
Clinical Review - FluLaval and FluLaval Quadrivalent · Clinical Reviewer: Sarah K. Browne, MD STN:...
43
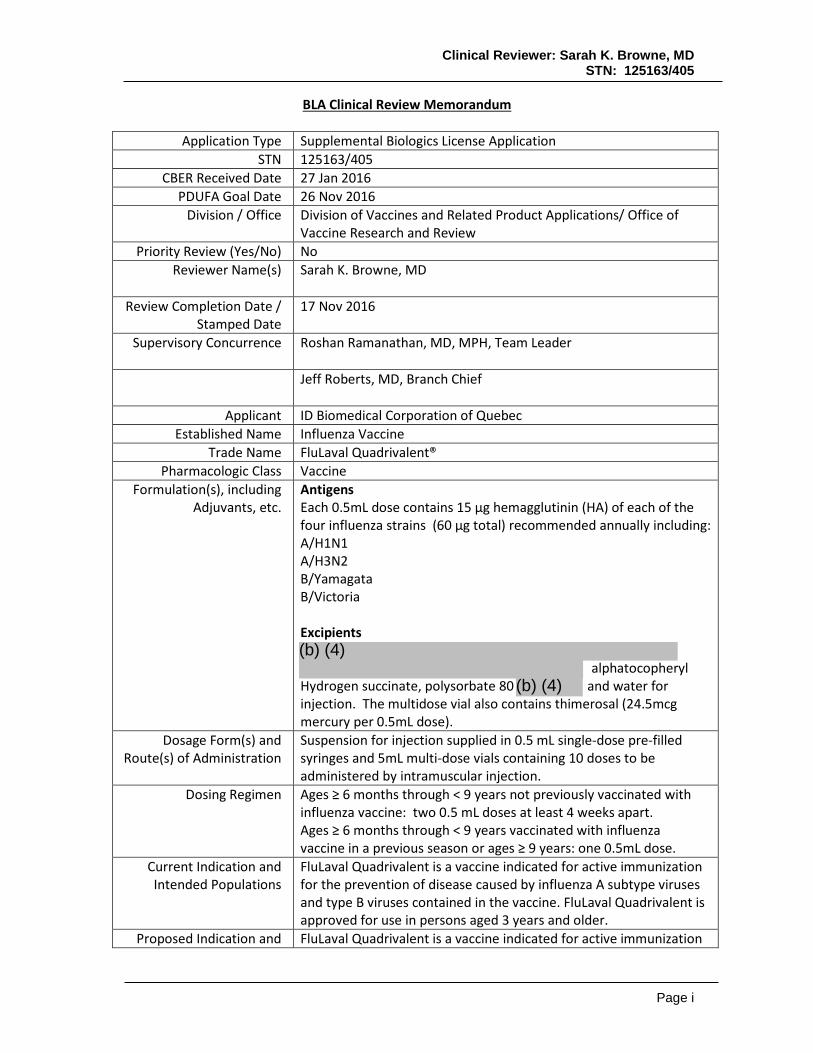
Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405 Page i BLA Clinical Review Memorandum Application Type Supplemental Biologics License Application STN 125163/405 CBER Received Date 27 Jan 2016 PDUFA Goal Date 26 Nov 2016 Division / Office Division of Vaccines and Related Product Applications/ Office of Vaccine Research and Review Priority Review (Yes/No) No Reviewer Name(s) Sarah K. Browne, MD Review Completion Date / Stamped Date 17 Nov 2016 Supervisory Concurrence Roshan Ramanathan, MD, MPH, Team Leader Jeff Roberts, MD, Branch Chief Applicant ID Biomedical Corporation of Quebec Established Name Influenza Vaccine Trade Name FluLaval Quadrivalent® Pharmacologic Class Vaccine Formulation(s), including Adjuvants, etc. Antigens Each 0.5mL dose contains 15 µg hemagglutinin (HA) of each of the four influenza strains (60 µg total) recommended annually including: A/H1N1 A/H3N2 B/Yamagata B/Victoria Excipients alphatocopheryl Hydrogen succinate, polysorbate 80 and water for injection. The multidose vial also contains thimerosal (24.5mcg mercury per 0.5mL dose). Dosage Form(s) and Route(s) of Administration Suspension for injection supplied in 0.5 mL single-dose pre-filled syringes and 5mL multi-dose vials containing 10 doses to be administered by intramuscular injection. Dosing Regimen Ages ≥ 6 months through < 9 years not previously vaccinated with influenza vaccine: two 0.5 mL doses at least 4 weeks apart. Ages ≥ 6 months through < 9 years vaccinated with influenza vaccine in a previous season or ages ≥ 9 years: one 0.5mL dose. Current Indication and Intended Populations FluLaval Quadrivalent is a vaccine indicated for active immunization for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine. FluLaval Quadrivalent is approved for use in persons aged 3 years and older. Proposed Indication and FluLaval Quadrivalent is a vaccine indicated for active immunization (b) (4) (b) (4)
Transcript of Clinical Review - FluLaval and FluLaval Quadrivalent · Clinical Reviewer: Sarah K. Browne, MD STN:...
Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page i
BLA Clinical Review Memorandum
Application Type Supplemental Biologics License Application STN 125163/405
CBER Received Date 27 Jan 2016 PDUFA Goal Date 26 Nov 2016
Division / Office Division of Vaccines and Related Product Applications/ Office of Vaccine Research and Review
Priority Review (Yes/No) No Reviewer Name(s) Sarah K. Browne, MD
Review Completion Date /
Stamped Date 17 Nov 2016
Supervisory Concurrence Roshan Ramanathan, MD, MPH, Team Leader
Jeff Roberts, MD, Branch Chief
Applicant ID Biomedical Corporation of Quebec Established Name Influenza Vaccine
Trade Name FluLaval Quadrivalent® Pharmacologic Class Vaccine
Formulation(s), including Adjuvants, etc.
Antigens Each 0.5mL dose contains 15 µg hemagglutinin (HA) of each of the four influenza strains (60 µg total) recommended annually including: A/H1N1 A/H3N2 B/Yamagata B/Victoria Excipients
alphatocopheryl
Hydrogen succinate, polysorbate 80 and water for injection. The multidose vial also contains thimerosal (24.5mcg mercury per 0.5mL dose).
Dosage Form(s) and Route(s) of Administration
Suspension for injection supplied in 0.5 mL single-dose pre-filled syringes and 5mL multi-dose vials containing 10 doses to be administered by intramuscular injection.
Dosing Regimen Ages ≥ 6 months through < 9 years not previously vaccinated with influenza vaccine: two 0.5 mL doses at least 4 weeks apart. Ages ≥ 6 months through < 9 years vaccinated with influenza vaccine in a previous season or ages ≥ 9 years: one 0.5mL dose.
Current Indication and Intended Populations
FluLaval Quadrivalent is a vaccine indicated for active immunization for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine. FluLaval Quadrivalent is approved for use in persons aged 3 years and older.
Proposed Indication and FluLaval Quadrivalent is a vaccine indicated for active immunization
(b) (4)
(b) (4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page ii
Intended Polulations for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine. FluLaval Quadrivalent is approved for use in persons aged 6 months and older.
Orphan Designated (Yes/No)
No
TABLE OF CONTENTS
GLOSSARY ....................................................................................................................... 3
1. EXECUTIVE SUMMARY ................................................................................................... 4
1.1 Demographic Information: Subgroup Demographics and Analysis Summary .................................. 6
2. CLINICAL AND REGULATORY BACKGROUND ................................................................... 6
2.1 Disease Studied ................................................................................................................................. 6 2.2 Currently Available, Pharmacologically Unrelated Treatment(s)/Intervention(s) for the Proposed
Indication(s) ..................................................................................................................................... 7 2.3 Safety and Efficacy of Pharmacologically Related Products .............................................................. 7 2.4 Previous Human Experience with the Product (Including Foreign Experience) ................................ 8 2.5 Summary of Pre- and Post-submission Regulatory Activity Related to the Submission ................... 8
3. SUBMISSION QUALITY AND GOOD CLINICAL PRACTICES ................................................. 9
3.1 Submission Quality and Completeness ............................................................................................. 9 3.2 Compliance With Good Clinical Practices And Submission Integrity ................................................. 9 3.3 Financial Disclosures.......................................................................................................................... 9
4. SIGNIFICANT EFFICACY/SAFETY ISSUES RELATED TO OTHER REVIEW DISCIPLINES ........ 11
4.1 Chemistry, Manufacturing, and Controls ........................................................................................ 11 4.2 Assay Validation .............................................................................................................................. 11 4.3 Nonclinical Pharmacology/Toxicology ............................................................................................. 11 4.4 Clinical Pharmacology ..................................................................................................................... 11
4.4.1 Mechanism of Action ............................................................................................... 11 4.5 Statistical ......................................................................................................................................... 11 4.6 Pharmacovigilance .......................................................................................................................... 11
5. SOURCES OF CLINICAL DATA AND OTHER INFORMATION CONSIDERED IN THE REVIEW ... 12
5.1 Review Strategy ............................................................................................................................... 12 5.2 BLA/IND Documents That Serve as the Basis for the Clinical Review ............................................. 12 5.3 Table of Studies/Clinical Trials ......................................................................................................... 12 5.4 Consultations ................................................................................................................................... 13 5.5 Literature Reviewed ........................................................................................................................ 13
6. DISCUSSION OF INDIVIDUAL STUDIES/CLINICAL TRIALS ................................................. 14
6.1 Study FLU Q-QIV-022 ....................................................................................................................... 14 6.1.1 Objectives ................................................................................................................ 14 6.1.2 Design Overview ..................................................................................................... 15 6.1.3 Population ................................................................................................................ 16 6.1.4 Study Treatments or Agents Mandated by the Protocol ......................................... 17 6.1.5 Directions for Use .................................................................................................... 18

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page iii
6.1.6 Sites and Centers .................................................................................................... 18 6.1.7 Surveillance/Monitoring ........................................................................................... 18 6.1.8 Endpoints and Criteria for Study Success ............................................................... 19 6.1.9 Statistical Considerations & Statistical Analysis Plan ............................................. 20 6.1.10 Study Population and Disposition .......................................................................... 21 6.1.11 Efficacy Analyses .................................................................................................. 23 6.1.12 Safety Analyses ..................................................................................................... 27 6.1.13 Study Summary and Conclusions ......................................................................... 33
6.2 Trial #2 FLU Q-QIV-021 .................................................................................................................... 33 6.3 Trial #3 FLU Q-QIV-013 .................................................................................................................... 35 6.4 Trial #4 FLU Q-QIV-003 .................................................................................................................... 37
7. INTEGRATED OVERVIEW OF EFFICACY .......................................................................... 38
8. INTEGRATED OVERVIEW OF SAFETY ............................................................................. 38
8.1 Safety Assessment Methods ........................................................................................................... 38 8.2 Safety Database ............................................................................................................................... 38 8.3 Caveats Introduced by Pooling of Data Across Studies ................................................................... 38 8.4 Safety Results .................................................................................................................................. 38
8.4.1 Deaths ..................................................................................................................... 38 8.4.2 Nonfatal Serious Adverse Events ............................................................................ 38 8.4.3 Study Dropouts/Discontinuations ............................................................................ 39 8.4.4 Common Adverse Events ........................................................................................ 39 8.4.6 Systemic Adverse Events ........................................................................................ 39 8.4.7 Local Reactogenicity ............................................................................................... 39 8.4.8 Potentially Immune Mediated Diseases .................................................................. 39
8.6 Safety Conclusions ........................................................................................................................... 39
9. ADDITIONAL CLINICAL ISSUES ..................................................................................... 40
9.1 Special Populations ......................................................................................................................... 40 9.1.1 Human Reproduction and Pregnancy Data............................................................. 40 9.1.2 Use During Lactation ............................................................................................... 40 9.1.3 Pediatric Use and PREA Considerations ................................................................ 40
10. CONCLUSIONS .......................................................................................................... 40
11. RISK-BENEFIT CONSIDERATIONS AND RECOMMENDATIONS ......................................... 40
11.1 Risk-Benefit Considerations........................................................................................................... 40 11.2 Risk-Benefit Summary and Assessment ........................................................................................ 42 11.4 Recommendations on Regulatory Actions .................................................................................... 42 11.5 Labeling Review and Recommendations ....................................................................................... 42 11.6 Recommendations on Postmarketing Actions .............................................................................. 43
GLOSSARY
CI Confidence interval FDA Food and Drug Administration GMT geometric mean titers HA Hemagglutinin antigen HI Hemagglutinin inhibition LL Lower limit QIV Quadrivalent influenza vaccine

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 4
SCR Seroconversion rate sBLA Supplemental Biologics Licensing Application TIV Trivalent influenza vaccine UL Upper limit USPI United States package insert
1. Executive Summary
A supplemental Biologics License Application (sBLA) was submitted by GlaxoSmithKline Biologicals (GSK) to the Food and Drug Administration (FDA) for a seasonal quadrivalent split-virion, inactivated influenza virus vaccine (FluLaval Quadrivalent). The vaccine includes a total dose of 60 µg (15 µg per strain) of hemagglutinin antigen (HA) prepared from virus propagated in the allantoic cavity of embryonated hens’ eggs. The product is currently approved for active immunization for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine in persons ages 3 years and older. The sBLA is intended to extend the indication to include persons ages 6 months and older. The sBLA includes immunogenicity and safety data from one phase 3 clinical trial conducted in children ages > 6 to < 36 months (study FLU Q-QIV-022), which was designed to provide data to support approval for use of FluLaval Quadrivalent (QIV) in this age group. Study FLU Q-QIV-022 was a randomized, active-controlled, observer-blind, multicenter clinical trial that compared the safety and immunogenicity of FluLaval-QIV to Fluzone-QIV (a quadrivalent inactivated influenza subunit vaccine licensed in the U.S. in persons ages ≥ 6 months) in subjects ages > 6 to < 36 months. Subjects were randomly allocated in a 1:1 ratio to receive FluLaval-QIV (n = 1207) or Fluzone-QIV (n= 1217). FluLaval was administered as a 0.5mL dose containing 60µg of HA (15µg of each of the four vaccine strains) which is the US licensed formulation for persons ≥ 3 years of age. The US licensed formulation of Fluzone for persons ages ≥6 to < 36 months, which served as the comparator arm, was a 0.25mL volume containing 30µg of HA (7.5µg of each of the four vaccine strains). The primary immunogenicity objective was immunologic noninferiority of FluLaval-QIV compared to Fluzone-QIV for vaccine strains at 28 days after completion of the vaccination series. Even as FluLaval Quadrivalent contains twice the antigen content for each strain, as in accordance with the 2007 Guidance for Industry Clinical Data Needed to Support Licensure of Seasonal Inactivated Influenza Vaccines, its effectiveness can be established by immunogenicity non-inferiority criteria if the upper limit (UL) of the 95% confidence interval for the geometric mean titers (GMT) ratios (Fluzone-QIV: FluLaval-QIV) were <1.5 and the difference in seroconversion rates (Fluzone-QIV – FluLaval-QIV) were <10% for each of the 4 influenza vaccine strains. The pre-specified criteria for immunologic noninferiority of FluLaval-QIV relative to Fluzone-QIV were met for all four vaccine strains. The safety evaluation in study FLU Q-QIV-022 included collection of local and systemic solicited adverse events (AEs) captured via diary card for 7 days post vaccination; unsolicited adverse events, serious adverse events (SAEs), potentially immune mediated diseases (pIMDs), medically attended AEs, and deaths were collected for the 180-day study duration. For both vaccines injection site pain was the most commonly reported local AE (40.3% and 37.4% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively). Grade 3 injection site pain was reported for 2.9% and 1.7% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. Overall, irritability/ fussiness was the most frequently reported solicited general

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 5
AE (54.4% and 50.5% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively) followed by drowsiness (40.6% and 40.9% of subjects, in the FluLaval QIV and Fluzone-QIV groups, respectively) and loss of appetite (33.7% and 33.4% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively). Grade 3 irritability/fussiness was reported for 5.3% and 3.9% of subjects, respectively. Grade 3 drowsiness was reported for 3.1% and 3.0% of subjects, respectively. Grade 3 loss of appetite was reported for 2.2% and 1.6% of subjects, respectively. Rates of fever were similar across treatment arms. During the 7-day (Day 0-6) follow-up post-vaccination, fever (≥38°C) was reported for 7.9% and 7.5% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. Grade 3 or higher fever (>39°C) was reported for 2.2% and 1.5% of subjects, in the FluLaval-QIV and Fluzone-QIV groups, respectively. As noted above, FluLaval-QIV contains twice the antigen load (60 µg; 15 µg of each of the four HA antigens) as Fluzone-QIV (30 µg; 7.5 µg of each of the four HA antigens) and therefore is expected to be more reactogenic although this was not indicated by the rates of observed AE’s. The statistical reviewer noted that approximately 2% of the fever observations were ≤35°C, attributed by the Applicant to be due to “mishandling by the parents”. Whether this “mishandling by the parents” was restricted to fever observations ≤35C or was a systematic problem across all fever observations is not verifiable. However, no significant difference in the distribution of fever between the two groups was observed. While the study was underpowered to detect statistical differences in rates of febrile seizures, no imbalances were detected between the treatment arms. During the 28-day post-vaccination period, at least one unsolicited AE was reported for 45.5% and 44.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. At least one unsolicited AE with a medically attended visit during the entire study period was reported for 60.2% and 59.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. A total of 56 non-fatal SAEs were reported for 43 subjects during the entire study period. Of these, 29 SAEs were experienced by 22 subjects (1.8%) in the FluLaval-QIV group and 28 SAEs were reported for 21 subjects (1.7%) in the Fluzone-QIV group. Nature and severity of unsolicited AEs, MAEs, and SAEs were similar between treatment groups. One of the 4 pIMDs reported (kawasaki’s disease) occurred in the FluLaval-QIV group. There were no deaths reported. Multidisciplinary review of the data submitted for this supplement did not reveal new issues about the product that required the opinion of an independent panel of experts including the Vaccines and Related Biological Products Advisory Committee (VRBPAC) or other external consultative groups.
The Pediatric Research Equity Act (PREA) requires that for any product approved for use in adults, that the safety and effectiveness of the product be evaluated in children (ages 0 to 17 years). However, the Applicant may provide an evidence-based rationale to support a request that evaluation of the product be waived (the possible bases for such a waiver are included in the statute. FluLaval and FluLaval-QIV are currently approved for use in persons ages 3 years and older. The manufacturer received a partial waiver for infants 0 to <6 months of age based on the reasoning that FluLaval (trivalent formulation) and FluLaval Quadrivalent would provide no meaningful therapeutic benefit over vaccination beginning at 6 months of age, and these vaccines are unlikely to be used by a substantial number of infants 0 to <6 months of age (Section 505B(a)(4)(A)iii of the Food Drug and Cosmetic Act). Thus, if approved, the Applicant will fulfill the remaining PREA-postmarketing requirement to evaluate FluLaval Quadrivalent in

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 6
children ages ≥ 6 to < 36 months. Because the manufacturing process is the same for the trivalent formulation of FluLaval, except that it does not contain one of the two influenza B strains contained in the quadrivalent formulation, the conclusions of this supplement, including fulfillment of the PREA requirement, can be applied to both products. Based on distribution data, it is estimated that over 11.7 million doses have been administered, over of which were distributed in the US. There are currently no postmarketing requirements or postmarketing commitments based on safety signals observed in the pre- or post-licensure setting. The routine pharmacovigilance plan is adequate. No changes to the submitted pharmacovigilance plan for FluLaval Quadrivalent are recommended based on the information contained in this application. The data submitted by the Applicant in this sBLA support approval of FluLaval and FluLaval Quadrivalent for active immunization of children ages 6 months and older against influenza disease caused by influenza subtypes A and type B contained in the vaccine.
1.1 Demographic Information: Subgroup Demographics and Analysis Summary
Post hoc subgroup analyses of immunogenicity and safety were performed by age, sex, ethnicity, and country. The subgroup analyses of immunogenicity and safety by age, sex, ethnicity, and country generally were shown to be consistent with the overall immunogenicity and safety results.
2. Clinical and Regulatory Background
2.1 Disease Studied
Influenza is an acute, highly contagious, respiratory disease condition caused by influenza viruses, mainly spread through respiratory droplets. The illness is accompanied by fever and variable degrees of other systemic symptoms, ranging from mild fatigue to respiratory failure and even death. Influenza occurs in annual epidemics that are associated with significant morbidity and mortality and have substantial public health impact. During seasonal epidemics, 5-15% of the worldwide population is typically infected, resulting in 35 million cases of severe illness and a quarter to half a million excess deaths annually (1). The highest risk of complications occur among young children and in particular children younger than 2 years, adults aged 65 years or older, pregnant women, and people of any age with underlying chronic conditions that put them at risk for influenza disease (1). The highest influenza burden in terms of pediatric respiratory admissions is seen in infants 6 to 11 months of age (2) and rates of illness in children younger than 2 years of age are substantially higher than those in children 2 years of age or older (3, 4). Children also play an important role in the spread of the disease (5), possibly because of their high levels of virus shedding. Since annual influenza vaccination is currently the most effective means of controlling influenza and preventing its complications and mortality (6), it is recommended for all persons ages 6 months and older.
(b) (4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 7
Influenza A H1N1, A H3N2 and B viruses have co-circulated in the community since the late 1970s, and from that time seasonal influenza vaccines have contained three influenza strains, one from each A subtype and one type B virus (7). Since 1985, two antigenically distinct lineages of influenza B viruses (Victoria or Yamagata lineages) have co-circulated globally and have caused extensive illness, particularly in children, as limited cross protection is provided against strains in the B lineage not contained in the trivalent vaccine (7, 8). Because of difficulty predicting which influenza B lineage will be predominantly circulating resulting in frequent seasonal mismatches for the influenza B strain, quadrivalent influenza vaccines (QIV) have been developed which include both influenza B lineages .
2.2 Currently Available, Pharmacologically Unrelated Treatment(s)/Intervention(s) for the Proposed Indication(s)
Currently, four FDA-licensed antiviral drugs are available for use in the United States (Tamiflu®, Relenza®, Symmetrel® and Flumadine®). Of these, only the neuraminidase inhibitors Tamiflu and Relenza are currently recommended for use by the Centers for Disease Control and Prevention. Use of adamantine class derivatives (Symmetrel and Flumadine) is no longer recommended because many strains of influenza, including the 2009 H1N1 influenza, are now resistant to this class of drugs. Although neuraminidase inhibitors are currently effective against most seasonal influenza viruses, resistance to drugs in this class has developed sporadically (9) with most of the benefit derived when given prophylactically or early in the disease course. However, none of these drugs are indicated for the prevention of influenza.
2.3 Safety and Efficacy of Pharmacologically Related Products
Inactivated whole-virus influenza vaccines have been commercially available since the 1940s. Currently, eight inactivated split-virus influenza vaccines are licensed in the U.S. Of these, only four are approved for individuals less than 18 years of age. However, only Fluzone and Fluzone Quadrivalent are approved for children 6 through 35 months of age. A recent meta-analysis of 31 studies conducted between 1967 and 2011 calculated a pooled efficacy of 59% in healthy adults against laboratory-confirmed influenza illness (10). Data regarding the efficacy of vaccination against influenza-related hospitalization and other severe outcomes also indicate that some protection is conferred (11). The most frequent adverse events after seasonal inactivated influenza vaccination are local adverse reactions, resulting in pain, erythema and induration in up to 65% of individuals. Serious adverse events associated with influenza vaccination are uncommon. Anaphylaxis has been reported after influenza vaccination, but occurs rarely (0-10 per million doses of vaccine (11). Increased rates of Guillain-Barré syndrome (GBS) were reported during the swine influenza virus vaccination campaign of 1976. Observational studies since then have identified an increased risk of at most 1 additional GBS case per million vaccinated persons associated with seasonal influenza vaccines. Influenza vaccination has also been associated in passive surveillance studies with an increased rate of febrile seizures in children, potentially related to co-administration with pneumococcal conjugate vaccine (Prevnar 13)(12). A live, cold-adapted, attenuated influenza virus vaccine is currently indicated for use in persons 2 through 49 years of age. The efficacy of FluMist® has been demonstrated in clinical studies of

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 8
children; however, the use of FluMist in children is limited by the increased risk of wheezing in very young children.
2.4 Previous Human Experience with the Product (Including Foreign Experience)
FluLaval was first licensed in Canada in 1992 and was subsequently approved in the US in 2006. Flulaval Quadrivalent was approved in the US on 8 Oct 2013 and is also available in Canada and Mexico. Based on distribution data, it is estimated that over 11.7 million doses have been administered, over of which were distributed in the US. Routine pharmacovigilance monitoring of these products has not identified any safety signals.
2.5 Summary of Pre- and Post-submission Regulatory Activity Related to the Submission
FluLaval was licensed on 5 October 2006 for the prevention of influenza subtypes A and type B contained in the vaccine under the accelerated approval regulations. The approval was based on the immune response elicited by FluLaval in clinical studies in adults. Since products approved under the accelerated approval regulations (21 CFR 601.41) require further studies that are adequate and well controlled to verify and describe clinical benefit, a clinical endpoint efficacy study (IDB-707-106; NCT00216242) was conducted during the 2005-2006 and 2006-2007 influenza seasons in adults 18 through 49 years of age. In this study, the efficacy against culture-confirmed, antigenically matched strains was 46.3%, with a lower limit of the one-sided 97.5% confidence interval (CI) of 9.8%. Because the pre-specified success criterion for the lower limit of the CI was ≥35%, vaccine efficacy was not demonstrated according to the pre-defined criteria. It was noted however, that the 1.2% attack rate in the placebo group for culture-confirmed, antigenically matched strains was lower than expected, contributing to a wide confidence interval for the estimate of vaccine efficacy. Thus, based on the results of the study IDB-707-106, approval was not granted. After discussions with CBER, a randomized, controlled, observer-blind, clinical endpoint study in 5200 children 3 through 8 years of age demonstrated absolute efficacy of FluLaval QIV for prevention of reverse-transcriptase polymerase chain reaction (RT-PCR)-confirmed influenza A and/or B disease presenting as influenza like illness (ILI) caused by community acquired influenza strains (reviewed in sBLA 125163/253). The study estimated an absolute vaccine efficacy of 55.4% (LL of 95% CI was 39%), which satisfied the pre-specified criterion for demonstration of effectiveness (LL 95% CI > 30%). Concurrently, because the original approval was based on evaluation of adults, a PREA-required safety and immunogenicity study was conducted which demonstrated safety and immunologic noninferiority of FluLaval TIV compared with Fluzone TIV in children ages ≥3 to < 18 years (reviewed in sBLA125163/254). On 15 August 2013 ‘traditional’ approval was granted on the basis that these studies supported satisfied the requirement under accelerated approval to confirm clinical benefit of both FluLaval and FluLaval QIV for persons ≥ 3 years of age because the products are manufactured according to the same process. The Applicant was required to conduct a PREA postmarketing study in infants and children ≥ 6 to <36 months of age according to PREA. A waiver was granted for children < 6 months of age based on the rationale that vaccination in this age group provides no meaningful therapeutic benefit over initiating vaccination at 6 months of age, and this vaccine is not likely to be used in a substantial number of infants under 6 months of age. A description of the 3 completed
(b) (4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 9
supportive studies FLU Q-QIV-003, Flu Q-QIV-013 and FLU Q-QIV-022 was provided (see Table 5 for overview). To fulfill PREA requirements with regard to children 6 to 35 months of age the Applicant submitted for CBER review study Flu Q-QIV-022 (described in Section 6.1). It was agreed during the Type C meeting between CBER and eth Applicant on 18 March 2016 that the sample size and safety and immunogenicity endpoints were acceptable as proposed. However, CBER requested and the Applicant agree to perform a descriptive analyses of occurrence of febrile seizures.
3. SUBMISSION QUALITY AND GOOD CLINICAL PRACTICES
3.1 Submission Quality and Completeness
The submission was adequately organized to accommodate the conduct of a complete clinical review without difficulty.
3.2 Compliance With Good Clinical Practices And Submission Integrity
Bioresearch monitoring (BIMO) inspections were conducted for one domestic and one foreign clinical investigator study site for the primary study submitted to this sBLA, FLU Q-QIV-022. The study sites inspected enrolled a total of 205 subjects ages 6 to less than 36 months of age, which represented approximately 8.5 percent of all subjects (N=2,424) that were enrolled in the United States and Mexico. The inspections revealed no issues that would impact the data submitted in this BLA. For full details please refer to the BIMO Review Memo dated 16 September 2016.
3.3 Financial Disclosures
Financial disclosures for the studies evaluated in this sBLA are listed below in Tables 1-4. Table 1. Financial Disclosures for Study FLU-Q-QIV-022 (NCT02242643)
Covered clinical study (name and/or number): FLU-Q-QIV-022 (NCT02242643)
Was a list of clinical investigators provided:
Yes No (Request list from applicant)
Total number of investigators identified: 503
Number of investigators who are sponsor employees (including both full-time and part-time employees): 0 Number of investigators with disclosable financial interests/arrangements (Form FDA 3455): 0*
*Data not obtained from 12 investigators. Table 2. Financial Disclosures for Study FLU-Q-QIV-021 (NCT01974895)
Covered clinical study (name and/or number): FLU-Q-QIV-021 (NCT01974895)
Was a list of clinical investigators provided: Yes No (Request list from

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 10
applicant)
Total number of investigators identified: 98
Number of investigators who are sponsor employees (including both full-time and part-time employees): 0 Number of investigators with disclosable financial interests/arrangements (Form FDA 3455): 0*
*Data not obtained from 2 investigators. Table 3. Financial Disclosures for Study FLU-Q-QIV-013 (NCT01711736)
Covered clinical study (name and/or number): FLU-Q-QIV-013 (NCT01711736)
Was a list of clinical investigators provided:
Yes No (Request list from applicant)
Total number of investigators identified: 25
Number of investigators who are sponsor employees (including both full-time and part-time employees): 0 Number of investigators with disclosable financial interests/arrangements (Form FDA 3455): 0
Table 4. Financial Disclosures for Study FLU-Q-QIV-003 (NCT01198756)
Covered clinical study (name and/or number): FLU-Q-QIV-003 (NCT01198756) Was a list of clinical investigators provided:
Yes No (Request list from applicant)
Total number of investigators identified: 181
Number of investigators who are sponsor employees (including both full-time and part-time employees): 0 Number of investigators with disclosable financial interests/arrangements (Form FDA 3455): 0*
*Data not obtained from 2 investigators. Despite due diligence efforts outlined by GSK’s standard operating procedures (up to 3 documented efforts to contact and collect information from each investigator), the Applicant was unable to obtain financial disclosure information from 15 out of a total of 807 investigators. Reviewer comment: Given the large number of investigators (503) and sites (69) involved in the study, it is unlikely that the 15 investigators who did not provide financial disclosures would have significantly impacted the integrity of the data. Furthermore, all principal investigators for each of 108 sites submitted their financial disclosures; none had any conflicts to report.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 11
4. SIGNIFICANT EFFICACY/SAFETY ISSUES RELATED TO OTHER REVIEW DISCIPLINES
4.1 Chemistry, Manufacturing, and Controls
A formal chemistry, manufacturing, and controls review was not conducted for this sBLA since this product is currently licensed and no formulation changes were made.
4.2 Assay Validation
The hemagglutinin inhibition (HI) methods were reviewed and found to be acceptable. For full details please refer to the review memo from the Division of Antiviral Products dated 13 October 2016.
4.3 Nonclinical Pharmacology/Toxicology
A formal nonclinical pharmacology/toxicology review was not conducted for this sBLA since this product is currently licensed.
4.4 Clinical Pharmacology
This section is not applicable to vaccines.
4.4.1 Mechanism of Action
Vaccination against influenza results in an immune response that can be quantified by elevation in serum HI titers. Some studies and meta-analyses associate HI titers ≥ 1:40 with 50% reduction in the risk of contracting influenza, based on controlled, influenza challenge studies in adults (13).
4.5 Statistical
Statistical review confirmed immunologic noninferiority by GMT ratios and SCR differences for all four strains contained in the vaccine. Although no imbalances in safety were identified, it noted that approximately 2% of the temperature observations were ≤ 35°C, attributed by the Applicant to “mishandling by the parents”. It was not verifiable whether this was restricted to fever observations ≤35°C or was a systematic problem across all fever observations. However, no significant difference in the distribution of fever between the two groups was observed. For full details of the statistical review please refer to the review memo from the Office of Biostatistics and Epidemiology dated 24 Oct 2016.
4.6 Pharmacovigilance
No changes were recommended to the routine pharmacovigilance plan proposed for FluLaval Quadrivalent. No postmarketing safety studies or risk evaluation and mitigation strategies (REMS) were recommended. For full review of the Applicant’s pharmacovigilance plan please refer to the review memo from the Office of Biostatistics and Epidemiology dated 31 Oct 2016.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 12
5. SOURCES OF CLINICAL DATA AND OTHER INFORMATION CONSIDERED IN THE REVIEW
5.1 Review Strategy
A single phase 3 Study, FLU Q-QIV-022, was submitted to this BLA to serve as the primary basis for licensure and is described in detail in Section 6.1. Three additional studies, FLU Q-QIV-021, FLU Q-QIV-013, and FLU Q-QIV-003 are described briefly in sections 6.2, 6.3, and 6.4, respectively. Their data, however, will be evaluated in the integrated summary of safety, primarily for important safety signals such as SAEs and deaths, and will not be included in will not be included in the integrated summary of efficacy because:
• they were small descriptive studies that did not evaluate the same primary immunogenicity endpoints as study FLU Q-QIV-022;
• studies FLU-Q-QIV-021 and FLU-Q-QIV-013 used different active comparators from study FLU Q-QIV-022, both of which were trivalent formulations (Fluzone and Fluarix); and
• the sub-study of FLU-Q-QIV-003 which enrolled children ages ≥6 to < 36 months was single-arm and open labeled.
The four studies are summarized below in Table 5.
5.2 BLA/IND Documents That Serve as the Basis for the Clinical Review
The following files served as the basis for the clinical review of STN 125163/405: STN 125163/405.0
• m1.3 Financial Disclosures • m1.14 Labeling • m2.5 Clinical Overview • m2.7 Clinical Overview • m5 Clinical Study Reports • Amendments 1 through 9
5.3 Table of Studies/Clinical Trials
Four clinical studies were submitted to this BLA as outlined in Table 5. All studies were conducted under US IND 14466 with the exception of study FLU Q-QIV-013 for which the clinical study report was provided after study completion. Table 5. Summary of Primary Study FLU Q-QIV-022 and 3 Supportive Studies Evaluating FluLaval-QIV in Healthy Children Ages 6 through 35 Months.
Study number (NCT number)
Countries (number of sites) Years
Study design1 Treatment arms (N2)
FLU Q-QIV-022 (NCT02242643)
Mexico (2) US (67) 2014-2015
Phase 3, double-blind, randomized, active-controlled
FluLaval-QIV3 (1207) Fluzone-QIV (1217)
FLU Q-QIV-021 US (12) Phase 2, observer- FluLaval-QIV (158)
Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 13
(NCT01974895) 2013-2014 blind, randomized, active-controlled study
Fluzone (156)
FLU Q-QIV-013 (NCT01711736)
Canada (6), Dominican Republic (1), Honduras (1) 2013-2014
Phase 3, double-blind, randomized, active-controlled
FluLaval-QIV (299) Fluarix (302)
FLU Q-QIV-003 (NCT01198756)
Canada (9), Mexico (2), Spain (4), Taiwan (2), US (16) 2011-2012
Open label7 FluLaval-QIV (301)
Source: Adapted from BLA 125163/405.0, m2.5 Clinical Overview, Table 1 1In all studies primed subjects received a single intramuscular (IM) dose of the study product on day 0 and unprimed subjects (those who had not received prior seasonal influenza vaccination) received two IM doses of study product 28 days apart. 2N: total vaccinated cohort 3Q: quadrivalent
5.4 Consultations
Multidisciplinary review of the data submitted for this supplement did not reveal new issues about the product that required the opinion of an independent panel of experts including the Vaccines and Related Biological Products Advisory Committee (VRBPAC). As required by the FDA Amendments Act of 2007, a review of pediatric safety was presented to the Pediatric Advisory Committee on April 12, 2016. The review, which included the period from approval (August 15, 2013) through June 30, 2015, did not identify any new safety concerns. The PAC voted unanimously to continue FDA’s routine monitoring.
5.5 Literature Reviewed
1. WHO Fact Sheet N°211 March 2014. At http://www.who.int/mediacetre/factsheets/fs211/en/
2. Schanzer D, Langley J, Tam T. Hospitalization Attributable to Influenza and Other Viral Respiratory Illnesses in Canadian Children. Pediatr Infect Dis J 2006;25:795-800.
3. CDC (Centers for Disease Control and Prevention). Influenza vaccination coverage among children aged 6-23 months--United States, 2005-06 influenza season. MMWR 2007;56(37):959-63
4. Poehling KA, Edwards KM, Weinberg GA, Szilagyi P et al, for the New Vaccine Surveillance Network. The under-recognized burden of influenza in young children. NEJM 2006;355:31-40.
5. Brownstein JS, Mandl KD. Pediatric population size is associated with local timing and rate of influenza and other acute respiratory infections among adults. Ann Emerg Med 2008;52:63-8
6. Barr IG, McCauley J, Cox N et al. Writing Committee of the World Health Organization Consultation on Northern Hemisphere Influenza Vaccine Composition for 2009–2010. Epidemiological, antigenic and genetic characteristics of seasonal influenza A (H1N1), A (H3N2) and B influenza viruses: Basis for the WHO recommendation on the composition of

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 14
influenza vaccines for use in the 2009-2010 Northern Hemisphere season. Vaccine 2010;28(5) :1156-67. Online version of manuscript accessed for Table (Dec 2009)
7. Ambrose CS, Levin MJ. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother. 2012;8(1):81–8
8. Belshe RB, Coelingh K, Ambrose CS, et al. Efficacy of live attenuated influenza vaccine in children against influenza B viruses by lineage and antigenic similarity. Vaccine. 2010;28(9):2149–56
9. Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. Influenza-associated hospitalizations in the United States. JAMA. Sep 15, 2004; 292(11):1333-40
10. Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis. 2012 Jan;12(1):36-44.
11. Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine. 2012 Mar 2;30(11):1993-8.
12. Talbot HK, Griffin MR, Chen Q, Zhu Y, Williams JV, Edwards KM. Effectiveness of seasonal vaccine in preventing confirmed influenza-associated hospitalizations in community dwelling older adults. J Infect Dis. 2011 Feb 15;203(4):500-8.
13. Hobson D, Curry RL, Beare AS, Ward- Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J. Hyg. (Lond.) 70(4): 767–777 (1972).
14. Reber A, Immunological assessment of influenza vaccines and immune correlates of protection. Expert Rev Vaccines 12 (5):519-36 (2013).
15. Ohmit SE, Petrie JG, Cross RT, Johnson E, Monto AS. Influenza hemagglutination inhibition antibody titer as a correlate of vaccine-induced protection. J. Infect. Dis. 204(12): 1879–85 (2011). 16. Febrile Seizures Fact Sheet.
http://www.ninds.nih.gov/disorders/febrile_seizures/detail_febrile_seizures.htm
6. DISCUSSION OF INDIVIDUAL STUDIES/CLINICAL TRIALS
6.1 Study FLU Q-QIV-022
The primary study for safety and immunogenicity of FluLaval, FLU Q-QIV-022 was entitled, “A Phase III, observer-blind, randomized, controlled, multi-center study to evaluate the immunogenicity and safety of GSK Biologicals’ quadrivalent influenza vaccine candidate, FluLaval, compared to Sanofi Pasteur’s quadrivalent influenza vaccine Fluzone Quadrivalent, administered intramuscularly to children 6 to 35 months of age”. The first subject was enrolled in the study on 01 October 2014 and the last study contact was on 23 June 2015. The data lock point (date of database freeze) occurred on 18 August 2015.
6.1.1 Objectives
Primary objective To demonstrate the immunologic non-inferiority of FLU Q-QIV versus Fluzone Quadrivalent (in terms of geometric mean titers [GMTs] and SCRs) approximately 28 days after completion of dosing (Day 28 and Day 56 for vaccine primed and vaccine-unprimed subjects, respectively). Selected secondary immunogenicity objectives

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 15
• If the primary objective is met, the first secondary objective will be to evaluate the percent of subjects who demonstrate seroconversion (defined as a prevaccination HI titer <10 and postvaccination HI titer ≥ 40, or at least a 4-fold increase in HI titer from prevaccination titer > 10) and the percent of subjects who demonstrate postvaccination HI titers ≥ 1:40 at 28 days after completion of dosing (Day 28 and Day 56 for vaccine primed and vaccine-unprimed subjects, respectively).
• To describe the immunogenicity of FluLaval-QIV and Fluzone-QIV for each of the four strains, overall, by age group (6-17 and 18-35 months of age) and by priming status (vaccine-primed and vaccine-unprimed).
Safety objectives • To describe the reactogenicity and safety of FluLaval-QIV and Fluzone-QIV overall, by
age group (6-17 and 18-35 months of age) and by priming status (vaccine-primed and vaccine-unprimed) in terms of: o Solicited local and general adverse events (AEs) during the 7-day post vaccination
follow-up period (day of vaccination and six subsequent days). o Unsolicited AEs during the 28-day post-vaccination follow-up period (day of
vaccination and 27 subsequent days). o Serious adverse events (SAEs), medically attended adverse events (MAEs) and
potential immune-mediated diseases (pIMDs) during the entire study period. • To evaluate the relative risk of fever after administration of FluLaval-QIV compared to
Fluzone-QIV during the 2-day post-vaccination follow-up period (day of vaccination and one subsequent day).
6.1.2 Design Overview
This was a phase 3, randomized, active-controlled, observer-blind, multicenter study in subjects ages ≥ 6 to < 36 months. Subjects were randomly allocated in a 1:1 ratio to receive either FluLaval-QIV or a US licensed comparator, Fluzone-QIV. The randomization of supplies within blocks were performed at GSK Biologicals, using
a program developed for use in Statistical Analysis System (SAS ) by GSK Biologicals. Entire blocks were shipped to the study centers /warehouse(s). The randomization algorithm used a minimization procedure accounting for age (6-17 and 18-35 months), study center, and the pre-study influenza vaccine priming status of the subjects to ensure balanced representation of the combination of the minimization factors in the two study groups. The study aimed to enroll at least 40%, but no more than 50%, of the total subjects in the age group of 6-17 months of age. Allocation of the subject to a study group at the investigator site was performed using an internet-based randomization system. Data was to be collected in an observer-blind manner. By observer-blind, it is meant that during the course of the study, the subject, subject‘s parent(s)/LAR(s), and those responsible for the evaluation of any study endpoint (e.g. safety, reactogenicity) were all to be unaware of the treatment assignments. Therefore, vaccine preparation and administration were be done by authorized medical personnel who were not to participate in any of the study clinical evaluation assays.
(b) (4)(b) (4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 16
The laboratory in charge of the laboratory testing was to be blinded to the treatment, and codes were used to link the subject and study (without any link to the treatment attributed to the subject) to each sample. Blood samples for immunogenicity testing were collected prevaccination on Day 0 and postvaccination on Day 28 after completion of the vaccination series (day 28 for primed subjects and day 56 for unprimed subjects) to evaluate the primary and secondary immunogenicity endpoints. Subjects were followed for solicited AEs by diary card through Day 7 post vaccination. Unsolicited AEs were collected at the Day 28 clinical visit. Medically attended adverse events (MAEs), AEs leading to study withdrawal, potentially immune mediated diseases (pIMDs), and SAEs including deaths were monitored for 180 days following vaccination. Reviewer comment: Design strategies utilized to minimize bias included randomization and blinding and to this end the described procedures appear appropriate.
6.1.3 Population
Primed and unprimed children were eligible. Vaccine-primed subjects included all subjects who have received a total of two or more doses of seasonal influenza vaccine since 01 July 2010 or at least 1 dose of the 2013 2014 seasonal influenza vaccine. Vaccine-unprimed subjects included all subjects who have never received any seasonal influenza vaccine or have received only one dose of seasonal influenza vaccine since 01 July 2010, but did not receive any 2013-2014 seasonal influenza vaccine. Inclusion criteria
• Males and females ages ≥ 6 to < 36 months • Written informed consent obtained from legal guardian • Able to attend scheduled visits, receive phone calls, and adhere to study procedures
Exclusion criteria
• Use of any investigational or non-registered product (drug or vaccine) other than the study vaccine within 30 days preceding the first dose of study vaccine, or planned use during the study period. Routine registered childhood vaccinations are permitted.
• Placed under control of an agency, such as the courts, or those who are institutionalized or in foster care
• Chronic administration (defined as more than 14 days in total) of immunosuppressants or other immune-modifying drugs within six months prior to the first vaccine dose. For corticosteroids, this meant a dose equivalent to either > 2 mg/kg/day of body weight, or to ≥ 20 mg/day of prednisone for persons who weighed ≥ 10 kg, when administered for more than 2 weeks. Inhaled and topical steroids were allowed.
• Prior receipt of any seasonal or pandemic influenza vaccine (registered or investigational) within six months preceding the first dose of study vaccine, or planned use during the study period. Administration of immunoglobulins and/or any blood products within the three months preceding the first dose of study vaccine or planned administration during the study period.
• History of Guillain-Barré syndrome within six weeks of receipt of prior influenza vaccine.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 17
• Any known or suspected allergy to any constituent of influenza vaccines (including egg proteins); a history of anaphylactic-type reaction to consumption of eggs; or a history of severe adverse reaction to a previous influenza vaccine.
• Acute disease and/or fever at the time of enrolment. o Fever was defined as temperature ≥38.0°C/100.4°F by any route. o Subjects with a minor illness (such as mild diarrhea, mild upper respiratory
infection) without fever could be enrolled at the discretion of the investigator. • Any significant disorder of coagulation or treatment with warfarin derivatives or
heparin. • Any confirmed or suspected immunosuppressive or immunodeficient condition, based
on medical history and physical examination (no laboratory testing required). • Any other condition which, in the opinion of the investigator, prevented the subject
from participating in the study.
6.1.4 Study Treatments or Agents Mandated by the Protocol
Subjects were randomly assigned to receive FluLaval-QIV or Fluzone QIV in a 1:1 ratio. Unprimed subjects Product information and lot numbers are provided in Table 6 below. Table 6. Vaccines used in study FLU Q-QIV-022 Vaccine Composition (0.5 mL) Investigational product: Active ingredients: Excipients: Lot numbers:
FluLaval Quadrivalent (Influenza Virus Vaccine) 15 µg HA of each of the 4 strains2 (0.5mL): A/Christchurch/16/2010 (H1N1); A/Texas/50/2012 (H3N2); B/Massachusetts/02/2012 (Yamagata lineage); B/Brisbane/60/2008 (Victoria lineage)
, alphatocopheryl
Hydrogen succinate, polysorbate 80 and water for injection AFLHVA821A
Comparator product: Active ingredients: Excipients: Lot number:
Fluzone Quadrivalent® (Influenza Virus Vaccine) 7.5 µg HA of each of the 4 strains2 (0.25 mL): A/Christchurch/16/2010 (H1N1); A/Texas/50/2012 (H3N2); B/Massachusetts/02/2012 (Yamagata lineage); B/Brisbane/60/2008 (Victoria lineage)
DLOCA143A
Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Tables 5
(b) (4)
(b) (4)
(b) (4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 18
1HA: Hemagglutinin Antigen 2Strains to be included in the each vaccine were in accordance with WHO recommendations for the Northern Hemisphere – Season 2014-2015.
Reviewer comment: As noted in the above Table 6, the volume administered and antigen content is 2-fold higher for FluLaval compared with Fluzone, the only US licensed seasonal inactivated influenza vaccine for ages ≥ 6 to < 36 months. Thus, this study represents a departure from previous clinical studies conducted in this age group, and from the currently approved formulation for this population. With regard to volume, other vaccines such as Diphtheria and Tetanus Toxoids and Acellular Pertussis Vaccines (DTaP), Haemophilus b Conjugate Vaccines (HIB), and Pneumococcal 13-valent Conjugate Vaccine (PCV13) are approved as a 0.5mL dose to infants ages ≥ 2 months and there is no statutory specification of a maximum volume that can be administered in this age group. With regard to antigen content, FluLaval and Fluzone are two unique products with their own individual formulations. Because children ages 6-35 months were not included in the comparative efficacy study supporting Traditional Approval (See Section 2.5 for the regulatory history of this product), it is a reasonable approach to evaluate effectiveness in this population based on appropriate immunogenicity endpoints (e.g., a non-inferiority immunogenicity study comparing a new vaccine to a U.S. licensed seasonal vaccine). Thus, the antigen content the Applicant chooses for their formulation is at their discretion provided the safety and immunogenicity data are supportive (see CBER ‘s “Guidance for Industry Clinical Data Needed to Support the Licensure of Seasonal Inactivated Influenza Vaccines”).
6.1.5 Directions for Use
Vaccine-primed subjects were to receive a single 0.5 mL dose of FLU Q-QIV or a single 0.25 mL dose of Fluzone-QIV administered IM on Day 0. Vaccine-unprimed subjects were to receive two 0.5 mL doses of FluLaval-QIV or two 0.25 mL doses of Fluzone-QIV administered IM on Days 0 and 28. The vaccines were to be administered into the anterolateral region of the thigh (subjects < 12 months of age) or in the deltoid region (subjects ≥12 months of age. See Section 6.1.3 for definition of priming.
6.1.6 Sites and Centers
The study was conducted at 67 sites in the US enrolling 2,232 (92.1%) of subjects, and 2 sites in Mexico enrolling 192 (7.9%) of subjects.
6.1.7 Surveillance/Monitoring
Monitoring procedures for study FLU Q-QIV-022 are described in Table 7. Unprimed subjects had an additional visit compared with primed subjects (see Table footer for definition) because they received a second vaccination at postvaccination ay 28 with their immunogenicity evaluation occurring at postvaccination day 56.
Table 7. Schedule of Procedures for Study FLU Q-QIV-022
Time points Day 0 Day 28 for unprimed1
only
Day 28 for primed2 or Day 56 for unprimed
Day 180 Site/Phone
contact3
Informed consent and eligibility assessment X

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 19
Collect demographic data (including weight and height) X
Medical history and history of influenza vaccination4 X Physical examination X X5 X5 Check contraindications to vaccination X X Pre-vaccination body temperature X X Randomization - Study group and treatment number allocation X
Treatment number allocation for subsequent doses X Blood sampling immunogenicity X X Study vaccine administration and observation for 30 minutes postvaccination X X
Distribution of diary cards for postvaccination recording of solicited AEs6 daily (Days 0-6) and unsolicited AEs (Days 0-27)
X X
Return of diary cards X X Record any concomitant medication/vaccination/intercurrent medical conditions
X X X X
Recording of SAEs, MAEs, pIMDs7 X X X X Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Tables 1 and 2 1 Vaccine-unprimed subjects included all subjects who have never received any seasonal influenza vaccine or have received only one dose of seasonal influenza vaccine since 01 July 2010, but did not receive any 2013-2014 seasonal influenza vaccine. 2 Vaccine-primed subjects included all subjects who have received a total of two or more doses of seasonal influenza vaccine since 01 July 2010 or at least 1 dose of the 2013 2014 seasonal influenza vaccine. 3Site visit preferred
4 Recorded prior influenza vaccinations for the previous three influenza seasons (2013/2014, 2012/2013, 2011/2012), including the vaccine type (inactivated versus live intranasal). 5Targetted exam as deemed appropriate by the investigator 6AE: adverse events; 7SAE: serious adverse events; MAE: medically attended adverse event; pIMD: potentially immune-mediated disease
6.1.8 Endpoints and Criteria for Study Success
Primary Endpoints Immunogenicity of FluLaval-QIV was evaluated at Day 28 for primed subjects and at Day 56 for unprimed subjects. Noninferiority of FluLaval-QIV compared with Fluzone-QIV was demonstrated if:
• the upper limit (UL) of the two-sided 95% CI for the GMT ratio (Fluzone-QIV/FluLaval-QIV) ≤ 1.5 for each of the four strains, and
• the UL of the two-sided 95% CI for SCR difference (Fluzone –Q minus FluLaval-QIV) ≤ 10% for each of the four strains.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 20
Secondary Immunogenicity Endpoints Humoral immune response to each strain, overall, by age group (6-17 and 18-35 months of age) and by priming status (vaccine-primed and vaccine-unprimed). Serum HI antibody on Day 0 and/or 28 days after the last vaccine dose from both groups will be used to calculate:
• GMTs on Day 0 and 28 days after the last vaccine dose • Percent of subjects with HI titer ≥ 1:40 at baseline and 28 days after the last vaccine
dose • SCRs and percent of subjects achieving an HI titer ≥ 1:40 at 28 days after the last vaccine
dose Success criteria were met if the lower limit (LL) of the two-sided 95% CI for SCR was ≥40% and the LL of the two-sided 95% CI for all subjects with an HAI titer of ≥ to 1:40 (regardless of baseline serostatus) was ≥70% for each strain. Safety Endpoints Each of the following categories will be described for each vaccine group overall as well as by age (≥ 6 to < 18 months and ≥18 to < 36 months) and by priming status (primed and unprimed; definitions provided in Section 6.1.3)
• Solicited local and general AEs summarized by incidence rate, intensity, duration and relationship to vaccination for 7 days postvaccination
• Unsolicited AEs summarized by incidence rate, intensity, and relationship to vaccination for 28 days postvaccination
• SAEs, MAEs, and pIMDs for the entire 180 day study period • Occurrence of any fever (≥ 38°C) or Grade 3 fever or higher (> 39°C) for 2 days
postvaccination • Relative risk of fever after administration of FluLaval-QIV compared to Fluzone-QIV
during the 2-day post-vaccination follow-up period (day of vaccination and one subsequent day)
Reviewer comment: A specific endpoint comparing rates of fever in each vaccine arm was an important evaluation because for this age group (≥ 6 to < 36 months) the antigenic load is 2-fold higher in FluLaval-QIV compared with Fluzone-QIV (see Section 6.1.4 for description of the investigational products). A higher antigen content in the formulation might lead to higher rates of fever.
6.1.9 Statistical Considerations & Statistical Analysis Plan
Primary hypotheses addressed the endpoints are described above in Section 6.1.8. The total target sample size of the study was approximately 2400 subjects divided evenly; with 1200 each to receive either FluLaval-QIV or Fluzone-QIV. Assuming a GMT ratio of 1.0 and an SCR difference of 0%, it was determined that 1020 (85%) evaluable subjects per group would be needed to achieve a global statistical power of 99%. Each of the calculations assumed a type I of 0.025. Please see the statistical review for detailed description of the statistical analysis.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 21
6.1.10 Study Population and Disposition
6.1.10.1 Populations Enrolled/Analyzed Table 8. Analysis populations for study FLU QIV-Q-022 Parameter Fluzone-QIV1
n (%)2 FluLaval-QIV
n (%)
Total n (%)
Total Cohort 1220 (100) 1209 (100%) 2430 (100) Number of subjects vaccinated 1217 (99.8) 1207 (99.8) 2424 (99.8)
Administration of vaccine(s) forbidden in the protocol 7 (0.6) 9 (0.7) 16 (0.6) Randomization failure 1 (0.1) 2 (0.2) 3 (0.1) Study vaccine dose not administered according to protocol 1 (0.1) 0 (0) 1 (< 0.1) Vaccine temperature deviation 3 (0.2) 2 (0.2) 5 (0.2)
According to Protocol cohort for safety 1205 (98.8) 1194 (99.0) 2399(99.0) Protocol violation (inclusion/exclusion criteria) 1 (0.1) 0 (0) 1 (< 0.1) Administration of any medication forbidden by the protocol 2 (0.2) 4 (0.3) 6 (0.2) Underlying medical condition forbidden by the protocol 1 (0.1) 1 (0.1) 2 (0.1) Noncompliance with vaccination schedule
11 (0.9) 13 (1.1) 24 (1.0)
Noncompliance with blood sampling schedule
41 (3.4) 38 (3.1) 79 (3.3) Essential serological data missing 120 (9.8) 122 (10.1) 242 (10.0) Others 1 (0.1) 3 (0.2) 4 (0.2)
According to Protocol cohort for immunogenicity 1028 (84.3) 1013 (83.8) 2041 (84.0) Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Table 19 1QIV: quadrivalent influenza vaccine 2n (%): total number and percentage of subjects within each treatment cohort As noted in Table 8, 192 subjects in the Fluzone-QIV group and 196 subjects in the FluLaval-QIV group were excluded from the ATP analysis group. The majority of these exclusions were because essential serological data were missing (120 and 122 subjects in FluLaval-QIV- and Fluzone-QIV groups, respectively). The other rason was noncompliance with the blood sampling schedule or protocol 52 and 51 122 subjects in FluLaval-QIV- and Fluzone-QIV groups, respectively). Reviewer comment: For sample size calculations, the assumed attrition was 15% (the actual attrition rate was ~16% per group) and the assumed number of evaluable subjects was 1020 per grroup (1028 subjects were included in the Fluzone-QIV ATP analysis group but only 1013 subjects were included om the FluLaval-QIV ATP analysis group). However, the global estimated power for the primary endpoints for comparison of GMT ratios and SCR differences was calculated at 99% or higher, suggesting that the sample size was adequate. The reasons for elimination were balanced between groups and seem typical of the reasons that might be anticipated. 6.1.1.1.1 Demographics Table 9. Summary of Demographic Characteristics in the Total Vaccinated Cohort for Study FLU Q-QIV-022
Parameter Fluzone-QIV1 N2 = 1207
n (%)3
FluLaval-QIV N = 1207
n (%)
Total N = 2424
n (%) Age (Mean ± SD; months) 19.5 ± 8.9 19.4 ± 8.7 19.5 ± 8.8

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 22
Sex: Female 582 (47.8) 547 (54.3) 1129 (46.6) Male 635 (52.2) 660 (54.7) 1295 (53.4)
Age Cohorts: ≥6 to < 18 months 502 (41.2) 500 (41.1) 1002 (41.3) ≥18 to < 36 months 715 (58.8) 707 (58.6) 1422 (58.7)
Ethnicity: American Hispanic or Latino 302 (24.8) 305 (25.3) 607 (25.0) Non-American Hispanic or Latino 915 (75.2) 902 (74.7) 1817 (75.0)
Geographic Ancestry: African / African American 187 (15.4) 190 (15.7) 377 (15.6) American Indian or Alaskan Native 24 (2.0) 29 (2.4) 53 (2.2) Asian 39 (3.2) 26 (2.1) 65 (2.6) Native Hawaiian or Pacific Islander 10 (0.8) 4 (0.3) 14 (0.6) White - Arabic / North African 4 (0.3) 5 (0.4) 9 (0.4) White - Caucasian / European 781 (64.2) 770 (63.8) 1551 (64.0) Other 172 (14.1) 183 (15.2) 355 (14.6)
Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Table 20 1QIV: quadrivalent influenza vaccine 2N: total number of subjects in the cohort 3n (%): total number and percentage of subjects within each treatment cohort Reviewer comment: In preBLA negotiations between CBER and the Applicant it was agreed that at least 40% of subjects ages ≥6 to < 18 months would be enrolled. As noted in Table 9, the applicant met this accrual goal. The demographics of the study population seem generally consistent with those of the United States. 6.1.10.1.2 Medical/Behavioral Characterization of the Enrolled Population Baseline medical history of subjects indicating the presence of at least one risk factor that could predispose a subject to complications of influenza infection was reported in 6.8% and 6.2% of all subjects in the Q-QIV and F-QIV groups respectively (Table 10). The most frequent risk factor was chronic pulmonary disorder, including asthma (4.5% and 5.2% of subjects in the Q-QIV and F-QIV groups, respectively). Rates of baseline HI titers of ≥ 1:10 (seropositive) and baseline HI titers of ≥ 1:40 (‘seroprotective’) were comparable across treatment groups for all 4 vaccine strains. Table 10. Incidence of risk factors for complications from Influenza infections in the Total Vaccinated Cohort for Study FLU Q-QIV-022
Condition Fluzone-QIV N = 1217
n (%)
FluLaval-QIV1 N2 = 1207
n (%)3 At least one risk factor 75 (6.2) 82 (6.8)
Chronic pulmonary disorder including Asthma 63 (5.2) 54 (4.5) Chronic hepatic disorder 0 (0) 0 (0) Chronic renal disorder 1 (0.1) 2 (0.2)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 23
Chronic cardiovascular disorder 8 (0.7) 10 (0.8) Chronic neurologic/neuromuscular disorder 2 (0.2) 5 (0.4) Chronic hematologic disorder 3 (0.2) 9 (0.7) Chronic metabolic disorder 1 (0.1) 4 (0.3) Receiving long term aspirin therapy 0 (0) 0 (0) Morbid obesity 0 (0) 0 (0)
Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Table 19 1QIV: quadrivalent influenza vaccine 2N: total number of subjects in the cohort 3n (%): total number and percentage of subjects within each treatment cohort 6.1.10.1.3 Subject Disposition Table 11. Subjects disposition for study FLU QIV-Q-022 Parameter Fluzone-QIV
n (%) FluLaval-QIV1
n (%)2
Total n (%)
Total Cohort 1220 (100) 1209 (100) 2430 (100) Subjects randomized but not vaccinated 3 (0.1) 2 (0.2) 63 (0.2) Total Vaccinated Cohort 1217 (99.8) 1207 (99.8) 2424 (99.8) Number of subjects completed 1139 (93.4) 1132 (93.6) 2271 (93.5) Number of subjects withdrawn 78 (6.4) 75 (6.2) 153 (6.3) Reasons for withdrawal:
Serious Adverse Event 0 (0) 0 (0) 0 (0) Non-Serious Adverse Event 0 (0) 0 (0) 0 (0) Protocol violation 2 (0.2) 1 (0.1) 3 (0.2) Consent withdrawal (not due to an adverse event) 10 (0.8) 15 (1.2) 25 (2.0) Migrated/moved from study area 3 (0.2) 1 (0.1) 4 (0.3) Lost to follow-up (subjects with incomplete vaccination
16 (1.3) 11 (0.9) 27 (2.2)
Lost to follow-up (subjects with complete vaccination course) 45 (3.7) 43 (3.6) 88 (7.2) Sponsor study termination 0 (0) 0 (0) 0 (0) Other 2 (0.2) 4 (0.3) 5 (0.4)
Source: Adapted from BLA 125163/405.0; Clinical Study Report FLU Q-QIV-022 Table 18 1QIV: quadrivalent influenza vaccine 2n (%): total number and percentage of subjects within each treatment cohort 3One subjects was enrolled but not randomized to a group Reviewer comment: Ethnic and sex distribution were balanced across cohorts with similar percentages of males and females enrolled (53.4% and 46.6%, respectively, Table 9). In general, most subjects appeared healthy and the rates of chronic medical conditions were balanced across groups (6.8 versus 6.2 for FluLaval and Fluzone respectively; Table 10). The study nearly met its sample size goals for immunogenicity in the ATP cohort for immunogenicity. The accrual goal was 1020 per arm and actual enrollment was 1013 and 1028 for FluLaval and Fluzone, respectively (Table 11). Given that the power for this sample size was calculated to be 99% and the study met non-inferiority criteria by a large margin (see Tables 12 and 13) the sample size was acceptable.
6.1.11 Efficacy Analyses
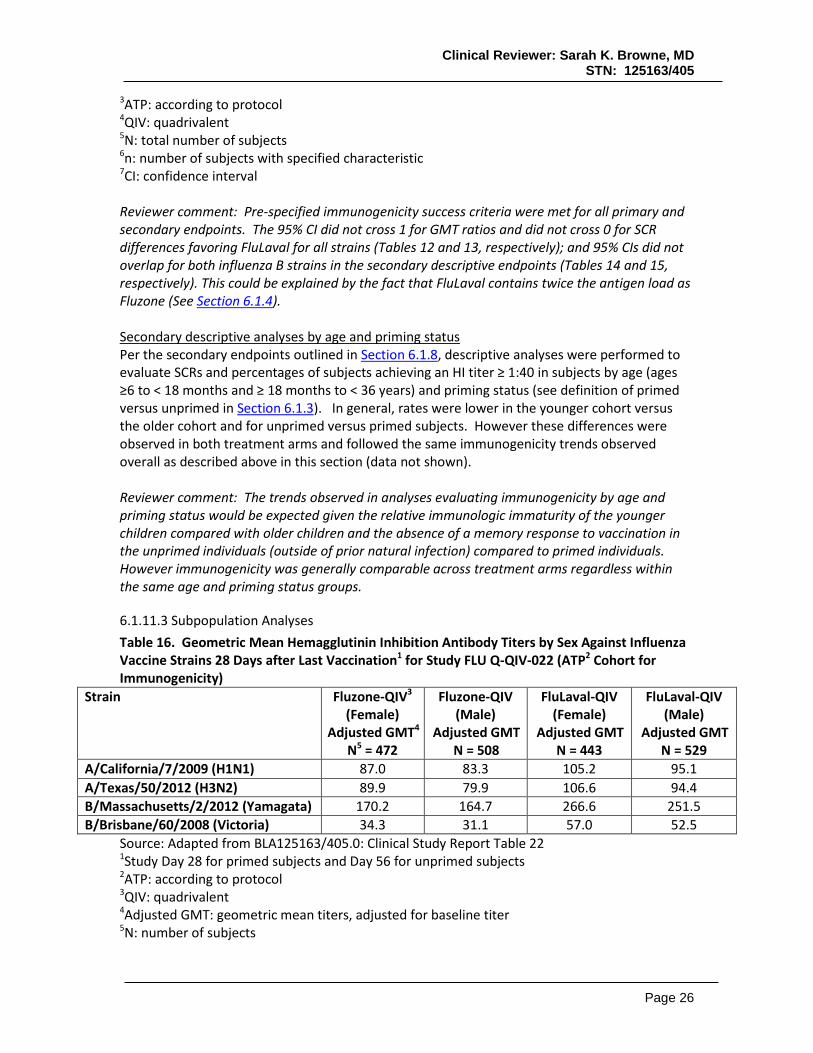
6.1.11.1 Analyses of Primary Endpoint Non-inferiority of FluLaval-QIV compared to Fluzone-QIV, (per criteria outlined in Section 6.1.8), was demonstrated by GMTs and SCRs for all four vaccine strains (Tables 12 and 13).

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 24
Table 12. Non-Inferiority1 Comparison of Geometric Mean Hemagglutinin Inhibition Antibody Titers Against Influenza Vaccine Strains 28 Days after Last Vaccination2 for Study FLU Q-QIV-022 (ATP3 Cohort for Immunogenicity)
Strain Fluzone-QIV4 Adjusted GMT5
N6 = 972
FluLaval-QIV Adjusted GMT
N = 980
Ratio Fluzone-QIV: FluLaval-
QIV (95% CI7) A/California/7/2009 (H1N1) 85.1 99.6 0.85 (0.77, 0.95) A/Texas/50/2012 (H3N2) 84.6 99.8 0.85 (0.77, 0.94) B/Massachusetts/2/2012 (Yamagata) 167.3 258 0.65 (0.59, 0.71) B/Brisbane/60/2008 (Victoria) 33.7 54.5 0.62 (0.56, 0.69)
Source: Adapted from BLA125163/405.0: Clinical Study Report Table 22 1Non-inferiority (GMTs): upper limit of 95%CI for ratio of Fluzone-QIV: FluLaval-QIV ≤ 1.5 2Study Day 28 for primed subjects and Day 56 for unprimed subjects
3ATP: according to protocol 4QIV: quadrivalent 5Adjusted GMT: geometric mean titers, adjusted for baseline titer 6N: number of subjects 7CI: confidence interval Table 13. Non-Inferiority1 Comparison of Seroconversion Rates2 for Influenza Vaccine Strains 28 Days after Last Vaccination3 for Study FLU Q-QIV-022 (ATP4 Cohort for Immunogenicity)
Strain Fluzone-QIV5 SCR
N6 = 972 N7 (%)
FluLaval-QIV SCR
N = 980 n (%)
SCR difference Fluzone-QIV- FluLaval-QIV
(95% CI8)
A/California/7/2009 (H1N1) 660 (67.3) 716 (73.7) -6.32 (-10.35, -2.27) A/Texas/50/2012 (H3N2) 680 (69.4) 740 (76.1) -6.74 (-10.68, -2.80) B/Massachusetts/2/2012 (Yamagata) 475 (48.5) 631 (64.9) -16.38 (-20.68, -12.02) B/Brisbane/60/2008 (Victoria) 723 (73.8) 833 (85.5) -11.75 (-15.28, -8.21)
Source: Adapted from BLA125163/405.0: Clinical Study Report Table 22 1Non-inferiority (SCRs): lower bound of 95%CI for ratio of Fluzone-QIV minus FluLaval-QIV ≤ 10% 2SCR: Seroconversion rate; defined as a prevaccination HI titer <10 and postvaccination HI titer ≥ 40, or at least a 4-fold increase in HI titer from prevaccination titer > 10
3Study Day 28 for primed subjects and Day 56 for unprimed subjects
4ATP: according to protocol 5QIV: quadrivalent 6N: total number of subjects 7n: number of subjects with specified characteristic 8CI: confidence interval
6.1.11.2 Analyses of Secondary Endpoints The first secondary endpoint, contingent on demonstration of immunologic noninferiority of FluLaval-QIV compared to Fluzone-QIV (see above Tables 12 and 13) evaluated seroconversion rates (Table 14) and overall rates of HI titers that were ≥ 1:40 in subjects who received FluLaval (Table 15).

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 25
Table 14. Seroconversion Rates1,2 for Each Vaccine Strain 28 Days After Receipt of Last Vaccine Dose3 for Study FLU Q-QIV-022 (ATP4 Cohort for Immunogenicity)
Strain Fluzone-QIV5 SCR
N6 = 972 N7 (%)
[95%CI8]
FluLaval-QIV SCR
N = 980 n (%)
[95% CI] A/California/7/2009 (H1N1) 660 (67.3)
[64.3, 70.3] 716 (73.7)
[70.8, 76.4] A/Texas/50/2012 (H3N2) 680 (69.4)
[66.4, 72.3] 740 (76.1)
[73.3, 78.8] B/Massachusetts/2/2012 (Yamagata) 723 (73.8)
[70.9, 76.5] 833 (85.5)
[83.2, 87.7] B/Brisbane/60/2008 (Victoria) 475 (48.5)
[45.3, 51.6] 631 (64.9)
[61.8, 67.9] Source: Adapted from BLA125163/405.0: Clinical Study Report Table 24 1SCR: Seroconversion rate; defined as a prevaccination HI titer <10 and postvaccination HI titer ≥ 40, or at least a 4-fold increase in HI titer from prevaccination titer > 10
2 Success criteria were met if the lower limit (LL) of the two-sided 95% CI for SCR was ≥40%
3Study Day 28 for primed subjects and Day 56 for unprimed subjects
4ATP: according to protocol 5QIV: quadrivalent 6N: total number of subjects 7n: number of subjects with specified characteristic 8CI: confidence interval
Table 15. Rates of HI Titers ≥ 1:401 for Each Vaccine Strain 28 Days After Receipt of Last Vaccine Dose1 for Study FLU Q-QIV-022 (ATP3 Cohort for Immunogenicity)
Strain Fluzone-QIV4
Prevaccination N5 = 980
n6 (%) [95%CI7]
Fluzone-QIV Postvaccination
N = 1013 n (%)
[95%CI7]
FluLaval-QIV Prevaccination
N = 1028 n (%)
[95% CI]
FluLaval-QIV Postvaccination
N = 972 n (%)
[95% CI] A/California/7/2009 (H1N1) 190 (19.4)
[17.0, 22.0] 775 (75.4)
[72.6, 78.0] 191 (19.7)
[17.2, 22.3] 814 (80.4)
[77.8, 82.8] A/Texas/50/2012 (H3N2) 140 (14.4)
[12.2, 16.6] 800 (77.8)
[75.2, 80.3] 135 (13.9)
[11.8, 16.2] 833 (82.2)
[79.7, 84.5] B/Massachusetts/2/2012 (Yamagata) 336 (34.3)
[31.3, 37.4] 911 (88.6)
[86.5, 90.5] 324 (33.3)
[30.3, 36.3] 983 (97.0)
[95.8, 98.0] B/Brisbane/60/2008 (Victoria) 46 (4.7)
[3.5, 6.2] 512 (49.8)
[46.7, 52.9] 40 (4.1)
[3.0, 5.6] 669 (66.0)
[63.0, 69.0] Source: Adapted from BLA125163/405.0: Clinical Study Report Table 24 1 Success criteria were met if the lower limit (LL) of the two-sided 95% CI for all subjects with an HAI titer of ≥ to 1:40 (regardless of baseline serostatus) was ≥70% for each strain. 2Study Day 28 for primed subjects and Day 56 for unprimed subjects
Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 26
3ATP: according to protocol 4QIV: quadrivalent 5N: total number of subjects 6n: number of subjects with specified characteristic 7CI: confidence interval Reviewer comment: Pre-specified immunogenicity success criteria were met for all primary and secondary endpoints. The 95% CI did not cross 1 for GMT ratios and did not cross 0 for SCR differences favoring FluLaval for all strains (Tables 12 and 13, respectively); and 95% CIs did not overlap for both influenza B strains in the secondary descriptive endpoints (Tables 14 and 15, respectively). This could be explained by the fact that FluLaval contains twice the antigen load as Fluzone (See Section 6.1.4). Secondary descriptive analyses by age and priming status Per the secondary endpoints outlined in Section 6.1.8, descriptive analyses were performed to evaluate SCRs and percentages of subjects achieving an HI titer ≥ 1:40 in subjects by age (ages ≥6 to < 18 months and ≥ 18 months to < 36 years) and priming status (see definition of primed versus unprimed in Section 6.1.3). In general, rates were lower in the younger cohort versus the older cohort and for unprimed versus primed subjects. However these differences were observed in both treatment arms and followed the same immunogenicity trends observed overall as described above in this section (data not shown). Reviewer comment: The trends observed in analyses evaluating immunogenicity by age and priming status would be expected given the relative immunologic immaturity of the younger children compared with older children and the absence of a memory response to vaccination in the unprimed individuals (outside of prior natural infection) compared to primed individuals. However immunogenicity was generally comparable across treatment arms regardless within the same age and priming status groups.
6.1.11.3 Subpopulation Analyses Table 16. Geometric Mean Hemagglutinin Inhibition Antibody Titers by Sex Against Influenza Vaccine Strains 28 Days after Last Vaccination1 for Study FLU Q-QIV-022 (ATP2 Cohort for Immunogenicity)
Strain Fluzone-QIV3 (Female)
Adjusted GMT4 N5 = 472
Fluzone-QIV (Male)
Adjusted GMT N = 508
FluLaval-QIV (Female)
Adjusted GMT N = 443
FluLaval-QIV (Male)
Adjusted GMT N = 529
A/California/7/2009 (H1N1) 87.0 83.3 105.2 95.1 A/Texas/50/2012 (H3N2) 89.9 79.9 106.6 94.4 B/Massachusetts/2/2012 (Yamagata) 170.2 164.7 266.6 251.5 B/Brisbane/60/2008 (Victoria) 34.3 31.1 57.0 52.5
Source: Adapted from BLA125163/405.0: Clinical Study Report Table 22 1Study Day 28 for primed subjects and Day 56 for unprimed subjects
2ATP: according to protocol 3QIV: quadrivalent 4Adjusted GMT: geometric mean titers, adjusted for baseline titer 5N: number of subjects
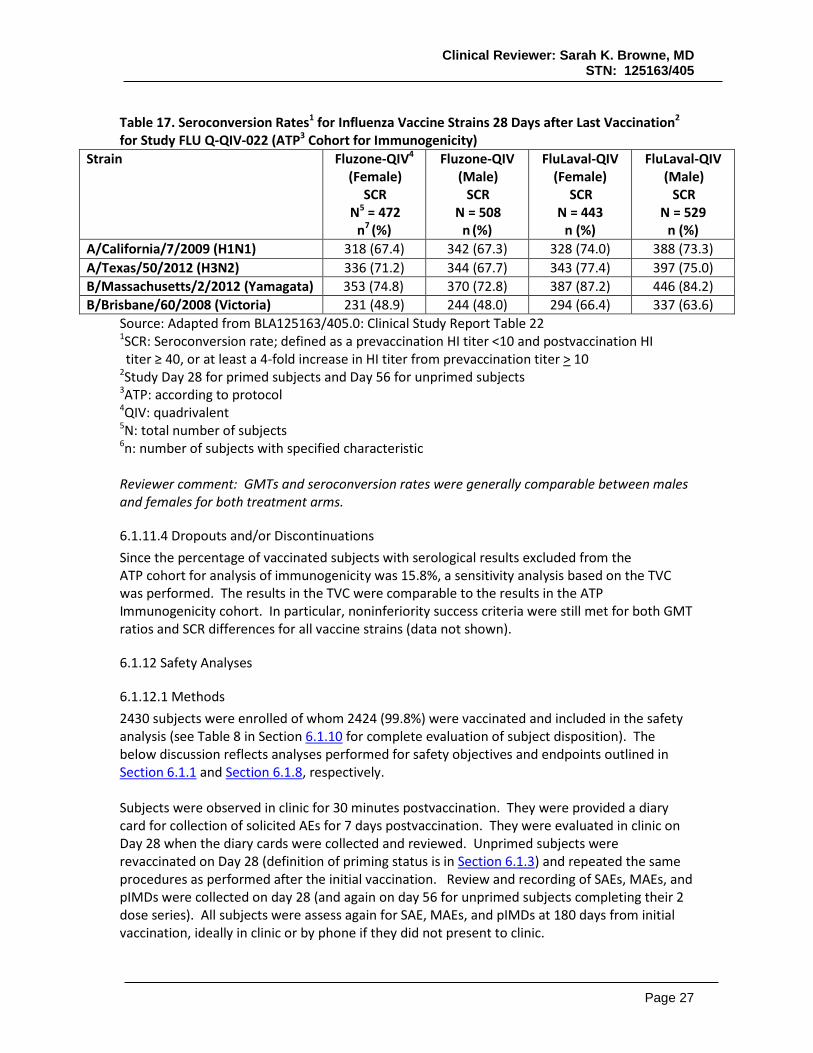
Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 27
Table 17. Seroconversion Rates1 for Influenza Vaccine Strains 28 Days after Last Vaccination2 for Study FLU Q-QIV-022 (ATP3 Cohort for Immunogenicity)
Strain Fluzone-QIV4 (Female)
SCR N5 = 472
n7 (%)
Fluzone-QIV (Male)
SCR N = 508
n (%)
FluLaval-QIV (Female)
SCR N = 443
n (%)
FluLaval-QIV (Male)
SCR N = 529
n (%) A/California/7/2009 (H1N1) 318 (67.4) 342 (67.3) 328 (74.0) 388 (73.3) A/Texas/50/2012 (H3N2) 336 (71.2) 344 (67.7) 343 (77.4) 397 (75.0) B/Massachusetts/2/2012 (Yamagata) 353 (74.8) 370 (72.8) 387 (87.2) 446 (84.2) B/Brisbane/60/2008 (Victoria) 231 (48.9) 244 (48.0) 294 (66.4) 337 (63.6)
Source: Adapted from BLA125163/405.0: Clinical Study Report Table 22 1SCR: Seroconversion rate; defined as a prevaccination HI titer <10 and postvaccination HI titer ≥ 40, or at least a 4-fold increase in HI titer from prevaccination titer > 10
2Study Day 28 for primed subjects and Day 56 for unprimed subjects
3ATP: according to protocol 4QIV: quadrivalent 5N: total number of subjects 6n: number of subjects with specified characteristic Reviewer comment: GMTs and seroconversion rates were generally comparable between males and females for both treatment arms.
6.1.11.4 Dropouts and/or Discontinuations Since the percentage of vaccinated subjects with serological results excluded from the ATP cohort for analysis of immunogenicity was 15.8%, a sensitivity analysis based on the TVC was performed. The results in the TVC were comparable to the results in the ATP Immunogenicity cohort. In particular, noninferiority success criteria were still met for both GMT ratios and SCR differences for all vaccine strains (data not shown).
6.1.12 Safety Analyses
6.1.12.1 Methods 2430 subjects were enrolled of whom 2424 (99.8%) were vaccinated and included in the safety analysis (see Table 8 in Section 6.1.10 for complete evaluation of subject disposition). The below discussion reflects analyses performed for safety objectives and endpoints outlined in Section 6.1.1 and Section 6.1.8, respectively. Subjects were observed in clinic for 30 minutes postvaccination. They were provided a diary card for collection of solicited AEs for 7 days postvaccination. They were evaluated in clinic on Day 28 when the diary cards were collected and reviewed. Unprimed subjects were revaccinated on Day 28 (definition of priming status is in Section 6.1.3) and repeated the same procedures as performed after the initial vaccination. Review and recording of SAEs, MAEs, and pIMDs were collected on day 28 (and again on day 56 for unprimed subjects completing their 2 dose series). All subjects were assess again for SAE, MAEs, and pIMDs at 180 days from initial vaccination, ideally in clinic or by phone if they did not present to clinic.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 28
6.1.12.2 Overview of Adverse Events At least one solicited AE was reported within 7 days postvaccination for 74.1% and 71.6% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. At least one grade 3 solicited AE was reported for 11.0% and 8.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. Local solicited AEs, systemic solicited AEs, unsolicited AEs and MAEs will be addressed separately below. Local solicited AEs within 7 days postvaccination Injection site pain was the most frequently reported solicited local AE (44.0% and 40.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively). Grade 3 injection site pain was reported for 2.9% and 1.7% of subjects, respectively (Table 18). After Dose 1, the incidence of injection site pain was 40.3% and 37.4% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. After Dose 2, the incidence of injection site pain was 28.2% and 29.8% of subjects, respectively. Redness at injection site was reported for 1.4% of subjects in each of the FluLaval-QIV and Fluzone-QIV groups. Swelling at injection site was reported for 1.0% and 0.4% of subjects in the FluLaval-QIV and Fluzone-QIV group, respectively. There were no reports of grade 3 redness or swelling. The median duration of any solicited local adverse events was between 1.0-2.0 days (range 1-7 days). Table 18. Solicited Local AEs by Type and Maximum Severity Occurring within 7 Days of Vaccination with Dose 1 for Study FLU Q-QIV-022 (Total Vaccinated Cohort)
Subjects experiencing at least one local AE1 by maximum intensity
Fluzone-QIV N2 = 1146
n3 (%)
FluLaval-QIV N = 1151
n (%) At least one local AE 435 (38.0) 467 (40.6)
Pain: Total 429 (37.4) 464 (40.3) Grade 24 or 35 127 (11.1) 150 (13.0) Grade 3 16 (1.4) 28 (2.4) Medical advice 3 (0.3) 0 (0) Redness: Total 15 (1.3) 15 (1.3) > 50 mm 4 (0.3) 5 (0.4) > 100 mm 0 (0) 0 (0)
Medical advice 2 (0.2) 0 (0) Swelling: Total 5 (0.4) 11 (1.0) > 50 mm 0 (0) 2 (0.2) > 100 mm 0 (0) 0 (0)
Medical advice 0 (0) 0 (0) Source: Adapted from BLA125163/405.0: Clinical Study Report Table 32 1AE: adverse event 2N: total number of subjects 3n: number of subjects per group 4Grade 2: cries/protests on touch

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 29
5Grade 3: cries when limb is moved/spontaneously painful Systemic solicited AEs within 7 days postvaccination Overall, irritability/ fussiness was the most frequently reported solicited general AE (54.4% and 50.5% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively) followed by drowsiness (40.6% and 40.9% of subjects, in the FluLaval QIV and Fluzone-QIV groups, respectively) and loss of appetite (33.7% and 33.4% of subjects in the FluLaval- QIV and Fluzone-QIV groups, respectively). Grade 3 irritability/fussiness was reported for 5.3% and 3.9% of subjects, respectively. Grade 3 drowsiness was reported for 3.1% and 3.0% of subjects, respectively. Grade 3 loss of appetite was reported for 2.2% and 1.6% of subjects, respectively. During the 7-day (Day 0-6) follow-up, fever (≥38°C) was reported for 7.9% and 7.5% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. Grade 3 or higher fever (>39°C) was reported for 2.2% and 1.5% of subjects, in the FluLaval-QIV and Fluzone –QIV groups, respectively. The relative risk of any fever (≥38°C) for the subjects in the FluLaval-QIV group compared to the subjects in the Fluzone-QIV group, during a 2-day (48 hours) follow-up period was 0.97 (overall/subject, 3.6% for FluLaval-QIV vs. 3.7% for Fluzone-QIV) with a 95% CI of [0.62; 1.52]. Of note, after dose 1 there were more subjects numerically in the FluLaval-QIV treatment group than in the Fluzone-QIV treatment group with temperatures ≥ 39.5°C (11 versus 4 subjects, respectively) and ≥ 40°C (2 and 0 subjects, respectively), however, this difference did not meet statistical significance. Overall, the relative risk of grade 3 or above fever (>39.0°C) for subjects in the FluLaval-QIV group compared to the subjects in the Fluzone-QIV group, during a 2-day (48 hours) follow-up period was 1.49 (overall/subject, 0.8% for FluLaval-QIV vs. 0.5% for Fluzone-QIV) with a 95% CI of [0.47; 5.09]. After Dose 1, the incidence of irritability/fussiness was 49.4% and 45.9% of subjects for FluLaval-QIV and Fluzone-QIV, respectively (Table 19). After Dose 2, the incidence of irritability/fussiness was 43.1% and 43.2%, for FluLaval-QIV and Fluzone-QIV, respectively (Table 19). The median duration of solicited general adverse events was between 1.0-2.0 days (range 1 to 7 days) Table 19. Solicited Systemic AEs by Type and Maximum Severity Occurring within 7 Days of Vaccination with Dose 1 for Study FLU Q-QIV-022 (Total Vaccinated Cohort)
Subjects experiencing at least one local AE1 by maximum intensity
Fluzone-QIV N2 = 1148
n3 (%)
FluLaval-QIV N = 1155
n (%) At least one systemic AE 698 (60.8) 707 (61.2)
Drowsiness: Total 424 (36.9) 424 (36.7) Grade 2 or 34 143 (12.5) 132 (11.4) Grade 3 30 (2.6) 31 (2.7) Medical advice 7 (0.6) 12 (1.0) Fever: Total 147 (12.8) 146 (12.6)
≥ 38°C 67 (5.8) 65 (5.6) ≥ 38.5°C 30 (2.6) 33 (2.9) ≥ 39.0°C 11 (1.0) 16 (1.4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 30
Subjects experiencing at least one local AE1 by maximum intensity
Fluzone-QIV N2 = 1148
n3 (%)
FluLaval-QIV N = 1155
n (%) ≥ 39.5°C 4 (0.3) 11 (1.0) ≥ 40°C 0 (0) 2 (0.2) Medical Advice 10 (0.9) 17 (1.5)
Irritability/Fussiness: Total 527 (45.9) 570 (49.4) Grade 2 or 35 192 (16.7) 224 (19.4) Grade 3 34 (3.0) 44 (3.8) Medical advice 12 (1.0) 17 (1.5)
Loss of appetite: Total 328 (28.6) 334 (28.9) Grade 2 or 36 92 (8.0) 83 (7.2) Grade 3 15 (1.3) 19 (1.6) Medical advice 9 (0.8) 14 (1.2)
Source: Adapted from BLA125163/405.0: Clinical Study Report Tables 28 and 33 1AE: adverse event 2N: total number of subjects 3n: number of subjects per group 4Grade 2 drowsiness interferes with normal activity and grade 3 prevents normal activity 5Grade 2 crying more than usual or interferes with normal activity and grade 3 cannot be comforted 6Grade 2 eating less than usual or interferes with normal activity and grade 3 not eating at all Reviewer comment: Statistical review determined that in approximately 2% of subjects a temperature of ≤ 35°C was recorded (see Section 4.5). The Applicant attributed this to be due to mishandling by the parents. The aberrant recordings were distributed evenly across treatment arms due to randomization. Thus while fever rates might be higher that reported, it is likely that this observation would be balanced across arms. Unsolicited AEs During the 28-day post-vaccination period, at least one unsolicited AE was reported for 45.5% and 44.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. Upper respiratory tract infection (9.2% of subjects) was the most frequently reported AE in the FluLaval-QIV group followed by cough (5.8% of subjects), diarrhea and nasopharyngitis (both in 5.5% of subjects), and otitis media (5.1% of subjects). The Fluzone-QIV group followed a similar pattern, where upper respiratory tract infection (8.4% of subjects) was the most frequently reported AE followed by cough (6.3% of subjects), rhinorrhea (6.2% of subjects), pyrexia (4.6% of subjects), diarrhea and nasopharyngitis (both in 4.4% of subjects), and otitis media (4.0% of subjects). At least one grade 3 unsolicited AE was reported for 5.8% and 6.2% subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively. MAEs At least one unsolicited AE with a medically attended visit during the entire study period was reported for 60.2% and 59.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 31
Upper respiratory tract infection was the most frequently reported MAE in both groups (20.1% and 19.1% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively) followed by otitis media (16.1% and 18.2% of subjects in the FluLaval-QIV and Fluzone-QIV groups, respectively). Reviewer comment: Local and systemic solicited reactions were similar across treatment arms. In particular the relative risk of fever was similar for any fever. The point estimate for fever > 39.0°C was higher 1.49 for FluLaval-QIV compared to Fluzone-QIV, however the 95% CI included 1. Furthermore, although the study would not be powered to detect difference in rates of febrile seizures, and febrile seizures were detected (4 in subjects who received FluLaval-QIV and 5 in subjects who received Fluzone-QIV), none were in proximity to vaccination (see below description of SAEs in Section 6.1.12.4). Review of the unsolicited AEs, SAEs and MAEs did not reveal imbalances or an unusual distribution of events within or related to a particular system organ class.
6.1.12.3 Deaths No fatal events were reported during the entire study period.
6.1.12.4 Nonfatal Serious Adverse Events A total of 56 non-fatal SAEs were reported for 43 subjects during the entire study period. Of these, 29 SAEs were experienced by 22 subjects (1.8%) in the FluLaval-QIV group and 28 SAEs were reported for 21 subjects (1.7%) in the Fluzone-QIV group (Table 20). None of the SAEs was assessed by the investigator to be causally related to vaccination. All SAEs in the FluLaval-QIV group were reported as “resolved/recovered,” with the exception of one case of Kawasaki’s disease and a case of croup in subject 2077, which were reported as “resolving/recovering” at the time of this report. All SAEs in the Fluzone-QIV group were also reported as resolved/recovered at the time of this report, with the exception of 4 SAEs in 3 subjects (B precursor type acute leukemia; failure to thrive; and developmental delay and hemiplegia). Of note, in the System Organ Class for nervous system disorders 9 febrile convulsions occurred 4 and 5 times in the FluLaval and Fluzone groups, respectively. One febrile convulsion occurred within 7 days of vaccination in a subject who receive FluLaval-QIV (study day 4). The subjects had been afebrile the day prior and had a temperature of > 40°C associated with influenza infection on study day 5 at the time of the seizure. The remaining seizures occurred greater than one-month postvaccination (ranges 50-168 days and 39-178 days in the FluLaval-QIV and Fluzone-QIV arms, respectively) suggesting underlying etiologies other than vaccination. Table 20. Subjects in the Total Vaccinated Cohort with SAE1s Through Day 180 by System Organ Class for Study FLU Q-QIV-022 Primary System Organ Class Fluzone-QIV2
N3 = 1217 n (%)
FluLaval-QIV N = 1207
n (%) Any SAE 22 (1.7) 22 (1.8) Gastrointestinal disorders 2 (0.2) 0 (0)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 32
General disorders and administration site conditions 1 (<0.1) 0 (0) Infections and infestations 11 (0.9) 11 (0.9) Injury, poisoning and procedural complications 0 (0) 4 (0.3) Metabolism and nutrition disorders 4 (0.3) 3 (0.2) Neoplasms benign, malignant and unspecified 1 (< 0.1) 0 (0) Nervous system disorders 6 (0.5) 5 (0.4) Renal and urinary disorders 1 (<0.1) 0 (0) Respiratory, thoracic and mediastinal disorders 1 (<0.1) 4 (0.3) Social circumstances 0 (0) 1 (< 0.1) Vascular disorders 0 (0) 1 (< 0.1) Adapted from BLA125163/405.0: Clinical Study Report Table 38 1SAE: serious adverse event 2QIV: quadrivalent 3N: total number of subjects 7n (%): number and percent of subjects with specified characteristic Reviewer comment: The reviewer agrees with the investigators’ assessments of relatedness. Although one episode of febrile seizure occurred within 7 days of vaccination in the FluLaval-QIV group, this subject had been previously afebrile and was found to be positive for influenza. Thus, it is likely that the febrile seizure was related to the influenza –associated fever and not vaccine associated fever. Overall, it is reassuring that no increased rates of febrile seizure were observed in the FluLaval group, given the higher antigen content; seizures were detected as might be expected in this age group suggesting if they had occurred the study would have successfully captured the data.
6.1.12.5 Potentially Immune Mediated Diseases (pIMDs) There were two cases of pIMDs reported during the entire study period and both occurred after the first vaccination dose, but neither was assessed by the investigator as causally related to vaccination. One pIMD was in the FluLaval-QIV group (Kawasaki’s disease, also reported as an SAE; recovering/resolving at the time of this report) and the other in the Fluzone-QIV group (erythema multiforme; reported recovered/resolved at the time of this report). Reviewer comment: Given that only one case each occurred of 2 generally rare events, one per arm, it is difficult to establish relatedness or evaluate causality. However, these are conditions occasionally observed in the general pediatric population (some estimates suggest a rate of 2 to 5% of children under the age of 5[16]) and therefore might be expected to be observed sporadically in the study setting as well.
6.1.12.6 Clinical Test Results No laboratory or other clinical testing was routinely performed for safety monitoring purposes.
6.1.12.7 Dropouts and/or Discontinuations No AEs or SAEs leading to premature discontinuation of study vaccine and/or study were reported in this study.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 33
6.1.13 Study Summary and Conclusions
Study FLU Q-QIV-022 was a Phase 3, randomized, controlled, multi-center, observer-blind study conducted in the US and Mexico that enrolled 2430 subjects, to compare the safety and immunogenicity of FluLaval-QIV with Fluzone-QIV in children ages ≥6 to < 36 months. Overall, 92.1% of subjects were from the United States and 7.9% of subjects were from Mexico. The demographic characteristics were similar between the 2 treatment groups. Of the 2430 subjects who were enrolled, 2424 were vaccinated, of which 1207 received FluLaval-QIV and 1217 received Fluzone-QIV. Primed subjects received a single IM dose of study vaccine whereas unprimed subjects received 2 doses 28 days apart. All subjects had blood drawn at Day 0 (baseline) and Day 28 (primed subjects) or Day 56 (unprimed subjects) for evaluation of HI titers. Local and systemic reactogenicity was captured by diary card for 7 days postvaccination; SAEs, MAEs, and pIMDs were assessed at either day 28 (primed subjects) or days 28 and 58 (unprimed subjects) as well as at study completion (study day 180). The primary endpoint of immunologic noninferiority for all four vaccine strains was demonstrated for FluLaval-QIV compared to Fluzone-QIV by GMT ratios and seroconversion rate differences. Local and systemic reactogenicity, including fever and rates of grade 3 AEs, and medically attended AEs were balanced between treatment arms. There were no withdrawals due to adverse events reports. There were two pIMDs reported, one case of Kawasaki’s disease in the FluLaval-QIV group and one case of erythema multiforme in the Fluzone-QIV group; with only one case each a causal relationship cannot be established. There were no imbalances in deaths, or other serious adverse events between the two study arms. The available safety and immunogenicity data support extending the indication of FluLaval-QIV, for active immunization for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine, from use in persons ages 3 years and older to use in persons ages 6 months and older.
6.2 Trial #2 FLU Q-QIV-021
Design overview Trial FLU Q-QIV-021 was a phase 2, randomized, active-controlled, observer-blind, multicenter study in subjects ages ≥ 6 to < 36 months. Subjects were randomly allocated in a 1:1 ratio to receive either FluLaval-QIV or a US licensed comparator (Fluzone-TIV). The study enrolled consented eligible subjects with stable health between the age of 6 and 35 months, and for whom the investigator determined that their guardian could and would comply with the requirements of the protocol. Standard eligibility criteria were applied. Primed subjects received one IM dose of study product and unprimed subjects received 2 doses of study product 28 days apart. Blood samples for immunogenicity testing were collected prevaccination on Day 0 and postvaccination on Day 28 after completion of the vaccination series (day 28 for primed subjects and day 56 for unprimed subjects) to evaluate the primary and secondary immunogenicity endpoints. Subjects were followed for solicited AEs by diary card through Day 7 post vaccination. Unsolicited AEs were collected at the Day 28 clinical visit.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 34
Medically attended adverse events (MAEs), AEs leading to study withdrawal, potentially immune mediated diseases (pIMDs), and SAEs including deaths were monitored for 180 days following vaccination. The primary immunogenicity endpoint was a descriptive evaluation of postvaccination HI titer and seroconversion rates (see secondary immunogenicity endpoints for study FLU Q-QIV-022 in Section 6.1.8). Safety endpoints were the same as for study FLU Q-QIV-022 (Section 6.1.8). Results The first subject was enrolled on 23 October 2013 and the last subject completed the study on 03 July 2014. The total vaccinated cohort included 314 subjects; 158 received FluLaval-QIV and 156 subjects received Fluzone-TIV. Two hundred eighty-four subjects completed their 6 month study visit (143 [90.5%] subjects in the FluLaval-QIV group and 141 [90.4%] subjects in the Fluzone-TIV group. With regard to the primary immunogenicity endpoint, at day 28 after completion of the vaccination series with FluLaval-QIV, the LL of the two-sided 95% CI for SCR was ≥ 40% against all four strains (range 58.1% to 79.2%); the LL for the 2 sided 95% CI for those achieving an HI titer of ≥ 1:40 overall was ≥ 70% for all four strains (range 70.6% to 94.4%). Safety results are summarized as follows:
• Solicited local AEs: Overall, injection site pain was the most frequently reported solicited local AE (31.8% and 32.4% of subjects in the FluLaval-QIV and Fluzone-TIV groups, respectively). Grade 3 injection site pain was reported for 2.6% and 0.7% of subjects, respectively.
• Solicited general AEs: Overall, irritability/fussiness was the most frequently reported solicited general AE (50.3% and 45.3% of subjects in the FluLaval-QIV and Fluzone-TIV groups, respectively). Grade 3 irritability/fussiness was reported for 8.6% and 4.1% of subjects, respectively. Fever(≥38°C) was reported for 6.6% and 6.8% of subjects in the FluLaval-QIV and Fluzone-TIV groups, respectively. Grade 3 or higher fever (>39.0°C) was reported for 1.3% and 2.0% of subjects, respectively.
• Relative risk of fever: The relative risk of any fever (≥38°C) for FluLaval-QIV compared to Fluzone-TIV during a 4-day follow-up period was 0.86 with a 95% CI of [0.33; 2.23]. The relative risk of grade 3 or above fever (>39.0°C) for Q-QIV compared to Fluzone-TIV during a 4-day follow-up period was 0.00 (grade 3 fever was reported for none of the subjects in the Q-QIV group, and for one subject in the TIV-YB group post-dose 1) with a 95% CI of [0.00; 3.76].
• Unsolicited AEs: During the 28-day post-vaccination period, at least one unsolicited AE was reported for 48.7% and 48.1% of subjects in the FluLaval-QIV and Fluzone-TIV groups, respectively. There were no imbalances in type or severity of AEs.,
• MAEs: At least one unsolicited AE with a medically attended visit during the entire study period was reported for 48.7% and 57.1% of subjects in the FluLaval-QIV and Fluzone-TIV groups, respectively. Otitis media was the most frequently reported MAE in both groups (14.6% and 19.2% of subjects, respectively).
• pIMDs: No pIMDs were reported in the study.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 35
• SAEs: A total of 9 non-fatal SAEs were reported for 9 subjects [5 (3.2%) subjects in the FluLaval-QIV group and 4 (2.6%) subjects in the Fluzone-TIV group] during the entire study period. No fatal SAEs were reported.
Conclusions Study FLU Q-QIV-021 was a phase 2, randomized, active controlled, observer-blind multi-centered study evaluating safety and immunogenicity of FluLaval Quadrivalent (n = 158 for the total vaccinated cohort) with Fluzone (trivalent) (n = 156 for the total vaccinated cohort) in children ages ≥6 to < 36 months. The study met its descriptive immunogenicity endpoints for immunogenicity of FluLaval Quadrivalent at 28 days after completion of the vaccination series (SCRs were > 40% for all 4 vaccine strains and HI titers of ≥ 1:40 were achieved in > 70% of subjects overall. There were no imbalances observed in local or systemic reactogenicity through study day 7 or unsolicited AEs through study day 28. There were no imbalances in MAEs, or SAEs and there were no pIMDs or deaths reported during the 180 day study period. Reviewer comment: The point estimates for relative risk of any fever or ≥ grade 3 fever was lower for FluLaval-QIV containing 60µg total HA than it was for Fluzone-TIV containing 22.5µg total HA with confidence intervals crossing 1.
6.3 Trial #3 FLU Q-QIV-013
Design overview Trial FLU Q-QIV-013 was a phase 3, randomized, active-controlled, observer-blind, multicenter study in subjects ages ≥ 6 to < 36 months. Subjects were randomly allocated in a 1:1 ratio to receive either FluLaval-QIV or a US licensed comparator (Fluarix-TIV). The study enrolled consented healthy subjects between the age of 6 and 35 months, and for whom the investigator determined that their guardian could and would comply with the requirements of the protocol. Standard eligibility criteria were applied. Primed subjects received one IM dose of study product and unprimed subjects received 2 doses of study product 28 days apart. Blood samples for immunogenicity testing were collected prevaccination on Day 0 and postvaccination on Day 28 after completion of the vaccination series (day 28 for primed subjects and day 56 for unprimed subjects) to evaluate the primary and secondary immunogenicity endpoints. Subjects were followed for solicited AEs by diary card through Day 7 post vaccination. Unsolicited AEs were collected at the Day 28 clinical visit. Medically attended adverse events (MAEs), AEs leading to study withdrawal, potentially immune mediated diseases (pIMDs), and SAEs including deaths were monitored for 180 days following vaccination. The primary immunogenicity endpoint was a descriptive evaluation of postvaccination HI titer and seroconversion rates (see secondary immunogenicity endpoints for study FLU Q-QIV-022 in Section 6.1.8). Safety endpoints were the same as for study FLU Q-QIV-022 (Section 6.1.8). Results The first subject was enrolled on 1 November 2012 and the last subject completed the study on 19 June 2013. The total vaccinated cohort included 601 subjects; 299 received FluLaval-QIV and 302 subjects received Fluarix-TIV. Five hundred eighty subjects completed their 6 month study

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 36
visit (286 [95.7%] subjects in the FluLaval-QIV group and 294 [97.4%] subjects in the Fluarix-TIV group. With regard to the primary immunogenicity endpoint, at day 28 after completion of the vaccination series with FluLaval-QIV, the LL of the two-sided 95% CI for SCR was ≥ 40% against all four strains (range 66.6% to 81.3%); the LL for the 2 sided 95% CI for those achieving an HI titer of ≥ 1:40 overall was ≥ 70% for all four strains (range 76.3% to 85.3%). Safety results are summarized as follows:
• Solicited local AEs: Injection site pain was the most frequently reported solicited local AE (32.6% and 30.6% of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively). Grade 3 injection site pain was reported for 7 (2.4%) and 3 (1.0%) subjects, respectively.
• Solicited general AEs: Irritability/fussiness was the most frequently reported solicited general AE (40.7% and 41.6% of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively). Grade 3 irritability/fussiness was reported for 5.2% and 4.7% of subjects, respectively.
• The relative risk of any fever for FluLaval-QIV compared to Fluarix-TIV during a 4-day follow-up period was 1.12 with a 95% CI of [0.76; 1.64] (p-value = 0.6439). The relative risk of Grade 3 or higher fever for FluLaval-QIV compared to Fluarix-TIV during a 4-day follow-up period was 2.04 with a 95% CI of [0.91; 4.60].
• Unsolicited AEs: During the 28-day post-vaccination period, at least one unsolicited AE was reported for 47.5% and 54.6% of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively. There were no imbalances in types or severity of AEs.
• MAEs: At least one unsolicited AE with a medically attended visit during the entire study period was reported for 52.2 % and 51.7 % of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively. Diarrhea (8.0% and 9.6% of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively) and pharyngitis (5.7% and 2.6% of subjects in the FluLaval-QIV and Fluarix-TIV groups, respectively) were the only unsolicited MAEs reported by more than 5.0% of subjects in any study group.
• pIMDs: 2 pIMDs were reported, both of which occurred in the Fluarix-TIV arm (alopecia areata and ulcerative colitis)
• SAEs: A total of 25 non-fatal SAEs were reported for 17 subjects [9 (3.0%) subjects in the FluLaval-QIV group and 8 (2.6%) subjects in the Fluarix-TIV group] during the entire study period. There were no imbalances noted in type of SAE. One SAE in the FluLaval-QIV arm was a febrile seizure which occurred on the day of vaccination in an 18-month old male. No fatal SAEs were reported.
Conclusions Study FLU Q-QIV-013 was a phase 3, randomized, active controlled, observer-blind multi-centered study evaluating safety and immunogenicity of FluLaval Quadrivalent (n = 299 for the total vaccinated cohort) with Fluarix (trivalent) (n = 302 for the total vaccinated cohort) in children ages ≥6 to < 36 months. The study met its descriptive immunogenicity endpoints for immunogenicity of FluLaval Quadrivalent at 28 days after completion of the vaccination series (SCRs were > 40% for all 4 vaccine strains and HI titers of ≥ 1:40 were achieved in > 70% of subjects overall. There were no imbalances observed in local or systemic reactogenicity through

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 37
study day 7 or unsolicited AEs through study day 28. There were no imbalances in MAEs, or SAEs and there were no deaths reported during the 180 day study period.
6.4 Trial #4 FLU Q-QIV-003
Design overview FLU Q-QIV-003 was a Phase III, randomized, double-blind, multi-country (Canada, Mexico, Spain, Taiwan, and the United States) study conducted in 2010-2011 to evaluate the immunogenicity and safety of Q-QIV in children 6 months to 17 years of age. The study included an open-label arm or FluLaval-QIV administered to children ages ≥ 6 to < 36 months of age in Canada and the US, the data from which was reviewed for this BLA. The safety follow-up in this study was for 6 months from the first dose. There were no primary objectives. Descriptive secondary immunogenicity and safety endpoints were similar to the secondary immunogenicity and safety endpoints described above for study Flu Q-QIV-022 in Section 6.1.8. The relative risk of fever could not be assessed for subjects ages ≥ 6 to < 36 months because there was no comparator arm. Results The first subject was enrolled on 10 October 2010 and the last subject completed the study on 6 July 2011. The total vaccinated cohort included 301 subjects who received FluLaval-QIV of whom 259 completed the study. With regard to the secondary descriptive immunogenicity endpoint, at day 28 after completion of the vaccination series with FluLaval-QIV, the LL of the two-sided 95% CI for SCR was ≥ 40% against all four strains (range 67.1% to 90.2%). The LL for the 2 sided 95% CI for those achieving an HI titer of ≥ 1:40 overall was ≥ 70% for 3 of the 4 vaccine strains (85.2% for A/California/7/2009 (H1N1); 68.8% for A/Victoria/210/2009 (H3N2); 83.4% for B/Brisbane/60/2008 (Victoria-lineage); and 93.5% for, B/Florida/4/2006 (Yamagata-lineage). Safety results are summarized as follows:
• Solicited local AEs: Injection site pain was the most frequently reported solicited local AE in 131 (44.6 %) subjects. Grade 3 injection site pain was reported in 69 (2.3%) subjects.
• Solicited general AEs: Irritability/fussiness was the most frequently reported solicited general AE occurring in 120 (41.1%) subjects. Grade 3 irritability/fussiness was reported in 8 (2.7%) subjects.
• Unsolicited AEs: During the 28-day post-vaccination period, at least one unsolicited AE was reported in 160 (53.2%) subjects. Cough was the most frequently reported AE (11.3%)
• MAEs: At least one MAE was reported for 147 (48.8%) subjects. The most frequently reported MAE was cough (13.3%).
• pIMDs: There were no pIMDs reported • SAEs: A total of 10 non-fatal SAEs were reported for 7 (2.3%) subjects. One non-
fever associated seizure occurred on study day 0 in a 12-month old female. One fever associated seizure occurred on study day 24-month old male. The remaining events all had a plausible etiology other than vaccination. There were no deaths reported.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 38
7. INTEGRATED OVERVIEW OF EFFICACY
Not applicable. See Section 5.1 for discussion of the review strategy applied to the studies submitted to this sBLA.
8. INTEGRATED OVERVIEW OF SAFETY
8.1 Safety Assessment Methods
The integrated summary of safety and safety section of clinical study reports for supportive studies FLU Q-QIV-021, FLU Q-QIV-013, and FLU Q-QIV-003 were evaluated for deaths, SAEs and pIMDs.
8.2 Safety Database
See above Section 8.1.
8.3 Caveats Introduced by Pooling of Data Across Studies
See discussion in Review Strategy, Section 5.1.
8.4 Safety Results
8.4.1 Deaths
There were no deaths reported across the four studies.
8.4.2 Nonfatal Serious Adverse Events
SAEs were generally balanced between treatment arms in the 3 studies with comparator arms. In the primary study FLU Q-QIV-022 there were 29 SAEs reported in 22 subjects (1.8%) in the FluLaval-QIV group and 28 SAEs in 21 subjects (1.7%) in the Fluzone-QIV group. In supportive study FLU Q-QIV-021 there were 5SAEs in 5 subjects (3.2%) in the FluLaval-QIV group and 4 SAEs ion 4 subjects in the Fluzone-TIV group. In supportive study FLU Q-QIV-013, there were 9 SAEs in 9 subjects (3%) in the FluLaval-QIV group and 8 SAEs in 8 subjects (2.6%) in the Fluarix-TIV group. Supportive study FLU Q-QIV-003 there were 10 subjects (2.3%) in the FluLaval-QIV group experienced an SAE; there was no comparator arm but this rate of SAEs is consistent with the other studies. There were 13 reports of convulsions in 13 subjects, 11 of which were associated with fever. Nine of these events occurred in the primary study FLU Q-QIV-022 (5 subjects in the FluLaval-QIV group and 4 subjects in the Fluzone-QIV group) and are described in Section 6.1.12.4. The remaining reported seizures are described below:
• 1 febrile seizure occurred on study day 0 post vaccination in an 18-month old male who received FluLaval-QIV (see discussion and reviewer comment in Section 6.3 describing Study FLU Q-QIV0913)
• 1 febrile seizure occurred on study day 18 in a subject receiving FluLaval-QIV in the single arm open label study, FLU Q-QIV-003 (described in Section 6.4)

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 39
• 2 seizures were not fever-associated (one on study day 70 in a subject receiving Fluzone-TIV and one on study day 0 in a subject receiving FluLaval-QIV, again in study FLU Q QIV-003.
Reviewer comment: Presumably rate and/or severity of reactogenicity would be proportionally increased if these episodes of febrile seizures represented a vaccine-associated safety signal; it is reassuring that this was not observed. Overall, the other reported SAEs were reasonably attributed to etiologies other than vaccination and the nature of SAEs reported were consistent with events that might occur in the age range being studied, for example, respiratory infections, asthma, gastroenteritis, trauma.
8.4.3 Study Dropouts/Discontinuations
Among the 4 studies, one subject was withdrawn subsequent to a non-serious adverse event (moderate fever) which was reported as resolved. This subject was enrolled in the study Q-QIV-003.
8.4.4 Common Adverse Events
The sample size for the primary supportive study, FLU Q-QIV-022, was sufficient to adequately characterize local and systemic reactogenicity for FluLaval-QIV and is described in Section 6.1.12.2. Given that each study had different comparators (One with Fluzone-TIV, one with Fluarix-TIV and one single-arm and open-label), the integrated safety analysis focuses on rare and serious adverse events (deaths, SAEs, pIMDs).
8.4.6 Systemic Adverse Events
See above Section 8.4.4.
8.4.7 Local Reactogenicity
See above Section 8.4.4.
8.4.8 Potentially Immune Mediated Diseases
During the entire study period, 4 cases of pIMD were reported in 4 subjects across the 4 studies. Two cases of pIMD were reported in study Q-QIV-022. One pIMD was in the Q- QIV group (Kawasaki’s disease which was also reported as an SAE) and the other in the F-QIV group (Erythema multiforme). Both were reported as recovered/resolved at the time of the study report (see Section 6.1.12.5). Two cases of pIMD were reported in study Q-QIV-013 (alopecia areata and ulcerative colitis) but both cases in the comparator group (Fluarix-TIV). Both were reported as resolved at the time of the study report.
8.6 Safety Conclusions
Evaluation of the integrated summary of safety focused on deaths, SAEs, MAEs and pIMDs for studies FLU Q-QIV-022, FLU Q-QIV-021, FLU Q-QIV-013 and FLU Q-QIV-003. There were no deaths reported during any of the four study periods. There were no imbalances noted in number or nature of SAEs or MAEs. There were 4 pIMDs reported, only one of which occurred in a FluLaval-QIV study group (Kawasaki’s disease). Overall, the integrated summary of safety does not raise safety concerns.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 40
9. ADDITIONAL CLINICAL ISSUES
9.1 Special Populations
9.1.1 Human Reproduction and Pregnancy Data
There are insufficient data to establish whether there is a vaccine-associated risk with FluLaval Quadrivalent in pregnant women.
9.1.2 Use During Lactation
There is no information available on the presence of FluLaval Quadrivalent in human milk, the effects on the breastfed infant, or the effects on milk production.
9.1.3 Pediatric Use and PREA Considerations
FluLaval and FluLaval-QIV are currently approved for use in persons ages 3 years and older. The purpose of this sBLA 125163/405 is to extend the age indication to ages 6 months and older. The manufacturer received a partial waiver for infants <6 months of age based on the reasoning that FluLaval (trivalent formulation) and FluLaval Quadrivalent would provide no meaningful therapeutic benefit over vaccination beginning at 6 months of age, and these vaccines are unlikely to be used by a substantial number of infants <6 months of age (Section 505B(a)(4)(A)iii of the Food Drug and Cosmetic Act). Thus, if approved, this sBLA will fulfill the PREA-postmarketing requirement for both FluLaval (trivalent formulation) and FluLaval Quadrivalent.
10. CONCLUSIONS
In subjects ages ≥ 6 to < 36 months, FluLaval-QIV met criteria for immunologic noninferiority against all four vaccine strains at 28 days after completing the vaccination series (one dose in primed subjects and 2 doses 28 days apart in unprimed subjects) by GMT ratios and SCR differences when compared with Fluzone-QIV. No imbalances in safety were noted in the primary study FLU Q-QIV-022 or in evaluation of the safety data for supportive studies FLU Q-QIV-021, FLU Q-QIV-013, and FLU Q-QIV-003.
11. RISK-BENEFIT CONSIDERATIONS AND RECOMMENDATIONS
11.1 Risk-Benefit Considerations
Table 21. Summary of Risk-Benefit Analysis for FluLaval Quadrivalent Decision Factor
Evidence and Uncertainties Conclusions and Reasons

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 41
Decision Factor
Evidence and Uncertainties Conclusions and Reasons
Analysis of Condition
• Influenza virus infection is a major cause of morbidity and mortality.
• Children are a high-risk group for developing complications associated with influenza virus infection.
• Influenza vaccination has been shown to be effective in reducing the incidence of influenza-like illness (ILI), hospitalization for influenza/pneumonia/other respiratory conditions, acute complications among high-risk patients, and mortality from all causes.
• Influenza virus infection is a potentially life-threatening disease.
• Influenza virus infection is a serious condition, particularly in children who are high-risk for developing complications including death .
Unmet Medical Need
• Only Fluzone and Fluzone Quadrivalent are approved for children ages ≥ 6 to < 36 months
• Live attenuated influenza vaccine (FluMist) is approved for persons ages ≥ 2 to < 50 years.
• Vaccine shortages could lead to delays or lapses in annual vaccination in children.
• Additional licensed products in in this age range will help meet the need for effective prevention of influenza.
Clinical Benefit
• One clinical trial in children ages ≥ 6 to < 36 months conducted under IND (FLU Q-QIV-022) demonstrated immunologic noninferiority compared to the US licensed comparator Fluzone-QIV with regard to Day 28 HAI titers expressed as GMT ratios and SCR rate differences.
• Demonstration of immunologic non-inferiority compared with Fluzone-QIV supports clinical effectiveness of FluLaval-QIV.
• Prevention of influenza illness in the children ages ≥ 6 to < 36 months reduces morbidity and mortality associated with influenza infection in this population.
Risk
• The most substantial risks of vaccination with FluLaval were mild local and systemic reactogenicity.
• No other safety signals were apparent in evaluation of the primary and 3 additional supportive safety studies conducted in children ages ≥ 6 to < 36 months.
• FluLaval was first licensed in Canada in 1992 and approved in the US in 2006. Flulaval Quadrivalent was approved in the US on 8 Oct 2013 and is also available in Canada and Mexico. Based on distribution data, it is estimated that over 11.7 million doses have been administered. No other safety signals have been identified in postmarketing surveillance.
• All the evidence indicates that the risk of vaccination with FluLaval-QIV is minimal.

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 42
Decision Factor
Evidence and Uncertainties Conclusions and Reasons
Risk Management
• The most substantial risks of vaccination with FluLaval-QIV are associated with inflammation (pain) produced at the injection site. However, the most injection site reactions are mild in severity, and they resolve relatively quickly and without sequelae.
• The package insert and the current pharmacovigilance plan are adequate to manage these risks.
11.2 Risk-Benefit Summary and Assessment
Data submitted to sBLA125163/405 establish a substantial likelihood of benefit for prevention of laboratory-confirmed influenza caused by any influenza viral type/subtype included in the vaccine. The risks of vaccination with FluLaval-QIV in children ages ≥ 6 to < 36 months have been found to be minimal in association with a substantial likelihood of benefit in the prevention of influenza disease caused by vaccine types/subtypes contained in the vaccine. Thus, the overall risk-benefit profile of this product is favorable in this young age group .
11.4 Recommendations on Regulatory Actions
FluLaval (trivalent) and FluLaval-QIV are recommended for approval in children ages ≥ 6 to < 36 months for active immunization for the prevention of disease caused by influenza A subtype viruses and type B viruses contained in the vaccine.
11.5 Labeling Review and Recommendations
Negotiations and CBER recommendations resulted in the following changes to the current labels for both FluLaval (trivalent) and FluLaval-QIV:
• In Section 6.1, Clinical Trials Experience, CBER requested that the Applicant expand the safety tables 2 through 5, describing overall rates of local and systemic reactogenicity for each age range so that they specifically described rates of grade 3 adverse reactions. CBER considered the severe reactions to be clinically important information for prescribers.
• In Section 6.2 entitled Postmarketing Experience, the Applicant distinguished between safety reports occurring for FluLaval (trivalent) and FluLaval Quadrivalent. This resulted in only one event being reported for FluLaval Quadrivalent, allergic reactions under the subheading Immune System Disorders. In clinical trials and postmarketing surveillance, the AEs reported for FluLaval Quadrivalent have been similar to those for FluLaval (trivalent), and to influenza vaccines in general. This similarity would be anticipated because the formulation of these products is identical except for the one additional influenza B strain contained in the quadrivalent formulation. Given the limitations of passive surveillance, including under-reporting, distinguishing between AEs reported for FluLaval (trivalent) and FluLaval Quadrivalent in this section could be misleading and imply that FluLaval is associated with fewer AEs than the TIV version or other, similar influenza vaccines. Thus, CBER requested that adverse events be consolidated and to state that the listed events have been reported with either FluLaval (trivalent) or Quadrivalent.
• Section 8.1 entitled Pregnancy was updated in compliance with the Pregnancy and Lactation Labeling Rule (PLLR). CBER Toxicology reviewed developmental toxicology studies evaluating FluLaval Quadrivalent and recommended language indicating that, “no adverse

Clinical Reviewer: Sarah K. Browne, MD STN: 125163/405
Page 43
effects on pre-weaning development up to post-natal Day 25 were observed [and] there were no fetal malformations or variations observed due to the vaccine.“ The section also states that clinical data were insufficient to women to inform risks of FluLaval and FluLaval Quadrivalent in pregnant or lactating women.
11.6 Recommendations on Postmarketing Actions
No changes to the submitted pharmacovigilance plan for FluLaval (trivalent) or FluLaval- QIV are recommended based on the information contained in this application.







![[Product Monograph Template - Schedule D] · FLULAVAL TETRA should under no circumstances be administered intravascularly. General It is good clinical practice to precede vaccination](https://static.fdocuments.net/doc/165x107/5e74f0fe9210e1759e2d74ee/product-monograph-template-schedule-d-flulaval-tetra-should-under-no-circumstances.jpg)










