
CLPTM1L Promotes Growth and Enhances Aneuploidy...
12
Molecular and Cellular Pathobiology CLPTM1L Promotes Growth and Enhances Aneuploidy in Pancreatic Cancer Cells Jinping Jia 1 , Allen D. Bosley 6 , Abbey Thompson 1 , Jason W. Hoskins 1 , Adam Cheuk 2 , Irene Collins 1 , Hemang Parikh 1 , Zhen Xiao 6 , Kris Ylaya 3 , Marta Dzyadyk 1 , Wendy Cozen 9 , Brenda Y. Hernandez 10 , Charles F. Lynch 11 , Jadranka Loncarek 7 , Sean F. Altekruse 4 , Lizhi Zhang 12 , Christopher J. Westlake 8 , Valentina M. Factor 5 , Snorri Thorgeirsson 5 , William R. Bamlet 12 , Stephen M. Hewitt 3 , Gloria M. Petersen 12 , Thorkell Andresson 6 , and Laufey T. Amundadottir 1 Abstract Genome-wide association studies (GWAS) of 10 different cancers have identified pleiotropic cancer predis- position loci across a region of chromosome 5p15.33 that includes the TERT and CLPTM1L genes. Of these, susceptibility alleles for pancreatic cancer have mapped to the CLPTM1L gene, thus prompting an investigation of the function of CLPTM1L in the pancreas. Immunofluorescence analysis indicated that CLPTM1L localized to the endoplasmic reticulum where it is likely embedded in the membrane, in accord with multiple predicted transmembrane domains. Overexpression of CLPTM1L enhanced growth of pancreatic cancer cells in vitro (1.3–1.5–fold; P DAY7 < 0.003) and in vivo (3.46-fold; P DAY68 ¼ 0.039), suggesting a role in tumor growth; this effect was abrogated by deletion of two hydrophilic domains. Affinity purification followed by mass spectrometry identified an interaction between CLPTM1L and non-muscle myosin II (NMM-II), a protein involved in main- taining cell shape, migration, and cytokinesis. The two proteins colocalized in the cytoplasm and, after treatment with a DNA-damaging agent, at the centrosomes. Overexpression of CLPTM1L and depletion of NMM-II induced aneuploidy, indicating that CLPTM1L may interfere with normal NMM-II function in regulating cytokinesis. Immunohistochemical analysis revealed enhanced staining of CLPTM1L in human pancreatic ductal adenocar- cinoma (n ¼ 378) as compared with normal pancreatic tissue samples (n ¼ 17; P ¼ 1.7 10 4 ). Our results suggest that CLPTM1L functions as a growth-promoting gene in the pancreas and that overexpression may lead to an abrogation of normal cytokinesis, indicating that it should be considered as a plausible candidate gene that could explain the effect of pancreatic cancer susceptibility alleles on chr5p15.33. Cancer Res; 74(10); 2785–95. Ó2014 AACR. Introduction Risk variants in the TERT-CLPTM1L gene region on chro- mosome 5p15.33 have been reported in genome-wide associ- ation studies (GWAS) for 10 cancer types, including bladder, breast, glioma, lung, melanoma, non-melanoma skin cancer, ovarian, pancreas, prostate, and testicular germ cell cancer (1– 13). The TERT gene encodes the catalytic subunit of the telomerase reverse transcriptase complex known for its role in maintaining telomere ends and the increased telomerase activity often seen in human cancers (14). The CLPTM1L gene encodes the cleft lip and palate-associated transmem- brane 1-like protein (CLPTM1L) and was originally identified in a screen for genes conferring resistance to cisplatin in ovarian cancer cells (15). When overexpressed in ovarian cancer cell lines, CLPTM1L induced apoptosis in cisplatin- sensitive cells, giving rise to its original name: cisplatin resis- tance–related protein (CRR9; ref. 15). CLPTM1L was later shown to protect lung cancer cells from apoptosis after treat- ment with DNA-damaging agents via Bcl-xL (16). Gain of chromosome 5p is one of the most recurrent chromosomal abnormalities in human cancers (17). Although most commonly seen in thyroid, lung, and cervical cancer, 5p gain is also frequent in other cancers, including gastric, ovar- ian, colorectal, hepatocellular, esophageal, bladder, and pan- creatic adenocarcinoma (17–19). The most common event in early stages of non–small cell lung cancer is gain at 5p15.33 involving both TERT (78%) and CLPTM1L (53%; ref. 20). How- ever, a recent study of cervical cancer noted that CLPTM1L, but Authors' Affiliations: 1 Laboratory of Translational Genomics, Division of Cancer Epidemiology and Genetics; 2 Pediatric Oncology Branch; 3 Labo- ratory of Pathology; 4 Division of Cancer Control and Population Sciences; 5 Laboratory of Experimental Carcinogenesis, National Cancer Institute, NIH, Department of Health and Human Services, Bethesda; 6 Laboratory of Proteomics and Analytical Technologies, Leidos Biomedical Research, Frederick National Laboratory for Cancer Research; 7 Laboratory of Protein Dynamics and Signaling and 8 Laboratory of Cell & Developmental Signal- ing, NCI-Frederick, Frederick, Maryland; 9 Keck School of Medicine, Uni- versity of Southern California, Los Angeles, California; 10 University of Hawaii Cancer Center, Honolulu, Hawaii; 11 Department of Epidemiology, College of Public Health, University of Iowa, Iowa City, Iowa; and 12 Depart- ment of Health Sciences Research, Mayo Clinic, Rochester, Minnesota Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). Corresponding Author: Laufey T. Amundadottir, Laboratory of Transla- tional Genomics, Division of Cancer Epidemiology and Genetics, NIH, 8717 Grovemont Circle, Gaithersburg, MD 20877. Phone: 301-594-8131; Fax: 301-402-3134; E-mail: [email protected] doi: 10.1158/0008-5472.CAN-13-3176 Ó2014 American Association for Cancer Research. Cancer Research www.aacrjournals.org 2785 on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
Transcript of CLPTM1L Promotes Growth and Enhances Aneuploidy...

Molecular and Cellular Pathobiology
CLPTM1L Promotes Growth and Enhances Aneuploidy inPancreatic Cancer Cells
Jinping Jia1, Allen D. Bosley6, Abbey Thompson1, Jason W. Hoskins1, Adam Cheuk2, Irene Collins1,Hemang Parikh1, Zhen Xiao6, Kris Ylaya3, Marta Dzyadyk1, Wendy Cozen9, Brenda Y. Hernandez10,Charles F. Lynch11, Jadranka Loncarek7, Sean F. Altekruse4, Lizhi Zhang12, Christopher J. Westlake8,Valentina M. Factor5, Snorri Thorgeirsson5, William R. Bamlet12, Stephen M. Hewitt3, Gloria M. Petersen12,Thorkell Andresson6, and Laufey T. Amundadottir1
AbstractGenome-wide association studies (GWAS) of 10 different cancers have identified pleiotropic cancer predis-
position loci across a region of chromosome 5p15.33 that includes the TERT and CLPTM1L genes. Of these,susceptibility alleles for pancreatic cancer havemapped to theCLPTM1L gene, thus prompting an investigation ofthe function of CLPTM1L in the pancreas. Immunofluorescence analysis indicated that CLPTM1L localized tothe endoplasmic reticulum where it is likely embedded in the membrane, in accord with multiple predictedtransmembrane domains. Overexpression of CLPTM1L enhanced growth of pancreatic cancer cells in vitro(1.3–1.5–fold; PDAY7 < 0.003) and in vivo (3.46-fold; PDAY68 ¼ 0.039), suggesting a role in tumor growth; this effectwas abrogated by deletion of two hydrophilic domains. Affinity purification followed by mass spectrometryidentified an interaction between CLPTM1L and non-muscle myosin II (NMM-II), a protein involved in main-taining cell shape, migration, and cytokinesis. The two proteins colocalized in the cytoplasm and, after treatmentwith a DNA-damaging agent, at the centrosomes. Overexpression of CLPTM1L and depletion of NMM-II inducedaneuploidy, indicating that CLPTM1L may interfere with normal NMM-II function in regulating cytokinesis.Immunohistochemical analysis revealed enhanced staining of CLPTM1L in human pancreatic ductal adenocar-cinoma (n¼ 378) as compared with normal pancreatic tissue samples (n¼ 17; P¼ 1.7� 10�4). Our results suggestthat CLPTM1L functions as a growth-promoting gene in the pancreas and that overexpression may lead to anabrogation of normal cytokinesis, indicating that it should be considered as a plausible candidate gene that couldexplain the effect of pancreatic cancer susceptibility alleles on chr5p15.33.Cancer Res; 74(10); 2785–95.�2014 AACR.
IntroductionRisk variants in the TERT-CLPTM1L gene region on chro-
mosome 5p15.33 have been reported in genome-wide associ-ation studies (GWAS) for 10 cancer types, including bladder,
breast, glioma, lung, melanoma, non-melanoma skin cancer,ovarian, pancreas, prostate, and testicular germ cell cancer (1–13). The TERT gene encodes the catalytic subunit of thetelomerase reverse transcriptase complex known for its rolein maintaining telomere ends and the increased telomeraseactivity often seen in human cancers (14). The CLPTM1Lgene encodes the cleft lip and palate-associated transmem-brane 1-like protein (CLPTM1L) and was originally identifiedin a screen for genes conferring resistance to cisplatin inovarian cancer cells (15). When overexpressed in ovariancancer cell lines, CLPTM1L induced apoptosis in cisplatin-sensitive cells, giving rise to its original name: cisplatin resis-tance–related protein (CRR9; ref. 15). CLPTM1L was latershown to protect lung cancer cells from apoptosis after treat-ment with DNA-damaging agents via Bcl-xL (16).
Gain of chromosome 5p is one of the most recurrentchromosomal abnormalities in human cancers (17). Althoughmost commonly seen in thyroid, lung, and cervical cancer, 5pgain is also frequent in other cancers, including gastric, ovar-ian, colorectal, hepatocellular, esophageal, bladder, and pan-creatic adenocarcinoma (17–19). The most common event inearly stages of non–small cell lung cancer is gain at 5p15.33involving both TERT (78%) and CLPTM1L (53%; ref. 20). How-ever, a recent study of cervical cancer noted that CLPTM1L, but
Authors' Affiliations: 1Laboratory of Translational Genomics, Division ofCancer Epidemiology and Genetics; 2Pediatric Oncology Branch; 3Labo-ratory of Pathology; 4Division of Cancer Control and Population Sciences;5Laboratory of Experimental Carcinogenesis, National Cancer Institute,NIH, Department of Health and Human Services, Bethesda; 6Laboratory ofProteomics and Analytical Technologies, Leidos Biomedical Research,Frederick National Laboratory for Cancer Research; 7Laboratory of ProteinDynamics and Signaling and 8Laboratory of Cell & Developmental Signal-ing, NCI-Frederick, Frederick, Maryland; 9Keck School of Medicine, Uni-versity of Southern California, Los Angeles, California; 10University ofHawaii Cancer Center, Honolulu, Hawaii; 11Department of Epidemiology,College of Public Health, University of Iowa, Iowa City, Iowa; and 12Depart-ment of Health Sciences Research, Mayo Clinic, Rochester, Minnesota
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Laufey T. Amundadottir, Laboratory of Transla-tionalGenomics, Division ofCancer Epidemiology andGenetics, NIH, 8717Grovemont Circle, Gaithersburg, MD 20877. Phone: 301-594-8131; Fax:301-402-3134; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-13-3176
�2014 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 2785
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

not TERT, was among themultiple genes on 5p (33%) that wereboth amplified and overexpressed (21, 22).
The most significant GWAS risk variants on 5p15.33 forpancreatic cancer lie in intron 13 of the CLPTM1L gene and arelocated approximately 27 kb from the transcriptional start ofTERT (11). Although this does not exclude TERT as a plausiblecandidate gene explaining this pancreatic cancer risk allele,CLPTM1L should be considered a potential target gene. Thus,to explore a possible function for CLPTM1L in pancreaticcancer, we examined its role in growth control in vitro andin vivo, and searched for interacting proteins that could provideclues to its function.
Materials and MethodsCell lines and antibodies
The human embryonic kidney cell line HEK293T, humanpancreatic cancer cell line PANC-1, and mouse kidney cell lineIMCD3 (all from the American Type Culture Collection) weremaintained in Dulbecco's Modified Eagle Medium (DMEM;Mediatech Inc) supplemented with 10% FBS (Life Technologies).
Commercial antibodies used included those for endogenousCLPTM1L (HPA014791; Sigma), FLAG-tagged CLPTM1L (M2F1804; Sigma), the endoplasmic reticulum (ER) marker Cal-nexin, (C4731; Sigma), a mitochondrial marker (MTC02,ab3298; Abcam), the centrosome marker g-tubulin (T5192;Sigma), the Golgi marker GM130 (G7295; Sigma), a-tubulin(ab7291; Abcam), and MYH9/MYH10 (sc-33729; Santa CruzBiotechnology). Secondary antibodies were Alexa Fluor594 or488 donkey anti-mouse or anti-rabbit IgG (HþL; A21202,A21203, A21206, and A21207; Life Technologies).
Generation of CLPTM1L expression plasmids andcreation of stable cell lines
Full-length human CLPTM1L cDNA (Invitrogen, UltimateORF Clone ID: IOH13343) was cloned into pDest-737 (a Gate-way-adapted version of Sigma's p3xFLAG-CMV10) using theGateway system (Invitrogen). Three constructs were generat-ed from the full-length human CLPTM1L cDNA (RefSeqNM_030782.3) and verifiedbySanger sequencing:WTCLPTM1L(full-length CLPTM1L), CLPTM1L-DLoop [missing amino acids36–280 between transmembrane (TM) domains 1 and 2] andCLPTM1L-DCterm (lacking amino acids 455–538). The design-ed constructs include three tandem FLAG tags at the aminoterminus of CLPTM1L. A fourth construct contained a FLAGtag at the C-terminus of CLPTM1L, to compare the effect ofFLAG tags on the N- or C- termini on growth rates in vitro.
Stable cell lines were generated by transfecting PANC-1 cells(Lipofectamine 2000, 11668-019; Life technologies) with selec-tion in G418 (Mediatech; 30-234-CI;). The constructs above,expressing wild-type (WT) or mutant CLPTM1L from thecytomegalovirus (CMV) promoter, were used to generate thefollowing stable lines: PANC1-vo (empty vector), PANC1-CLPTM1L (full-length CLPTM1L), PANC1-CLPTM1L-DLoop(loop deletion), or PANC1-CLPTM1L-DCterm (C-terminaldeletion). Transient transfections for HEK293T and mIMCD3cells were performed with the same constructs.
Prediction of the topology of WT CLPTM1L wasassessed using: TMHMM v.2.0, TMPred, and TopPred2
(http://www.cbs.dtu.dk/services/TMHMM-2.0/; http://www.ch.embnet.org/software/TMPRED_form.html; http://www.sbc.su.se/~erikw/toppred2/; refs. 23–26).
In vitro and in vivo growth assaysCell proliferation was measured in vitro by seeding PANC-1
stably expressing CLPTM1L (full-length or deletion mutants)at 3 � 103 cells per well in 96-well plates. Time points weretaken every 2 days (days 1, 3, 5, and 7) and cell growth wasassessed using the WST-1 reagent (Roche Applied Science) for30 minutes. The optical density change created by the metab-olizing of the reagent was evaluated in a spectrophotometer(Tecan) at 450 nm. Absorbance at the reference wavelength of600 nm was subtracted from the A450 values.
CLPTM1L knockdownwas performed using the DharmaconDharmaFECT siRNA transfection reagent (Thermo ScientificDharmacon; #T-2001-01) according to the manufacturer'sinstructions. DharmaconON-TARGETplus SMARTpool siRNAspecifically targetingCLPTM1L (L-015661-02-0005) or a controlnontarget siRNA (D-001810-02-05) was purchased fromThermo Scientific Dharmacon. Cell proliferation experimentswere performed 48 hours after transfection with 100 nmol/LsiRNA. The efficiency of CLPTM1L knockdownwas assessed byisolating RNA from PANC-1 cells, using the mirVana RNA kit(ABI). Briefly, 1 mg RNA (RIN scores > 9.0) was reverse tran-scribed using SuperScript III reverse transcriptase (Invitrogen).Quantitative real-time PCR (qRT-PCR) was performed on a7900HT system (ABI) using TaqMan gene expression assays forCLPTM1L (Hs00363947_m1) and B2M (Hs00187842_m1) fromLife Technologies. Each reaction was run in quadruplicate andanalyzed according to the DDCt method using B2M as thehousekeeping gene.
Tumor growth was measured in vivo using a xenograftmouse model. Female nude mice (8- to 10-weeks old) werepurchased from the Animal Production Area, NCI, Frederick,MD, and housed in a pathogen-free environment. Briefly, 106
PANC-1 cells, stably transfected with different CLPTM1Lconstructs or the vector control, were injected subcutane-ously into the flank of each mouse. Tumor size was measuredby a caliper three times a week for up to 77 days using theformula of length � width � width/2 to estimate tumorvolumes in mm3, or when protocol experimental end pointswere reached (tumor diameter reached 2 cm). For eachgroup, 5 mice were injected per stable cell line per experi-ment. After seeing similar results for two independent con-structs expressing WT CLPTM1L with FLAG tags at eitherend, the CLPTM1L constructs tagged on the N-terminus werechosen for further work. Final results were pooled from threeindependent experiments: two that were performed withPANC1-vo (empty vector) and PANC1-CLPTM1L cells, anda third experiment that used PANC1-vo, PANC1-CLPTM1L,PANC1-CLPTM1L-DLoop, and PANC1-CLPTM1L-DCtermcells. The difference in growth rates was analyzed by com-paring tumor volumes at day 68 using the Mann–Whitney Utest. Animal care and experimental procedures were approv-ed by the NIH Animal Care and User Committees (PB-047 M1to Dr. Javed Khan, Pediatric Oncology Branch, National Can-cer Institute, NIH, Bethesda, MD).
Jia et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2786
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

Affinity purification of protein complexes, trypticdigestion, and fractionationHEK293T cells were grown to approximately 60% con-
fluency and transfected with 3xFLAG-tagged WT CLPTM1L(experimental analysis) or vector only (control) in a complexof polymer PEI (polyethyleneimine; Polysciences Inc) andDNA at a ratio of 5:2. After 48 hours, cells were harvested onice in RIPA buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl,0.1% SDS, 0.5% sodium deoxycholate, and 1.0% NP-40) with1� protease inhibitor cocktail (Roche). Lysates were incu-bated with 25 mL of anti-FLAG M2 agarose (Sigma) for 2hours followed by washing and elution via a 3x-FLAGpeptide as previously described (Das PMID: 20968308). Theeluates were subjected to overnight digestion with trypsin (1mg) at 37�C followed by lyophilization, reconstitution, andfractionation using strong cation exchange liquid chroma-tography (LC) and mass spectrometry analysis as previouslydescribed (27). Proteomics data analysis was performed aspreviously described (27).
Validation of CLPTM1L-MYH9 interaction by co-immunoprecipitationHEK293T cells were transfected with 3xFLAG-tagged WT
CLPTM1L expression plasmids or the vector control only, andcollected 48 hours after transfection. PANC-1 cells stablyexpressing WT CLPTM1L, CLPTM1L-DLoop, or CLPTM1L-DCterm were cultured to a confluence of approximately80%. Cells were washed with ice-cold PBS and harvested inlysis buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 0.5mmol/L EDTA, 1.0% NP-40, with 1� protease inhibitor cock-tail). Immunoprecipitation of FLAG-tagged CLPTM1L wasperformed by incubating the supernatant with anti-FLAGM2 agarose for 2 hours at 4�C. Affinity complexes were washedand interacting complexes released from FLAG beads byboiling in 2� LDS sample buffer (Invitrogen) and resolved ona 3% to 8% Tris Acetate gel (Invitrogen). Samples were sub-jected to Western blot analysis using antibodies to FLAG M2,Myosin 9/10 and b-actin followed by appropriate secondarymouse or rabbit antibodies (Thermo Scientific) before detec-tion of the signal with ECL (Thermo Scientific).
Cell cycle and DNA content analysisThe effect of CLPTM1L and MYH9 on cell-cycle progression
was studied by flow cytometric analysis of DNA content andBrdUrd incorporation using the FITC BrdU Flow Kit (557891;BD Pharmingen BioSciences) according to the manufacturer'srecommendation. Briefly, logarithmically growing cells (tran-siently transfected HEK293T or stably transfected PANC-1cells) were labeled with 10 mmol/L BrdUrd for 30 minutes at37�C, fixed, permeabilized, and treated with DNase. A fluores-cein isothiocyanate (FITC)–conjugated anti-BrdUrd antibodywas used for staining DNA with incorporated BrdUrd, and 7-amino-actinomycin D (7-AAD) for total DNA staining. Fluo-rescence of 30,000 cells was acquired on a FACSCalibur instru-ment (Becton Dickinson). The resulting DNA histograms werequantified by using the CellQuest Pro software (Becton Dick-inson) and the percentage of G0–G1, S, G2–M and aneuploidy(>4N) cells determined.
For knockdown experiments, cells were plated in six-wellplates 24 hours before transfectionwith siRNA, so they reached80% confluency at the time of transfection. The DharmaconDharmaFECT siRNA transfection reagent (Thermo ScientificDharmacon; #T-2001-01) was used for transfection accordingto the manufacturer's instructions. Dharmacon ON-TARGETplus SMARTpool siRNA specifically targetingMYH9 (L-007668-00-0005) or a control nontarget siRNA (D-001810-02-05) waspurchased from Thermo Scientific Dharmacon. Twenty-fourhours after transfection with 100 nmol/L siRNA, the mediawere replaced with fresh complete media. Assays were per-formed 48 hours after transfection.
Efficiency of MYH9 knockdown was assessed by isolatingRNA from PANC-1 cells, using the mirVana RNA kit (ABI).Briefly, 1 mg RNA (RIN scores > 9.0) from cell lines was reversetranscribed using SuperScript III reverse transcriptase (Invitro-gen). qRT-PCR was performed on a 7900HT system (ABI) usingTaqMan gene expression assays for MYH9 (Hs00159522_ml)and B2M (Hs00187842_m1) from Life Technologies. Each reac-tion was run in quadruplicate and analyzed according to theDDCt method using B2M as the housekeeping gene.
Immunofluorescence and in situ proximity ligationassay
To visualize the subcellular localization of the endogenousor FLAG-tagged CLPTM1L proteins, cells were grown on glasscoverslips, fixed with 4% paraformaldehyde or 100% methanoland stained with antibodies against endogenous CLPTM1L(1:500), FLAG-tagged CLPTM1L (1:500), calnexin (1:500), mito-chondrial marker (1:500), GM130 (1:500), a-tubulin (1:1,000),and g-tubulin (1:1,000) followed by visualization by confocalmicroscopy (Zeiss LSM 510 Meta). Assessment of colocaliza-tion with MYH9 was performed in the same manner usingantibodies against MYH9 (1:500). Secondary antibodies usedare Alexa Fluor594 or 488 donkey anti-mouse or anti-rabbitIgG (HþL; Invitrogen). Cells were counterstained with 406-diamidino-2-phenylindole (DAPI).
The Duolink in situ Proximity Ligation Assay (DPLA; in situred starter kit; #92101 from Olink Bioscience) was used accord-ing to the manufacturer's protocol to evaluate interactionsbetween CLPTM1L and other proteins. Briefly, cells were grownon coverslips and fixed with 4% paraformaldehyde or 100%methanol for 10 minutes at room temperature and air-dried,followed by blocking and labeling with antibodies to endoge-nous CLPTM1L (Sigma; HPA014791) or FLAG-tagged CLPTM1L(Sigma; F1804), MYH9/10 (Santa Cruz Biotechnology; SC-33729), at a dilution of 1:500 in a preheated humidity chamberat 37�C for 30 minutes. After washing, slides were incubatedwith anti-rabbit and anti-mouse secondary antibodies withattached DPLA probes (dilution 1:5) for 30 minutes in a pre-heated humidity chamber at 37�C. Finally, cells were counter-stained by DAPI and signals were observed by confocal micros-copy (Zeiss LSM 510 Meta); a red fluorescence signal indicatesthat two proteins are separated by less than 40 nm (28).
Cells were treated with 50 mmol/L of the DNA-damagingagent cisplatin (ALX-400-040-M250, Enzo Life Sciences) for 0,24, or 48 hours for immunofluorescence and Duolink experi-ments as indicated.
CLPTM1L in Pancreatic Cancer
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2787
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

ImmunohistochemistryTissue microarrays (TMA) with normal- and tumor-derived
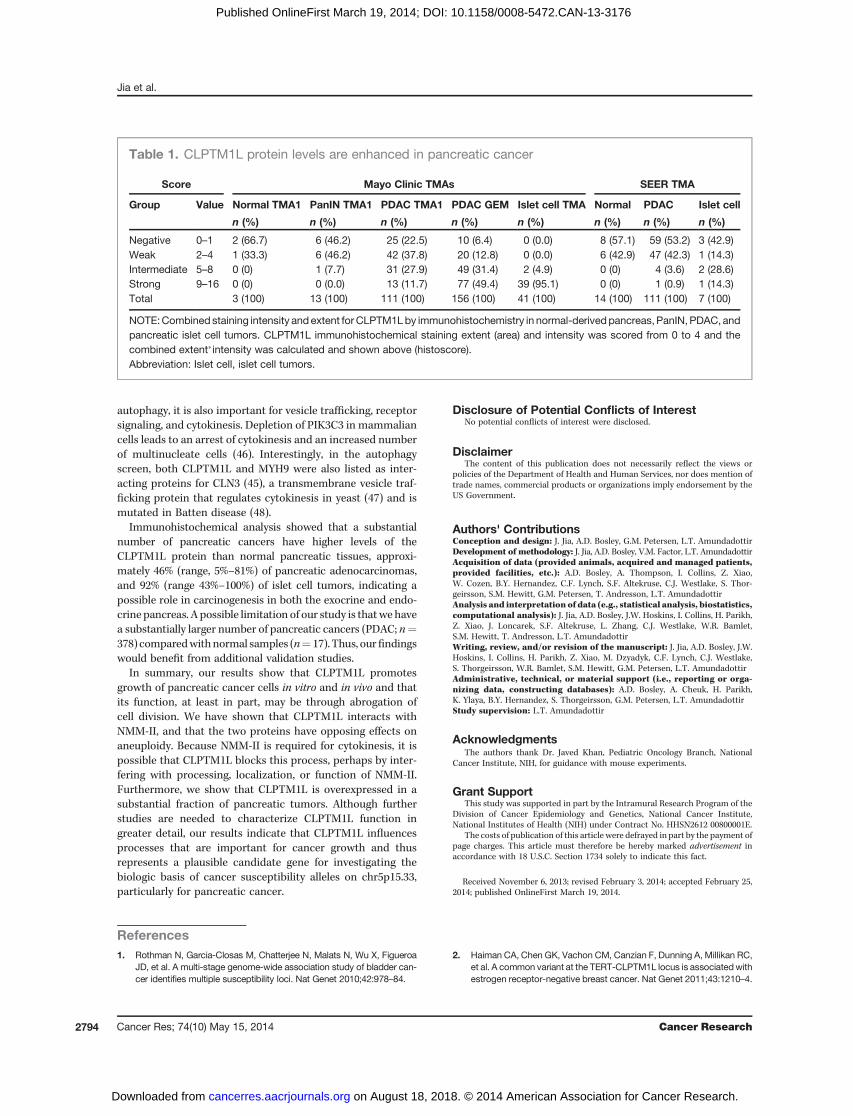
pancreatic tissue samples were obtained from the Mayo Clinicand from the Surveillance, Epidemiology, and End Results(SEER) registries (29, 30). The TMAs from the Mayo Clinic[four TMAs were used: PDAC (pancreatic ductal adenocarci-noma) TMA1 (n ¼ 111), PDAC GEM (gemcitibine; n ¼ 156),PanIN (pancreatic intraepithelial neoplasia) TMA (n¼ 13), andpancreatic islet cell tumors (Islet cell TMAs; n¼ 41) along withthree cores of tissues from normal pancreas that could beevaluated for CLPTM1L expression. The SEER TMA contained14 cores from normal pancreas (adjacent to tumor) in additionto PDAC (n ¼ 111) and islet cell tumors (n ¼ 7).
Immunohistochemistry for CLPTM1Lwas performedwith arabbit polyclonal antibody (HPA014791; Sigma) at 1:1,600dilution. Briefly, slides were deparaffinized in xylene andgraded alcohols, and subject to antigen retrieval in a pressurecooker with citrate buffer (pH 6.0) for 20 minutes. Endogenousenzyme activity was blocked with 3% hydrogen peroxide inmethanol with an additional block in 2% nonfat milk used toreduce nonspecific reactions. Subsequently, slides were incu-bated with primary antibody for 60 minutes at room temper-ature, detected antigen–antibody reaction with DakoEnVisionþ Dual Link system-HRP (Dako), visualized in 3,30-diaminobenzidine (DAB), counterstained with hematoxylin,dehydrated, and mounted for visualization.
CLPTM1L staining intensity was scored on a scale from 0–4as follows: 0, negative; 1, background; 2, weak; 3, positive; and 4,strong. Staining extent of cells was also scored on a scale of 0–4:0, negative; 1, <10%; 2, 10% to 25%; 3, 25% to 75%; and 4, >75%.Combined scores (histoscore) were calculated by multiplyingscores for intensity and extent (individual range, 0–4; com-bined range, 0–16). For statistical analysis, CLPTM1L stainingwas classified as negative/weak (combined scores, 0–4; inter-mediate/strong, 6–16). A 2 � 2 x2 test was used to assess thedifference in staining between normal- and tumor-derivedsamples (df¼ 1). A Pearson uncorrected P value was reported.
ResultsSubcellular localization of CLPTM1L
The protein product of the CLPTM1L gene is predicted tocontain six transmembrane domains and two large hydrophilicdomains: a loop between the first and second transmembranedomains, and a C-terminal tail. It is highly conserved inprimates and relatively well conserved in flies and nematodes(31). We assessed the subcellular localization of endogenousCLPTM1L in pancreatic cancer cell lines by immunofluores-cence analysis and demonstrated perinuclear cytoplasmicstaining and a punctate pattern of CLPTM1L over the nucleus,indicating possible nuclear or nuclearmembrane staining (Fig.1A). Colocalization with markers for cytoplasmic organellesshowed that CLPTM1L localized mainly to the ER (Fig. 1B) inaccord with its six predicted transmembrane domains. Nostaining was detected inmitochondria (Fig. 1C) despite carefulassessment of colocalization with a mitochondrial marker in200-nm optical sections in XY, XZ, and YZ projections, inapparent contradiction to recent work showing mitochondriallocalization in 95-D lung cancer cells (32). No staining was
detected in the Golgi apparatus (Fig. 1D). To confirm that theER localization of overexpressed WT FLAG-tagged CLPTM1Lobserved was not due to defects in polarized sorting, wetransiently expressed CLPTM1L in mouse kidney IMCD3 epi-thelial cells and observed colocalization with the ER markercalnexin (Supplementary Fig. S1). Two of the three programsused to predict the topology of CLPTM1L indicated that theN- and C-termini of the protein protrude into the lumen of theER and other organelles it resides in (TMPred and TMHMM),but one (TopPred) predicted an opposite topology with bothends on the cytoplasmic face of the ER (23). The hydrophilicloop structure between TM1 and TM2 is expected to be onthe opposite side of the membrane as compared with theN- and C-termini.
CLPTM1L overexpression enhances cell proliferationin vitro and in vivo
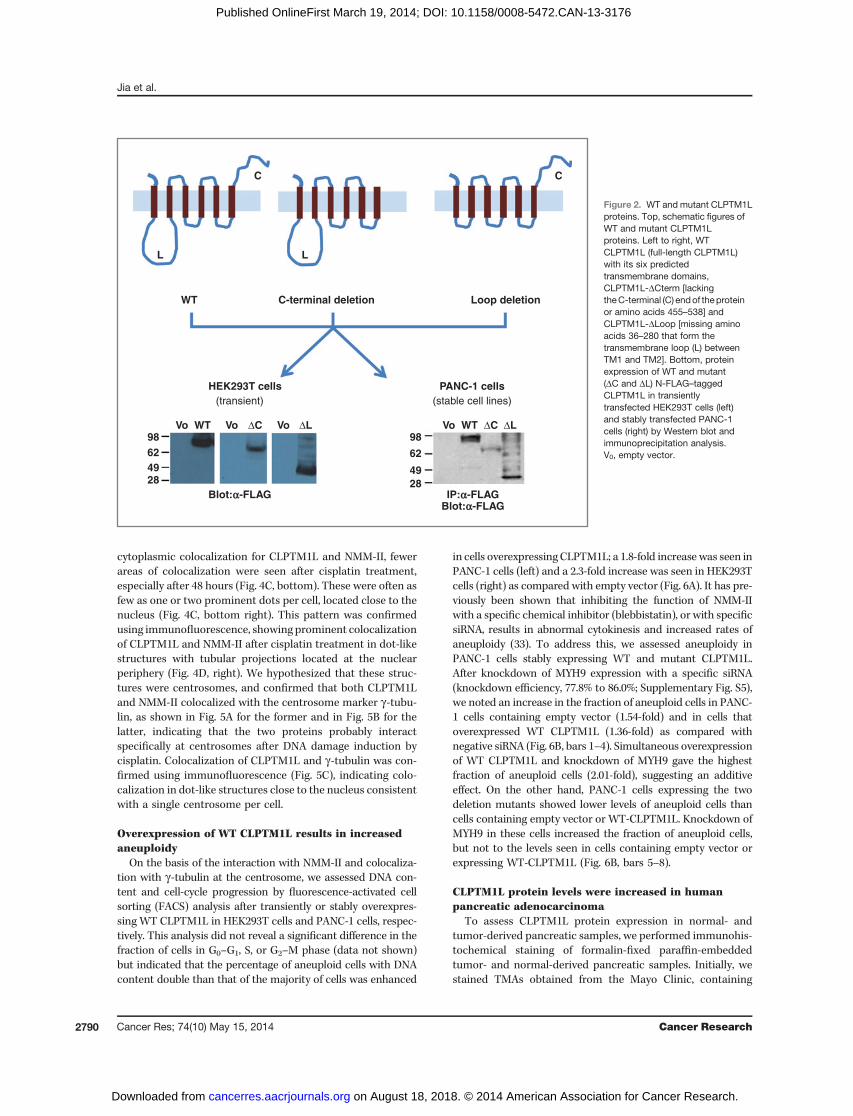
To investigate whether CLPTM1L influenced growth, weoverexpressed full-length or mutant CLPTM1L in HEK293T(transient transfection) and PANC-1 (stable transfection) cells.These included four FLAG epitope–tagged cDNA expressionconstructs: WT full-length CLPTM1L (tagged on either C- or N-termini), and two deletion mutants tagged at the N-termini:CLPTM1L-DCterm (hydrophilic C-terminal domain after thelast transmembrane domain deleted) and CLPTM1L-DLoop(hydrophilic loop between the first and second transmembranedomains deleted). Figure 2 shows a schematic figure of theresulting proteins (top) as well as protein expression of the threedifferent forms of CLPTM1L (WT and twomutant) in transientlytransfected HEK293T cells (by Western blotting, bottom left)and in PANC-1 cells stably expressing the proteins (by FLAGimmunoprecipitation and Western blotting, bottom right).
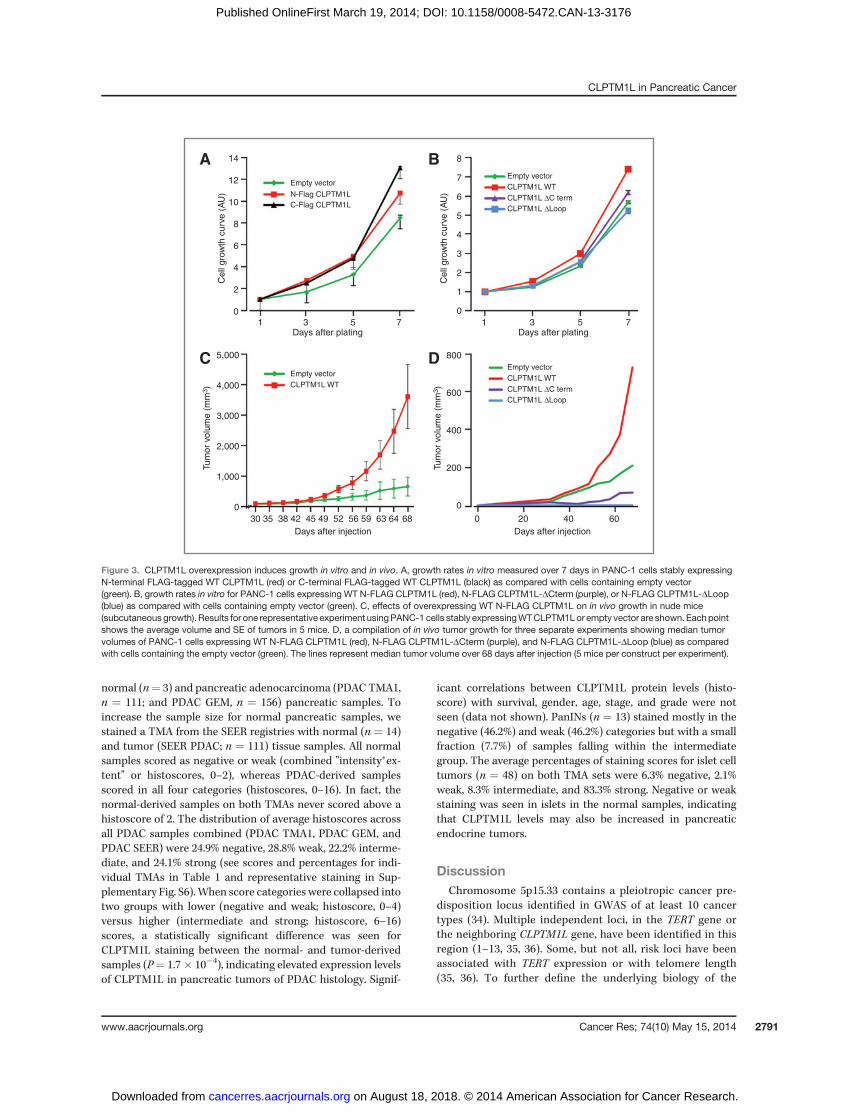
We assessed cell proliferation in vitro and observed that cellsoverexpressing full-length WT CLPTM1L grew faster than cellscontaining empty vector (Fig. 3A). Two sets of PANC-1 cellsstably transfected with CLPTM1L (with N- or C-terminal FLAGepitope tags) showed similar results (1.3–1.5–fold increase atday 7; P ¼ 0.0019 and P ¼ 0.0027, respectively), indicating thatthe FLAG tag does not influence the growth-promoting func-tion of the protein. This effect was abolished by the twomutants, CLPTM1L-DLoop and CLPTM1L-DCterm (Fig. 3B),and inhibited by siRNA targeting CLPTM1L (SupplementaryFig. S2). These results prompted an investigation of whether thesame growth effect was mediated by CLPTM1L in vivo. PANC-1cells stably overexpressing WT CLPTM1L generated largertumors in nude mice in vivo than those containing emptyvector (average tumor volume is shown for one representativeexperiment in Fig. 3C; median tumor volume is shown for allthree experiments combined in Fig. 3D). A significantly largertumor size was observed in vivo for cells overexpressingCLPTM1L as compared with the empty vector (3.46-fold atday 68; P ¼ 0.039). The two deletion mutants did not inducegrowth as compared with the empty vector, indicating thatthese two domains are critical for the growth-promoting func-tion of the CLPTM1L protein. In fact, PANC-1 cells expressingCLPTM1LDLoop and CLPTM1LDCterm grew slower than con-trols, indicating a possible dominant negative effect (Fig. 3D).This effect was significant for the CLPTM1L construct lacking
Jia et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2788
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

the Loop structure (P¼ 0.012) but not for the construct lackingthe C terminus (P ¼ 0.69). To rule out the possibility that thelack of growth promotion by the mutant CLPTM1L proteins iscaused by abrogated subcellular localization, we evaluated thelocalization of the two mutants in PANC-1 cells. Both mutantslocalized to the ER (Supplementary Fig. S3), indicating thatmislocalization does not explain the reduced biologic effect.However, a defect in folding of the mutants within the mem-branes cannot be ruled out.
WT CLPTM1L interacts with non-muscle myosin II inpancreatic cancer cellsTo investigate further the molecular function of the
CLPTM1L protein, we searched for interacting proteins usingaffinity purification, followed by liquid chromatography andmass spectrometry analysis. Because CLPTM1L is predicted tobe highly hydrophobic, we performed the screen in RIPA bufferto facilitate solubilization of membrane-associated proteins.The most frequently observed peptides (Supplementary TableS1) belonged to Myosin-9 (MYH9) and Myosin-10 (MYH10),two heavy-chain non-musclemyosin type II (NMM-II) proteinsinvolved in maintaining cell shape, migration, secretion, and
cytokinesis. PANC-1 cells express approximately 25-fold higheramounts of MYH9 compared with MYH10 at the RNA level(Supplementary Fig. S4), suggesting that this interaction maybe primarily between CLPTM1L and MYH9. Interestingly, theER marker calnexin was identified as a potential CLPTM1L-interacting protein (Supplementary Table S1), consistent withthe observed ER localization shown above using immunoflu-orescence analysis.
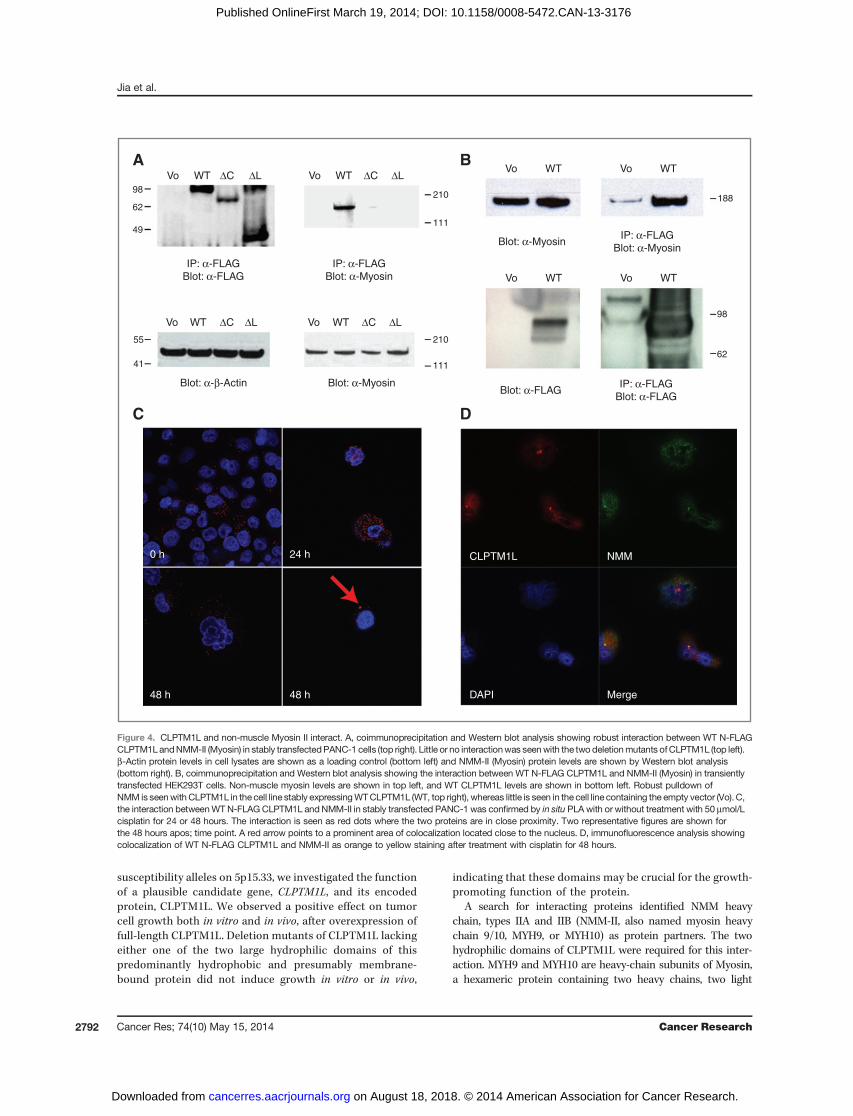
The interaction between CLPTM1L and NMM-II wasconfirmed by coimmunoprecipitation in both PANC-1(Fig. 4A) and 293T cells (Fig. 4B). An interaction between thetwo CLPTM1L deletion mutants and NMM-II was absent orvery low (Fig. 4A), indicating that the loop and C-terminaldomains are required for the interaction between the twoproteins. To assess where in the cells CLPTM1L and NMM-IIinteract, we performed colocalization experiments by DPLA(Fig. 4C) and immunofluorescence (Fig. 4D) and confirmed theinteraction. Because CLPTM1L has been shown to protect cellsfrom cisplatin-induced apoptosis (16, 32), we treated PANC-1stably transfected cells with 50 mmol/L cisplatin for 0, 24, or 48hours and observed a shift in the extent of colocalization after48 hours of cisplatin treatment (Fig. 4C). Instead of widespread
CLPTM1L
CLPTM1L
DAPI Merge DAPI Merge
CLPTM1LMitochondria Golgi
α-Tubulin CLPTM1L ER
DAPI Merge DAPI Merge
A B
C D
Figure 1. Localization ofendogenous CLPTM1L inpancreatic cells and colocalizationof CLPTM1L with cytoplasmicorganelles. A, localization ofendogenous CLPTM1L in PANC-1pancreatic cancer cells.Cytoplasmic and punctate nuclear,or nuclear membrane, staining isseen (red). Counter staining wasperformed for a-tubulin (green). B,colocalization of WT CLPTM1L(red) with the ER marker calnexin(green) in PANC-1 cells stablytransfected with WT CLPTM1L.Colocalization of WT CLPTM1L(red) with a mitochondrial marker(green; C), or a Golgi marker(GM130 in green; D), was not seenin PANC-1 cells stably transfectedwith WT CLPTM1L. Cells werecounterstained with DAPI (blue).
CLPTM1L in Pancreatic Cancer
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2789
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
cytoplasmic colocalization for CLPTM1L and NMM-II, fewerareas of colocalization were seen after cisplatin treatment,especially after 48 hours (Fig. 4C, bottom). These were often asfew as one or two prominent dots per cell, located close to thenucleus (Fig. 4C, bottom right). This pattern was confirmedusing immunofluorescence, showing prominent colocalizationof CLPTM1L and NMM-II after cisplatin treatment in dot-likestructures with tubular projections located at the nuclearperiphery (Fig. 4D, right). We hypothesized that these struc-tures were centrosomes, and confirmed that both CLPTM1Land NMM-II colocalized with the centrosome marker g-tubu-lin, as shown in Fig. 5A for the former and in Fig. 5B for thelatter, indicating that the two proteins probably interactspecifically at centrosomes after DNA damage induction bycisplatin. Colocalization of CLPTM1L and g-tubulin was con-firmed using immunofluorescence (Fig. 5C), indicating colo-calization in dot-like structures close to the nucleus consistentwith a single centrosome per cell.
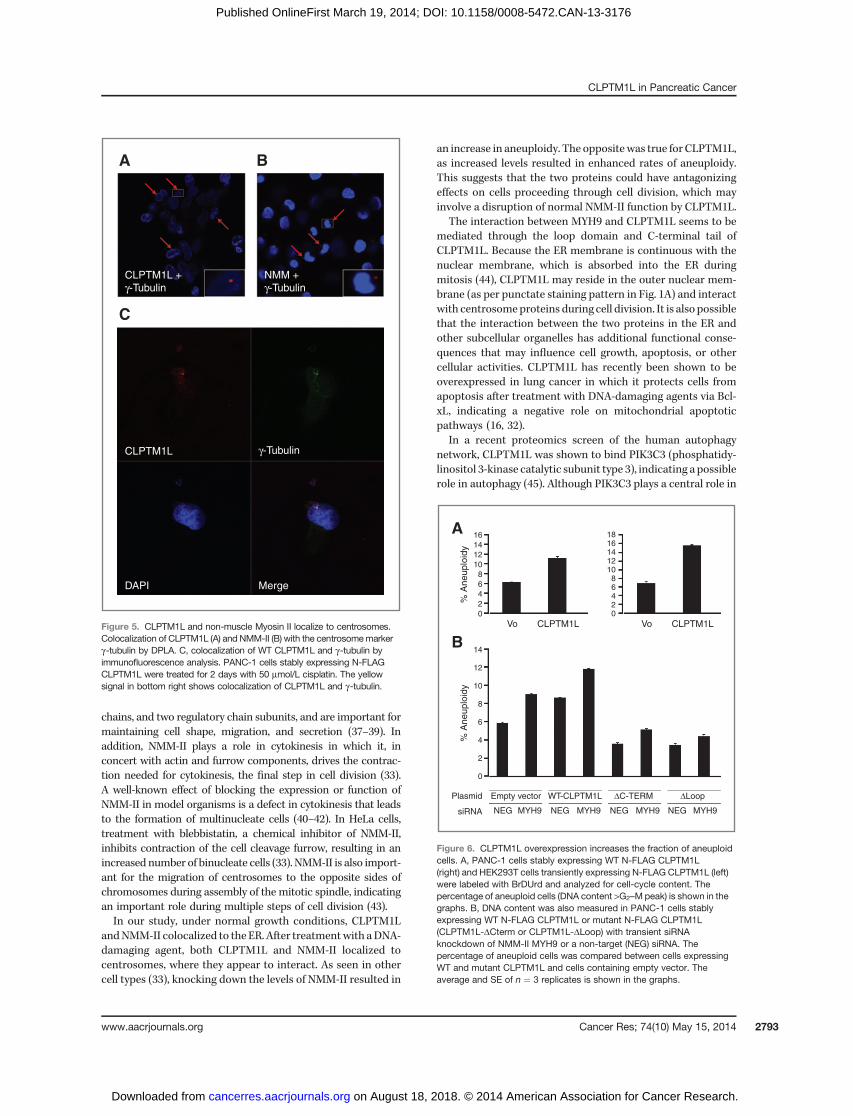
Overexpression of WT CLPTM1L results in increasedaneuploidy
On the basis of the interaction with NMM-II and colocaliza-tion with g-tubulin at the centrosome, we assessed DNA con-tent and cell-cycle progression by fluorescence-activated cellsorting (FACS) analysis after transiently or stably overexpres-sing WT CLPTM1L in HEK293T cells and PANC-1 cells, respec-tively. This analysis did not reveal a significant difference in thefraction of cells in G0–G1, S, or G2–M phase (data not shown)but indicated that the percentage of aneuploid cells with DNAcontent double than that of the majority of cells was enhanced
in cells overexpressing CLPTM1L; a 1.8-fold increase was seen inPANC-1 cells (left) and a 2.3-fold increase was seen in HEK293Tcells (right) as compared with empty vector (Fig. 6A). It has pre-viously been shown that inhibiting the function of NMM-IIwith a specific chemical inhibitor (blebbistatin), or with specificsiRNA, results in abnormal cytokinesis and increased rates ofaneuploidy (33). To address this, we assessed aneuploidy inPANC-1 cells stably expressing WT and mutant CLPTM1L.After knockdown of MYH9 expression with a specific siRNA(knockdown efficiency, 77.8% to 86.0%; Supplementary Fig. S5),we noted an increase in the fraction of aneuploid cells in PANC-1 cells containing empty vector (1.54-fold) and in cells thatoverexpressed WT CLPTM1L (1.36-fold) as compared withnegative siRNA (Fig. 6B, bars 1–4). Simultaneous overexpressionof WT CLPTM1L and knockdown of MYH9 gave the highestfraction of aneuploid cells (2.01-fold), suggesting an additiveeffect. On the other hand, PANC-1 cells expressing the twodeletion mutants showed lower levels of aneuploid cells thancells containing empty vector or WT-CLPTM1L. Knockdown ofMYH9 in these cells increased the fraction of aneuploid cells,but not to the levels seen in cells containing empty vector orexpressing WT-CLPTM1L (Fig. 6B, bars 5–8).
CLPTM1L protein levels were increased in humanpancreatic adenocarcinoma
To assess CLPTM1L protein expression in normal- andtumor-derived pancreatic samples, we performed immunohis-tochemical staining of formalin-fixed paraffin-embeddedtumor- and normal-derived pancreatic samples. Initially, westained TMAs obtained from the Mayo Clinic, containing
C C
L
WT
(transient) (stable cell lines)
C-terminal deletion Loop deletion
L
HEK293T cells
Blot:a-FLAGBlot:a-FLAGIP:a-FLAG
PANC-1 cells
Vo Vo VoΔC ΔCΔL ΔLWT Vo WT98624928
98
62
4928
Figure 2. WT and mutant CLPTM1Lproteins. Top, schematic figures ofWT and mutant CLPTM1Lproteins. Left to right, WTCLPTM1L (full-length CLPTM1L)with its six predictedtransmembrane domains,CLPTM1L-DCterm [lackingtheC-terminal (C) endof theproteinor amino acids 455–538] andCLPTM1L-DLoop [missing aminoacids 36–280 that form thetransmembrane loop (L) betweenTM1 and TM2]. Bottom, proteinexpression of WT and mutant(DC and DL) N-FLAG–taggedCLPTM1L in transientlytransfected HEK293T cells (left)and stably transfected PANC-1cells (right) by Western blot andimmunoprecipitation analysis.V0, empty vector.
Jia et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2790
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
normal (n¼ 3) and pancreatic adenocarcinoma (PDAC TMA1,n ¼ 111; and PDAC GEM, n ¼ 156) pancreatic samples. Toincrease the sample size for normal pancreatic samples, westained a TMA from the SEER registries with normal (n ¼ 14)and tumor (SEER PDAC; n ¼ 111) tissue samples. All normalsamples scored as negative or weak (combined "intensity�ex-tent" or histoscores, 0–2), whereas PDAC-derived samplesscored in all four categories (histoscores, 0–16). In fact, thenormal-derived samples on both TMAs never scored above ahistoscore of 2. The distribution of average histoscores acrossall PDAC samples combined (PDAC TMA1, PDAC GEM, andPDAC SEER) were 24.9% negative, 28.8% weak, 22.2% interme-diate, and 24.1% strong (see scores and percentages for indi-vidual TMAs in Table 1 and representative staining in Sup-plementary Fig. S6).When score categories were collapsed intotwo groups with lower (negative and weak; histoscore, 0–4)versus higher (intermediate and strong; histoscore, 6–16)scores, a statistically significant difference was seen forCLPTM1L staining between the normal- and tumor-derivedsamples (P¼ 1.7� 10�4), indicating elevated expression levelsof CLPTM1L in pancreatic tumors of PDAC histology. Signif-
icant correlations between CLPTM1L protein levels (histo-score) with survival, gender, age, stage, and grade were notseen (data not shown). PanINs (n ¼ 13) stained mostly in thenegative (46.2%) and weak (46.2%) categories but with a smallfraction (7.7%) of samples falling within the intermediategroup. The average percentages of staining scores for islet celltumors (n ¼ 48) on both TMA sets were 6.3% negative, 2.1%weak, 8.3% intermediate, and 83.3% strong. Negative or weakstaining was seen in islets in the normal samples, indicatingthat CLPTM1L levels may also be increased in pancreaticendocrine tumors.
DiscussionChromosome 5p15.33 contains a pleiotropic cancer pre-
disposition locus identified in GWAS of at least 10 cancertypes (34). Multiple independent loci, in the TERT gene orthe neighboring CLPTM1L gene, have been identified in thisregion (1–13, 35, 36). Some, but not all, risk loci have beenassociated with TERT expression or with telomere length(35, 36). To further define the underlying biology of the
14
12
10
8
6
4
2
01 3 5 7
Days after plating
Days after injection Days after injection
1 3 5 7Days after plating
Cell
gro
wth
curv
e (
AU
)
Tu
mor
volu
me (
mm
3)
Tu
mor
volu
me (
mm
3)
Cell
gro
wth
curv
e (
AU
)
8
7
6
5
4
3
2
1
0
800
600
400
200
0
0 20 40 60
5,000
4,000
3,000
2,000
1,000
0
30 35 38 42 45 49 52 56 59 63 64 68
Empty vector
A B
C DEmpty vector
CLPTM1L WT
N-Flag CLPTM1L
C-Flag CLPTM1L
Empty vector
CLPTM1L WT
CLPTM1L ΔC term
CLPTM1L ΔLoop
Empty vector
CLPTM1L WT
CLPTM1L ΔC term
CLPTM1L ΔLoop
Figure 3. CLPTM1L overexpression induces growth in vitro and in vivo. A, growth rates in vitro measured over 7 days in PANC-1 cells stably expressingN-terminal FLAG-tagged WT CLPTM1L (red) or C-terminal FLAG-tagged WT CLPTM1L (black) as compared with cells containing empty vector(green). B, growth rates in vitro for PANC-1 cells expressing WT N-FLAG CLPTM1L (red), N-FLAG CLPTM1L-DCterm (purple), or N-FLAG CLPTM1L-DLoop(blue) as compared with cells containing empty vector (green). C, effects of overexpressing WT N-FLAG CLPTM1L on in vivo growth in nude mice(subcutaneousgrowth). Results for one representative experiment usingPANC-1 cells stably expressingWTCLPTM1Lor empty vector are shown. Eachpointshows the average volume and SE of tumors in 5 mice. D, a compilation of in vivo tumor growth for three separate experiments showing median tumorvolumes of PANC-1 cells expressing WT N-FLAG CLPTM1L (red), N-FLAG CLPTM1L-DCterm (purple), and N-FLAG CLPTM1L-DLoop (blue) as comparedwith cells containing the empty vector (green). The lines represent median tumor volume over 68 days after injection (5 mice per construct per experiment).
CLPTM1L in Pancreatic Cancer
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2791
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
susceptibility alleles on 5p15.33, we investigated the functionof a plausible candidate gene, CLPTM1L, and its encodedprotein, CLPTM1L. We observed a positive effect on tumorcell growth both in vitro and in vivo, after overexpression offull-length CLPTM1L. Deletion mutants of CLPTM1L lackingeither one of the two large hydrophilic domains of thispredominantly hydrophobic and presumably membrane-bound protein did not induce growth in vitro or in vivo,
indicating that these domains may be crucial for the growth-promoting function of the protein.
A search for interacting proteins identified NMM heavychain, types IIA and IIB (NMM-II, also named myosin heavychain 9/10, MYH9, or MYH10) as protein partners. The twohydrophilic domains of CLPTM1L were required for this inter-action. MYH9 and MYH10 are heavy-chain subunits of Myosin,a hexameric protein containing two heavy chains, two light
Vo
IP: α-FLAG
Blot: α-FLAG
IP: α-FLAG
Blot: α-FLAG
Blot: α-β-Actin Blot: α-Myosin
Blot: α-Myosin
Blot: α-FLAG
IP: α-FLAG
Blot: α-Myosin
IP: α-FLAG
Blot: α-Myosin
98
62
49
55
41
WT ΔC ΔL Vo WT
A
C
B
D
Vo WT Vo WT
Vo WT Vo WT
ΔC ΔL
Vo WT
0 h CLPTM1L NMM
DAPI Merge
24 h
48 h 48 h
ΔC ΔL Vo WT ΔC ΔL
210 188
98
62
111
210
111
Figure 4. CLPTM1L and non-muscle Myosin II interact. A, coimmunoprecipitation and Western blot analysis showing robust interaction between WT N-FLAGCLPTM1LandNMM-II (Myosin) in stably transfectedPANC-1 cells (top right). Little or no interactionwas seenwith the two deletionmutants ofCLPTM1L (top left).b-Actin protein levels in cell lysates are shown as a loading control (bottom left) and NMM-II (Myosin) protein levels are shown by Western blot analysis(bottom right). B, coimmunoprecipitation and Western blot analysis showing the interaction between WT N-FLAG CLPTM1L and NMM-II (Myosin) in transientlytransfected HEK293T cells. Non-muscle myosin levels are shown in top left, and WT CLPTM1L levels are shown in bottom left. Robust pulldown ofNMM is seenwith CLPTM1L in the cell line stably expressingWTCLPTM1L (WT, top right), whereas little is seen in the cell line containing the empty vector (Vo). C,the interaction betweenWTN-FLAG CLPTM1L andNMM-II in stably transfected PANC-1 was confirmed by in situ PLA with or without treatment with 50 mmol/Lcisplatin for 24 or 48 hours. The interaction is seen as red dots where the two proteins are in close proximity. Two representative figures are shown forthe 48 hours apos; time point. A red arrow points to a prominent area of colocalization located close to the nucleus. D, immunofluorescence analysis showingcolocalization of WT N-FLAG CLPTM1L and NMM-II as orange to yellow staining after treatment with cisplatin for 48 hours.
Jia et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2792
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
chains, and two regulatory chain subunits, and are important formaintaining cell shape, migration, and secretion (37–39). Inaddition, NMM-II plays a role in cytokinesis in which it, inconcert with actin and furrow components, drives the contrac-tion needed for cytokinesis, the final step in cell division (33).A well-known effect of blocking the expression or function ofNMM-II in model organisms is a defect in cytokinesis that leadsto the formation of multinucleate cells (40–42). In HeLa cells,treatment with blebbistatin, a chemical inhibitor of NMM-II,inhibits contraction of the cell cleavage furrow, resulting in anincreased number of binucleate cells (33). NMM-II is also import-ant for the migration of centrosomes to the opposite sides ofchromosomes during assembly of the mitotic spindle, indicatingan important role during multiple steps of cell division (43).In our study, under normal growth conditions, CLPTM1L
andNMM-II colocalized to the ER. After treatmentwith aDNA-damaging agent, both CLPTM1L and NMM-II localized tocentrosomes, where they appear to interact. As seen in othercell types (33), knocking down the levels of NMM-II resulted in
an increase in aneuploidy. The oppositewas true for CLPTM1L,as increased levels resulted in enhanced rates of aneuploidy.This suggests that the two proteins could have antagonizingeffects on cells proceeding through cell division, which mayinvolve a disruption of normal NMM-II function by CLPTM1L.
The interaction between MYH9 and CLPTM1L seems to bemediated through the loop domain and C-terminal tail ofCLPTM1L. Because the ER membrane is continuous with thenuclear membrane, which is absorbed into the ER duringmitosis (44), CLPTM1L may reside in the outer nuclear mem-brane (as per punctate staining pattern in Fig. 1A) and interactwith centrosomeproteins during cell division. It is also possiblethat the interaction between the two proteins in the ER andother subcellular organelles has additional functional conse-quences that may influence cell growth, apoptosis, or othercellular activities. CLPTM1L has recently been shown to beoverexpressed in lung cancer in which it protects cells fromapoptosis after treatment with DNA-damaging agents via Bcl-xL, indicating a negative role on mitochondrial apoptoticpathways (16, 32).
In a recent proteomics screen of the human autophagynetwork, CLPTM1L was shown to bind PIK3C3 (phosphatidy-linositol 3-kinase catalytic subunit type 3), indicating a possiblerole in autophagy (45). Although PIK3C3 plays a central role in
CLPTM1L +
γ-Tubulin
CLPTM1L
NMM +
γ-Tubulin
γ-Tubulin
DAPI Merge
A
C
B
Figure 5. CLPTM1L and non-muscle Myosin II localize to centrosomes.Colocalization of CLPTM1L (A) and NMM-II (B) with the centrosomemarkerg-tubulin by DPLA. C, colocalization of WT CLPTM1L and g-tubulin byimmunofluorescence analysis. PANC-1 cells stably expressing N-FLAGCLPTM1L were treated for 2 days with 50 mmol/L cisplatin. The yellowsignal in bottom right shows colocalization of CLPTM1L and g-tubulin.
16
14
12
10
8
6
4
2
0
14
12
10
8
6
4
2
0
181614121086420
Vo
A
B
% A
neuplo
idy
% A
neuplo
idy
CLPTM1L
WT-CLPTM1LEmpty vectorPlasmid
siRNA NEG MYH9 NEG MYH9 NEG MYH9 NEG MYH9
ΔC-TERM ΔLoop
Vo CLPTM1L
Figure 6. CLPTM1L overexpression increases the fraction of aneuploidcells. A, PANC-1 cells stably expressing WT N-FLAG CLPTM1L(right) and HEK293T cells transiently expressing N-FLAG CLPTM1L (left)were labeled with BrDUrd and analyzed for cell-cycle content. Thepercentage of aneuploid cells (DNA content >G2–Mpeak) is shown in thegraphs. B, DNA content was also measured in PANC-1 cells stablyexpressing WT N-FLAG CLPTM1L or mutant N-FLAG CLPTM1L(CLPTM1L-DCterm or CLPTM1L-DLoop) with transient siRNAknockdown of NMM-II MYH9 or a non-target (NEG) siRNA. Thepercentage of aneuploid cells was compared between cells expressingWT and mutant CLPTM1L and cells containing empty vector. Theaverage and SE of n ¼ 3 replicates is shown in the graphs.
CLPTM1L in Pancreatic Cancer
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2793
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176
autophagy, it is also important for vesicle trafficking, receptorsignaling, and cytokinesis. Depletion of PIK3C3 in mammaliancells leads to an arrest of cytokinesis and an increased numberof multinucleate cells (46). Interestingly, in the autophagyscreen, both CLPTM1L and MYH9 were also listed as inter-acting proteins for CLN3 (45), a transmembrane vesicle traf-ficking protein that regulates cytokinesis in yeast (47) and ismutated in Batten disease (48).
Immunohistochemical analysis showed that a substantialnumber of pancreatic cancers have higher levels of theCLPTM1L protein than normal pancreatic tissues, approxi-mately 46% (range, 5%–81%) of pancreatic adenocarcinomas,and 92% (range 43%–100%) of islet cell tumors, indicating apossible role in carcinogenesis in both the exocrine and endo-crine pancreas. A possible limitation of our study is thatwe havea substantially larger number of pancreatic cancers (PDAC; n¼378) comparedwithnormal samples (n¼ 17). Thus, ourfindingswould benefit from additional validation studies.
In summary, our results show that CLPTM1L promotesgrowth of pancreatic cancer cells in vitro and in vivo and thatits function, at least in part, may be through abrogation ofcell division. We have shown that CLPTM1L interacts withNMM-II, and that the two proteins have opposing effects onaneuploidy. Because NMM-II is required for cytokinesis, it ispossible that CLPTM1L blocks this process, perhaps by inter-fering with processing, localization, or function of NMM-II.Furthermore, we show that CLPTM1L is overexpressed in asubstantial fraction of pancreatic tumors. Although furtherstudies are needed to characterize CLPTM1L function ingreater detail, our results indicate that CLPTM1L influencesprocesses that are important for cancer growth and thusrepresents a plausible candidate gene for investigating thebiologic basis of cancer susceptibility alleles on chr5p15.33,particularly for pancreatic cancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
DisclaimerThe content of this publication does not necessarily reflect the views or
policies of the Department of Health and Human Services, nor does mention oftrade names, commercial products or organizations imply endorsement by theUS Government.
Authors' ContributionsConception and design: J. Jia, A.D. Bosley, G.M. Petersen, L.T. AmundadottirDevelopment of methodology: J. Jia, A.D. Bosley, V.M. Factor, L.T. AmundadottirAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): A.D. Bosley, A. Thompson, I. Collins, Z. Xiao,W. Cozen, B.Y. Hernandez, C.F. Lynch, S.F. Altekruse, C.J. Westlake, S. Thor-geirsson, S.M. Hewitt, G.M. Petersen, T. Andresson, L.T. AmundadottirAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): J. Jia, A.D. Bosley, J.W. Hoskins, I. Collins, H. Parikh,Z. Xiao, J. Loncarek, S.F. Altekruse, L. Zhang, C.J. Westlake, W.R. Bamlet,S.M. Hewitt, T. Andresson, L.T. AmundadottirWriting, review, and/or revision of the manuscript: J. Jia, A.D. Bosley, J.W.Hoskins, I. Collins, H. Parikh, Z. Xiao, M. Dzyadyk, C.F. Lynch, C.J. Westlake,S. Thorgeirsson, W.R. Bamlet, S.M. Hewitt, G.M. Petersen, L.T. AmundadottirAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): A.D. Bosley, A. Cheuk, H. Parikh,K. Ylaya, B.Y. Hernandez, S. Thorgeirsson, G.M. Petersen, L.T. AmundadottirStudy supervision: L.T. Amundadottir
AcknowledgmentsThe authors thank Dr. Javed Khan, Pediatric Oncology Branch, National
Cancer Institute, NIH, for guidance with mouse experiments.
Grant SupportThis study was supported in part by the Intramural Research Program of the
Division of Cancer Epidemiology and Genetics, National Cancer Institute,National Institutes of Health (NIH) under Contract No. HHSN2612 00800001E.
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received November 6, 2013; revised February 3, 2014; accepted February 25,2014; published OnlineFirst March 19, 2014.
References1. Rothman N, Garcia-Closas M, Chatterjee N, Malats N, Wu X, Figueroa
JD, et al. A multi-stage genome-wide association study of bladder can-cer identifies multiple susceptibility loci. Nat Genet 2010;42:978–84.
2. Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC,et al. A common variant at the TERT-CLPTM1L locus is associatedwithestrogen receptor-negative breast cancer. Nat Genet 2011;43:1210–4.
Table 1. CLPTM1L protein levels are enhanced in pancreatic cancer
Score Mayo Clinic TMAs SEER TMA
Group Value Normal TMA1 PanIN TMA1 PDAC TMA1 PDAC GEM Islet cell TMA Normal PDAC Islet cell
n (%) n (%) n (%) n (%) n (%) n (%) n (%) n (%)
Negative 0–1 2 (66.7) 6 (46.2) 25 (22.5) 10 (6.4) 0 (0.0) 8 (57.1) 59 (53.2) 3 (42.9)Weak 2–4 1 (33.3) 6 (46.2) 42 (37.8) 20 (12.8) 0 (0.0) 6 (42.9) 47 (42.3) 1 (14.3)Intermediate 5–8 0 (0) 1 (7.7) 31 (27.9) 49 (31.4) 2 (4.9) 0 (0) 4 (3.6) 2 (28.6)Strong 9–16 0 (0) 0 (0.0) 13 (11.7) 77 (49.4) 39 (95.1) 0 (0) 1 (0.9) 1 (14.3)Total 3 (100) 13 (100) 111 (100) 156 (100) 41 (100) 14 (100) 111 (100) 7 (100)
NOTE:Combined staining intensity and extent for CLPTM1Lby immunohistochemistry in normal-derivedpancreas, PanIN, PDAC, andpancreatic islet cell tumors. CLPTM1L immunohistochemical staining extent (area) and intensity was scored from 0 to 4 and thecombined extent�intensity was calculated and shown above (histoscore).Abbreviation: Islet cell, islet cell tumors.
Jia et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2794
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

3. SheteS,HoskingFJ,Robertson LB,DobbinsSE, SansonM,MalmerB,et al. Genome-wide association study identifies five susceptibility locifor glioma. Nat Genet 2009;41:899–904.
4. Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A,et al. Common 5p15.33 and 6p21.33 variants influence lung cancerrisk. Nat Genet 2008;40:1407–9.
5. McKay JD, HungRJ, Gaborieau V, Boffetta P, Chabrier A, ByrnesG, et al.Lung cancer susceptibility locus at 5p15.33. Nat Genet 2008;40:1404–6.
6. Broderick P, Wang Y, Vijayakrishnan J, Matakidou A, Spitz MR, Eisen T,et al. Deciphering the impact of commongenetic variation on lung cancerrisk: a genome-wide association study. Cancer Res 2009;69:6633–41.
7. Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M,et al. A genome-wide association study of lung cancer identifies aregion of chromosome 5p15 associated with risk for adenocarcinoma.Am J Hum Genet 2009;85:679–91.
8. Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, SigurdssonA, et al. Sequence variants at the TERT-CLPTM1L locus associatewithmany cancer types. Nat Genet 2009;41:221–7.
9. Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G,Jakobsdottir M, et al. New common variants affecting susceptibility tobasal cell carcinoma. Nat Genet 2009;41:909–14.
10. Yang X, Yang B, Li B, Liu Y. Association between TERT-CLPTM1Lrs401681[C] allele and NMSC cancer risk: a meta-analysis including45,184 subjects. Arch Dermatol Res 2013;305:49–52.
11. Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Sol-omon RZ, Jacobs KB, et al. A genome-wide association study iden-tifies pancreatic cancer susceptibility loci on chromosomes 13q22.1,1q32.1 and 5p15.33. Nat Genet 2010;42:224–8.
12. Kote-Jarai Z, OlamaAA,GilesGG, Severi G, Schleutker J,WeischerM,et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet 2011;43:785–91.
13. Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D, et al.Variants near DMRT1, TERT and ATF7IP are associated with testiculargerm cell cancer. Nat Genet 2010;42:604–7.
14. KimNW, PiatyszekMA, Prowse KR, Harley CB,West MD, Ho PL, et al.Specific association of human telomerase activity with immortal cellsand cancer. Science 1994;266:2011–5.
15. YamamotoK,OkamotoA, Isonishi S,Ochiai K,OhtakeY. Anovel gene,CRR9, which was up-regulated in CDDP-resistant ovarian tumor cellline, was associated with apoptosis. Biochem Biophys Res Commun2001;280:1148–54.
16. James MA, Wen W, Wang Y, Byers LA, Heymach JV, Coombes KR,et al. Functional characterization of CLPTM1L as a lung cancer riskcandidate gene in the 5p15.33 locus. PLoS ONE 2012;7:e36116.
17. BaudisM.Genomic imbalances in 5918malignant epithelial tumors: anexplorative meta-analysis of chromosomal CGH data. BMC Cancer2007;7:226.
18. Hoglund M, Gisselsson D, Hansen GB, Mitelman F. Statistical dissec-tion of cytogenetic patterns in lung cancer reveals multiple modes ofkaryotypic evolution independent of histological classification. CancerGenet Cytogenet 2004;154:99–109.
19. Baudis M, Cleary ML. Progenetix.net: an online repository for molec-ular cytogenetic aberration data. Bioinformatics 2001;17:1228–9.
20. Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Gain at chromosomalregion 5p15.33, containing TERT, is themost frequent genetic event inearly stages of non-small cell lung cancer. Cancer Genet Cytogenet2008;182:1–11.
21. Vazquez-Mena O, Medina-Martinez I, Juarez-Torres E, Barron V,Espinosa A, Villegas-Sepulveda N, et al. Amplified genes may beoverexpressed, unchanged, or downregulated in cervical cancer celllines. PLoS ONE 2012;7:e32667.
22. LandoM, Holden M, Bergersen LC, Svendsrud DH, Stokke T, SundforK, et al. Gene dosage, expression, and ontology analysis identifiesdriver genes in the carcinogenesis and chemoradioresistance of cer-vical cancer. PLoS Genet 2009;5:e1000719.
23. PuntaM, Forrest LR, BigelowH, Kernytsky A, Liu J, Rost B. Membraneprotein prediction methods. Methods 2007;41:460–74.
24. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predictingtransmembrane protein topology with a hidden Markov model: appli-cation to complete genomes. J Mol Biol 2001;305:567–80.
25. Hofmann K, Stoffel W. TMbase - A database of membrane spanningproteins segments. Biol. Chem. Hoppe-Seyler 1993;374:166.
26. von Heijne G. Membrane protein structure prediction. Hydrophobicityanalysis and the positive-inside rule. J Mol Biol 1992;225:487–494.
27. DasS,Bosley AD,YeX,ChanKC,Chu I,Green JE, et al. Comparison ofstrong cation exchange and SDS-PAGE fractionation for analysis ofmultiprotein complexes. J Proteome Res 2010;9:6696–704.
28. FredrikssonS,GullbergM, Jarvius J,OlssonC,PietrasK,GustafsdottirSM, et al. Protein detection using proximity-dependent DNA ligationassays. Nat Biotechnol 2002;20:473–7.
29. Takikita M, Altekruse S, Lynch CF, Goodman MT, Hernandez BY,Green M, et al. Associations between selected biomarkers and prog-nosis in a population-based pancreatic cancer tissue microarray.Cancer Res 2009;69:2950–5.
30. Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC,Bamlet WR, et al. SOX2 promotes dedifferentiation and imparts stemcell-like features to pancreatic cancer cells. Oncogenesis 2013;2:e61.
31. NCBI Resource Coordinators. Database resources of the National Cen-ter for Biotechnology Information. Nucleic Acids Res 2014;42:D7–17.
32. Ni Z, Tao K, Chen G, Chen Q, Tang J, Luo X, et al. CLPTM1L isoverexpressed in lung cancer and associated with apoptosis. PLoSONE 2012;7:e52598.
33. Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR,et al. Dissecting temporal and spatial control of cytokinesis with amyosin II Inhibitor. Science 2003;299:1743–7.
34. Mocellin S, Verdi D, Pooley KA, Landi MT, Egan KM, Baird DM, et al.Telomerase reverse transcriptase locus polymorphisms and cancerrisk: a field synopsis and meta-analysis. J Natl Cancer Inst2012;104:840–54.
35. Kote-Jarai Z, Saunders EJ, Leongamornlert DA, Tymrakiewicz M,Dadaev T, Jugurnauth-Little S, et al. Fine-mapping identifies multipleprostate cancer risk loci at 5p15, one of which associates with TERTexpression. Hum Mol Genet 2013;22:2520–8.
36. Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, TyrerJP, et al. Multiple independent variants at the TERT locus are asso-ciated with telomere length and risks of breast and ovarian cancer. NatGenet 2013;45:371–84.
37. Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-musclemyosin II takes centre stage in cell adhesion and migration. Nat RevMol Cell Biol 2009;10:778–90.
38. Bond LM, Brandstaetter H, Sellers JR, Kendrick-Jones J, Buss F.Myosin motor proteins are involved in the final stages of the secretorypathways. Biochem Soc Trans 2011;39:1115–9.
39. WangA,Ma X, Conti MA, Adelstein RS. Distinct and redundant roles ofthe non-muscle myosin II isoforms and functional domains. BiochemSoc Trans 2011;39:1131–5.
40. Mabuchi I, Okuno M. The effect of myosin antibody on the division ofstarfish blastomeres. J Cell Biol 1977;74:251–63.
41. De Lozanne A, Spudich JA. Disruption of the Dictyostelium myosinheavy chain gene by homologous recombination. Science 1987;236:1086–91.
42. Urven LE, Yabe T, Pelegri F. A role for non-muscle myosin II function infurrow maturation in the early zebrafish embryo. J Cell Sci 2006;119:4342–52.
43. Rosenblatt J, Cramer LP, Baum B, McGee KM. Myosin II-dependentcortical movement is required for centrosome separation and posi-tioning during mitotic spindle assembly. Cell 2004;117:361–72.
44. English AR, Voeltz GK. Endoplasmic reticulum structure and inter-connections with other organelles. Cold Spring Harb Perspect Biol2013;5:a013227.
45. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization ofthe human autophagy system. Nature 2010;466:68–76.
46. Sagona AP, Nezis IP, Pedersen NM, Liestol K, Poulton J, Rusten TE,et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruit-ment of FYVE-CENT to the midbody. Nat Cell Biol 2010;12:362–71.
47. Codlin S, Haines RL, Burden JJ, Mole SE. Btn1 affects cytokinesis andcell-wall deposition by independent mechanisms, one of which islinked to dysregulation of vacuole pH. J Cell Sci 2008;121:2860–70.
48. Consortium TIBD. Isolation of a novel gene underlying Batten disease,CLN3. Cell 1995;82:949–57.
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2795
CLPTM1L in Pancreatic Cancer
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176

2014;74:2785-2795. Published OnlineFirst March 19, 2014.Cancer Res Jinping Jia, Allen D. Bosley, Abbey Thompson, et al. Pancreatic Cancer CellsCLPTM1L Promotes Growth and Enhances Aneuploidy in
Updated version
10.1158/0008-5472.CAN-13-3176doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/03/20/0008-5472.CAN-13-3176.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/74/10/2785.full#ref-list-1
This article cites 48 articles, 11 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/74/10/2785.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/74/10/2785To request permission to re-use all or part of this article, use this link
on August 18, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 19, 2014; DOI: 10.1158/0008-5472.CAN-13-3176


















