
Cloning,Expression,andCharacterizationofBacterial L ... ·...
15
Cloning, Expression, and Characterization of Bacterial L-Arabinose 1-Dehydrogenase Involved in an Alternative Pathway of L-Arabinose Metabolism * Received for publication, June 14, 2005, and inn revised form, November 28, 2005 Published, JBC Papers in Press, December 2, 2005, DOI 10.1074/jbc.M506477200 Seiya Watanabe ‡§¶ , Tsutomu Kodak § , and Keisuke Makino §¶1 From the ‡ Faculty of Engineering, Kyoto University, Kyotodaigakukatsura, Saikyo-ku, Kyoto 615-8530, § Institute of Advanced Energy, Kyoto University, Gokasyo, Uji, Kyoto 611-0011, ¶ International Innovation Center, Kyoto University, Yoshidahonmachi, Sakyo-ku, Kyoto 606-8501,and CREST, Japan Science and Technology Agency, Gokasyo, Uji, Kyoto 611-0011, Japan Azospirillum brasiliense converts L-arabinose to -ketoglutarate via five hypothetical enzymatic steps. We purified and character- ized L-arabinose 1-dehydrogenase (EC 1.1.1.46), catalyzing the con- version of L-arabinose to L-arabino--lactone as an enzyme respon- sible for the first step of this alternative pathway of L-arabinose metabolism. The purified enzyme preferred NADP to NAD as a coenzyme. Kinetic analysis revealed that the enzyme had high cat- alytic efficiency for both L-arabinose and D-galactose. The gene encoding L-arabinose 1-dehydrogenase was cloned using a partial peptide sequence of the purified enzyme and was overexpressed in Escherichia coli as a fully active enzyme. The enzyme consists of 308 amino acids and has a calculated molecular mass of 33,663.92 Da. The deduced amino acid sequence had some similarity to glucose- fructose oxidoreductase, D-xylose 1-dehydrogenase, and D-galac- tose 1-dehydrogenase. Site-directed mutagenesis revealed that the enzyme possesses unique catalytic amino acid residues. Northern blot analysis showed that this gene was induced by L-arabinose but not by D-galactose. Furthermore, a disruptant of the L-arabinose 1-dehydrogenase gene did not grow on L-arabinose but grew on D-galactose at the same growth rate as the wild-type strain. There was a partial gene for L-arabinose transport in the flanking region of the L-arabinose 1-dehydrogenase gene. These results indicated that the enzyme is involved in the metabolism of L-arabinose but not D-galactose. This is the first identification of a gene involved in an alternative pathway of L-arabinose metabolism in bacterium. L-Arabinose is a major constituent of some plant materials (1), and L-arabinose catabolism is therefore relevant for microorganisms using plant material as a carbon source. The metabolic pathway from L-arab- inose to D-xylulose 5-phosphate in bacterium (Fig. 1A) has been inves- tigated extensively. Many bacteria, including Escherichia coli, depend on protein products of the araBAD operon, which contains araB (ribu- lokinase, EC 2.7.1.16), araA (L-arabinose isomerase, EC 5.3.1.4), and araD (L-ribulose-phosphate 4-epimerase, EC 5.1.3.4) to convert L-arab- inose to D-xylulose 5-phosphate through L-ribulose and L-ribulose 5-phosphate (2). Richard et al. (3–5) recently reported a complete fun- gal pathway (Fig. 1B) containing the sequential action of four oxi- doreductases. In this pathway, NAD(P)H-dependent aldose reductase (EC 1.1.1.21) produces L-arabinitol; L-arabinitol 4-dehydrogenase (EC 1.1.1.12) produces L-xylulose; L-xylulose reductase (EC 1.1.1.10) pro- duces D-xylitol; and D-xylulose reductase (EC 1.1.1.9) produces D-xylu- lose. D-Xylulose is then phosphorylated by xylulokinase (EC 2.7.1.17) to yield D-xylulose 5-phosphate. It is believed that there are two alternative pathways for bacterial L-arabinose metabolism, which do not involve a phosphorylation reac- tion, in contrast to the known bacterial and fungal pathways (6 –14). In the first pathway, L-arabinose is oxidized to L-arabino--lactone by NAD(P) -dependent dehydrogenase. The lactone is cleaved by a lacto- nase to L-arabonate, followed by two successive dehydration reactions forming L-2-keto-3-deoxyarabonate (L-KDA) 2 and -ketoglutaric semi- aldehyde. The last step is the NAD(P) -dependent dehydrogenation of the semialdehyde to -ketoglutaric acid. The second pathway has the same initial three steps, but L-KDA is cleaved through an aldolase reac- tion to glycoaldehyde and pyruvate. No gene-encoding enzyme involved in these alternative pathways of L-arabinose metabolism has been iden- tified so far. Dehydrogenases for D-arabinose, D-glucose, D-xylose, and D-galac- tose are known in many organisms. D-Arabinose 1-dehydrogenase (EC 1.1.1.117) is involved in the biosynthesis of D-erythroascorbic acid (15). Glucose 1-dehydrogenase (GDH) is classified into two types, pyrrolo- quinoline-quinone-dependent GDH (EC 1.1.5.2) and NAD(P)-depend- ent GDH. According to primary structure analysis, these GDH types are not related. NAD(P)-dependent GDH is further classified into two dif- ferent protein families (16). One GDH (EC 1.1.118 (119)) belongs to a medium chain dehydrogenase/reductase family, which contains an active zinc ion (17); the other GDH is glucose-fructose oxidoreductase (GFOR, EC 1.1.99.28), which catalyzes the coupled intermolecular oxi- dation-reduction of D-glucose and D-fructose (18 –20). GFOR belongs to the Gfo/Idh/MocA family, which also contains D-xylose 1-dehydro- genase (EC 1.1.1.175 (179)) and D-galactose 1-dehydrogenase (EC 1.1.1.48 (120)) (21, 22). GDH is involved in the nonphosphorylative Entner-Doudoroff (ED) pathway of Archaea (17, 23–25) and Aspergillus fungi (26) (Fig. 1D). GDH also functions as “gluconolactonase” in this pathway and converts D-glucose to D-gluconate via D-glucono--lac- tone. It is interesting that enzymes of the nonphosphorylative ED path- way are used for the metabolism of both D-glucose and D-galactose in * This work was supported by the Center of Excellence program for the ”Establishment of Centers of Excellence on Sustainable Energy System,“ a grant-in-aid for scientific research, and grants for regional science and technology promotion from the Minis- try of Education, Science, Sports, and Culture, Japan, and by CREST of the Japan Science and Technology Corp. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/Gen- Bank TM /EBI Data Bank with accession number(s) AB211983. 1 To whom correspondence should be addressed. Tel.: 81-774-38-3517; Fax: 81-774-38- 3524; E-mail: [email protected]. 2 The abbreviations used are: L-KDA, L-2-keto-3-deoxyarabonate; GDH, glucose 1-dehy- drogenase; ED pathway, Entner-Doudoroff pathway; GFOR, glucose-fructose oxi- doreductase; CNBr, cyanogen bromide; DD, dihydrodiol dehydrogenase; ZmGFOR, GFOR from Z. mobilis; G6PDH, glucose-6-phosphate dehydrogenase; LmG6PDH, G6PDH from L. mesenteroides; CAPS, cyclohexylaminopropanesulfonic acid; HPLC, high pressure liquid chromatography; ORF, open reading frame; ABC, ATP-binding cassette. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 5, pp. 2612–2623, February 3, 2006 © 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. 2612 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006 by guest on April 24, 2019 http://www.jbc.org/ Downloaded from by guest on April 24, 2019 http://www.jbc.org/ Downloaded from by guest on April 24, 2019 http://www.jbc.org/ Downloaded from by guest on April 24, 2019 http://www.jbc.org/ Downloaded from
-
Upload
nguyenhanh -
Category
Documents
-
view
265 -
download
0
Transcript of Cloning,Expression,andCharacterizationofBacterial L ... ·...

Cloning, Expression, and Characterization of BacterialL-Arabinose 1-Dehydrogenase Involved in anAlternative Pathway of L-Arabinose Metabolism*
Received for publication, June 14, 2005, and inn revised form, November 28, 2005 Published, JBC Papers in Press, December 2, 2005, DOI 10.1074/jbc.M506477200
Seiya Watanabe‡§¶�, Tsutomu Kodak§�, and Keisuke Makino§¶�1
From the ‡Faculty of Engineering, Kyoto University, Kyotodaigakukatsura, Saikyo-ku, Kyoto 615-8530, §Institute of AdvancedEnergy, Kyoto University, Gokasyo, Uji, Kyoto 611-0011, ¶International Innovation Center, Kyoto University, Yoshidahonmachi,Sakyo-ku, Kyoto 606-8501,and �CREST, Japan Science and Technology Agency, Gokasyo, Uji, Kyoto 611-0011, Japan
Azospirillum brasiliense converts L-arabinose to �-ketoglutaratevia five hypothetical enzymatic steps. We purified and character-ized L-arabinose 1-dehydrogenase (EC 1.1.1.46), catalyzing the con-version of L-arabinose to L-arabino-�-lactone as an enzyme respon-sible for the first step of this alternative pathway of L-arabinosemetabolism. The purified enzyme preferred NADP� to NAD� as acoenzyme. Kinetic analysis revealed that the enzyme had high cat-alytic efficiency for both L-arabinose and D-galactose. The geneencoding L-arabinose 1-dehydrogenase was cloned using a partialpeptide sequence of the purified enzyme and was overexpressed inEscherichia coli as a fully active enzyme. The enzyme consists of 308amino acids and has a calculated molecular mass of 33,663.92 Da.The deduced amino acid sequence had some similarity to glucose-fructose oxidoreductase, D-xylose 1-dehydrogenase, and D-galac-tose 1-dehydrogenase. Site-directed mutagenesis revealed that theenzyme possesses unique catalytic amino acid residues. Northernblot analysis showed that this gene was induced by L-arabinose butnot by D-galactose. Furthermore, a disruptant of the L-arabinose1-dehydrogenase gene did not grow on L-arabinose but grew onD-galactose at the same growth rate as the wild-type strain. Therewas a partial gene for L-arabinose transport in the flanking region ofthe L-arabinose 1-dehydrogenase gene. These results indicated thatthe enzyme is involved in the metabolism of L-arabinose but notD-galactose. This is the first identification of a gene involved in analternative pathway of L-arabinose metabolism in bacterium.
L-Arabinose is a major constituent of some plant materials (1), andL-arabinose catabolism is therefore relevant for microorganisms usingplant material as a carbon source. The metabolic pathway from L-arab-inose to D-xylulose 5-phosphate in bacterium (Fig. 1A) has been inves-tigated extensively. Many bacteria, including Escherichia coli, dependon protein products of the araBAD operon, which contains araB (ribu-lokinase, EC 2.7.1.16), araA (L-arabinose isomerase, EC 5.3.1.4), andaraD (L-ribulose-phosphate 4-epimerase, EC 5.1.3.4) to convert L-arab-inose to D-xylulose 5-phosphate through L-ribulose and L-ribulose5-phosphate (2). Richard et al. (3–5) recently reported a complete fun-
gal pathway (Fig. 1B) containing the sequential action of four oxi-doreductases. In this pathway, NAD(P)H-dependent aldose reductase(EC 1.1.1.21) produces L-arabinitol; L-arabinitol 4-dehydrogenase (EC1.1.1.12) produces L-xylulose; L-xylulose reductase (EC 1.1.1.10) pro-duces D-xylitol; and D-xylulose reductase (EC 1.1.1.9) produces D-xylu-lose. D-Xylulose is then phosphorylated by xylulokinase (EC 2.7.1.17) toyield D-xylulose 5-phosphate.
It is believed that there are two alternative pathways for bacterialL-arabinose metabolism, which do not involve a phosphorylation reac-tion, in contrast to the known bacterial and fungal pathways (6–14). Inthe first pathway, L-arabinose is oxidized to L-arabino-�-lactone byNAD(P)�-dependent dehydrogenase. The lactone is cleaved by a lacto-nase to L-arabonate, followed by two successive dehydration reactionsforming L-2-keto-3-deoxyarabonate (L-KDA)2 and�-ketoglutaric semi-aldehyde. The last step is the NAD(P)�-dependent dehydrogenation ofthe semialdehyde to �-ketoglutaric acid. The second pathway has thesame initial three steps, but L-KDA is cleaved through an aldolase reac-tion to glycoaldehyde andpyruvate.No gene-encoding enzyme involvedin these alternative pathways of L-arabinose metabolism has been iden-tified so far.Dehydrogenases for D-arabinose, D-glucose, D-xylose, and D-galac-
tose are known in many organisms. D-Arabinose 1-dehydrogenase (EC1.1.1.117) is involved in the biosynthesis of D-erythroascorbic acid (15).Glucose 1-dehydrogenase (GDH) is classified into two types, pyrrolo-quinoline-quinone-dependent GDH (EC 1.1.5.2) and NAD(P)-depend-entGDH.According to primary structure analysis, theseGDH types arenot related. NAD(P)-dependent GDH is further classified into two dif-ferent protein families (16). One GDH (EC 1.1.118 (119)) belongs to amedium chain dehydrogenase/reductase family, which contains anactive zinc ion (17); the other GDH is glucose-fructose oxidoreductase(GFOR, EC 1.1.99.28), which catalyzes the coupled intermolecular oxi-dation-reduction of D-glucose and D-fructose (18–20). GFOR belongsto the Gfo/Idh/MocA family, which also contains D-xylose 1-dehydro-genase (EC 1.1.1.175 (179)) and D-galactose 1-dehydrogenase (EC1.1.1.48 (120)) (21, 22). GDH is involved in the nonphosphorylativeEntner-Doudoroff (ED) pathway of Archaea (17, 23–25) andAspergillusfungi (26) (Fig. 1D). GDH also functions as “gluconolactonase” in thispathway and converts D-glucose to D-gluconate via D-glucono-�-lac-tone. It is interesting that enzymes of the nonphosphorylative ED path-way are used for the metabolism of both D-glucose and D-galactose in
* This work was supported by the Center of Excellence program for the ”Establishment ofCenters of Excellence on Sustainable Energy System,“ a grant-in-aid for scientificresearch, and grants for regional science and technology promotion from the Minis-try of Education, Science, Sports, and Culture, Japan, and by CREST of the JapanScience and Technology Corp. The costs of publication of this article were defrayed inpart by the payment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/Gen-BankTM/EBI Data Bank with accession number(s) AB211983.
1 To whom correspondence should be addressed. Tel.: 81-774-38-3517; Fax: 81-774-38-3524; E-mail: [email protected].
2 The abbreviations used are: L-KDA, L-2-keto-3-deoxyarabonate; GDH, glucose 1-dehy-drogenase; ED pathway, Entner-Doudoroff pathway; GFOR, glucose-fructose oxi-doreductase; CNBr, cyanogen bromide; DD, dihydrodiol dehydrogenase; ZmGFOR,GFOR from Z. mobilis; G6PDH, glucose-6-phosphate dehydrogenase; LmG6PDH,G6PDH from L. mesenteroides; CAPS, cyclohexylaminopropanesulfonic acid; HPLC,high pressure liquid chromatography; ORF, open reading frame; ABC, ATP-bindingcassette.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 5, pp. 2612–2623, February 3, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
2612 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
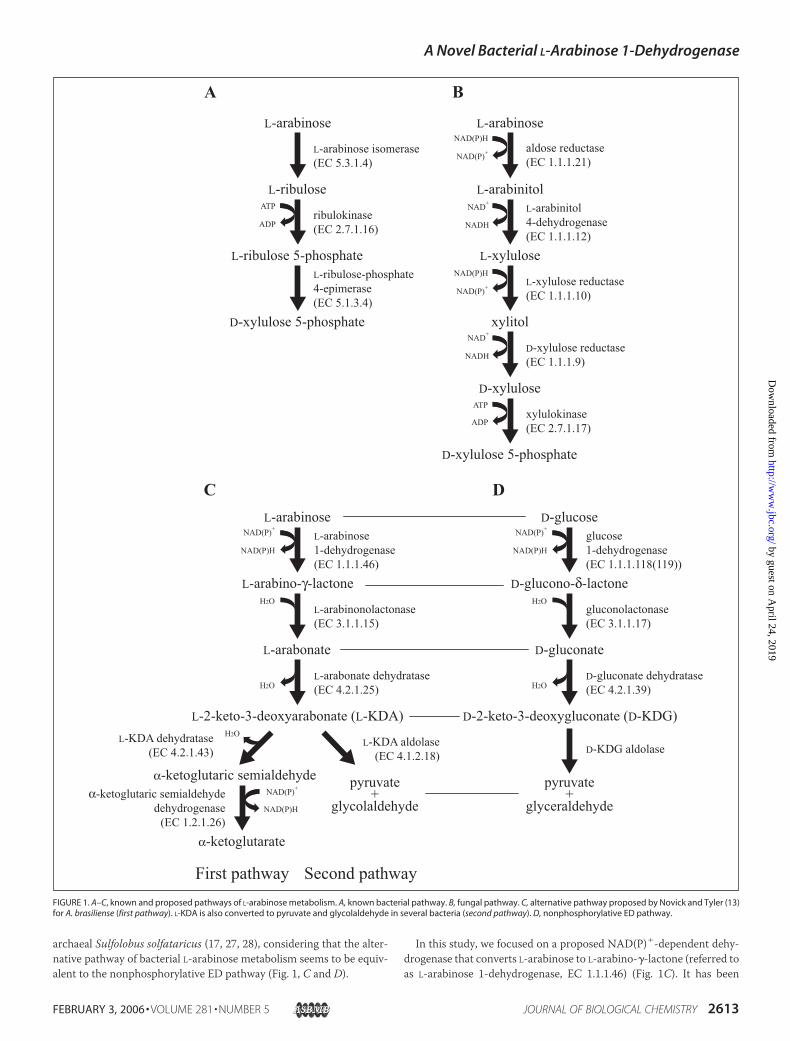
archaeal Sulfolobus solfataricus (17, 27, 28), considering that the alter-native pathway of bacterial L-arabinose metabolism seems to be equiv-alent to the nonphosphorylative ED pathway (Fig. 1, C and D).
In this study, we focused on a proposed NAD(P)�-dependent dehy-drogenase that converts L-arabinose to L-arabino-�-lactone (referred toas L-arabinose 1-dehydrogenase, EC 1.1.1.46) (Fig. 1C). It has been
FIGURE 1. A–C, known and proposed pathways of L-arabinose metabolism. A, known bacterial pathway. B, fungal pathway. C, alternative pathway proposed by Novick and Tyler (13)for A. brasiliense (first pathway). L-KDA is also converted to pyruvate and glycolaldehyde in several bacteria (second pathway). D, nonphosphorylative ED pathway.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2613
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

reported that Azospirillum brasiliense, a bacterium used in this study,can grow on L-arabinose as a sole carbon source and has the first alter-native pathway of L-arabinose metabolism (13). This is the first enzy-mological and molecular biological analysis of the proposed pathway ofL-arabinose metabolism.
EXPERIMENTAL PROCEDURES
Bacterial Strain, Culture Conditions, and Preparation of Cell-freeExtracts—A. brasiliense ATCC29145 was purchased from RIKENBioResource Center (Saitama, Japan) and cultured aerobically with vig-orously shaking at 30 °C for 24 h in a minimal medium (13) (pH 6.8),containing 4.0 g of KH2PO4, 6.0 g of K2HPO4, 0.2 g ofMgSO4�H2O, 0.1 gof NaCl, 0.026 g of CaSO4�2H2O, 1.0 g of NH4Cl, 0.01 g of FeCl3�6H2O,0.002 g of NaMoO4�2H2O, and 0.0001 g of biotin/liter supplementedwith 37 mM L-arabinose. L-Arabinose was sterilized separately by filtra-tion and added to the medium. The grown cells were harvested bycentrifugation at 30,000 � g for 20 min, washed with 20 mM potassiumphosphate buffer (pH 7.0), containing 2 mM MgCl2 and 10 mM 2-mer-captoethanol (referred to as Buffer A), and stored at �35 °C until used.The washed cells were suspended in Buffer A, disrupted by sonicationfor 20 min with appropriate intervals on ice using ASTRASON� Ultra-sonic Liquid Processor XL2020 (Misonix Inc., New York), and thencentrifuged at 108,000� g for 20min at 4 °C to obtain cell-free extracts.
PAGE—SDS-PAGE was performed as described by Laemmli (29).Nondenaturing PAGE was performed by omitting SDS and 2-mercap-toethanol from the solution used in SDS-PAGE. Proteins on the gelwerestained with Coomassie Brilliant Blue R-250 and destained with 7.5%(v/v) acetic acid in 25% methanol.
Enzyme Activity Assay—L-Arabinose 1-dehydrogenase activity wasassayed routinely in the direction of L-arabinose oxidation bymeasuringthe reduction of NAD(P)� at 340 nm at 30 °C using Jasco spectropho-tometer model V-550 (Japan Spectroscopic Co., Ltd., Tokyo, Japan).The standard assay mixture contained 10 mM L-arabinose in 100 mM
Tris-HCl (pH 9.0) buffer. The reaction was started by the addition of 10mMNAD(P)� solution (100�l) with a final reaction volume of 1ml. Thekinetic parameters, Km and kcat values, were calculated by Lineweaver-Burk plot. Protein concentrations were determined by the method ofLowry et al. (30) with bovine serum albumin as a standard.
Zymogram Staining Analysis—The cell-free extracts or purifiedenzyme were separated on nondenaturing PAGE with 12% gel at 4 °C.The gels were then soaked in 10ml of staining solution (31) consisting of100 mM Tris-HCl (pH 9.0), 100 mM L-arabinose, 0.25 mM nitro bluetetrazolium, 0.06 mM phenazine methosulfate, and 1 mM NAD(P)� atroom temperature for 15 min. The dehydrogenase activity appeared asa dark band.
Purification of L-Arabinose 1-Dehydrogenase—All purification stepswere performed below 4 °C. The cell-free extracts were fractionatedbetween 50 and 60% saturation of (NH4)2SO4. The precipitate was dis-solved in a small volume of Buffer A, and the solution was then dialyzedagainst a large volume of Buffer A containing 1.3 M (NH4)2SO4 over-night. All chromatography was carried out using an �KTA purifier sys-tem (Amersham Biosciences). After insoluble materials were removedby centrifugation, the supernatant was applied to a HiPrep 16/10 ButylFF column (1.6 � 10 cm, Amersham Biosciences) equilibrated withBuffer A containing 1.3 M (NH4)2SO4 and washed with the same buffer.Proteins were eluted using a reversed linear gradient of 1.3–0 M
(NH4)2SO4 in Buffer A (300 ml). The fractions with high enzymaticactivity were pooled and dialyzed overnight against a large volume ofBuffer A. The enzyme solution was loaded onto a column of HiPrep16/10 Q FF (1.6 � 10 cm, Amersham Biosciences) equilibrated with
Buffer A and washed thoroughly with the same buffer. The column wasdeveloped with 300 ml of linear gradient 0–1 M NaCl in Buffer A. Thefractions containing L-arabinose 1-dehydrogenase activity were com-bined and dialyzed against a large volume of 5mM potassium phosphate(pH 7.0), containing 10 mM 2-mercaptoethanol (referred to as Buffer B)overnight. The enzyme solution was applied to a column of CeramicHydroxyapatite Type I (1.6� 5 cm, Bio-Rad), equilibratedwith Buffer B.The column was washed thoroughly with the same buffer and devel-opedwith 200ml of linear gradient 0.005–0.5Mpotassiumphosphate inBuffer B. The fractions with high enzymatic activity were combined andconcentrated by ultrafiltration with Centriplus YM-30 (Millipore) at18,000 � g for �2 h. The enzyme solution was loaded onto a column ofHiLoad 16/60 Superdex 200 pg column (1.6 � 60 cm, Amersham Bio-sciences) equilibrated with Buffer A. The active fractions were pooled,concentrated, and re-loaded onto the same column. Proteins in thefractions containing high activity L-arabinose 1-dehydrogenase wereanalyzed by SDS-PAGE, and the fractions containing a single proteinwere collected, concentrated, dialyzed against Buffer C (100 mM Tris-HCl (pH 9.0), containing 2 mM MgCl2, 10 mM L-arabinose, 1 mM dithi-othreitol, and 50% (v/v) glycerol), and stored at �35 °C until used.The native molecular mass of L-arabinose 1-dehydrogenase was esti-
mated by gel filtration. The purified enzyme was loaded onto a HiLoad16/60 Superdex 200 pg column equilibrated with 20 mM potassiumphosphate (pH 7.0), containing 2 mM MgCl2 and 1 mM dithiothreitol.High and lowmolecularweight gel filtration calibration kits (AmershamBiosciences) were used as molecular markers.
Product Identification by HPLC—The product of the dehydrogena-tion reaction of L-arabinose was identified by HPLCwith aMultistationLC-8020model II system (Tosoh). L-Arabino-�-lactone was chemicallysynthesized with boiling potassium arabonate in 0.2 M HCl for 5 min.Potassium arabonate was prepared by the hypoiodite-in-methanol oxi-dization of L-arabinose (32). A solution containing 100 mM Tris-HCl(pH 9.0), 10 mM L-arabinose, 10 mM NADP�, and the purified enzyme(10�g)was incubated for 30min at 30 °C, and 100�l of this solutionwasthen analyzed. Samples were applied at 30 °C into an AminexHPX-87HOrganic Analysis column (300 � 7.8 mm, Bio-Rad) linked to an RID-8020 refractive index detector (Tosoh) and eluted with 5 mM H2SO4 ata flow rate of 0.6 ml/min.
Determination of N-terminal and Internal Amino Acid Sequences—Todetermine the N-terminal amino acid sequence of L-arabinose 1-de-hydrogenase, the purified enzyme was separated by SDS-PAGE with12% (w/v) gel, and then transferred to HybondTM-P (Amersham Bio-sciences) at 3 mA/cm2 for 0.5 h in a transfer buffer (10 mMCAPS (pH11) containing 10% (v/v) methanol) with a horizontal electro-phoretic blotting system (model AE-7500, Atto). After staining anddestaining the protein, an area of the membrane corresponding tothe protein band of L-arabinose 1-dehydrogenase was excised andanalyzed with a ProciseTM 492 HT protein sequencer (AppliedBiosystems).Chemical digestion with cyanogen bromide (CNBr) was carried out
to determine internal amino acid sequences (33). The purified L-arabi-nose 1-dehydrogenase (100 �g) was dialyzed overnight against deion-izedwater and lyophilized. The enzyme proteinwas digested chemicallyat room temperature in 70% (v/v) formic acid containing 1% (w/v)CNBr(100 �l) in the dark and under N2 overnight. The solution was dilutedwith 900 �l of deionized water, frozen with liquid N2, and lyophilized.The sample was dissolved in SDS-PAGE sample buffer (500 mM Tris-HCl (pH 6.8), containing 5% (w/v) SDS, 10% (v/v) glycerol, 0.25% (w/v)bromphenol blue, and 5% (v/v) 2-mercaptoethanol) and separated bySDS-PAGEwith 18% (w/v) gel. Peptide fragments on the gel were trans-
A Novel Bacterial L-Arabinose 1-Dehydrogenase
2614 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ferred to a HybondTM-P membrane as described above. After stainingand destaining, areas of the membrane corresponding to the two pep-tide fragments from the L-arabinose 1-dehydrogenase (see in Fig. 2B)were excised and sequenced.
Cloning of L-Arabinose 1-Dehydrogenase Gene—The N-terminal andinternal peptide sequences were used to design PCR primers for ampli-fication of a partial DNA fragment of the L-arabinose 1-dehydrogenasegene. Eight upstream primers (U1–U8, 26-mer) were designed from(M)SDQVSLGV, the N-terminal amino acid sequence, as follows:5�-ATG(TCN/AGY)GAYCARGTN(TCN/AGY) (CTN/TTR)GGN-GT-3�. Two downstream primers (D1 and D2, 26-mer) were designedfrom the internal amino acid sequence (M)LEKPPGAT, as follows:5�-GTNGCNCCNGGNGGYTTYTC(NAG/YAA)CAT-3�. A. brasil-iense genomicDNAwas prepared using aDNeasyTM tissue kit (Qiagen).PCRwas carried out using a PCR Thermal Cycler-Personal (Takara) for30 cycles in a 50-�l reaction mixture containing 10 pmol of primers,1.25 units of Ex Taq� DNA polymerase (Takara), and 300 ng of A.brasiliense genomic DNA under the following conditions: denaturationat 98 °C for 10 s, annealing at 50 °C for 30 s, and extension at 72 °C for30 s, each for 30 cycles. Based on the results of genomic PCR using eachset of primers, U6 andD1were chosen for cloning. The sequences of U6and D1 were 5�-ATGAGYGAYCARGTNTCNTTRGGNGT-3� and5�-GTNGCNCCNGGNGGYTTYTCNAGCAT-3�, respectively. A sin-gle PCR product with a length of �300 bp was purified, cloned into apGEM�-T vector (Promega) (referred to as pGEM1), and sequencedusing a Dual CyDyeTM terminator sequencing kit (Veritas) and appro-priate primers with Long-Read Tower, UBC DNA sequencer (Amer-sham Biosciences). The inserted fragment was amplified with U6 andD1 primers and with pGEM1 as a template DNA, and the PCR productwas purified and utilized as a probe for Southern and Northern blotanalysis and colony hybridization (34).For Southern blot analysis, �1.8 �g of A. brasiliense genomic DNA
was digested with six restriction enzymes, EcoRI, HindIII, NotI, PstI,SalI, and XbaI, separated on 1% (w/v) agarose gel and blotted toHybondTM-N (Amersham Biosciences) by capillary transfer using 10�SSC as a transfer buffer (1� SSC is 15 mM sodium citrate (pH 7.0), and0.15 M NaCl). The blotted filter was cross-linked in an UV cross-linkerCX-2000 (Ultra-Violet Products, Ltd.). A double-stranded probe DNAwas labeled with digoxigenin-11-dUTP and hybridized using a DIG-High Prime DNA labeling and detection starter kit (Roche Applied Sci-ence). Membrane was visualized using a nitro blue tetrazolium/5-bro-mo-4-chloro-3-indolyl phosphate reagent detection system (RocheApplied Science).A. brasiliense partial genomic library was prepared with genomic
DNAwithNotI based on the results of Southern blot analysis. TheDNAfragments corresponding to a positive band in size (�2.0 kbp of thelength) were ligated to a plasmid pBluescript� SK(�) (Stratagene). Col-ony hybridization was carried out under the same conditions as South-ern blot analysis except for the use of nylon membranes for colony andplaque hybridization (Roche Applied Science). The plasmid from a pos-itive clone (referred to as pBS1) was purified, and the inserted A. brasil-iense genome fragment was sequenced.
Northern Blot Analysis—A. brasiliense cells were cultured at 30 °C tothe mid-log phase (A600 � 0.6–0.8) in minimal medium supplementedwith 37 mM appropriate sugar (D-glucose, L-arabinose, D-galactose, orD-xylose) or nutrientmedium (10 g of peptone, 10 g ofmeat extract, and5.0 g of NaCl (pH 7.0–7.2)) and harvested by centrifugation. TotalRNAs fromA. brasiliensewere preparedwith an RNeasy�mini kit (Qia-gen) and subsequently treated with RNase-free DNase I. The isolatedRNA (4 �g) was subjected to electrophoresis on 1.2% (w/v) agarose gel
containing 0.66 M formaldehyde. The subsequent steps were performedusing the same methods as for Southern blot analysis.
Cloning of the L-Arabinose 1-Dehydrogenase Gene into ExpressionPlasmid Vector—To introduce the restriction site for BglII at the 5�-endand PstI at the 3�-end of the L-arabinose 1-dehydrogenase gene, PCRwas carried out using pBS1 as a template and the following two primers(lowercase letters indicate additional bases for introducing digestionsites of BglII and PstI (underlined letters)): 5�-caccatagaTCTGATCAG-GTTTCGCTGGGTG-3� (HISBglII) and 5�-gcttggctgcagTCAGCGGC-CGAACGCGTCG-3� (HISPstI). The amplified DNA fragment wasintroduced into BamHI-PstI sites in pQE-80L (Qiagen), a plasmid vec-tor for conferring N-terminal His6 tag on the expressed proteins, toobtain pHISWT.
Site-directed Mutagenesis—The following sense and antisense prim-ers were designed to introduce themutations into the L-arabinose 1-de-hydrogenase gene (the mutated regions are underlined): to substituteAla for Asp168 (D168A), 5�-CGGCGTGTTCGCGCCGGGCATC-3�
(D168AS) and 5�-GATGCCCGGCGCGAACACGCCG-3� (D168AAS);to substitute Ala for Asn172 (N172A), 5�-CCCGGGCATCGCG-GCGCTGTCG-3� (N172AS) and 5�-CGACAGCGCCGCGATGC-CCGGG-3� (N172AAS). The mutations were introduced by sequentialsteps of PCR (35) with small modifications. In the first round, two reac-tions, I and II, were performed with the appropriate primers andpHISWT as a template: reaction I, HISBglII and one of the antisense prim-ers containing the mutations; and reaction II, one of the sense primerscontaining the mutations and HISPstI. In the final amplification step,purified overlapping PCR products were used as templates and HISBglII
and HISPstI as primers. The final PCR products were cloned into pQE-80L to obtain plasmids pHISD168A and pHISN172A, respectively. Thecoding region of the mutated genes was confirmed by subsequentsequencing in both directions.
Functional Expression and Purification of His6-tagged L-Arabinose1-Dehydrogenase—E. coli DH5� harboring the expression plasmid forthe His6-tagged wild-type and mutated enzymes was grown at 37 °C toa turbidity of 0.6 at 600 nm in Super Broth medium (12 g of tryptone,24 g of yeast extract, 5 ml of glycerol, 3.81 g of KH2PO4, and 12.5 g ofK2HPO4/liter (pH 7.0)) containing 50 mg/liter ampicillin. After theaddition of 1 mM of isopropyl-�-D-thiogalactopyranoside, the culturewas further grown for 6 h to induce the expression of His6-tagged L-a-rabinose 1-dehydrogenase protein. Cells were harvested and resus-pended in Buffer D (50 mM sodium phosphate containing 2 mMMgCl2,300mMNaCl, 1mM L-arabinose, 10mM2-mercaptoethanol, and 10mM
imidazole (pH 8.0)). The cells were then disrupted by sonication, andthe solutionwas centrifuged. The supernatant was loaded onto a nickel-nitrilotriacetic acid spin column (Qiagen) equilibrated with Buffer D.The column was washed three times with Buffer E (Buffer D containing10% (v/v) glycerol and 50mM imidazole instead of 10mM imidazole (pH8.0)). The enzymes were then eluted with Buffer F (Buffer E containing250 mM imidazole instead of 50 mM imidazole (pH 8.0)). The elutantwas dialyzed against Buffer C and stored at �35 °C until use.
WesternBlotAnalysis ofHis6-tagged L-Arabinose 1-Dehydrogenase—ForWestern blot analysis, the purified L-arabinose 1-dehydrogenasefrom A. brasiliense and/or recombinant His6-tagged L-arabinose1-dehydrogenase from E. coli was separated by SDS-PAGE, and theproteins on the gels were transferred onto a nitrocellulose mem-brane (HybondTM-ECL; Amersham Biosciences). Western blot anal-ysis was carried out using the ECLTM Western blotting analysis sys-tem (Amersham Biosciences) and RGS�His horseradish peroxidaseantibody, a horseradish peroxidase-fused mouse monoclonal anti-
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2615
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

body against Arg-Gly-Ser-His6 in the N-terminal additional peptideof the expressed recombinant proteins (Qiagen).
Disruptant Construction—The overall scheme of the plasmid con-struction for disruption of the L-arabinose 1-dehydrogenase gene isshown in Fig. 9A. The Tn5-derived 1.3-kb BamHI kanamycin resistance(Kmr) cassette of pUC4K (AmershamBiosciences) was inserted into thesingle BamHI site in the coding sequence of the L-arabinose 1-dehydro-genase gene of pHISWT to yield pHISWT::Km. To introduce the restric-tion site for MfeI at the 5�- and 3�-end of the DNA fragment containingtheKmr gene in the L-arabinose 1-dehydrogenase gene, PCRwas carriedout using pHISWT::Km as a template and the following two primers (low-ercase letters indicate additional bases for introducing digestion sites ofMfeI (underlined letters)): 5�-caccatcaattgGATCAGGTTTCGCTGG-GTGTCGTCGGCATCG-3� (MfeI-up) and 5�-gcttggcaattgTCAGCG-GCCGAACGCGTCGGTCTGCACGCGC-3� (MfeI-down). The 2.3-kbpMfeI DNA fragment was subcloned into the EcoRI site in the chloram-phenicol resistance (Cmr) cassette of the suicide vector pSUP202 (36) toyield pSUPWT::Km.E. coli S17-1 (36) was transformed with pSUPWT::Km, and then the
transformant was further mobilized to A. brasiliense by biparental mat-ing. The transconjugants were selected on aminimalmedium agar platesupplemented with 5 g of sodium malate and 25 �g of kanamycin/literusing Kmr (the presence of Kmr cassette) and TcS (loss of pSUP202)phenotypes. The construction was confirmed by genomic PCR andSouthern hybridization on total DNA digested with NotI. One of theresulting disruptants of A. brasiliense was named �ARA5034 and wasused in this study.
Amino Acid Sequence Alignment and Phylogenetic Analysis—Proteinsequence of L-arabinose 1-dehydrogenase from A. brasiliense was ana-lyzed using the Protein-BLAST and ClustalW program distributed byDDBJ (www.ddbj.nig.ac.jp). The phylogenetic tree was produced usingthe TreeView 1.6.1. program.
RESULTS
Purification of L-Arabinose 1-Dehydrogenase from A. brasiliense—NAD�- and NADP�-dependent enzymatic oxidization of L-arabinosewas found in cell-free extracts prepared from A. brasiliense cells grownon L-arabinose as a sole carbon source (Fig. 2C). The L-arabinose 1-de-hydrogenase was purified by ammonium fractionation and five chro-matographic steps. A typical result of purification is summarized inTable 1. During the purification procedure, the ratio of NADP�- andNAD�-linked activity remained almost constant, suggesting the pres-ence of only one protein as L-arabinose 1-dehydrogenase. The purifiedenzyme was electrophoretically homogeneous (Fig. 2, A and C) andshowed NAD�- and NADP�-dependent specific activity of 25 and 44units/mg protein, respectively (Table 1). SDS-PAGE revealed only onesubunit with an apparent value of 39.5 � 0.7 kDa (Fig. 2A). To estimatenative molecular mass, the enzyme was loaded onto a HiLoad 16/60Superdex 200 pg column. The value from the calibration curve withmarker proteins was �46.4 � 1.9 kDa (data not shown), indicating thatthe L-arabinose 1-dehydrogenase is monomeric. There was no signifi-cant increase in activity in the presence ofMgCl2,MnCl2, ZnCl2, CoCl2,NiCl2, orCaCl2 at final concentrations of 1mM (data not shown). Zymo-gram staining analysis showed a major active band in the cell-free
TABLE 1Summary of L-arabinose 1-dehydrogenase purification from A. brasiliense
Step Totalprotein
Total activitySpecificactivity Yield Purification
NAD� NADP� NADP�/NAD�
mg units units/mg protein % -foldCell-free extract 1488 248 373 1.50 0.25 100 1.0(NH4)2SO4 fractionation 1147 171 287 1.68 0.25 77 1.0HiPrep 16/10 Butyl FF 186 109 202 1.85 1.09 54 4.4HiPrep 16/10 Q FF 53 97 158 1.63 2.98 42 12CHT ceramic hydroxyapatite 7.8 70 124 1.77 15.9 33 64HiLoad 16/60 Superdex 200 pg 1st 4.2 51 91 1.78 21.7 24 87HiLoad 16/60 Superdex 200 pg 2nd 1.0 25 44 1.76 44.0 12 176
FIGURE 2. PAGE of L-arabinose 1-dehydrogenase. A, SDS-PAGE of purification. M is marker proteins. Lane 1, cell-free extracts (50 �g); lane 2, ammonium sulfate fractionation (50 �g);lane 3, HiPrep 16/10 Butyl FF (10 �g); lane 4, HiPrep 16/10 Q FF (10 �g); lane 5, CHT ceramic hydroxyapatite (10 �g); lane 6, HiLoad 16/60 Superdex 200 pg 1st (10 �g); lane 7, HiLoad16/60 Superdex 200 pg 2nd (10 �g). B, SDS-PAGE of CNBr digestion. The purified enzyme (100 �g) was digested chemically with CNBr, and the solution corresponding to 5 �g ofprotein was applied. Two observed bands except undigested polypeptide were referred to as Fragments I and II with molecular masses of 26.4 and 0.92 kDa, respectively. C,nondenaturing PAGE (lane 1) and zymogram-staining analysis (lanes 2–5). The purified enzyme of 5 �g and cell-free extracts of 100 �g were applied. NAD� and NADP� in lanes 2 and4 and lanes 3 and 5, respectively, were used as a coenzyme.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
2616 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

extracts. The position corresponded to that of the purified enzyme (Fig.2C). A minor staining band with NAD� as a coenzyme may be derivedfrom the concomitant activity of other NAD�-dependent sugardehydrogenases.
Substrate Specificity and Kinetic Analysis—In addition to L-arabi-nose, various sugars were tested as substrates for dehydrogenase. In thefirst approach, the sugar concentrations were fixed at 10 mM. Activitywas observed with L-arabinose, D-galactose, D-xylose, and D-talose butnot with D-arabinose, D-glucose, D-ribose, L-xylose, L-mannose, L-lyx-ose, and D-fructose (less than 1% of the activity with L-arabinose). Theenzyme was subjected to further kinetic analysis with the active sub-strates, and the determined parameters are listed in Table 2. The cata-lytic efficiency (kcat/Km) values with L-arabinose, D-galactose, and D-xy-lose in the presence of NADP� were significantly higher than those inthe presence of NAD� because of the lower values of Km and the higher
values of kcat. Furthermore, when L-arabinose was used as a substrate,the enzyme showed 5.6-fold higher affinity with NADP� (Km �0.0095 � 0.0083 mM) than NAD� (Km � 0.053 � 0.020 mM). Thekinetic parameters of D-galactose were very similar to those of L-arabi-nose in the presence of either NAD� or NADP�, indicating that theenzyme functions not only as L-arabinose 1-dehydrogenase but also“D-galactose 1-dehydrogenase.” The enzyme showed 51- and 79-foldlower values of kcat/Km with D-talose in the presence of NAD� andNADP�, respectively, compared with those with L-arabinose. kcat/Km
values with D-xylose were lower than those with D-talose by 2 orders ofmagnitude in the presence of either NAD� or NADP� because ofremarkably high Km values and low kcat values.
Enzyme activities with these sugars, and the stereoconfiguration ofthese sugars are shown in Fig. 3. Active sugars for L-arabinose 1-dehy-drogenase belong not only to pentose(s) but also hexose(s), indicating
TABLE 2Kinetic parameters of L-arabinose 1-dehydrogenaseStocked L-arabinose 1-dehydrogenase was dialyzed against 100 mM Tris-HCl (pH 9.0) containing 2 mM MgCl2, overnight at 4 °C before enzyme activity was measured.Values are the means � S.D., n � 3.
Substrate Coenzyme Vmax Km kcat kcat/Km
units/mg protein mM min�1 min�1 mM�1
L-Arabinosea NAD� 25.0 � 0.2b 1.41 � 0.16c 1140 � 100c 806 � 17cNADP� 44.9 � 0.1c 0.255 � 0.016c 2000 � 40c 7860 � 330c
D-Galactosea NAD� 23.8 � 0.1b 1.49 � 0.14c 1190 � 90c 798 � 16cNADP� 35.6 � 0.1b 0.109 � 0.003d 1560 � 10d 14400 � 400d
D-Talosea NAD� 1.7 � 0.1b 3.95 � 0.09e 62.1 � 1.4e 15.7 � 0.1eNADP� 12.8 � 0.2b 5.87 � 0.20e 580 � 22e 98.9 � 0.4e
D-Xylosea NAD� 5.3 � 0.2f 210 � 49g 240 � 40g 1.16 � 0.07gNADP� 14.8 � 0.3f 72.0 � 7.6g 720 � 51g 10.0 � 0.3g
a Enzyme activity was measured with 100 mM Tris-HCl (pH 9.0) containing 1 mM NAD(P)�.b Ten mM sugar was used as a substrate.c Eleven different concentrations of sugar between 0.1 and 5.0 mM were used.d Ten different concentrations of D-galactose between 0.05 and 0.5 mM were used.e Five different concentrations of D-talose between 1 and 10 mM were used.f Five hundred mM D-xylose was used as a substrate.g Ten different concentrations of D-xylose between 0.1 and 1.0 M were used.
FIGURE 3. Relationship between catalytic efficiency (kcat/Km) and configuration of the substrate in L-arabinose 1-dehydrogenase. The kcat/Km values were taken from thevalues in Table 2 with NAD� (light gray bar) and NADP� (dark gray bar). Asterisks indicate the inactive substrate. Gray-shaded configuration is identical to that of L-arabinose.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2617
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

that the activity is not dependent on the C-5 and C-6 configuration.D-Galactose has the same configuration as L-arabinose at C-2, C-3, andC-4. The activity was also found with D-talose, C-1 epimer of D-galac-tose, but not with L-mannose and D-glucose, C-3 and C-4 epimer,respectively. Similarly, the activity with D-xylose, the C-4 epimer ofL-arabinose, decreased significantly, and no activity was observed withL-lyxose, the C-3 epimer of L-arabinose. It therefore appeared that theenzyme prefers the L-arabinose-specific configuration at C-3 and C-4.
It has been reported that the product of the enzyme reaction in thecell-free extract system is L-arabino-�-lactone by paper chromatogra-phy (10, 13, 14). We reinvestigated the product of the purified enzymeby HPLC as shown in Fig. 4. The retention time of L-arabino-�-lactonewas slightly earlier than that of L-arabinose, and the main product fromL-arabinose was L-arabino-�-lactone. The minor concomitant L-ar-abonate is probably due to spontaneous lactone hydrolysis (see Fig. 4B).These results indicated that the enzyme possessed only dehydrogenaseactivity with L-arabinose but not lactonase with L-arabino-�-lactone.
Cloning of the Gene Encoding L-Arabinose 1-Dehydrogenase and ItsFunctional Expression in E. coli—The N-terminal amino acid sequenceup to 15 amino acid residues of L-arabinose 1-dehydrogenase was asfollows: SDQVSLGVVGIGKIA. To determine the internal amino acidsequence, the purified enzyme was digested chemically by CNBr, whichdigests specifically at the C termini of methionyl residues in polypep-tides. As shown in Fig. 2B, two peptide bands with �26.4 and 0.92 kDaof molecular mass (referred to as fragments I and II, respectively) werefound on SDS-polyacrylamide gel. The 15-amino acid sequence of Frag-ment II was completely identical to the N-terminal sequence. Theamino acid sequence up to 15 amino acid residues of Fragment I was asfollows: (M)LEKPPGATLGEVAVL, suggesting that Fragment II islocated upstream of Fragment I in the protein sequences. Using syn-thetic DNA primers designed fromN-terminal and internal amino acidsequences, an �300-bp nucleotide fragment of the L-arabinose 1-dehy-drogenase gene was obtained by genomic PCR. Southern blot analysis
with this DNA fragment as a probe showed a single band on eachrestriction enzymedigestion ofA. brasiliense genomicDNAwith EcoRI,HindIII, NotI, PstI, SalI, and XbaI of �3.9, 15, 2.0, 4.5, 2.2, and 17 kbp(data not shown), revealing only a single copy of the gene on thegenome. Based on the results, we isolated a 1,805-bp NotI fragmentcontaining the full L-arabinose 1-dehydrogenase gene (Fig. 5). The G �C content of the entire sequence was significantly high (�69%), consist-ent with that of chromosomal DNA from A. brasiliense (70%) (37). TheL-arabinose 1-dehydrogenasewas 927 bp long, and a putative ribosome-binding site (Shine-Dalgarno sequence, AGGAG) was found 9–13bases upstreamof theATGcodon. The deduced amino acid sequence ofthe protein (polypeptide of 309 amino acids with a predicted mass of33,795.12 Da) agreed with the determined N-terminal amino acidsequences from the corresponding purified protein, except for removedformylmethionine. The internal amino acid sequences determined bythe digestion of purified protein with CNBr were found at positions89–103 of the protein sequences. The nucleotide sequence was submit-ted toGenBankTMwith accession numberAB211983. TwopartialORFswere found upstream and downstream of the L-arabinose 1-dehydro-genase gene (referred to as ORF1 and ORF2, respectively) (Fig. 5). ThededucedORF1 andORF2 proteins showed significant identitywith dihy-drodipicolinate synthase/N-acetylneuraminate lyase and periplasmicL-arabinose-binding proteins (involving in the ABC-type transport sys-tem) from many organisms, respectively.
L-Arabinose 1-dehydrogenase was overexpressed in E. coli cells byinduction with isopropyl-�-D-thiogalactopyranoside as a His6-taggedenzyme and purified homogeneously with a nickel-chelating affinitycolumn (Fig. 6A). The mobility of the recombinant enzyme in SDS-PAGE and zymogram-staining analysis (Fig. 6, A and C) was slightly
FIGURE 4. HPLC analysis of the reaction products. L-Arabinose (A), L-arabino-�-lactone(B), L-arabonate (C), and the enzyme reaction products (D) were loaded into an AminexHPX-87H Organic Analysis column and detected with a refractive index detector.
FIGURE 5. Comparison of a chromosomal region containing the L-arabinose 1-dehy-drogenase gene and its flanking region between A. brasiliense and B. capacia strainR18149. The NotI-NotI DNA fragment of A. brasiliense genome DNA was cloned in thisstudy.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
2618 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

later than that of the native enzyme purified from A. brasiliense cellsbecause of the additional 13 amino acid residues, including a His6 tag atthe N terminus. Western blot analysis with anti-His6 tag antibody con-firmed the His6 tag in the enzyme expressed in E. coli (Fig. 6B). Theenzyme showed similar NADP� preference and kinetic parameters forL-arabinose in the presence of NAD(P)� to those of the native enzyme(Tables 2 and 3), confirming that the isolated gene encodes L-arabinose1-dehydrogenase.
Amino Acid Sequence Analysis of L-Arabinose 1-Dehydrogenase—Protein-BLAST analysis revealed that L-arabinose 1-dehydrogenase is aputative member of the Gfo/Idh/MocA protein family, which includesGFOR (18–20), D-xylose 1-dehydrogenase (21), dimeric dihydrodioldehydrogenase (DD) (38), andmyo-inositol 2-dehydrogenase (39) (Fig.7A). The enzyme activity was not influenced by 1 mM EDTA (data notshown), supporting the hypothesis that the enzyme does not belong to amedium chain dehydrogenase/reductase family, which contains anactive zinc ion (see Introduction). It is noteworthy that GDH in thisfamily is a bifunctional enzyme with gluconolactonase (17, 23–25),whereas L-arabinose 1-dehydrogenase possesses no lactonase activitywith L-arabino-�-lactone (Fig. 4).
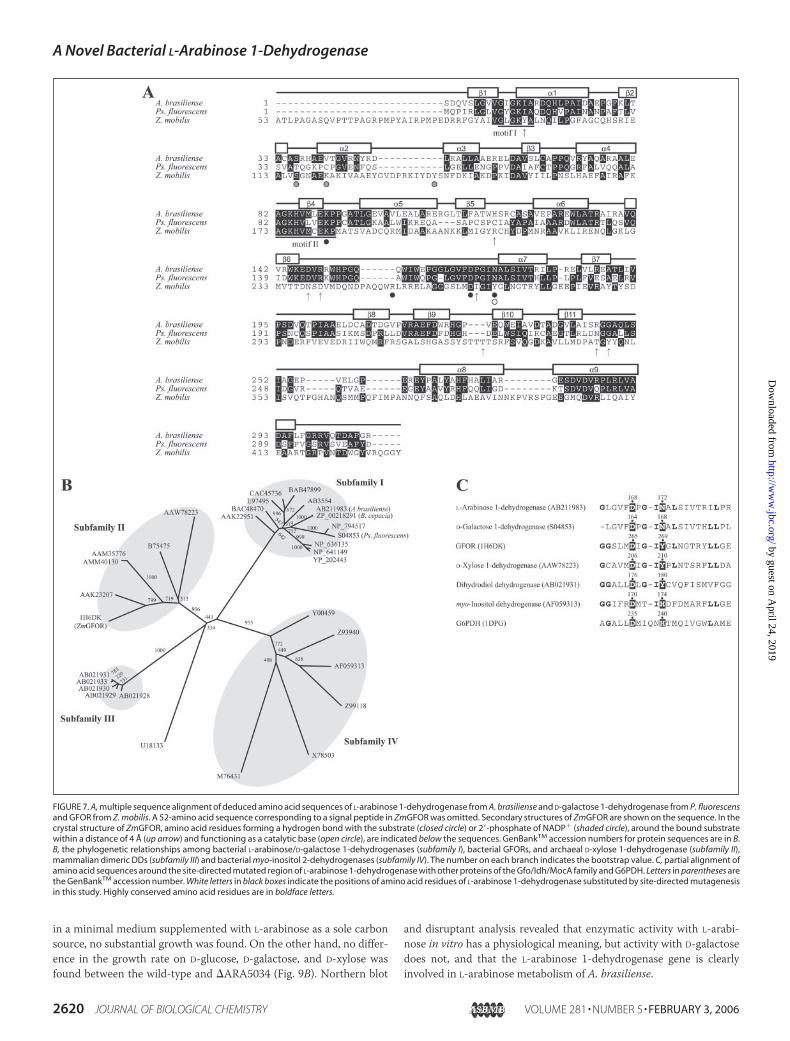
High degrees of similarity to L-arabinose 1-dehydrogenase (over 100bits of score) were found in bacterial (putative) oxidoreductases and
dehydrogenases, including D-galactose 1-dehydrogenase from Pseudo-monas fluorescens (22), confirming that L-arabinose 1-dehydrogenase isalso a D-galactose 1-dehydrogenase. In the unrooted phylogenetic tree,these enzymes containing L-arabinose 1-dehydrogenase formed a singlesubfamily (Fig. 7B, Subfamily I). L-Arabinose/D-galactose 1-dehydro-genase activities have been known to show NAD� preference (10, 13,14, 40–42). To our knowledge, this study is the first concerningNADP�-preferring L-arabinose and/or D-galactose 1-dehydrogenase.
Identification of Catalytic Amino Acid Residues—It is known thatAsp265 and Tyr269 in GFOR from Zymomonas mobilis (ZmGFOR) (20)and Asp176 and Tyr180 in dimeric DD (43, 44) are important for thecatalytic function. It was almost impossible to align the amino acidsequence of L-arabinose 1-dehydrogenase and ZmGFOR in the C-ter-minal domains because of weak sequential homology. Therefore, wecarried out multiple alignments with over 500 amino acid sequences ofGfo/Idh/MocA family enzymes, revealing thatAsp168–Asn172 in L-arab-inose 1-dehydrogenase corresponds to Asp265–Tyr269 in ZmGFOR andAsp176-Tyr180 in dimeric DD (Fig. 7,A andC). To obtain insight into thecatalytic mechanism of L-arabinose 1-dehydrogenase, Asp168 andAsn172 were substitutedwith alanine residues by site-directedmutagen-esis, as described under “Experimental Procedures,” to constructD168Aand N172A mutants, respectively. They were overexpressed in E. colicells as a His6-tagged enzyme and purified with the same procedures forthe wild-type enzyme (Fig. 6A). No activity of these mutants was foundin zymogram-staining analysis (Fig. 6B). Kinetic analysis showed thatthe N172A mutant decreased by 4 orders of magnitude in kcat/Km val-ues, comparedwith thewild-type enzyme (Table 3). TheD168Amutantshowed no activity under standard assay conditions. These results sug-gested that both Asp168 and Asn172 were important for catalytic func-tion in L-arabinose 1-dehydrogenase.
Expression of L-Arabinose 1-Dehydrogenase in A. brasiliense andMutant Analysis—L-Arabinose 1-dehydrogenase has been shown tohave dehydrogenase activities with other sugars such as D-galactose andD-xylose in vitro (Table 2). To estimate the physiological role of thesedehydrogenase activities, Northern blot analysis was carried out withRNAs isolated from A. brasiliense cells grown on different carbonsources as follows: L-arabinose, D-galactose, and D-xylose (active sub-strates in vitro) and D-glucose (inactive substrate in vitro) (Fig. 8A). Thesignificant expression of the L-arabinose 1-dehydrogenase gene wasonly found in cells grown on L-arabinose as a sole carbon source.Changes in L-arabinose 1-dehydrogenase activity in the cell-freeextracts prepared from cells grown on different carbon sources (Fig. 8,Band C) were analogous to those observed at the level of transcription.Furthermore, we constructed an A. brasiliense disruptant of the L-a-
rabinose 1-dehydrogenase gene, designated �ARA5034, by biparentalmating and double homologous recombination as described under“Experimental Procedures’’ (Fig. 9A). When �ARA5034 was cultivated
TABLE 3Kinetic parameters of recombinant L-arabinose 1-dehydrogenaseStocked L-arabinose 1-dehydrogenase was dialyzed against 100 mM Tris-HCl (pH 9.0) containing 2 mM MgCl2, overnight at 4 °C. Values are the means � S.D., n � 3.
Coenzyme Vmaxa Km kcat kcat/Km
units/mg protein mM min�1 min�1 mM�1
NAD� 14.9 � 0.1 0.785 � 0.048b 366 � 15b 467 � 10bNADP� 32.0 � 0.1 0.260 � 0.003b 989 � 11b 3810 � 10bNAD� 0.01 NDc ND NDNADP� 0.01 ND ND NDNAD� 0.042 � 0.001 42.5 � 2.7d 7.00 � 0.36d 0.164 � 0.002dNADP� 0.148 � 0.001 28.9 � 3.5d 19.7 � 1.9d 0.69 � 0.02d
a This was performed under standard assay conditions.b Ten different concentrations of L-arabinose between 0.1 and 1.0 mM were used.c No determination was made because of low activity.d Eleven different concentrations of L-arabinose between 1 and 100 mM were used.
FIGURE 6. SDS-PAGE, Western blot, and zymogram analysis of recombinant L-arab-inose 1-dehydrogenase. Lane 1, native enzyme purified from A. brasiliense cells; lanes2– 4, His6-tagged recombinant enzymes of wild-type (lane 2), D168A (lane 3), and N172A(lane 4); M, marker proteins. A, SDS-PAGE. Five �g of purified protein was applied on 12%(w/v) SDS-polyacrylamide gel. B, Western blot analysis. One �g of protein was electro-phoresed. Western blot analysis was carried out using anti-His6 antibody. C, zymogramstaining analysis. One �g of protein was applied. After electrophoresis, the gel wassoaked into staining solution in the presence of 100 mM L-arabinose and 1 mM NADP�.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2619
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
in a minimal medium supplemented with L-arabinose as a sole carbonsource, no substantial growth was found. On the other hand, no differ-ence in the growth rate on D-glucose, D-galactose, and D-xylose wasfound between the wild-type and �ARA5034 (Fig. 9B). Northern blot
and disruptant analysis revealed that enzymatic activity with L-arabi-nose in vitro has a physiological meaning, but activity with D-galactosedoes not, and that the L-arabinose 1-dehydrogenase gene is clearlyinvolved in L-arabinose metabolism of A. brasiliense.
FIGURE 7. A, multiple sequence alignment of deduced amino acid sequences of L-arabinose 1-dehydrogenase from A. brasiliense and D-galactose 1-dehydrogenase from P. fluorescensand GFOR from Z. mobilis. A 52-amino acid sequence corresponding to a signal peptide in ZmGFOR was omitted. Secondary structures of ZmGFOR are shown on the sequence. In thecrystal structure of ZmGFOR, amino acid residues forming a hydrogen bond with the substrate (closed circle) or 2�-phosphate of NADP� (shaded circle), around the bound substratewithin a distance of 4 Å (up arrow) and functioning as a catalytic base (open circle), are indicated below the sequences. GenBankTM accession numbers for protein sequences are in B.B, the phylogenetic relationships among bacterial L-arabinose/D-galactose 1-dehydrogenases (subfamily I), bacterial GFORs, and archaeal D-xylose 1-dehydrogenase (subfamily II),mammalian dimeric DDs (subfamily III) and bacterial myo-inositol 2-dehydrogenases (subfamily IV). The number on each branch indicates the bootstrap value. C, partial alignment ofamino acid sequences around the site-directed mutated region of L-arabinose 1-dehydrogenase with other proteins of the Gfo/Idh/MocA family and G6PDH. Letters in parentheses arethe GenBankTM accession number. White letters in black boxes indicate the positions of amino acid residues of L-arabinose 1-dehydrogenase substituted by site-directed mutagenesisin this study. Highly conserved amino acid residues are in boldface letters.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
2620 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
In this study, we reported a novel L-arabinose 1-dehydrogenaseinvolved in an alternative pathway of L-arabinosemetabolism. The puri-fied enzyme showed significant NADP�-preferring activity not onlywith L-arabinose but also D-galactose. The gene expression was onlyinduced by L-arabinose, suggesting that the physiological role is limitedin L-arabinose metabolism. L-Arabinose 1-dehydrogenase was a poten-tial member of the Gfo/Idh/MocA protein family and contained someunique catalytic amino acid residues.
Catalytic Insight into L-Arabinose 1-Dehydrogenase—Generally, sub-unit structures of the Gfo/Idh/MocA family are comprised of twodomains, the N-terminal and C-terminal domains. The N-terminaldomain (�120 amino acid residues) has a characteristic GXGXX(G/A)fingerprint motif (45) in the classical ��� dinucleotide binding pocket(46) and a recently postulated fingerprint motif for a novel class ofdehydrogenases with a consensus AGKHVXCEKP (where X is anyamino acid) (19). These motifs are also conserved in L-arabinose 1-de-hydrogenase with little deviation (Fig. 7A,motifs I and II). The C-termi-nal domain of Gfo/Idh/MocA family enzymes (�220 amino acid resi-dues) shows significant sequential divergence and contains someessential amino acid residues for enzyme catalysis and/or substratebinding (Fig. 7A, open and closed circles, respectively), probably becauseof inherent substrate specificity in each enzyme. The structural featuresand catalyticmechanisms ofZmGFORhave been extensively studied byenzymatic and crystallographic analysis (18–20). The enzyme containstightly bound coenzyme NADP�, and the strict NADP� specificity isrationalized by Ser116, Lys121, and Tyr139 (Fig. 7A, shaded circle) (47). Inparticular, the serine residue is important to distinguish betweenNAD�
and NADP�; the S116D mutant of ZmGFOR shows significant activitywithNAD�, andNAD�-dependentmyo-inositol 2-dehydrogenase pos-
sesses aspartate residue at the corresponding position (19).On the otherhand, NAD�-preferring D-galactose 1-dehydrogenase from P. fluore-scens (22, 41) possesses threonine residue but not aspartate at the cor-responding position to Ser116. No L-arabinose/D-galactose 1-dehydro-genase has amino acid residues equivalent to Lys121 and Tyr139. Thesecomparisons indicate that there are several modifications for determin-ing the coenzyme specificity between L-arabinose/D-galactose 1-dehy-drogenases and other Gfo/Idh/MocA family enzymes. Furthermore,compared with ZmGFOR, almost all putative amino acid residuesaround the substrate binding regions are substituted for those withfunctionally different characteristics in L-arabinose 1-dehydrogenase(Fig. 7A, up arrow), clearly creating a different environment at the activecenter.
ANovel Catalyst Type in L-Arabinose 1-Dehydrogenase—Glucose-6-phosphate dehydrogenase (G6PDH) is a structurally homologousenzyme with GFOR and has a very strong functional relationship withGFOR. In Leuconostoc mesenteroidesG6PDH (LmG6PDH) (48, 49), thecatalytic base (His240) removes a proton from the C-1 OH group ofGlc-6-P, andNAD(P)� abstracts a hydride ion (H�) from the C-1 atom.Asp235 forms a hydrogen bond with C-2 OH of Glc-6-P. In ZmGFOR,the structurally homologous Asp265–Tyr269 residues play equivalentroles (Fig. 7,A andC) (20). In enzymes of the Gfo/Idh/MocA family, thepair of Asp-(Tyr/His) is conserved completely (Fig. 7C). Multiple align-ments revealed that Asp168–Asn172 in L-arabinose 1-dehydrogenasecorresponds to Asp265–Tyr269 in ZmGFOR and Asp235–His240 inLmG6PDH (Fig. 7,A andC). It is very interesting to identify the involve-ment of Asn172 in the catalytic function because the LmG6PDHH240Nmutant shows a decreased kcat value by 4 orders of magnitude (48) anda 10-fold higher Km value for Glc-6-P. Therefore, we constructed twomutants of L-arabinose 1-dehydrogenase, D168A and N172A, by site-directed mutagenesis. When Asp168 was replaced with Ala, a D168Amutant of L-arabinose 1-dehydrogenase lacked sufficient activity fordetection in our standard assay conditions and zymogram staining anal-ysis (Table 3 and Fig. 6C). On the other hand, the N172A mutantretained some enzymatic activity (less than 1% of kcat/Km of the wild-type enzyme) (Table 3), whereas the activity was too weak to detect inzymogram staining analysis (Fig. 6C). These results suggest that Asn172
has a very important role in catalysis but is not absolutely necessary.This site-directed mutagenesis indicates that Asp168 and Asn172 aresurely involved in the catalytic function of L-arabinose 1-dehydrogen-ase, raising doubt as to whether Asn172 is a catalytic base. In the crystalstructure of wild-type LmG6PDH, the distance betweenAsp235 and C-1OHofGlc-6-P is 4.8Å,which is only slightly distant for a hydrogen bond(49). Considering that Asp168 in L-arabinose 1-dehydrogenase corre-sponds to Asp235 in LmG6PDH, we speculate that Asp168 functions as acatalytic base in L-arabinose 1-dehydrogenase. This novel mechanismevolved early from an ancestral Gfo/Idh/MocA enzyme possessing apair of Asp-(Tyr/His), such as ZmGFOR, because the asparagine resi-due equivalent to Asn172 in L-arabinose 1-dehydrogenase is only foundin the enzymes of subfamily I (Fig. 7, B and C).
Physiological Insight of L-Arabinose Metabolism in A. brasiliense—Wecould find many L-arabinose 1-dehydrogenase gene homologs on the bac-terial genomesequence (Fig. 7B,Subfamily I). It isnoteworthy that allmem-bers are from bacteria associating pathologically and/or parasitically withplants. In particular, the nucleotide sequences of the L-arabinose 1-dehy-drogenase gene and the flanking region ofA. brasiliense show high similar-ity (�91%) to part of the genomic sequence of Burkholderia cepacia strainR18149 (Fig. 5), whichwas isolated from forest soil. On the other hand, thecomparison of rRNA sequences revealed their far phylogenic relationship(�77% of identity) (data not shown). These results suggest that wide range
FIGURE 8. Effect of a carbon source on the intercellular expression level of L-arabi-nose 1-dehydrogenase. A, Northern blot analysis. Total RNAs (4 �g per lane) wereisolated from A. brasiliense cells grown in nutrient medium (lane 1) and synthetic mediumcontaining L-arabinose (lane 2), D-galactose (lane 3), D-xylose (lane 4), and D-glucose (lane5) at concentrations of 37 mM. B, zymogram-staining analysis using cell-free extracts.Fifty �g of protein was applied. After electrophoresis, the gel was soaked into stainingsolution in the presence of 100 mM L-arabinose and 1 mM NADP�. N is a purified nativeL-arabinose 1-dehydrogenase (1 �g). C, enzyme activity in cell-free extracts. L-Arabinose1-dehydrogenase activity was measured under standard assay conditions using NADP�
as a coenzyme. Values are the means � S.D., n � 3.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2621
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
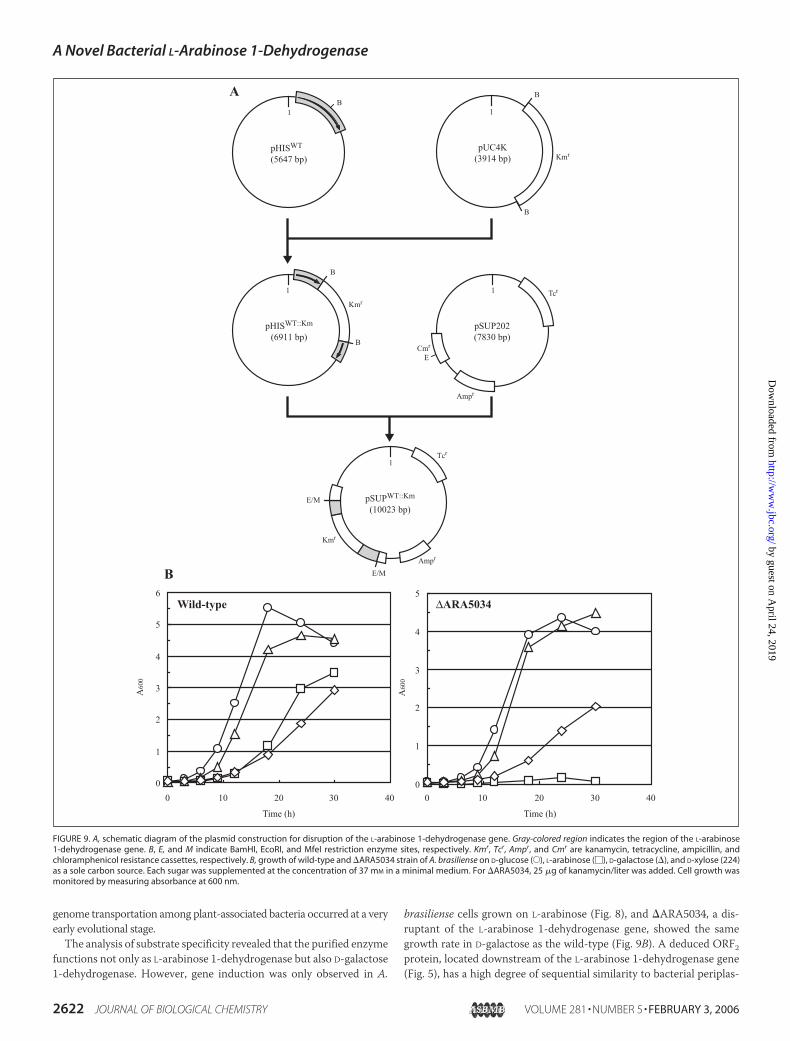
genome transportation among plant-associated bacteria occurred at a veryearly evolutional stage.The analysis of substrate specificity revealed that the purified enzyme
functions not only as L-arabinose 1-dehydrogenase but also D-galactose1-dehydrogenase. However, gene induction was only observed in A.
brasiliense cells grown on L-arabinose (Fig. 8), and �ARA5034, a dis-ruptant of the L-arabinose 1-dehydrogenase gene, showed the samegrowth rate in D-galactose as the wild-type (Fig. 9B). A deduced ORF2protein, located downstream of the L-arabinose 1-dehydrogenase gene(Fig. 5), has a high degree of sequential similarity to bacterial periplas-
FIGURE 9. A, schematic diagram of the plasmid construction for disruption of the L-arabinose 1-dehydrogenase gene. Gray-colored region indicates the region of the L-arabinose1-dehydrogenase gene. B, E, and M indicate BamHI, EcoRI, and MfeI restriction enzyme sites, respectively. Kmr, Tcr, Ampr, and Cmr are kanamycin, tetracycline, ampicillin, andchloramphenicol resistance cassettes, respectively. B, growth of wild-type and �ARA5034 strain of A. brasiliense on D-glucose (E), L-arabinose (�), D-galactose (�), and D-xylose (224)as a sole carbon source. Each sugar was supplemented at the concentration of 37 mM in a minimal medium. For �ARA5034, 25 �g of kanamycin/liter was added. Cell growth wasmonitored by measuring absorbance at 600 nm.
A Novel Bacterial L-Arabinose 1-Dehydrogenase
2622 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 5 • FEBRUARY 3, 2006
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

mic L-arabinose-binding proteins, which belong to ATP-binding cas-sette (ABC)-type sugar transporters. Generally, this type of bacterialtransporter consists of a periplasmic substrate-binding protein, anATP-binding complex (ATPase), and a membrane-spanning protein(permease). On the genome of B. cepacia strain R18149, the corre-sponding three genes are clustered downstream of the putative L-arab-inose 1-dehydrogenase gene (Fig. 5). This may also appear in A. brasil-iense, considering the sequence similarity of these two organisms. Theenzymatic and genetic evidence indicates that the L-arabinose 1-dehy-drogenase gene is only involved in themetabolic pathway of L-arabinoseand not D-galactose. Recently, Moore et al. (50) reported that twohomologous genes to a putative transcriptional regulator(ZP_00218288) and dehydrogenase (ZP_00218291) of B. cepacia strainR18149 (Fig. 5) are absolutely necessary for growth on L-arabinose inBurkholderia thiailandensis, a close relative bacteria of B. cepacia strainR18149; deletion mutants of one of these two genes showed an L-arab-inose-negative phenotype as well as �ARA5034 (Fig. 9B). It should benoted that these two Burkholderia strains possess the same L-arabinosemetabolic pathway as A. brasiliense, and the clustered genes, includingL-arabinose 1-dehydrogenase, comprise a transcriptional unit.
Van Bastelaere et al. (51) identified a plant root exudate-inducible40-kDa protein from A. brasiliense (designated as SbpA (sugar-bindingprotein A)). The deduced amino acid sequences are very similar tovirulent ChvE protein from Agrobacterium tumefaciens, a putativeperiplasmic ABC-type transporter (52). The SbpA protein is involved inthe uptake of D-galactose, L-arabinose, and D-fructose, whereas residualuptake of these sugars is found in the sbpA mutant. These findingssuggest the existence of multiple transport systems in A. brasiliense.Because the deduced ORF2 protein from A. brasiliense shows no simi-larity to SbpA, it can be speculated that this protein is an alternativeL-arabinose transporter(s). Further investigation of the region sur-rounding the L-arabinose 1-dehydrogenase gene is important for theelucidation of this alternative L-arabinose metabolism.
Acknowledgments—We thankDr. KiwamuMinamisawa (TohokuUniversity)and Dr. Masayuki Inui (Research Institute of Innovative Technology for theEarth) for the gifts of the plasmid pSUP202 and E. coli S17-1, respectively. Wealso thank Dr. Makoto Hidaka (Tokyo University) for technical advice in theconstruction of A. brasiliense disruptant. We are grateful to Dr. YasuhiroTakada and Tomohiro Hirose (Hokkaido University) for their help in thedetermination of amino acid sequences.
REFERENCES1. Zaldivar, J., Nielsen, J., and Olsson, L. (2001) Appl. Microbiol. Biotechnol. 56, 17–342. Stryer, L. (1988)Biochemistry, 3rd Ed., pp. 805–807,W.H. Freeman&Co., NewYork3. Richard, P., Londesborough, J., Putkonen, M., Kalkkinen, N., and Penttila, M. (2001)
J. Biol. Chem. 276, 40631–406374. Richard, P., Putkonen, M., Vaananen, R., Londesborough, J., and Penttila, M. (2002)
Biochemistry 41, 6432–64375. Verho, R., Putkonen, M., Londesborough, J., Penttila, M., and Richard, P. (2004)
J. Biol. Chem. 279, 14746–147516. Welmberg, R., and Doudoroff, M. (1955) J. Biol. Chem. 217, 607–6247. Welmberg, R. (1961) J. Biol. Chem. 236, 629–6358. Dagley, S., and Trudgill, P. W. (1965) Biochem. J. 95, 48–589. Dahms, A. S., and Anderson, R. L. (1969) Biochem. Biophys. Res. Commun. 36,
809–81410. Pedrosa, F. O., and Zancan, G. T. (1974) J. Bacteriol. 119, 336–33811. Duncan, M. J. (1979) J. Gen. Microbiol. 113, 177–17912. Duncan, M. J., and Fraenkel, D. G. (1979) J. Bacteriol. 137, 415–419
13. Novick, N. J., and Tyler, M. E. (1982) J. Bacteriol. 149, 364–36714. Mathias, A. L., Rigo, L. U., Funayama, S., and Pedrosa, F. O. (1989) J. Bacteriol. 171,
5206–520915. Kim, S. T., Huh,W.K., Lee, B. H., andKang, S. O. (1998)Biochim. Biophys. Acta 1429,
29–3916. Edwards, K. J., Barton, J. D., Rossjohn, J., Thorn, J. M., Taylor, G. L., and Ollis, D. L.
(1996) Arch. Biochem. Biophys. 328, 173–18317. Lamble, H. J., Heyer, N. I., Bull, S. D., Hough, D. W., and Danson, M. J. (2003) J. Biol.
Chem. 278, 34066–3407218. Zachariou, M., and Scopes, R. K. (1986) J. Bacteriol. 167, 863–86919. Wiegert, T., Sahm, H., and Sprenger, G. A. (1997) J. Biol. Chem. 272, 13126–1313320. Nurizzo, D., Halbig, D., Sprenger, G. A., and Baker, E. N. (2001) Biochemistry 40,
13857–1386721. Johnsen, U., and Schonheit, P. (2004) J. Bacteriol. 186, 6198–620722. Sperka, S., Zehelein, E., Fiedler, S., Fischer, S., Sommer, R., and Buckel, P. (1989)
Nucleic Acids Res. 17, 540223. Bonete, M. J., Pire, C., Llorca, F. I., and Camacho, M. L. (1996) FEBS Lett. 383,
227–22924. Selig, M., Xavier, K. B., Santos, H., and Schonheit, P. (1997) Arch. Microbiol. 167,
217–23225. Angelov, A., Futterer, O., Valerius, O., Braus, G. H., and Liebl, W. (2005) FEBS J. 272,
1054–106226. Elzainy, T. A., Hassan, M. M., and Allam, A. M. (1973) J. Bacteriol. 114, 457–45927. Lamble, H. J., Milburn, C. C., Taylor, G. L., Hough, D. W., and Danson, M. J. (2004)
FEBS Lett. 576, 133–13628. Theodossis, A., Walden, H., Westwick, E. J., Connaris, H., Lamble, H. J., Hough,
D. W., Danson, M. J., and Taylor, G. L. (2004) J. Biol. Chem. 279, 43886–4389229. Laemmli, U. K. (1970) Nature 227, 680–68530. Lowry,O.H., Rosebrough,N. J., Farr, A. L., and Randall, R. J. (1951) J. Biol. Chem. 193,
265–27531. Gennady, P.M. (2002)Handbook of Detection of Enzymes on Electrophoretic Gels, 2nd
Ed., pp. 27–33, CRC Press, Inc., Boca Raton, FL32. Moore, S., and Link, K. P. (1940) J. Biol. Chem. 133, 293–31133. Gross, E. (1967)Methods Enzymol. 11, 238–25534. Sambrook, J., Fritsch, E. F., and Maniatis, T. (2001)Molecular Cloning: A Laboratory
Manual, 3rd Ed., pp. 6.33–7.45, Cold Spring Harbor Laboratory Press, Cold SpringHarbor, NY
35. Penning, T. M., and Jez, J. M. (2001) Chem. Rev. 101, 3027–304636. Simon, R., Priefer, U., and Puhler, A. (1983) Bio/Technology 1, 789–79137. Tarrand, J. J., Krieg, N. R., and Dobereiner, J. (1978) Can. J. Microbiol. 24, 967–98038. Arimitsu, E., Aoki, S., Ishikura, S., Nakanishi, K., Matsuura, K., and Hara, A. (1999)
Biochem. J. 342, 721–72839. Galbraith, M. P., Feng, S. F., Borneman, J., Triplett, E. W., de Bruijn, F. J., and Ross-
bach, S. (1998)Microbiology 144, 2915–292440. Wengenmayer, F., Ueberschar, K. H., Kurz, G., and Sund, H. (1973) Eur. J. Biochem.
40, 49–6141. Blachnitzky, E. O., Wengenmayer, F., and Kurz, G. (1974) Eur. J. Biochem. 47,
235–25042. Elshafei, A. M., and Abdel-Fatah, O. M. (2001) Enzyme Microb. Technol. 29, 76–8343. Asada, Y., Aoki, S., Ishikura, S. Usami, N., and Hara, A. (2000) Biochem. Biophys. Res.
Commun. 278, 333–33744. Aoki, S., Ishikura, S., Asada, Usami, N., and Hara, A. (2001)Chem. Biol. Interact. 130,
775–78445. Wierenga, R. K., De Maeyer, M. C. H., and Hol, W. G. J. (1985) Biochemistry 24,
1346–135746. Rossmann, M. G., Liljas, A., Branden, C.-I., and Banaszak, L. J. (1975) in The Enzymes
(Boyer, P. D., ed) 3rd Ed., Vol. 11, pp. 61–102, Academic Press, New York47. Lott, J. S., Halbig, D., Baker, H. M., Hardman, M. J., Sprenger, G. A., and Baker, E. N.
(2000) J. Mol. Biol. 304, 575–58448. Cosgrove, M. S., Naylor, C., Paludan, S., Adams, M. J., and Levy, H. R. (1998) Bio-
chemistry 37, 2759–276749. Cosgrove, M. S., Gover, S., Naylor, C. E., Vandeputte-Rutten, L., Adams, M. J., and
Levy, H. R. (2000) Biochemistry 39, 15002–1501150. Moore, R. A., Reckseidler-Zenteno, S., Kim, H., Nierman, W., Yu, Y., Tuanyok, A.,
Warawa, J., DeShazer, D., and Woods, D. E. (2004) Infect. Immun. 72, 4172–418751. Van Bastelaere, E., Lambrecht, M., Vermeiren, H., Van Dommelen, A., Keijers, V.,
Proost, P., and Vanderleyden, J. (1999)Mol. Microbiol. 32, 703–71452. Kemner, J. M., Liang, X., and Nester, E. W. (1997) J. Bacteriol. 179, 2452–2458
A Novel Bacterial L-Arabinose 1-Dehydrogenase
FEBRUARY 3, 2006 • VOLUME 281 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 2623
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Seiya Watanabe, Tsutomu Kodak and Keisuke Makino1-Dehydrogenase Involved in an Alternative Pathway of l-Arabinose Metabolism
Cloning, Expression, and Characterization of Bacterial l-Arabinose
doi: 10.1074/jbc.M506477200 originally published online December 2, 20052006, 281:2612-2623.J. Biol. Chem.
10.1074/jbc.M506477200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/281/5/2612.full.html#ref-list-1
This article cites 46 references, 19 of which can be accessed free at
by guest on April 24, 2019
http://ww
w.jbc.org/
Dow
nloaded from
VOLUME 280 (2005) 10001–10010
Activation of mitogen-activated protein kinase kinase (MKK) 3 and MKK6 by type I interferons.Yongzhong Li, Sandeep Batra, Antonella Sassano, Beata Majchrzak, David E. Levy, Matthias Gaestel, Eleanor N. Fish, Roger J. Davis, and Leonidas C. Platanias
PAGE 10005:
Due to an inadvertent error, the wrong immunoblots were included in Fig. 8, E and F. Fig. 8 should appear as shown below. The figure legend andtext remain unchanged.
FIGURE 8
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 15, pp. 10651–10652, April 14, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
APRIL 14, 2006 • VOLUME 281 • NUMBER 15 JOURNAL OF BIOLOGICAL CHEMISTRY 10651
ADDITIONS AND CORRECTIONS This paper is available online at www.jbc.org
We suggest that subscribers photocopy these corrections and insert the photocopies in the original publication at the location of the originalarticle. Authors are urged to introduce these corrections into any reprints they distribute. Secondary (abstract) services are urged to carrynotice of these corrections as prominently as they carried the original abstracts.

VOLUME 280 (2005) PAGES 19594 –19599
Vertebrate nonmuscle myosin II isoforms rescue smallinterfering RNA-induced defects in COS-7 cellcytokinesis.Jianjun Bao, Siddhartha S. Jana, and Robert S. Adelstein
PAGES 19594 –19595:
Under “Experimental Procedures,” subheading “DNA Constructsand RNAOligonucleotides,” the last sentence that begins on page 19594is incorrect. It should read as follows: “NMHC II-B siRNA duplexes(directed at both the human, RefSeq accession number NM_005964,and monkey sequence, AAGGAUCGCUACUAUUCAGGA) andNMHC II-C siRNA (directed at the human C1-inserted sequence,UCCGUCAGCACCGUGUCUUAU (S. S. Jana, unpublished data))were chemically synthesized by Dharmacon, Inc. (Lafayette, CO) andQiagen (Valencia, CA), respectively.”
VOLUME 281 (2006) 2612–2623
Cloning, expression, and characterization of bacterialL-arabinose 1-dehydrogenase involved in an alternativepathway of L-arabinose metabolism.Seiya Watanabe, Tsutomu Kodaki, and Keisuke Makino
PAGE 2612:
Dr. Kodaki’s last name was misspelled. The correct spelling is shownabove.
Additions and Corrections
10652 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 15 • APRIL 14, 2006


















