attraction between particles
is weakened as there is an attraction with the medium also. The combined or compositeHamaker constant ( A) can be estimated as the geometric mean of that of the particle
( Aparticle) and that of the medium ( Amedium) with respect to their values in vacuum, and it
is this that should be used in the calculation of the attractive potential:
A = (
Aparticle −
Amedium)2 (3.3)
Hamaker constants have values in the range of 10−20 J, and a selection of values are given in
Table 3.1.
3.2.2 Electrostatic repulsion
Electrical repulsion is an important stabilising mechanism for particles dispersed in aqueous
solutions or moderate polarity liquids like ethylene glycol.
The diffuse part of the electrical double layer extends in solution over distances charac-
terisedbytheDebyelength(1/κ). In practice we usethe experimentallyaccessibleζ -potential
(see Section 3.3.3) as a measure of the electrical potential at the Stern layer. The rate of de-
cay of this potential is governed by the reciprocal of the Debye length and is commonly
referred to as the double layer thickness (see Table 3.2). This defines the extent to which the
ionic atmosphere, which is different from the bulk ionic medium, extends from the particle
surface.
Table 3.2 The extent of the double layer thickness as a function
of electrolyte concentration
NaCl Concentration (mM) Double layer thickness (nm)
30 2
10 3
1 10
0.1 30

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 4/14
Colloid Stability 39
Figure 3.2 The overlap of electrical double layers in adjacent particles.
Whentwoparticlesapproacheachothertheionicatmospheresoverlap(seeFigure3.2)and
the local ion concentration midway between the particles can be estimated by summing the
contributions from each particle. The difference in this local mid-point ion concentration
and that in the bulk results in an osmotic pressure acting to force the particles apart.
Integration of this force with respect to distance gives us the energy.
When two charged particles come together there are two extreme cases which we can
envisage. If the ion adsorption equilibrium is maintained then either the surface charge
remains constant and the surface potential compensates (constant charge) or the surface
potential remains constant and the surface charge density changes to compensate (constant
potential). Hogg et al . [2] derived expressions which enable us to calculate the interaction
between non-identical spheres under both constant charge and constant potential condi-
tions.
V ψ
R =εa 1a 2
ψ2
01+ψ2
02
4(a 1 + a 2)
2ψ01
ψ02
ψ201+ψ2
02
ln
1+ exp(−κh)
1− exp(−κh)
+ ln(1− exp(−2κh))
(3.4)
V σ
R
=εa 1a 2ψ
201+ψ2
024(a 1 + a 2)
2ψ01ψ02
ψ201 +ψ202
ln1+ exp(−κh)
1− exp(−κh)− ln(1− exp(−2κh)) (3.5)
These reduce to the basic expressions
V ψ
R =εa ψ2
0
2ln(1 + exp(−κh)) (3.6)
V σ R = −εa ψ2
0
2ln(1 − exp(−κh)) (3.7)
for the interaction between identical particles with a radius a . The expressions are valid in
the regime where κa , the product of the Debye constant and the particle radius, is greater
than 10.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 5/14
40 Colloid Science: Principles, Methods and Applications
For conditions where κa is less than 3 the general expression is
V R = 2πεa ψ2δ exp(−κh) (3.8)
3.2.3 Effect of particle concentration
Whenever we add charged colloidal particles to a liquid we do two things:
r add counter-ions with each particle r reduce the solution volume available to the ions.
Both of these factors become important as the particle concentration increases and when
the background electrolyte concentration is low. We can expand the expression for κ from
Equation (2.18), Chapter 2, to take account of these extra effects. The expanded expression is
κ2 =e 2z 2
εk B T
2n0 +3|σ δ|φ
ae 1− φ
(3.9)
where z is the counter-ion valency, n0 the concentration of counter-ions in solution (added
electrolyte), a is the particle radius and e is the formal charge on an electron.
We recognise the first part of the expression from Equation (2.18) but now we have the
expression 3|σ δ |φae
which takes into account the counter-ions which are carried by the particle
through the surface charge density σ δ . We can see that this expression becomes important
when the particles are small and the volume fraction φ and surface charge density are high.The denominator of (1 – φ) takes into account the volume taken up by the particles in the
dispersion.
This effect is only important when the background electrolyte levels are low and where we
have high concentrations of small highly charged particles. We can see the effect in Figure 3.3
which shows the effect for different background electrolytes as a function of particle volume
fraction for 85 nm radius particles with a surface charge density of 0.15 µC cm−2.
3.2.4 Total potential
The linear addition of the electrostatic and dispersion potentials is the basis of the DLVO
[3, 4] theory for colloid stability. When we add the attractive potential to the repulsive
electrostatic potential we have the typical curve for charge stabilised colloidal particles:
V T = V A + V R (3.10)
This curve has a number of interesting and important features. The shape of the curve is the
consequence of the addition of the exponential decay of the repulsive term and the more
steeply decaying one-over-distance relationship of the attractive term.
This linear superposition leads to a maximum in the curve as seen in Figure 3.4 and is
known as the primary maximum. It is this maximum in the pair potential which provides

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 6/14
Colloid Stability 41
ϕ
0.0 0.1 0.2 0.3 0.4 0.5
2.0 × 10−9
1 / κ ( m
− 1 )
4.0 × 10
−9
6.0 × 10−9
8.0 × 10−9
1.0 × 10−8
1.2 × 10−8
1.4 × 10−8
5 × 10−4 M
10−3 M
[NaCl] = 10−2 M
Figure 3.3 The effect of the counter-ions associated with the particle surface on the Debye length 1/ κ .
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0
−60
−40
−20
0
20
40
60
80
100
120
140
V T
( k B
T )
κh (nm)
Figure 3.4 Examples of a total interaction potential curve for two charge stabilised systems.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 7/14
42 Colloid Science: Principles, Methods and Applications
the mechanism for stability of charged colloidal particles. It creates an effective activation
energy for aggregation. As two particles come together they must collide with sufficient
energy to overcome the barrier provided by the primary maximum.
It is important to realise that this barrier to aggregation only provides kinetic stability to
a dispersion. The thermodynamic drive is towards an aggregated, phase separated state. Wecan say that the larger the barrier the longer the system will remain stable.
Note that the potential is plotted in units of k B T . These thermal energy units help us
relate the height of the maximum with the energy of a Brownian collision, which will be of
the order of 1.5k B T .
Therefore, in order to pose a suitable barrier to aggregation this primary maximum
must be at least 1.5k B T . In practice we need to manipulate the system so that the primary
maximum is at least 20k B T in order to achieve a level of stability which can be relied upon
over an extended period of time.
3.3 Criteria for Stability
We need to define how the various factors that we put into our systems affect the stability
so that we can define threshold values.
We need to consider
r the effects of ion type and concentration r the value of the ζ -potential r the effect of particle size.
3.3.1 Salt concentration
The example given in Figure 3.5 shows the curve for titanium dioxide particles of 100 nm
radius and with a ζ -potential of –50 mV at different concentrations of sodium chloride.
The points to note are that:
r In each case there is a steep rise to the primary maximum. r At larger distances there is a long repulsive tail, most notable at lower electrolyte concen-
trations. r The range of the tail reduces as the electrolyte concentration increases (in line with the
decrease in the Debye length). r The height of the maximum decreases with increasing electrolyte concentration. r At some point (in this case around 10−2 M NaCl) a significant energy minimum develops
since the van der Waals dispersion term is insensitive to the electrolyte changes. The
attractive minimum is known as the secondary minimum. r Astheprimarymaximumfallstojustafew k B T , a significant fraction of colliding particles
will collide with at least that energy and stick. r As the primary maximum falls to below zero (above 3 × 10−2 M NaCl in this case), all
collisions will lead to aggregation as there is no barrier.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 8/14
Colloid Stability 43
0 10 20 30 40 50 60 70 80
−80
−40
0
40
80
120
160
200
V T
( k B
T )
h (nm)
[NaCl] = 10−4 M
10−3 M
10−2 M
3 × 10−2 M
Figure 3.5 The effect of salt concentration on the shape of the total interaction potential curve.
3.3.2 Counter-ion valency
The counter-ions are the dominant ions in the Stern and diffuse layers and hence the stability
ismoresensitivetothecounter-iontypethantheco-iontype.Thevalencyofthecounter-ions
is in fact of major importance in determining the stability of charged colloids.
We can estimate a critical coagulation concentration (ccc) from the pair potential exercise
by taking the condition that when there is no primary maximum barrier, the inter-particle
force is also zero and so the coagulation of the particles will be diffusion controlled (all
collisions result in coagulation).
Early observations noted a 6th power dependence of z on the ccc, and this was formalisedin the Shultz–Hardy rule [5–7]:
ccc ∝1
z n(3.11)
r n = 6 for high potentials (unusual for coagulation due to ion adsorption) r n = 2 for low potentials (the more common occurrence).
With many systems the adsorption of ions and resultant decrease in the Stern potential and
hence ζ - potential (see also Section 2.1 of Chapter 2) result in a power less than 6; however,
a trivalent counter-ion such as Al3+ is six times as effective a coagulant as Na+.
8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 9/14
44 Colloid Science: Principles, Methods and Applications
z
1 3
E l e c t r o l y t e C o n c e n t r a t i o n ( m M )
0.01
0.1
1
10
100
1000
10000
KNO 3Ba(NO3)2
La(NO3)3
AgI
Poly(styrene)
2
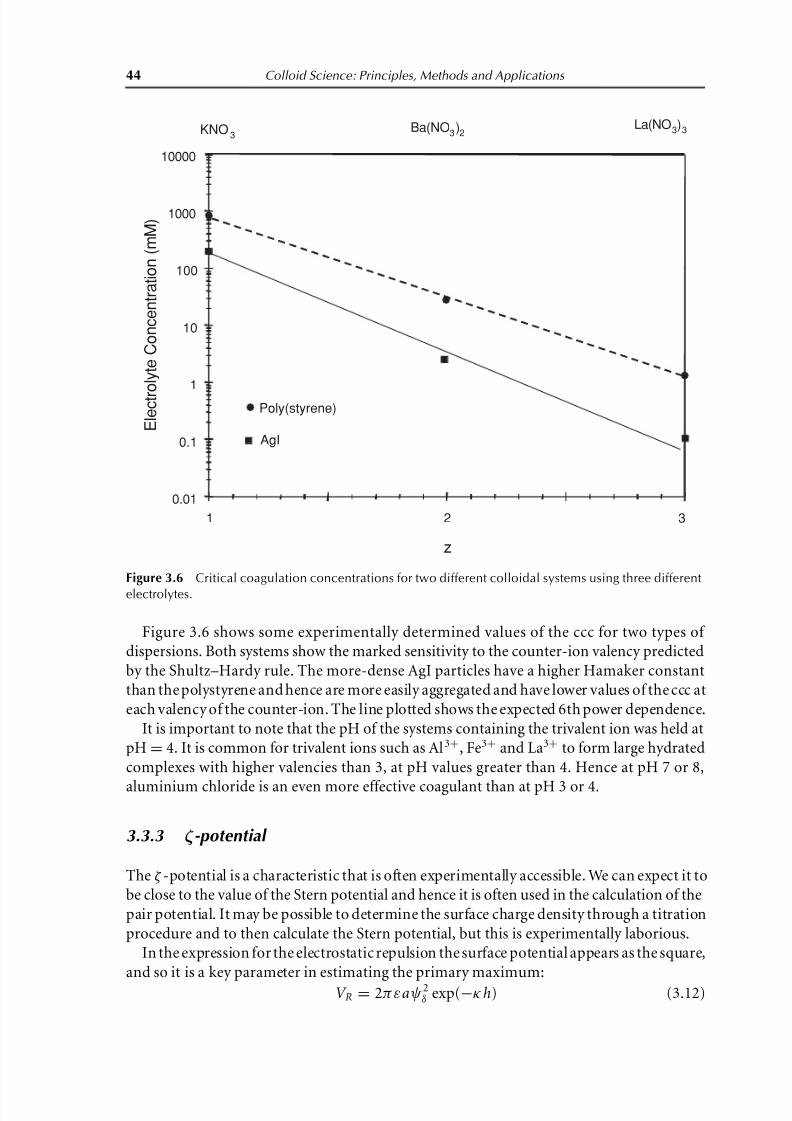
Figure 3.6 Critical coagulation concentrations for two different colloidal systems using three different
electrolytes.
Figure 3.6 shows some experimentally determined values of the ccc for two types of
dispersions. Both systems show the marked sensitivity to the counter-ion valency predicted
by the Shultz–Hardy rule. The more-dense AgI particles have a higher Hamaker constant
than the polystyrene and hence are more easily aggregated and have lower values of the ccc at
each valency of the counter-ion. The line plotted shows the expected 6th power dependence.
It is important to note that the pH of the systems containing the trivalent ion was held at
pH= 4. It is common for trivalent ions such as Al3+, Fe3+ and La3+ to form large hydrated
complexes with higher valencies than 3, at pH values greater than 4. Hence at pH 7 or 8,
aluminium chloride is an even more effective coagulant than at pH 3 or 4.
3.3.3 ζ -potential
The ζ -potential is a characteristic that is often experimentally accessible. We can expect it to
be close to the value of the Stern potential and hence it is often used in the calculation of the
pair potential. It may be possible to determine the surface charge density through a titration
procedure and to then calculate the Stern potential, but this is experimentally laborious.
In the expression for the electrostatic repulsion the surface potential appears as the square,
and so it is a key parameter in estimating the primary maximum:
V R = 2πεa ψ2δ exp(−κh) (3.12)

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 10/14
Colloid Stability 45
0
100
200
300
400
500
−1000 10 20 30 40 50
V T
( k B
T )
h (nm)
Figure 3.7 The effect of ζ -potential on the shape of the total interaction potential curve for a polystyrene
latex. From the bottom to top the ζ -potentials are −20 mV, −25 mV, −50 mV and −80 mV.
In the example given in Figure 3.7 for polymer latex particles in 1 mM NaCl, we need a value
in excess of 20 mV to produce a stable dispersion.
When the ζ -potential is−25 mV the value of V max is ∼40k B T . When we double that to
−50 mV we can see that V max ∼ 160k B T . So as expected, since V R is related to the square
of the ζ -potential, a doubling of the ζ -potential leads to a quadrupling of the value of V max.
In this example, when the ζ -potential reduces to less than −20 mV the value of V max
drops below 20k B T and significant aggregation will occur.
3.3.4 Particle size
Both the attractive and repulsive contributions are proportional to the particle radius. At
small sizes the value of V T is directly proportional to the particle size. However, at large sizes
the value of V T has a more complicated variation.
In all cases a larger particle radius leads to a higher energy barrier; in other words,
electrostatic stability increases with increasing particle radius (all other factors remaining
constant). For small particle sizes (<100 nm radius) the primary maximum is directly
proportional to the radius. However, the relationship breaks down at larger sizes and theheight of the primary maximum increases at a lower rate.
The shape of the curve of the attractive interaction is the important point here. Another
feature of this is that for particles with large radii, the attraction often dominates again at
long range giving rise to a secondary minimum at distances of the order of 5–10 nm. This
attraction is manifest as weak but reversible aggregation.
We can make a distinction here between two types of aggregation. Coagulation is the
rapid aggregation that happens in the absence of a primary maximum and leads to a strong
irreversible aggregated structure. Flocculation is a reversible aggregation that occurs in a
secondary minimum as described. Flocculation is reversible on the addition of energy to
the system, usually the application of a shear field by shaking, stirring or other mechanical
processes.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 11/14
46 Colloid Science: Principles, Methods and Applications
3.4 Kinetics of Coagulation
The rate of coagulation is used either directly or indirectly to determine the ccc. For example
if the rate is monitored, we find that it increases as the electrolyte concentration is increased
until it reaches a plateau value. If we just add particles to different electrolyte solutions in a
series of tubes, then we can check for the onset of aggregation after a fixed time. This may
be 5 min, or we may choose a little longer, but it is still the faster rate that we notice.
As we have already established that electrostatically stabilised dispersions are kinetically
stable and not thermodynamically stable, the key factor is the kinetics. If the rate is so slow
that we do not detect a significant change during our period of use, we would consider that
to be adequately stable.
3.4.1 Diffusion limited rapid coagulation
Recall that the diffusion constant is in terms of the flux through a unit area per second ( J p ):
J p = D 4πr 2dN
dr (3.13)
We can calculate the flow through a spherical surface around a reference particle (see
Figure 3.8). This gives us a differential equation that is easily solved to give the number of
collisions with that reference particle. Of course each particle is itself such a particle and so
the total number of collisions is that for one particle multiplied by the total particle number.
We must divide by 2 as particle A colliding with particle B is the same collision as particle Bcolliding with particle A.
We can allow for the fact that all particles are in motion by using the sum of the diffusion
constants of the two colliding particles. As we are assuming them all to be of the same size,
we multiply by 2. As each collision results in coagulation the coagulation rate is simply the
collision rate.
r
Figure 3.8 Theoretical spherical surface of influence surrounding a reference particle.
8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 12/14
Colloid Stability 47
φ
0.001 0.01 0.1
H
a l f - l i f e i n s e c o n d s f o r c o a g u l a t i o n
10−6
10−4
10−2
100
102
a = 20 nm
a = 100 nm
a = 500 nm
a = 1000 nm
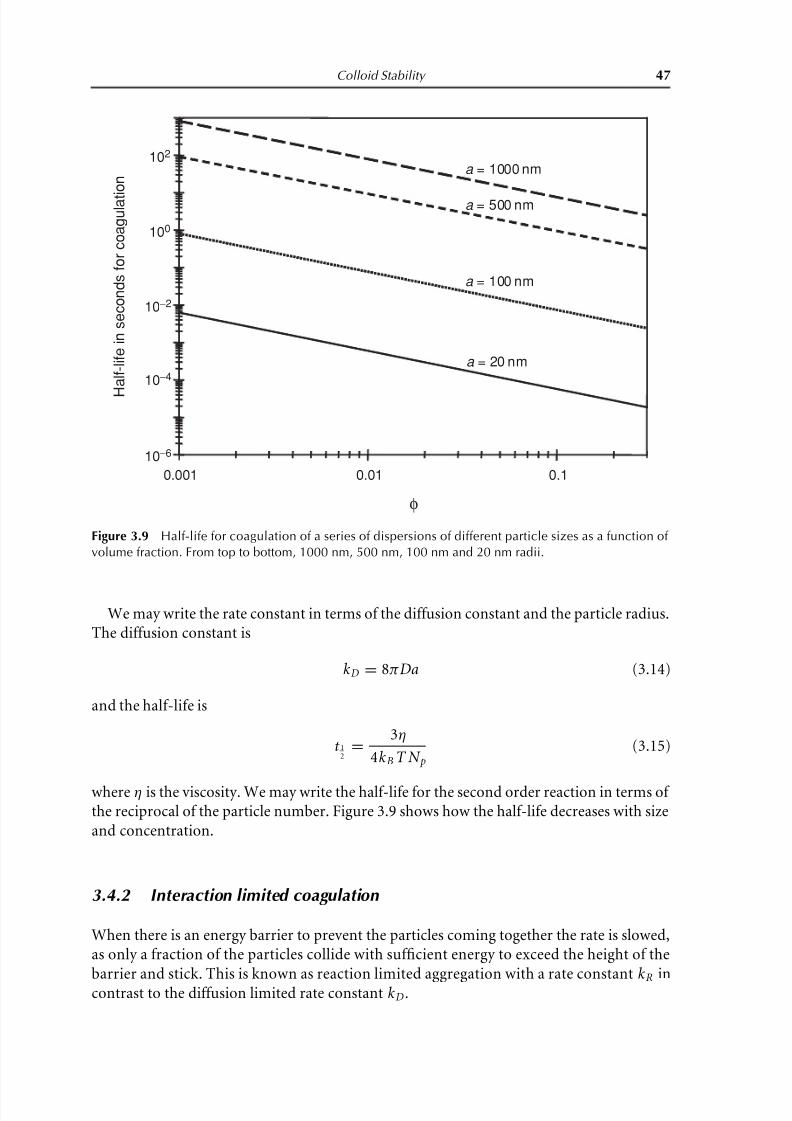
Figure 3.9 Half-life for coagulation of a series of dispersions of different particle sizes as a function of
volume fraction. From top to bottom, 1000 nm, 500 nm, 100 nm and 20 nm radii.
We may write the rate constant in terms of the diffusion constant and the particle radius.
The diffusion constant is
k D = 8πDa (3.14)
and the half-life is
t 12=
3η
4k B T N p(3.15)
where η is the viscosity. We may write the half-life for the second order reaction in terms of the reciprocal of the particle number. Figure 3.9 shows how the half-life decreases with size
and concentration.
3.4.2 Interaction limited coagulation
When there is an energy barrier to prevent the particles coming together the rate is slowed,
as only a fraction of the particles collide with sufficient energy to exceed the height of the
barrier and stick. This is known as reaction limited aggregation with a rate constant k R
in
contrast to the diffusion limited rate constant k D .

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 13/14
48 Colloid Science: Principles, Methods and Applications
Fuchs [8] defined the stability ratio W as the ratio of the rate constants so that the higher
the stability ratio the slower the rate:
W =
k D
k R (3.16)
To a good approximation the reaction limited diffusion rate constant is proportional to the
Boltzmann factor which gives the fraction of particles at any instant with energy in excess
of the primary maximum. The rate of aggregation drops rapidly with the increase in the
energy barrier, so that 10–20k B T gives us reasonable kinetic stability.
Overbeek [4] has shown that a reasonable approximation to the stability ratio is obtained
using the value of the primary maximum:
W =1
2κa
expV max
k B T (3.17)
from which we can show that
k R ≈ 16πκDa 2 exp
−V max
k B T
(3.18)
3.4.3 Experimental determination of the ccc
The ccc is the salt concentration at which there is a change from aggregation which is limitedby the presence of a primary maximum to aggregation which has no barrier. At this point
there is a distinct change in the coagulation rate. Measurement of the coagulation rate can
be made in two ways
r directly – by measuring the number of particles as a function of time by particle counting
(best for large particles) r indirectly – by light scattering (best for small particles).
In each case we are observing the change in the number of aggregates with time. This tells
us about the loss of primary particles from the system.
The simplest experiment is to make up a series of tubes with different electrolyte concen-trations and observe at which concentration aggregation becomes apparent after, say, 5 or
10 min. It can be quantified if the tubes are lightly centrifuged and a spectrophotometer is
used to measure the per cent transmission of the supernatant as a measure of the number
of particles in suspension.
More precise determination can be made if the rate of aggregation itself is measured.
At higher electrolyte concentrations the rate increases to the plateau value (as seen in Fig-
ure 3.10) representing the fast or diffusion limited rate.
Note that the analysis of the rate constant in terms of the diffusion of single particles
is strictly the initial rate. As we progress into the coagulation process the particle number
changes and the mechanism changes to one where the large much less mobile aggregates get
larger by adding singlets. Accurately describing the rates can then be quite complex.

8/3/2019 Colloid Stability
http://slidepdf.com/reader/full/colloid-stability 14/14
Colloid Stability 49
log [NaCl]
l o g k
Rapid coagulation W ~ 1
Slow coagulation
ccc
Figure 3.10 Rate of coagulation against salt concentration indicating the ccc.
3.5 Conclusions
In this chapter we have explored the basic theory surrounding the stability of systems
containing charged colloidal particles. The balance between the van der Waals interaction
and the repulsion between the electrical double layers surrounding charged particles can be
controlled to provide an energetic barrier to the coagulation of particles.We have seen that charge stabilisation can only be effective where significant surface
charge can be achieved and so is normally limited to systems in polar solvents.
The stability achieved is only a kinetic stability to coagulation and in the absence of any
other stabilising mechanism these systems will eventuallycoagulate. However, in the stability
ratio we have a method for evaluating the rate of coagulation compared with the theoretical
rate when there is no barrier to aggregation.
References
1. Hamaker, H.C. (1937) Physica , 4, 1058.
2. Hogg, R., Healey, T.W. and Fuerstenau, D.W. (1966) Trans. Faraday Soc ., 62, 1638.
3. Derjaguin, B.V. and Landau, L. (1941) Acta Physicochim. (URSS), 14, 633.
4. Verwey, E.J. and Overbeek, J.T.G. (1948) Theory of the Stability of Lyophobic Colloids . Elsevier,
Amsterdam.
5. Schulze, J. (1882) pr. Chem., 25, 471.
6. Schulze, J. (1883) pr. Chem., 27, 320.
7. Hardy, W.B. (1900) Proc. R. Soc., 66a, 110.
8. Fuchs, N. (1934) Z. Phys ., 89, 736.














![1 General Principles of Colloid Stability and the Role of ... · colloids and macrobodies: the microscopic [7] and the macroscopic approach [8]. ... 4 1 General Principles of Colloid](https://static.fdocuments.net/doc/165x107/5f01c8837e708231d4010350/1-general-principles-of-colloid-stability-and-the-role-of-colloids-and-macrobodies.jpg)



