
Clustering in Microarray Data-mining and Challenges Beyond Qing-jun Wang Center for Biophysics &...
28
Clustering in Microarray Data-mining and Challenges Beyond Qing-jun Wang Center for Biophysics & Computational Biology University of Illinois at Urbana-Champaign CS491jh presentation March 7, 2002
-
Upload
emil-martin-bruce -
Category
Documents
-
view
213 -
download
0
Transcript of Clustering in Microarray Data-mining and Challenges Beyond Qing-jun Wang Center for Biophysics &...

Clustering in Microarray Data-mining and Challenges Beyond
Qing-jun WangCenter for Biophysics & Computational Biology
University of Illinois at Urbana-Champaign
CS491jh presentation
March 7, 2002

Clustering
What?
Where?
How?
Challenges beyond clustering

Data AcquisitionExperimental design
-MIAME
-Replicates
-Single/multiple slides
Perform experiment
Collect data
Data ProcessingGrid alignment
Data quality e.g. bad data, S/N
Missing data
Normalization-Total intensity normalization
-Regression techniques
-Ratio statistics
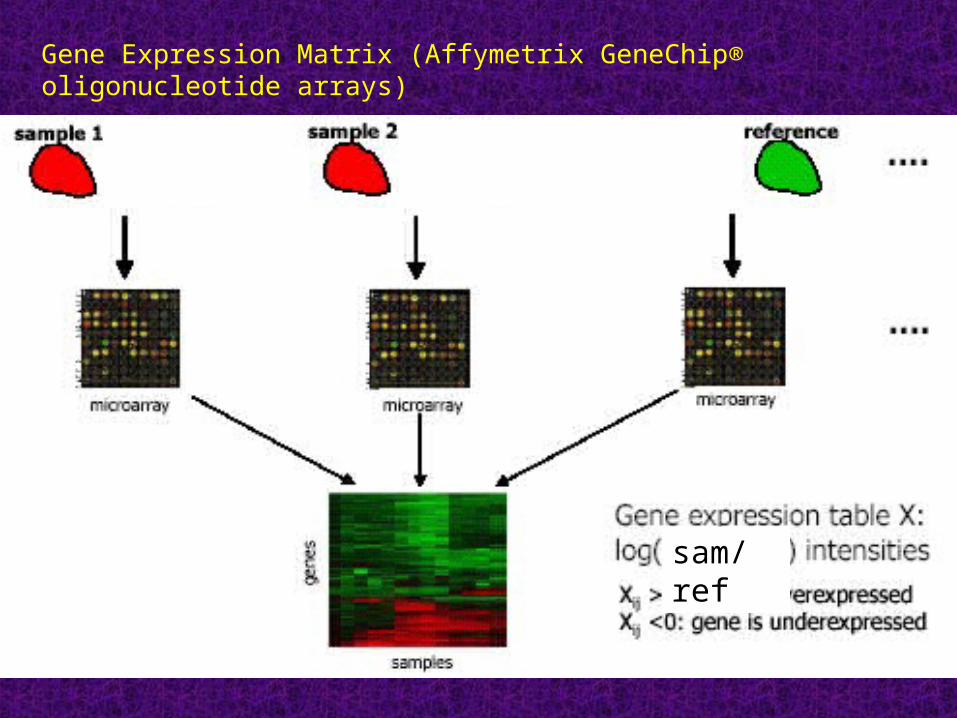
Gene Expression Matrix (Affymetrix GeneChip® oligonucleotide arrays)
sam/ref

Gene Expression Matrix (glass slides)
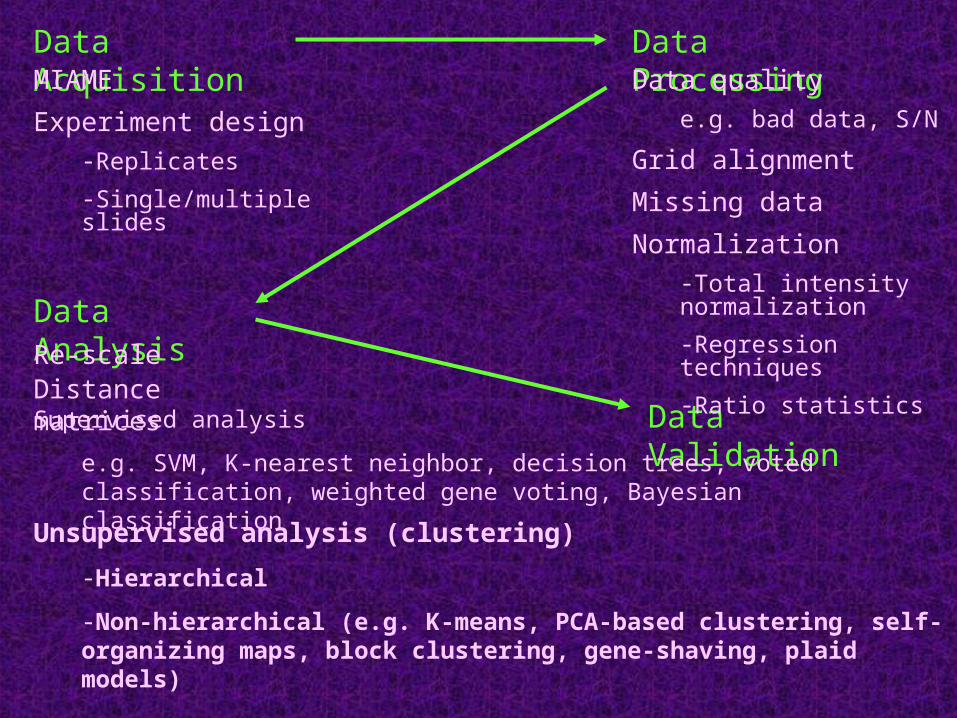
Data AcquisitionMIAME
Experiment design-Replicates
-Single/multiple slides
Data Validation
Data Analysis
Data Processing
Re-scale
Data quality e.g. bad data, S/N
Grid alignment
Missing data
Normalization-Total intensity normalization
-Regression techniques
-Ratio statisticsDistance matrices
Unsupervised analysis (clustering)
-Hierarchical
-Non-hierarchical (e.g. K-means, PCA-based clustering, self-organizing maps, block clustering, gene-shaving, plaid models)
Supervised analysis
e.g. SVM, K-nearest neighbor, decision trees, voted classification, weighted gene voting, Bayesian classification

Protocol 1. Calculate pairwise distance matrix
2. Find the two most similar genes or clusters
3. Merge the two selected clusters to produce a new cluster
4. Calculate pairwise distance matrix involving the new cluster
5. Repeat steps 2-4 until all objects are in one cluster
6. The clustering sequence is represented by a hierarchical tree – dendrogram.
Hierarchical clustering
Step 0 Step 1 Step 2 Step 3 Step 4
b
dc
e
a a b
d ec d e
a b c d e
Step 4 Step 3 Step 2 Step 1 Step 0
agglomerative(AGNES)
divisive(DIANA)

Hierarchical clustering
Variations – differ in how distances are calculated
Single-linkage clustering – minimum distance
Complete-linkage clustering – maximum distance
Average-linkage clustering (UPGMA)
Weighted pair-group average – use size of the clusters as the weights in computing averages
Within-groups clustering
Ward’s method – smallest possible increase in the sum of squared errors

Difficulties1. As clusters grow in size, the expression vector that represents the cluster might no longer represent any of the genes in the cluster – an artifact
2. If a bad assignment is made early on, it cannot be corrected
Hierarchical clustering
Bottom-up (agglomerative) approach
One-way clustering
Deterministic clustering
Produce a greater number of clusters than k-means clustering – valuable feature for discovery.
Produce an order for objects – informative for data display.

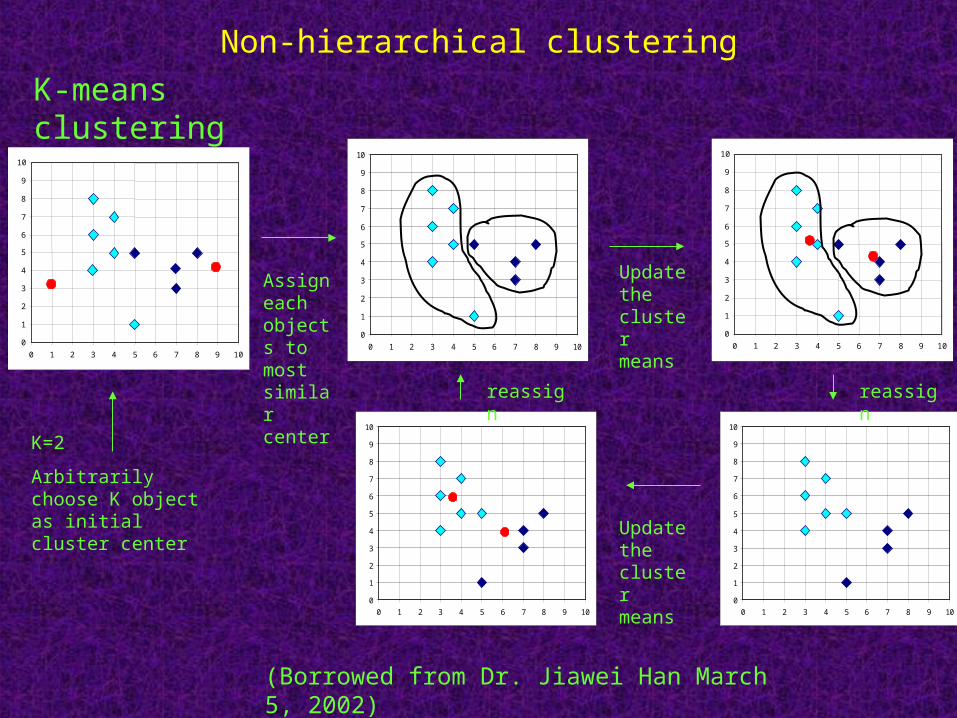
K-means clusteringTop-down (divisive) approach
Used when the number of clusters is known in advance
One-way clustering
Non-deterministic owing to the random initialization
Produce tighter clusters than hierarchical clustering
Protocol1. Initial reference vectors are assigned randomly or according to
previous knowledge
2. Assign each object to one of k clusters randomly
3. Calculate average expression vectors for each cluster (as reference vectors) and the distance between clusters
4. Iteratively move objects between clusters and the objects stay in the new cluster when they are closer to the new cluster than to the old cluster.
5. Repeat steps 3-4 until converge, i.e. moving any more objects would increase intra-cluster distances
Non-hierarchical clustering
K-means clustering
Non-hierarchical clustering
0
1
2
3
4
5
6
7
8
9
10
0 1 2 3 4 5 6 7 8 9 10
0
1
2
3
4
5
6
7
8
9
10
0 1 2 3 4 5 6 7 8 9 10
0
1
2
3
4
5
6
7
8
9
10
0 1 2 3 4 5 6 7 8 9 10
0
1
2
3
4
5
6
7
8
9
10
0 1 2 3 4 5 6 7 8 9 10
0
1
2
3
4
5
6
7
8
9
10
0 1 2 3 4 5 6 7 8 9 10
K=2
Arbitrarily choose K object as initial cluster center
Assign each objects to most similar center
Update the cluster means
Update the cluster means
reassignreassign
(Borrowed from Dr. Jiawei Han March 5, 2002)

Non-hierarchical clustering
Difficulty
How to determine whether there are really only k distinct clusters represented in the data or not.
Solutions
Use K-means clustering with principal component analysis (PCA), which allows visual estimation of the number of clusters represented in the data.
Try sequential k-means approach which finds number of clusters based on dataset.
K-means clustering

Self-organizing map clustering
Non-hierarchical clustering
Top-down (divisive) approach
One-way clustering
Neural-network-based clustering approach
Non-deterministic owing to the random order in which genes are used to move the reference vectors.
Similar to k-means clustering except that the cluster centers are restricted to lie in a one or two-dimensional manifold
Model the complexity within a dataset more effectively than k-means clustering.

Self-organizing map clustering
Non-hierarchical clustering
(Borrowed from Joshua Unger Feb. 28, 2002)
Protocol1. Define a geometric configuration
for the partitions, e.g. a 2D rectangular or hexagonal grid
2. Construct and assign random vectors to each partition
3. Pick a gene randomly; identify the reference vector that is closest to the gene
4. Adjust the reference vectors so that they are more similar to the gene vector
5. Repeat steps 3-4 until the reference vectors converge
6. Map genes to the relevant partitions based on the reference vectors to which they are most similar

One-way clustering – used to group genes with similar behavior across samples or samples with similar gene expression vectors
Two-way clustering – simultaneously cluster both genes and samples
Hierarchical clustering
K-means clustering
Self-organizing maps
Block clustering
Gene shaving
Plaid models
…
Non-hierarchical clustering

Blocking clustering
Protocol
Top-down approach
Two-way clustering
Produce a matrix with homogeneous blocks of the outcomes
Produce hierarchical clustering trees for the rows and columns
1. Begin with the entire matrix in one block
2. Sort rows and columns by row and column means
3. Find the row or column splits of all existing blocks, choosing the one that produces largest reduction in the total within-block-variance
4. If there are existing row/column splits that intersect the block, one of them must be used. Otherwise all split points are tried.
5. The splitting is continued until a large number of blocks are obtained
6. Apply weakest link pruning to recombine some of the blocks until the optimal number of blocks is obtained.
7. The optimal number of blocks is estimated by “maximum gap” approach
1
4
5
3
2
Gene
3 1 2
Sample
Non-hierarchical clustering
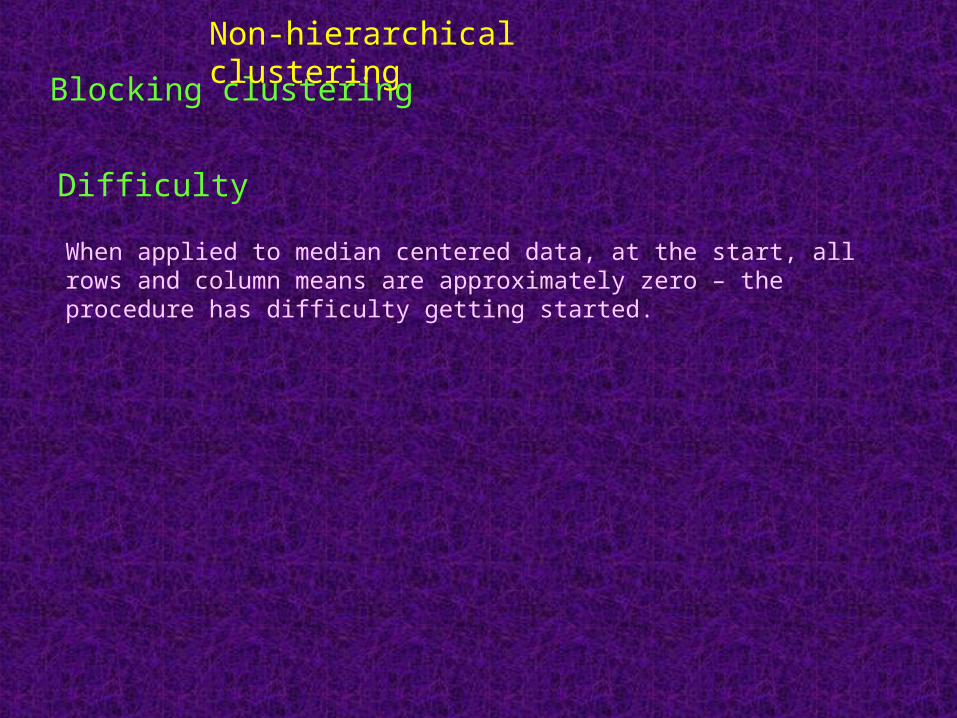
Blocking clustering
Difficulty
When applied to median centered data, at the start, all rows and column means are approximately zero – the procedure has difficulty getting started.
Non-hierarchical clustering

The two-way clustering approach seek a single re-ordering of the samples for all genes. However, one set of genes might cluster the samples in one way while another set of genes in a very different way.
Gene Shaving approach finds the linear combination of genes having maximal variation among samples. This linear combination of genes is viewed as a “super gene”.
The genes having lowest correlation with the “super gene” is removed (shaved). The process is continued until the subset of genes contains only one gene.
This process produces a sequence of gene blocks, each containing genes that are similar to one another and displaying large variance across samples.A statistical approach
Two-way clustering
Identifies subsets of genes with coherent expression patterns and large variation across conditions
Gene may belong to more than one cluster
Can be either un-supervised or supervised
Non-hierarchical clustering

Gene shaving
Protocol1. Start with all data in one block.
2. Find the first principal component of the genes
3. For each gene i, compute the absolute value of its correlation with the first principal component
4. Remove the fraction of genes having the smallest absolute correlation
5. Repeat steps 3~4 until only one gene remains
6. This procedure produces a set of nested gene groups G1G2 … G* … Gn, from which G* is selected as the optimal gene block (small ), where the optimal shave size is estimated using “maximum gap” method.
7. The rows of the gene expression matrix are orthogonalised with respect to the average of all genes in cluster G* to obtain a new gene expression matrix to encourage discovery of a different second cluster. Repeat steps 2-7 until no interesting gene shaves can be found.


A cellular process may involve a relatively small subset of genes in the dataset. The process may take place only in a small number of samples. Therefore, when the full dataset is analyzed, the signal of this process may be completely overwhelmed by the noise of vast majority of unrelated data.
Plaid models search for interpretable biological structures in microarray data, i.e. subsets of the genes/samples, one of which can be used to cluster the other to yield stable and significant partitions/layers.
Two-way clustering
Allows a gene to be in more than one cluster or in none at all
Allows a cluster of genes to be defined with respect to only a subset of samples, not necessarily all of them
Non-hierarchical clustering

Plaid models:
Non-hierarchical clustering
Ideal reordering:
Every gene and every sample are in exactly one cluster
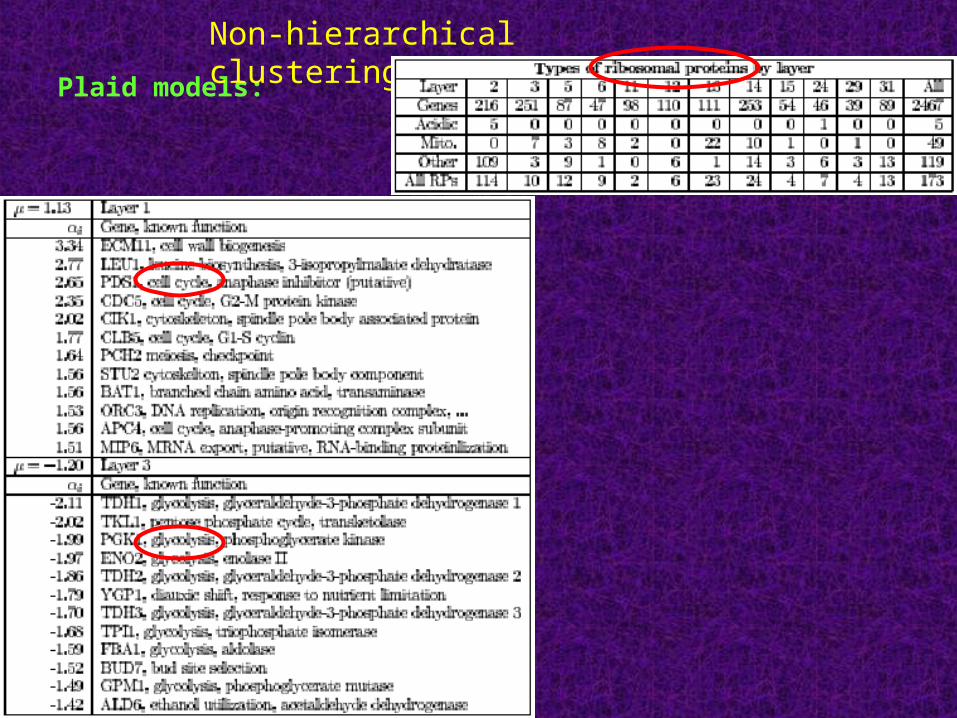
Plaid models:
Non-hierarchical clustering

Evaluate clustering
Clarity of cluster definitions
Computational cost
Robustness
Reproducibility Cancer researchCancer typing
Correlating whole-genome expression pattern with particular clinical implication
Diagnose malignant tissue from normal one
Drug effect studyPathway discoveryAssign functions of unknown genesGene network & regulation:
metabolism, photosynthesis, cell cycle, …

Challenges beyond clustering
Understand sources of noise and variations in microarray experiments
Combine expression data with other sources of information Published literature
DNA & protein sequence databases
Protein data bank
Phylogenetic profiles
Metabolic function
Annotated experimental functional studies

Clustering
Assumption: guilt-by-associationGenes that are contained in a particular pathway, or that respond to a common environmental challenge, should be co-regulated and consequently, should show similar patterns of expression.
This is a controversial hypothesis because the existence of
Convergent regulation
(similar temporal expression patterns, different control strategies)
&
Divergent regulation
(similar control regions, different ways to take effects)

Challenges beyond clustering
Understand sources of noise and variations in microarray experiments
Combine expression data with other sources of information
Reconstruct networks of genetic interactions to create integrated and systematic models of biological systems
Published literature
DNA & protein sequence databases
Protein data bank
Phylogenetic profiles
Metabolic function
Annotated experimental functional studies
Boolean networks
Linear modeling
Generic programming
Bayesian belief networks
References
1. Quackenbush (2001) Nature Reviews Genetics. 2:418-427
2. Altman & Raychaudhuri (2001) Curr. Opin. Struct. Biol. 11:340-347
3. Lazzeroni & Owen (2000) Tech. Report. Stanford Univ.
4. Aas (2001) SAMBA
5. Tibshirani et al. (1999) Tech. Report. Stanford Univ.
6. Hastie et al. (2000) Genome Biol. 1(2)


















