Clinical Practice Guidelines From the Cystic Fibrosis...
28
SPECIAL ARTICLE PEDIATRICS Volume 137, number 4, April 2016:e20151784 Clinical Practice Guidelines From the Cystic Fibrosis Foundation for Preschoolers With Cystic Fibrosis Thomas Lahiri, MD, a Sarah E. Hempstead, MS, b,c Cynthia Brady, DNP, d Carolyn L. Cannon, MD, PhD, e Kelli Clark, BS, f Michelle E. Condren, PharmD, g Margaret F. Guill, MD, c,h R. Paul Guillerman, MD, i Christina G. Leone, MSW, LCSW, j Karen Maguiness, MS, RD, CSP, k Lisa Monchil, RRT-NPS, CCRC, AE-C, l Scott W. Powers, PhD, ABPP, m Margaret Rosenfeld, MD, MPH, n Sarah Jane Schwarzenberg, MD, o Connie L. Tompkins, PhD, p Edith T. Zemanick, MD, q Stephanie D. Davis, MD k Cystic fibrosis (CF) clinical care guidelines exist for the care of infants up to age 2 years and for individuals ≥6 years of age. An important gap exists for preschool children between the ages of 2 and 5 years. This period marks a time of growth and development that is critical to achieve optimal nutritional status and maintain lung health. Given that disease often progresses in a clinically silent manner, objective and sensitive tools that detect and track early disease are important in this age group. Several challenges exist that may impede the delivery of care for these children, including adherence to therapies. A multidisciplinary committee was convened by the CF Foundation to develop comprehensive evidence-based and consensus recommendations for the care of preschool children, ages 2 to 5 years, with CF. This document includes recommendations in the following areas: routine surveillance for pulmonary disease, therapeutics, and nutritional and gastrointestinal care. abstract a Pediatric Pulmonology, University of Vermont Children’s Hospital and Department of Pediatrics, University of Vermont College of Medicine, Burlington, Vermont; b The Dartmouth Institute for Health Policy and Clinical Practice, Lebanon, New Hampshire; c Geisel School of Medicine at Dartmouth, Lebanon, New Hampshire; d Children’s Respiratory and Critical Care Specialists and Children’s Hospitals and Clinics of Minnesota, Minneapolis, Minnesota; e Texas A&M Health Science Center, College Station, Texas; f Department of Pediatrics, University of North Carolina, Charlotte, North Carolina; g University of Oklahoma College of Pharmacy and School of Community Medicine, Tulsa, Oklahoma; h Allergy and Pediatric Pulmonology, Dartmouth- Hitchcock Medical Center, Lebanon, New Hampshire; i Department of Radiology, Baylor College of Medicine and Department of Pediatric Radiology, Texas Children’s Hospital, Houston, Texas; j Cystic Fibrosis Center, Children’s Hospital Colorado, Aurora, Colorado; k Section of Pediatric Pulmonology, Allergy and Sleep Medicine, Department of Pediatrics, Riley Hospital for Children, Indiana University School of Medicine, Indianapolis, Indiana ; l Armond V. Mascia, MD Cystic Fibrosis Center, Maria Fareri Children’s Hospital at Westchester Medical Center, Valhalla, New York; m Department of Pediatrics and Cincinnati Children’s Research Foundation, University of Cincinnati College of Medicine and Division of Behavioral Medicine and Clinical Psychology, Cincinnati Children’s Hospital, Cincinnati, Ohio; n Division of Pulmonary Medicine, Seattle Children’s Hospital and Department of Pediatrics, University of Washington School of Medicine, Seattle, Washington; o Pediatric Gastroenterology, Hepatology and Nutrition, University of Minnesota Masonic Children’s Hospital, Minneapolis, Minnesota; p Department of Rehabilitation and Movement Sciences, University of Vermont College of Nursing and Health Sciences, Burlington, Vermont; and q Department of Pediatrics, University of Colorado School of Medicine, Aurora, Colorado Drs Lahiri and Davis reviewed selected articles, drafted the initial manuscript, and revised the final manuscript; Ms Hempstead performed a literature Early nutritional intervention and monitoring for respiratory and gastrointestinal disease in infants with cystic fibrosis (CF) is vital to improve long-term outcomes. Clinical care guidelines specific to infants with CF, 1 and nutrition and pulmonary guidelines for children > 6 years of age have been published by the CF Foundation. 2,3 However, a gap exists in clinical care recommendations pertaining to preschoolers with CF, children ages 2 to 5 years. In addition to typical developmental milestones, preschoolers with CF and their families face complex, time- consuming treatment regimens to maintain lung health and achieve optimal growth. It is well established that airway inflammation, infection, airway obstruction, and structural damage exist during preschool years in children with CF, often in the absence of overt respiratory symptoms. 4–7 For families of preschoolers with CF, it can be challenging to institute appropriate mealtime behaviors, which can lead to poor nutritional outcomes 8–10 and decreased survival at age 18 years when weight-for-age is ≤10th percentile for age and gender at age 4 years. 11 Supporting preschoolers with CF and their families is critical to establish habits that promote normal growth and development and an active lifestyle (ie, exercise) to prevent pulmonary decline. METHODS In January 2014, the CF Foundation convened a committee of 16 CF pediatric experts and parents to develop clinical care guidelines for To cite: Lahiri T, Hempstead SE, Brady C, et al. Clinical Practice Guidelines From the Cystic Fibrosis Foundation for Preschoolers With Cystic Fibrosis. Pediatrics. 2016;137(4):e20151784 by guest on October 12, 2018 www.aappublications.org/news Downloaded from
Transcript of Clinical Practice Guidelines From the Cystic Fibrosis...

SPECIAL ARTICLEPEDIATRICS Volume 137 , number 4 , April 2016 :e 20151784
Clinical Practice Guidelines From the Cystic Fibrosis Foundation for Preschoolers With Cystic FibrosisThomas Lahiri, MD, a Sarah E. Hempstead, MS, b, c Cynthia Brady, DNP, d Carolyn L. Cannon, MD, PhD, e Kelli Clark, BS, f Michelle E. Condren, PharmD, g Margaret F. Guill, MD, c, h R. Paul Guillerman, MD, i Christina G. Leone, MSW, LCSW, j Karen Maguiness, MS, RD, CSP, k Lisa Monchil, RRT-NPS, CCRC, AE-C, l Scott W. Powers, PhD, ABPP, m Margaret Rosenfeld, MD, MPH, n Sarah Jane Schwarzenberg, MD, o Connie L. Tompkins, PhD, p Edith T. Zemanick, MD, q Stephanie D. Davis, MDk
Cystic fibrosis (CF) clinical care guidelines exist for the care of infants
up to age 2 years and for individuals ≥6 years of age. An important gap
exists for preschool children between the ages of 2 and 5 years. This
period marks a time of growth and development that is critical to achieve
optimal nutritional status and maintain lung health. Given that disease
often progresses in a clinically silent manner, objective and sensitive tools
that detect and track early disease are important in this age group. Several
challenges exist that may impede the delivery of care for these children,
including adherence to therapies. A multidisciplinary committee was
convened by the CF Foundation to develop comprehensive evidence-based
and consensus recommendations for the care of preschool children, ages
2 to 5 years, with CF. This document includes recommendations in the
following areas: routine surveillance for pulmonary disease, therapeutics,
and nutritional and gastrointestinal care.
abstract
aPediatric Pulmonology, University of Vermont Children’s
Hospital and Department of Pediatrics, University of
Vermont College of Medicine, Burlington, Vermont; bThe
Dartmouth Institute for Health Policy and Clinical Practice,
Lebanon, New Hampshire; cGeisel School of Medicine
at Dartmouth, Lebanon, New Hampshire; dChildren’s
Respiratory and Critical Care Specialists and Children’s
Hospitals and Clinics of Minnesota, Minneapolis, Minnesota; eTexas A&M Health Science Center, College Station, Texas; fDepartment of Pediatrics, University of North Carolina,
Charlotte, North Carolina; gUniversity of Oklahoma College
of Pharmacy and School of Community Medicine, Tulsa,
Oklahoma; hAllergy and Pediatric Pulmonology, Dartmouth-
Hitchcock Medical Center, Lebanon, New Hampshire; iDepartment of Radiology, Baylor College of Medicine
and Department of Pediatric Radiology, Texas Children’s
Hospital, Houston, Texas; jCystic Fibrosis Center, Children’s
Hospital Colorado, Aurora, Colorado; kSection of Pediatric
Pulmonology, Allergy and Sleep Medicine, Department of
Pediatrics, Riley Hospital for Children, Indiana University
School of Medicine, Indianapolis, Indiana ; lArmond V.
Mascia, MD Cystic Fibrosis Center, Maria Fareri Children’s
Hospital at Westchester Medical Center, Valhalla, New
York; mDepartment of Pediatrics and Cincinnati Children’s
Research Foundation, University of Cincinnati College of
Medicine and Division of Behavioral Medicine and Clinical
Psychology, Cincinnati Children’s Hospital, Cincinnati, Ohio; nDivision of Pulmonary Medicine, Seattle Children’s Hospital
and Department of Pediatrics, University of Washington
School of Medicine, Seattle, Washington; oPediatric
Gastroenterology, Hepatology and Nutrition, University
of Minnesota Masonic Children’s Hospital, Minneapolis,
Minnesota; pDepartment of Rehabilitation and Movement
Sciences, University of Vermont College of Nursing and
Health Sciences, Burlington, Vermont; and qDepartment
of Pediatrics, University of Colorado School of Medicine,
Aurora, Colorado
Drs Lahiri and Davis reviewed selected articles,
drafted the initial manuscript, and revised the fi nal
manuscript; Ms Hempstead performed a literature
Early nutritional intervention and
monitoring for respiratory and
gastrointestinal disease in infants
with cystic fibrosis (CF) is vital to
improve long-term outcomes. Clinical
care guidelines specific to infants with
CF, 1 and nutrition and pulmonary
guidelines for children > 6 years of
age have been published by the CF
Foundation.2, 3 However, a gap exists
in clinical care recommendations
pertaining to preschoolers with CF,
children ages 2 to 5 years.
In addition to typical developmental
milestones, preschoolers with CF and
their families face complex, time-
consuming treatment regimens to
maintain lung health and achieve
optimal growth. It is well established
that airway inflammation, infection,
airway obstruction, and structural
damage exist during preschool years in
children with CF, often in the absence
of overt respiratory symptoms.4–7 For
families of preschoolers with CF, it can
be challenging to institute appropriate
mealtime behaviors, which can lead
to poor nutritional outcomes8–10
and decreased survival at age 18
years when weight-for-age is ≤10th
percentile for age and gender at age
4 years.11 Supporting preschoolers
with CF and their families is critical to
establish habits that promote normal
growth and development and an
active lifestyle (ie, exercise) to prevent
pulmonary decline.
METHODS
In January 2014, the CF Foundation
convened a committee of 16 CF
pediatric experts and parents to
develop clinical care guidelines for
To cite: Lahiri T, Hempstead SE, Brady C, et al.
Clinical Practice Guidelines From the Cystic
Fibrosis Foundation for Preschoolers With Cystic
Fibrosis. Pediatrics. 2016;137(4):e20151784
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
preschool-aged children with CF.
The treatment of this population
has become increasingly important,
as earlier diagnoses are made,
primarily through newborn
screening. Committee members
participated in 1 of 3 workgroups.
Each group developed population,
intervention, comparison, and
outcome (PICO) questions, 12 which
were reviewed and approved by
the wider committee. Dartmouth
College investigators conducted a
Medline literature search using PICO
questions and additional medical
search headings provided by the
committee. Committee members
conducted hand searches for
additional literature and existing
guidelines.
In May 2014 the committee convened
to review draft recommendation
statements and supporting evidence.
Unfortunately, the evidence is
lacking for most treatments and
monitoring tools in the 2- to 5-year-
old age group. Whenever possible,
statements were developed and
graded by using the US Preventive
Services Task Force grade
definitions (Supplemental Table
1). The committee decided to make
recommendations for CF care that
would guide both CF clinicians and
primary care providers (PCPs).
Therefore, questions for which
evidence was limited or absent were
then presented and discussed by
committee members. Use of existing
evidence from older children and
adults, as well as clinical experience,
was then used as the basis for
consensus recommendations. An
80% approval by the committee was
agreed on a priori, and required for
all statements. Revisions and voting
were managed by using an online
survey. All recommendations were
approved by the committee, and
had a final consensus rate of at least
87.5%.
A draft manuscript was distributed
by the CF Foundation to all
accredited care centers for a 2-week
public comment period. Feedback
was collected by using an online
survey and the guidelines were
revised accordingly. The committee
recognizes the limitations of these
guidelines, which are the first step
in standardization of preschool
CF care. We fully expect these
recommendations to evolve over
time, as randomized controlled
trials in these younger patients
may provide evidence in favor of or
against consensus recommendations.
RESULTS
In total, 10 427 articles were
retrieved. Review articles, case
reports, letters, nonhuman studies,
and studies not related to the PICO
questions were removed. A total
of 344 articles were retained for
review. Additional details on the
review process can be found in the
Supplemental Information. Guideline
recommendations are summarized in
Table 1.
DISCUSSION
Health Maintenance
Preschoolers with CF should
receive routine well-child care from
a PCP. Collaboration among the
family, PCP, and a CF Foundation
accredited care center is essential,
and information about CF and the
child’s CF care should be provided
to the PCP.1 Children should receive
age-appropriate immunizations and
annual seasonal influenza vaccination
along with family members and
caregivers.13–15 They should receive
the first dose of pneumococcal
polysaccharide vaccine (PPSV23),
given at least 8 weeks after the last
pneumococcal conjugate vaccine
(PCV13) dose. Environmental
tobacco smoke exposure should be
avoided; providers should routinely
assess for environmental smoke
exposure and offer caregivers
information about smoking cessation
and interventions to limit exposure.16
Table 2 outlines routine monitoring
care for preschoolers with CF
including primary care visits. Table 1:
Recommendations 1–5.
Caregiver Engagement
Parenting a preschooler with CF
can be demanding and stressful.
Families can face significant obstacles
accessing CF care, including the high
cost of insurance and prescription
medications, and delays in, or denial
of, coverage. Families may need
guidance to model behaviors to
handle refusals to take medications
and to navigate mealtime struggles.
To promote normal growth and
development, it is essential to
institute and maintain CF care
routines in partnership with families,
especially during preschool-age
development. Parents and CF
providers should collaborate to
develop individualized treatment
goals and plans that address barriers
to care. Supplemental Table 2 lists
some risk factors for poor adherence
and Supplemental Table 3 outlines
questions and topics to help
caregiver engagement with
treatment adherence. Table 1:
Recommendation 6.
SURVEILLANCE FOR PULMONARY DISEASE
Pulse Oximetry
Few studies have evaluated oxygen
saturation levels in CF. In older
children and adolescents with CF,
correlations between nocturnal O2
saturation and lung function indices,
chest computed tomography (CT),
and chest radiograph scores have
been reported17, 18; hypoxia during
sleep has been demonstrated to be
associated with low forced expiratory
volume in 1 second (FEV1).19
In infants and preschoolers, no
significant difference was observed
in nocturnal O2 saturation in those
with CF versus controls.20 Given
the paucity of data in preschoolers
with CF, the committee concluded
2 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016 3
TABLE 1 Recommendation Statements
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Health Maintenance 1. For children with CF, ages 2 through 5 y, the
CF Foundation recommends routine well-child
care at PCP following AAP guidelines.
Consensus Recommendation AAP, Preventative Health Care (2014)
Health Maintenance 2. The CF Foundation recommends that children
with CF, ages 2 through 5 y, receive all routine
immunizations, following the recommended
vaccination schedule per the AAP.
Consensus Recommendation AAP, Immunization Schedule (2014)
Health Maintenance 3. The CF Foundation recommends that children
with CF, ages 2 through 5 y, family members,
and caregivers should receive annual seasonal
infl uenza vaccination.
Consensus Recommendation AAP, Recommendations for Prevention
and Control of Infl uenza in Children,
2013–2014 (2013)
Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus Recommendation
Health Maintenance 4. The CF Foundation recommends that children
with CF, ages 2 through 5 y, receive the fi rst
dose of the pneumococcal polysaccharide
vaccine (PPSV23), given at least 8 wk after last
pneumococcal conjugate (Prevnar) vaccine
dose.
Consensus Recommendation AAP Immunization Schedule (2014)
Health Maintenance 5. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that a smoke-
free environment be provided and that all
caregivers are informed that cigarette smoke
exposure harms children with CF.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus Recommendation
AAP-Tobacco Use: A Pediatric Disease
(2009)
Caregiver Engagement 6. For children with CF ages, 2 through 5 y, the CF
Foundation recommends that parents and a
CF health care professional review treatment
goals and individualized care plans quarterly
to assess and address barriers to CF care.
Consensus Recommendation
Screening and Monitoring:
Pulse Oximetry
7. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against the use
of pulse oximetry routinely as an adjunctive
tool to detect lung disease.
Grade: I; Certainty: Low Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Grade: I, Certainty: Low, Benefi t: Small
Screening and Monitoring:
Spirometry
8. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that spirometry
should be attempted as early as age 3,
depending on the developmental stage of the
individual child.
Consensus Recommendation
Screening and Monitoring:
Spirometry
9. For children with CF, ages 3 and older, the CF
Foundation recommends the use of spirometry
for identifying pulmonary exacerbations and
monitoring response to therapy in those
children able to perform acceptable and
reproducible maneuvers.
Consensus Recommendation
Screening and Monitoring:
Bronchodilator
10. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against routine
monitoring of bronchodilator responsiveness.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: I
Certainty: Low
Screening and Monitoring:
Multiple Breath Washout
11. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against routine
monitoring of multiple breath washout.
Grade: I; Certainty: Low
Screening and Monitoring:
Chest Imaging
12. For children with CF, ages 2 through 5 y, the
CF Foundation recommends chest radiographs
be obtained at a minimum every other year to
monitor progression of lung disease.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus Recommendation
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al 4
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Cystic Fibrosis Adult Care (2004)
Consensus Recommendation
Screening and Monitoring:
Chest Imaging
13. For children with CF, ages 2 through 5 y, the
CF Foundation recommends consideration of
chest CT as an alternative to chest radiograph
to monitor progression of lung disease. If
chest CT is performed, it should replace chest
radiograph, be performed every 2–3 y, and use
the lowest radiation dose possible.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus Recommendation
Screening and Monitoring:
Microbiology
14. For children with CF, ages 2 through 5 y, the CF
Foundation recommends routine monitoring of
airway microbiology by oropharyngeal cultures
at least quarterly.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis
(2009) Consensus Recommendation,
Certainty: Low, Benefi t: Moderate
Screening and Monitoring:
Microbiology
15. For children with CF, ages 2 through 5 y, the CF
Foundation recommends against routine use of
bronchoscopy to obtain lower airway cultures.
Grade: D; Certainty: Moderate;
Benefi t: Negative
Cystic Fibrosis Foundation Pulmonary
Guideline: Pharmacologic
Approaches to Prevention and
Eradication of Initial Pseudomonas
aeruginosa Infection (2014) Grade: D,
Certainty: Moderate, Benefi t: Zero
Therapeutics: Exacerbations 16. For children with CF, ages 2 through 5 y, the
CF Foundation recommends the use of oral,
inhaled, and/or intravenous antibiotics to treat
pulmonary exacerbations.
Consensus Recommendation
Therapeutics: Airway
Clearance
17. For children with CF, ages 2 through 5 y, the
CF Foundation recommends the use of daily
airway clearance to improve lung function and
reduce exacerbations.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis
(2009) Consensus Recommendation
Certainty: Low Benefi t: Moderate
Cystic Fibrosis Pulmonary Guidelines:
Airway Clearance Therapies (2009)
Grade B, Certainty Fair, Benefi t:
Moderate
Therapeutics: Airway
Clearance
18. For children with CF, ages 2 through 5 y,
the CF Foundation recommends increasing
frequency and/or duration of airway clearance
treatments for children diagnosed with
pulmonary exacerbations.
Consensus Recommendation Cystic Fibrosis Pulmonary Guidelines:
Airway Clearance Therapies (2009)
Grade B
Therapeutics: Bronchodilators 19. For children with CF, ages 2 through 5 y, the
CF Foundation concludes that the evidence
is insuffi cient to recommend for or against
the chronic use of inhaled bronchodilators
to improve lung function and quality of life or
reduce exacerbations.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013), Grade: I,
Certainty: Low
Therapeutics: Hypertonic
saline
20. For children with CF, ages 2 through 5 y, the
CF Foundations recommends that hypertonic
saline be selectively offered to patients based
on individual circumstances.
Grade C; Certainty: Moderate;
Benefi t: Low
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: B,
Certainty: Moderate, Benefi t:
Moderate
Therapeutics: Dornase alfa 21. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that dornase alfa
be selectively offered to patients based on
individual circumstances.
Grade C; Certainty: Moderate;
Benefi t: Low
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Moderate to
severe disease: Grade: A, Certainty:
High, Benefi t: Substantial. Mild
disease: Grade: B. Certainty: High,
Benefi t: Moderate
Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
In symptomatic infants: Consensus
Recommendation, Certainty: Low,
Benefi t: Moderate
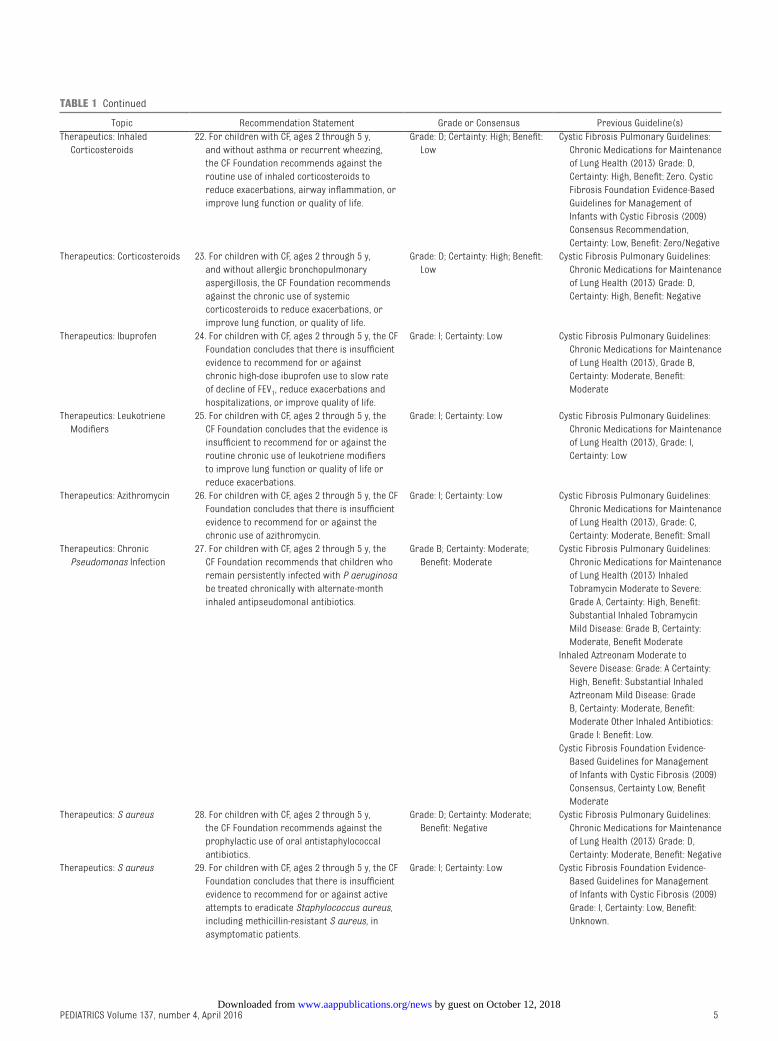
TABLE 1 Continued
by guest on October 12, 2018www.aappublications.org/newsDownloaded from
PEDIATRICS Volume 137 , number 4 , April 2016 5
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Therapeutics: Inhaled
Corticosteroids
22. For children with CF, ages 2 through 5 y,
and without asthma or recurrent wheezing,
the CF Foundation recommends against the
routine use of inhaled corticosteroids to
reduce exacerbations, airway infl ammation, or
improve lung function or quality of life.
Grade: D; Certainty: High; Benefi t:
Low
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: D,
Certainty: High, Benefi t: Zero. Cystic
Fibrosis Foundation Evidence-Based
Guidelines for Management of
Infants with Cystic Fibrosis (2009)
Consensus Recommendation,
Certainty: Low, Benefi t: Zero/Negative
Therapeutics: Corticosteroids 23. For children with CF, ages 2 through 5 y,
and without allergic bronchopulmonary
aspergillosis, the CF Foundation recommends
against the chronic use of systemic
corticosteroids to reduce exacerbations, or
improve lung function, or quality of life.
Grade: D; Certainty: High; Benefi t:
Low
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: D,
Certainty: High, Benefi t: Negative
Therapeutics: Ibuprofen 24. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against
chronic high-dose ibuprofen use to slow rate
of decline of FEV1, reduce exacerbations and
hospitalizations, or improve quality of life.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013), Grade B,
Certainty: Moderate, Benefi t:
Moderate
Therapeutics: Leukotriene
Modifi ers
25. For children with CF, ages 2 through 5 y, the
CF Foundation concludes that the evidence is
insuffi cient to recommend for or against the
routine chronic use of leukotriene modifi ers
to improve lung function or quality of life or
reduce exacerbations.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013), Grade: I,
Certainty: Low
Therapeutics: Azithromycin 26. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against the
chronic use of azithromycin.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013), Grade: C,
Certainty: Moderate, Benefi t: Small
Therapeutics: Chronic
Pseudomonas Infection
27. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that children who
remain persistently infected with P aeruginosa
be treated chronically with alternate-month
inhaled antipseudomonal antibiotics.
Grade B; Certainty: Moderate;
Benefi t: Moderate
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Inhaled
Tobramycin Moderate to Severe:
Grade A, Certainty: High, Benefi t:
Substantial Inhaled Tobramycin
Mild Disease: Grade B, Certainty:
Moderate, Benefi t Moderate
Inhaled Aztreonam Moderate to
Severe Disease: Grade: A Certainty:
High, Benefi t: Substantial Inhaled
Aztreonam Mild Disease: Grade
B, Certainty: Moderate, Benefi t:
Moderate Other Inhaled Antibiotics:
Grade I: Benefi t: Low.
Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus, Certainty Low, Benefi t
Moderate
Therapeutics: S aureus 28. For children with CF, ages 2 through 5 y,
the CF Foundation recommends against the
prophylactic use of oral antistaphylococcal
antibiotics.
Grade: D; Certainty: Moderate;
Benefi t: Negative
Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: D,
Certainty: Moderate, Benefi t: Negative
Therapeutics: S aureus 29. For children with CF, ages 2 through 5 y, the CF
Foundation concludes that there is insuffi cient
evidence to recommend for or against active
attempts to eradicate Staphylococcus aureus,
including methicillin-resistant S aureus, in
asymptomatic patients.
Grade: I; Certainty: Low Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Grade: I, Certainty: Low, Benefi t:
Unknown.
TABLE 1 Continued
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al 6
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Therapeutics: Staphylococcus
aureus
30. For children with CF, ages 2 through 5 y, and
with Staphylococcus aureus persistently
present in cultures of the airways, the CF
Foundation concludes that the evidence is
insuffi cient to recommend for or against
the chronic use of oral antistaphylococcal
antibiotics to improve lung function or quality
of life or reduce exacerbations.
Grade: I; Certainty: Low Cystic Fibrosis Pulmonary Guidelines:
Chronic Medications for Maintenance
of Lung Health (2013) Grade: I,
Certainty: Low
Therapeutics: Ivacaftor 31. For children with CF, ages 2 through 5 y, the
Preschool Guidelines Committee recommends
the routine use of ivacaftor in those with
specifi c gating mutations* and a consideration
for those with a confi rmed diagnosis of CF and
a R117H mutation.
Consensus Recommendation Chronic Medications (2013) Grade: A,
Certainty: Substantial, Benefi t: High
*The mutations are G551D, G1244E, G1349D, G178R,
G551S, S1251N, S1255P, S549N, and S549R.
Nutrition, Behavior, and
Gastrointestinal: Nutrition
32. For children with CF, ages 2 through 5 y, the CF
Foundation recommends that weight-for-age be
maintained at ≥10th percentile.
Grade: A; Certainty: High; Benefi t:
Substantial
Nutrition, Behavior, and
Gastrointestinal: Nutrition
33. For children with CF, ages 2 through 5 y, the
CF Foundation recommends weight-for-stature
assessments use the BMI% method on the
Centers for Disease Control and Prevention
growth charts and a BMI ≥50th percentile be
maintained.
Grade: B; Certainty: High; Benefi t:
Moderate
Evidence-Based Practice
Recommendations for Nutrition-
Related Management of Children
and Adults with Cystic Fibrosis and
Pancreatic Insuffi ciency: Results of
a Systematic Review (2008) Registry
Data-based Recommendation
Nutrition, Behavior, and
Gastrointestinal: Nutrition
34. For children with CF, ages 2 through 5 y, who
are meeting optimal nutritional thresholds, the
CF Foundation recommends ≥90–110 kcal/kg
per day and protein intake based on dietary
reference intakes and dietary guidelines
recommendations: ≥13 g protein/d 2–3 y old,
≥19 g protein/d 4–5 y old.
Grade: A; Certainty: High; Benefi t:
Substantial
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
35. For children with CF, ages 2 through 5 y,
the CF Foundation recommends evaluation
and more intensive management of children
demonstrating any of these criteria of
nutritional risk:
Grade: B; Certainty: High; Benefi t:
Moderate
• BMI <50th percentile, or rate of weight gain
<50th percentile expected for age (≥6
g/d), or weight-for-age <10th percentile, or
inappropriate weight loss
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
36. For children with CF, ages 2 through 5 y,
and at nutritional risk, the CF Foundation
recommends patients be seen in 8 wk or
sooner. These visits should include medical,
behavioral, and nutritional assessment;
education; and intervention. Nutritional
intervention should aim at achieving the
patient’s target goal for both weight-for-age
and BMI.
Consensus Recommendation Consensus Report on Nutrition for
Pediatric Patients with Cystic
Fibrosis (2002) Defi nition of
Nutritional Failure and Patients at
Risk
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
37. For children with CF, ages 2 through 5 y,
and at nutritional risk, the CF Foundation
recommends energy intake 10% to 20% above
baseline with continued incremental upward
adjustments of 10% to 20% as needed up to
200% to achieve weight gain.
Grade: B; Certainty: Moderate;
Benefi t: Moderate
Evidence-Based Practice
Recommendations for Nutrition-
Related Management of Children
and Adults with Cystic Fibrosis and
Pancreatic Insuffi ciency: Results of a
Systematic Review (2008) Grade: B
TABLE 1 Continued
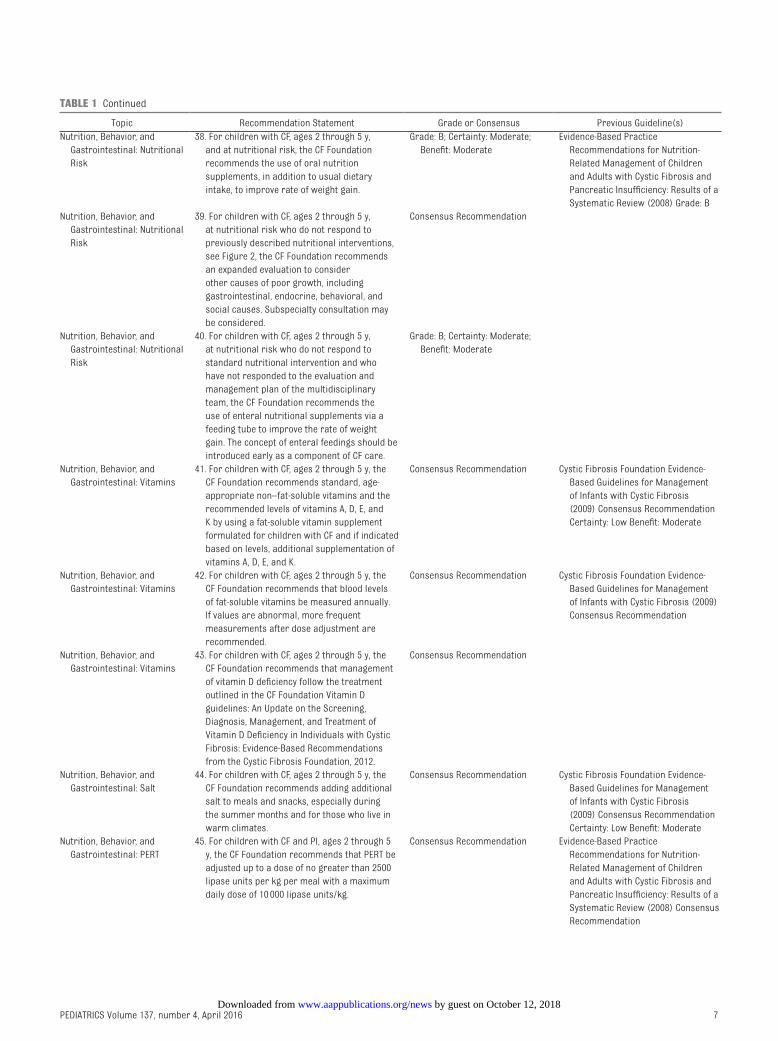
by guest on October 12, 2018www.aappublications.org/newsDownloaded from
PEDIATRICS Volume 137 , number 4 , April 2016 7
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
38. For children with CF, ages 2 through 5 y,
and at nutritional risk, the CF Foundation
recommends the use of oral nutrition
supplements, in addition to usual dietary
intake, to improve rate of weight gain.
Grade: B; Certainty: Moderate;
Benefi t: Moderate
Evidence-Based Practice
Recommendations for Nutrition-
Related Management of Children
and Adults with Cystic Fibrosis and
Pancreatic Insuffi ciency: Results of a
Systematic Review (2008) Grade: B
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
39. For children with CF, ages 2 through 5 y,
at nutritional risk who do not respond to
previously described nutritional interventions,
see Figure 2, the CF Foundation recommends
an expanded evaluation to consider
other causes of poor growth, including
gastrointestinal, endocrine, behavioral, and
social causes. Subspecialty consultation may
be considered.
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal: Nutritional
Risk
40. For children with CF, ages 2 through 5 y,
at nutritional risk who do not respond to
standard nutritional intervention and who
have not responded to the evaluation and
management plan of the multidisciplinary
team, the CF Foundation recommends the
use of enteral nutritional supplements via a
feeding tube to improve the rate of weight
gain. The concept of enteral feedings should be
introduced early as a component of CF care.
Grade: B; Certainty: Moderate;
Benefi t: Moderate
Nutrition, Behavior, and
Gastrointestinal: Vitamins
41. For children with CF, ages 2 through 5 y, the
CF Foundation recommends standard, age-
appropriate non–fat-soluble vitamins and the
recommended levels of vitamins A, D, E, and
K by using a fat-soluble vitamin supplement
formulated for children with CF and if indicated
based on levels, additional supplementation of
vitamins A, D, E, and K.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis
(2009) Consensus Recommendation
Certainty: Low Benefi t: Moderate
Nutrition, Behavior, and
Gastrointestinal: Vitamins
42. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that blood levels
of fat-soluble vitamins be measured annually.
If values are abnormal, more frequent
measurements after dose adjustment are
recommended.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis (2009)
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal: Vitamins
43. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that management
of vitamin D defi ciency follow the treatment
outlined in the CF Foundation Vitamin D
guidelines: An Update on the Screening,
Diagnosis, Management, and Treatment of
Vitamin D Defi ciency in Individuals with Cystic
Fibrosis: Evidence-Based Recommendations
from the Cystic Fibrosis Foundation, 2012.
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal: Salt
44. For children with CF, ages 2 through 5 y, the
CF Foundation recommends adding additional
salt to meals and snacks, especially during
the summer months and for those who live in
warm climates.
Consensus Recommendation Cystic Fibrosis Foundation Evidence-
Based Guidelines for Management
of Infants with Cystic Fibrosis
(2009) Consensus Recommendation
Certainty: Low Benefi t: Moderate
Nutrition, Behavior, and
Gastrointestinal: PERT
45. For children with CF and PI, ages 2 through 5
y, the CF Foundation recommends that PERT be
adjusted up to a dose of no greater than 2500
lipase units per kg per meal with a maximum
daily dose of 10 000 lipase units/kg.
Consensus Recommendation Evidence-Based Practice
Recommendations for Nutrition-
Related Management of Children
and Adults with Cystic Fibrosis and
Pancreatic Insuffi ciency: Results of a
Systematic Review (2008) Consensus
Recommendation
TABLE 1 Continued
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
that there is insufficient evidence to
recommend for or against routine use
of pulse oximetry as an adjunctive
tool to detect lung disease. Table 1:
Recommendation 7.
Spirometry
Spirometry is the most important
and widely used tool to assess lung
function in CF. Several studies have
demonstrated that spirometry
is feasible in preschoolers with
CF, and has the potential to
detect pulmonary exacerbations
and airway obstruction despite
minimal symptoms.6, 21, 22 In a
cross-sectional, multicenter
study, FEV in 0.5 second and flow-
related volumes were found to
be more sensitive than FEV1 for
identifying abnormal lung function
in asymptomatic, preschool-
aged children.23 Another study
demonstrated improvement of
spirometric indices in both
school-aged children and 10
preschoolers who were treated
for pulmonary exacerbations.24
Because there is no risk to
performing spirometry in
preschool-aged children, and
they benefit from practicing
with technicians, the committee
concluded that spirometry should
be attempted as early as 3 years
of age, depending on the child’s
8
Topic Recommendation Statement Grade or Consensus Previous Guideline(s)
Nutrition, Behavior, and
Gastrointestinal: Behavior
46. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that the CF team
members, working in concert with the family,
set energy-intake goals and assess progress
on a regular basis.
Grade: B; Certainty: Moderate;
Benefi t: Substantia
Nutrition, Behavior, and
Gastrointestinal: Behavior
47. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that all families
are regularly assessed for the presence
of mealtime behavior challenges and are
provided with proactive behavioral assistance
when needed.
Grade: A; Certainty: High; Benefi t:
Substantial
Nutrition, Behavior, and
Gastrointestinal: Behavior
48. For children with CF, ages 2 through 5 y, who
are at nutritional risk, or exhibiting challenging
mealtime behaviors, or not meeting energy-
intake goals, behavioral therapy provided
by knowledgeable team members should
accompany nutritional therapy.
Grade: A; Certainty: High; Benefi t:
Substantial
Evidence-Based Practice
Recommendations for Nutrition-
Related Management of Children
and Adults with Cystic Fibrosis and
Pancreatic Insuffi ciency: Results of a
Systematic Review (2008) Grade: B
Nutrition, Behavior, and
Gastrointestinal:
Gastrointestinal
49. The CF Foundation recommends that
all providers be aware of the presenting
symptoms of the following gastrointestinal
tract disorders: constipation,
gastroesophageal refl ux disease, small bowel
overgrowth, distal intestinal obstruction
syndrome, and celiac disease.
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal:
Gastrointestinal
50. For children with CF, ages 2 through 5 y, the
CF Foundation recommends that children
and their parents be questioned regarding
abdominal pain at each visit, and that pain is
investigated if persistent or recurrent.
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal:
Gastrointestinal
51. For children with CF, ages 2 through 5 y, and
who are PS, the CF Foundation recommends
that children are reevaluated annually for
the conversion to PI with fecal elastase
measurement, particularly if genetic testing
reveals 2 mutations potentially associated
with PI.
Consensus Recommendation Consensus Report on Nutrition for
pediatric Patients with Cystic
Fibrosis (2002) Issues Related to
Pancreatic Insuffi ciency: Identifying
Pancreatic Insuffi ciency
Nutrition, Behavior, and
Gastrointestinal:
Gastrointestinal
52. For children with CF, ages 2 through 5 y,
and who are PS with severe abdominal pain,
particularly if associated with vomiting, the
CF Foundation recommends measurement of
lipase and amylase to determine if pancreatitis
is present.
Consensus Recommendation
Nutrition, Behavior, and
Gastrointestinal:
Gastrointestinal
53. For children with CF, ages 2 through 5 y, who
had terminal ileal bowel resection, the CF
Foundation recommends annual measurement
of serum vitamin B12 concentration.
Consensus Recommendation
AAP, American Academy of Pediatrics;
TABLE 1 Continued
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
developmental level. Table 1:
Recommendations 8–9.
Bronchodilator Responsiveness
Three articles demonstrated
bronchodilator responsiveness
in <20% of children with CF.
Bronchodilator responsiveness
may be associated with certain
polymorphisms.25–27 Although there
is insufficient evidence to recommend
for or against routine monitoring
of bronchodilator responsiveness,
evaluation may be considered if there
are concerns or symptoms suggestive
of airway hyperreactivity. Table 1:
Recommendation 10.
Multiple-Breath Washout
Multiple-breath washout (MBW)
testing measures regional ventilation
heterogeneity and appears to be
more sensitive than spirometry
in detecting pulmonary function
abnormalities in young children
with CF. Two prospective studies
9
TABLE 2 Routine Monitoring and Care
Age at Visit 2 y 3 y 4 y 5 y
Intervention
Visit Done 1 2 3 4 1 2 3 4 1 2 3 4 1 2 3 4
Care issues
Continue high-salt diet, supplement
additional salt in hot weather.
X X X X
History and physical with weight and
height
X X X X X X X X X X X X X X X X
Teach and assess airway clearance,
review airway clearance techniques
X C C C X C C C X C C C X C C C
Assess weight gain, caloric intake, and
PERT dosing
X X X X X X X X X X X X X X X X
Continue vitamins designed for patients
with CF
X X X X X X X X X X X X X X X X
Introduce chronic dornase alfa and/
or HS
C C C C
Seasonal infl uenza vaccination X X X X
Pneumococcal polysaccharide
vaccination
X
Diagnostic testing
Sweat test and genotyping confi rmed
documentation
X C C C
Pancreatic functional status testing C C C C
Respiratory culture X X X X X X X X X X X X X X X X
Chest radiograph or CT X C X C
Vitamin levels A, D, E, prothrombin time X X X X
Serum electrolytes, SUN, creatinine,
glucose
X X X X
Complete blood count X X X X
AST/ALT/GGT/bilirubin, albumin, ALP X X X X
Spirometry A A A A A A A A X X X X
Abdominal pain questioning X X X X X X X X X X X X X X X X
Education
Teach and assess infection control X C C C X C C C X C C C X C C C
Document CF Foundation patient
registry consent
X X X X
Discuss clinical research C C C C
Referrals to community resources C C C C
Tobacco smoke exposure avoidance
education
X X X X
Patient and caregiver engagement
assessment
X C C C
Set energy goals and assess progress X C C C X C C C X C C C X C C C
Assess for presence of mealtime
behavior challenges and provide
proactive behavioral assistance
X C C C X C C C X C C C X C C C
Annual health supervision visits with
PCP
X X X X
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyltransferase; SUN, serum urea nitrogen.
A single letter under one of the 4 yearly headings indicates the task should be performed, considered or attempted once a year. Further letters appear under quarterly visits when the
task should be performed, considered, or attempted at >1 visit per year: A, attempt this task; C, consider this task; X, perform this task.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
that included preschool-aged
children reported that MBW indices,
specifically lung clearance index
(LCI), was more sensitive than
spirometry in detecting abnormal
lung function5, 28 and was generally
abnormal in preschoolers with CF
compared with healthy controls.20
The utility of MBW in the clinical
setting and what constitutes a
clinically significant change in the
LCI has not yet been determined.
The committee concluded that there
is currently insufficient evidence
to recommend for or against the
routine use of MBW in CF. Table 1:
Recommendation 11.
Chest Radiographs
Chest imaging can be performed at
all ages and can identify structural
changes that may be regional,
early, and presymptomatic. Chest
radiographs are relatively insensitive
and nonspecific compared with CT
scans, but require less radiation
exposure, expense, and patient
cooperation. Chest radiographs have
been shown to detect the existence
and progression of CF lung disease
in infants, 29 and preschool-aged30
and school-aged children.31 Chest
radiographs demonstrate a higher
correlation than FEV1/forced
vital capacity with Pseudomonas
acquisition in young children with
CF.32 Chest radiograph scores
are superior to FEV1 in detecting
concurrent abnormalities on chest
CT scans, 33 and in predicting future
FEV1 and chest radiograph scores.34
To monitor disease progression,
chest radiographs should be obtained
at a minimum of every other year
following a baseline radiograph at
diagnosis. Use of a scoring system, such
as Brasfield, 35 Chrispin-Norman, 36
or Wisconsin37 scores, should be
considered to monitor changes over
time. Chest radiographs also should
be considered in preschoolers with
CF with a change in respiratory
status that does not respond to
appropriate interventions. Table 1:
Recommendation 12.
Chest CT
Chest CT is a sensitive tool for
monitoring early CF lung disease, as
it detects early airway wall thickening,
bronchiectasis, and gas trapping in
infants and preschoolers, 4, 38, 39
without symptoms or abnormal
pulmonary function.40, 41
Bronchiectasis in this age group
often persists or progresses.42
The rate of structural lung disease
progression on CT in preschoolers
with CF is not well defined; however,
CT scans have revealed progressive
bronchiectasis in older children
over a 2-year period, despite stable
pulmonary function testing.40 In
children <4 years of age experiencing
a pulmonary exacerbation, CT
detected regional differences in
airway inflammation and scores
improved after treatment.43 Although
the carcinogenic risk of radiation
exposure is small and continues
to decrease with technological
innovations, 44 concerns remain
regarding the risk of radiation
exposure and expense. Controversy
also persists regarding the
significance of early changes seen on
CT45 and the preferred methodology
and reproducibility of scoring
systems, especially in early or mild
disease.46 There are currently no
valid surrogates to replace CT. In a
cohort of patients 5 to 19 years of
age, a normal LCI almost excluded
CT abnormalities41; in a younger
cohort (mean age 7.8 years), LCI
and high-resolution CT had similar
sensitivity to detecting CF lung
disease but provided complementary
information.47 If routine monitoring
is performed, it should replace chest
radiographs, be performed every 2 to
3 years, and use the lowest radiation
dose possible needed to obtain
diagnostic-quality images. If used,
consideration should be given to
using a scoring system to standardize
interpretation of repeated scans over
time. Table 1: Recommendation 13.
Microbiology
Quarterly respiratory cultures are
the standard of care for patients
with CF.48 For infants1 and other
nonexpectorating patients, an
oropharyngeal (OP) swab is the usual
respiratory sample, despite limited
diagnostic accuracy compared with
bronchoalveolar lavage (BAL).49
BAL is the only means by which to
directly sample the lower respiratory
tract and provides measures of
lower airway inflammation that
may have important prognostic
value, 4 but is invasive and costly. A
randomized controlled trial in infants
and preschoolers with CF50, 51 found
no difference in outcome at age 5
years when therapy was directed
by OP culture as opposed to BAL.
Induced sputum may have greater
yield than OP culture, 52–54 but it is
time-consuming and impractical in
standard clinic settings. Respiratory
microbiology monitoring by OP
cultures should be performed at
least quarterly and routine airway
monitoring by bronchoscopy is
not recommended. However,
bronchoscopy with BAL should be
considered in patients with a change
in respiratory status that does not
respond to antibiotics directed
toward pathogens isolated from OP
swabs. Table 1: Recommendations
14–15.
PULMONARY THERAPEUTICS
Pulmonary Exacerbations
Based on our current understanding
of early disease progression,
the association of pulmonary
exacerbations with worse outcomes,
and evidence of improvement
with antibiotic treatment, 43,
55–66 the recommendation for
preschoolers with CF experiencing
an exacerbation is oral, inhaled,
and/or intravenous antibiotic
treatment. Defining a pulmonary
10 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
exacerbation is beyond the scope of
this article; however, Fig 1 outlines
the recommended approach to
preschool-aged children with
increased respiratory symptoms.
There are no randomized clinical
trials of pulmonary exacerbation
treatment in preschool-aged children
with CF; thus, these guidelines
for management were based on
consensus recommendations, and
management decisions should be
individualized. Oral antibiotics
targeting bacteria detected on
airway cultures are recommended
for mild to moderate pulmonary
exacerbations, whereas intravenous
antibiotics are recommended for
moderate to severe pulmonary
exacerbations or mild exacerbations
unresponsive to oral antibiotics.
Inhaled antibiotics are recommended
based on standard guidelines for
treatment of new and chronic
Pseudomonas aeruginosa infection.2, 67
Table 1: Recommendation 16.
Airway Clearance Therapy
Airway clearance therapy (ACT)
is an important maintenance
treatment and is recommended for
all individuals with CF.1, 68 With early
airway disease noted in the youngest
population, ACT has the potential to
prevent development of irreversible
disease. Continuing ACT through
preschool years will encourage
maintenance throughout childhood.
An active lifestyle consisting of
vigorous physical activity and
exercise, important components
of maintaining lung health, should
be encouraged and initiated in this
age group. In preschoolers with
CF, daily ACT is recommended
and increased frequency and/or
duration is recommended during
pulmonary exacerbations. Table 1:
Recommendations 17–18.
Bronchodilators
No studies were found that
address bronchodilator efficacy
in the absence of asthma or
bronchial hyperresponsiveness
in CF; therefore, the evidence is
insufficient to recommend for or
against the chronic use of inhaled
bronchodilators in preschoolers.
However, viral-triggered wheezing or
asthma in preschoolers may respond
to bronchodilator therapy. Table 1:
Recommendation 19.
Hypertonic Saline
Several studies have demonstrated
safety and tolerability of 7%
hypertonic saline (HS) in infants
and young children.69–71 Unlike a
study in older individuals with CF, 72
a randomized controlled trial of
344 children <5 years failed to
show a reduction in the primary
endpoint of pulmonary exacerbation
rate.73 However, in 2 small studies
that were part of this larger trial,
infant lung function and the LCI
did demonstrate improvement in
subjects receiving 7% HS.73, 74 Given
these findings, the CF Foundation
recommends that HS be offered
to patients based on individual
circumstances, either for chronic
use or during acute pulmonary
exacerbation. Further studies may
alter this recommendation. Table 1:
Recommendation 20.
Dornase Alfa
Routine use of dornase alfa is
associated with reduced pulmonary
exacerbations, improved lung
function, and decreased rate of
lung function decline among older
children and adults with CF.75–81
Dornase alfa has been shown to have
positive effects on CT changes and
LCI82–84 and improved health-related
quality-of-life scores in children >6
years.85 Safety and tolerability of
dornase alfa has been demonstrated
in children ages 3 months to 5
years.86, 87 Potential benefits include
its effect on mucous plugging, air
trapping, and lung health in CF that
may result in delayed pulmonary
disease progression. Based on
moderate evidence that dornase
alfa is safe and effective, and the
potential benefit is at least small,
the CF Foundation recommends
that dornase alfa be offered to
patients based on individual
circumstances, either for chronic
use or during acute pulmonary
exacerbation. Further studies may
alter this recommendation. Table 1:
Recommendation 21.
Systemic and Inhaled Corticosteroids
With the exception of treatment
of allergic bronchopulmonary
aspergillosis, systemic
corticosteroids are not recommended
for routine use in children with CF,
as potential harm outweighs any
benefit. Inhaled corticosteroids are
not recommended for management
of CF lung disease, as no clear
benefit has been identified.2 Table 1:
Recommendation 22–23.
Ibuprofen
High-dose ibuprofen is recommended
for chronic use in individuals with
CF older than 6 years with mild lung
disease.2 We found no prospective
trials that support its use in children
younger than 6 years and conclude
there is insufficient evidence to
recommend for or against its use
in preschoolers with CF. Table 1:
Recommendation 24.
Leukotriene Modifi ers
In the absence of comorbid
conditions, such as asthma
or recurrent wheezing, there
is insufficient evidence to
recommend for or against the
chronic use leukotriene modifiers
for preschoolers with CF. Table 1:
Recommendation 25.
Azithromycin
Routine use of azithromycin is
recommended for individuals with CF
>6 years with persistent P aeruginosa
infection.2 Azithromycin is safe,
reduces lower airway inflammation
and exacerbations, and improves
11 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
lung function and weight gain in
older children with mild CF lung
disease.88, 89 There are conflicting
data regarding the potential for
higher nontuberculous mycobacterial
infection rates in individuals with
CF on chronic azithromycin.60, 90–92
There is insufficient evidence to
recommend for or against the chronic
use of azithromycin in preschoolers
with CF. Table 1: Recommendation
26.
Chronic P aeruginosa Infection
The use of cycled inhaled tobramycin
for the management of infants,
children, and adults with persistent
P aeruginosa infection has been
12
FIGURE 1Approach to preschool-aged children with increased respiratory symptoms.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
previously recommended.1, 2
Additional inhaled antibiotics, such as
aztreonam, also have been effective
and safe in improving lung function
and reducing exacerbations outside
of the infant and preschool age
range.93–96 Microbiologic efficacy and
safety of inhaled tobramycin has been
demonstrated in infants and young
children.97–100 The use of alternate-
month inhaled antipseudomonal
antibiotics for preschoolers with
persistent P aeruginosa infection
is recommended. Table 1:
Recommendation 27.
Staphylococcus aureus Prophylaxis and Eradication
In keeping with previous guide-
lines, 4, 42, 43, 101 the prophylactic use
of oral antistaphylococcal antibiotics
is not recommended given the
risk of increased P aeruginosa
isolation.2, 102–106 Several studies have
addressed the utility of eradication
attempts after first acquisition of
S aureus, particularly methicillin-
resistant isolates, but none have been
randomized or focused on young
children with CF.107–110 Prophylactic
use of oral antistaphylococcal
antibiotics is not recommended;
evidence is insufficient to
recommend for or against attempts
to eradicate S aureus and for chronic
use of oral antistaphylococcal
antibiotics in preschoolers with CF
who persistently culture S aureus.
Table 1: Recommendations 28–30.
Ivacaftor
Ivacaftor has been shown to improve
lung function, sweat chloride
values, weight gain, and quality
of life in people 6 years and older
with at least 1 copy of the G551D
mutation.2, 111–113 This therapy has
recently been approved for use
in patients with additional cystic
fibrosis transmembrane conductance
regulator (CFTR) gating mutations:
G1244E, G1349D, G178R, G551S,
S1251N, S1255P, S549N, S549R,
and R117H. A forthcoming study
in preschoolers has demonstrated
safety of ivacaftor in the preschool-
aged child.114, 115 The availability
of oral granules now allows for
appropriate dose adjustment in
young children. Monitoring of liver
function abnormalities will be
important. Based on data in older
subjects, forthcoming safety data, and
recent Food and Drug Administration
approval for this age group, the use
of ivacaftor for preschoolers with
specific gating mutations and R117H
is a consensus recommendation
of this committee. Table 1:
Recommendation 31.
CLINICAL AND BEHAVIORAL NUTRITION AND GASTROINTESTINAL CARE
Nutrition
Normal ranges of weight-for-age,
height-for-age, and BMI percentile
are associated with better pulmonary
function, height closer to the 50th
percentile for age and gender,
and survival.3 A recent study
demonstrated that weight-for-age
≥10th percentile for age and gender
at age 4 years was associated with
superior survival at 18 years.11
Therefore, it is recommended that
weight-for-age of preschoolers with
CF be maintained at ≥10th percentile.
Table 1: Recommendation 32.
Measurement of height and weight,
with calculation of BMI percentile
using Centers for Disease Control and
Prevention growth charts, should
be performed to assess weight-for-
stature.116, 117 Weight-for-height and
BMI must be evaluated in the context
of the child’s height. Stunting is a risk
in CF and can obscure nutritional
risk in a child. Preschoolers
with CF should maintain a BMI
≥50th percentile. Recommended
monitoring of growth and nutrition
in this age group is found in Table 3.
Table 1: Recommendations 33.
Energy and protein requirements
should be assessed in preschoolers
with CF.118, 119 Energy and protein
recommendations of ≥90 to 110
kcal/kg per day, and protein intake
based on dietary reference intakes120
and dietary guidelines recommend
≥13 g protein per day for children
aged 2 to 3 years and ≥19 g protein
per day for children aged 4 to 5
years to meet optimal nutritional
thresholds. Table 4 outlines general
energy guidelines by age and gender
for preschoolers with CF. Table 1:
Recommendation 34.
Nutritional Risk
Nutritional risk is defined as a BMI
<50th percentile, or rate of weight
gain <50th percentile expected for
age (≥6 g per day), or weight-for-age
<10th percentile, or inappropriate
weight loss. Families of preschoolers
with CF at nutritional risk and CF
13
TABLE 3 Routine Monitoring and Nutritional Care of Preschoolers with CF
Every visit
Measure and evaluate weight and height by using standard guidelines (American Academy of
Pediatrics).
Assess rate of weight gain in g/d since the last encounter.
Assess PERT dose for lipase units/kg per meal and lipase units/kg per day.
Assess for any issues that impede PERT administration as prescribed.
Encourage a well-balanced diet, with emphasis on calorically dense food sources.
Inquire if any eating behaviors need to be addressed.
Inquire if any changes in routine are expected to occur (preschool, daycare, baby sitter) for
anticipatory guidance.
Yearly
Serum levels of fat-soluble vitamins.
As appropriate
Referrals to community food resources (Supplemental Nutrition Program for Women, Infants,
and Children; food stamps; food pantries).
Information on nutrition resources via enzyme assistance programs.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
health care professionals should
establish a multifaceted care plan
that addresses medical, behavioral,
and nutritional issues and schedule
more frequent follow-up to ensure
rapid establishment of normal
growth. Interventions to improve
nutritional status can be guided
by the algorithm in Figs 2A, 2B,
2C, and 2D. Collaboration with a
PCP or a visiting nurse to obtain
weight and height measurement is
an option for preschoolers needing
more frequent evaluation. Table 1:
Recommendations 35–36.
Increased energy intake results in
improved weight gain3; however,
fewer data support improved height
with increased energy intake.121, 122
There is conflicting evidence
regarding the use of nutritional
supplements to increase energy
intake.122, 123 Oral supplements may
be beneficial to preschoolers with
CF at nutritional risk. Supplements
may increase protein and energy at
a time when eating behaviors may
interfere with daily food intake.
The use of supplements should be
integrated with other nutritional
and behavioral approaches for this
age group. Families and CF health
care professionals should be aware
of the substantial impact of behavior
on optimal nutrition.124–127 Children
who continue to be at nutritional
risk despite having addressed
pulmonary, social, and dietary factors
should be referred to pediatric
gastroenterologists, endocrinologists,
and behavioral specialists for
further evaluation and management.
Gastrostomy tube feedings have
been shown in older children and
adults with CF to improve weight and
pulmonary function.128–131 Table 1:
Recommendations 37–40.
Vitamins
CF-specific multivitamins are
recommended, in a form that
the child will best accept, and at
recommended doses based on
manufacturer guidelines.1 Patients
with low serum levels of a specific
fat-soluble vitamin(s) should
continue to receive CF-specific
multivitamins in addition to
supplementation of the specific fat-
soluble vitamin associated with low
serum levels. Serum levels should
be remeasured to ensure adequate
response to therapy. Frequency
of remeasurement should depend
on the level of deficiency, the
dose used for treatment, and the
risk associated with deficiency or
toxicity of the vitamin. Specific CF
guidelines for vitamin D management
are published132; there has been
limited evaluation of the efficacy
of these guidelines.133 Table 1:
Recommendations 41–43.
Salt
Preschoolers with CF are advised to
continue a high-salt diet, especially
in the summer, and for those who
live in warmer climates. For those
who may not select foods with a
high salt content, supplementation
with at least one-quarter teaspoon
of salt per day (581 mg sodium) may
be necessary. Caregivers should be
advised to avoid overuse of salt.134
Table 1: Recommendation 44.
Pancreatic Enzyme Replacement Therapy
For preschoolers with CF who
are pancreatic insufficient (PI),
consistent administration of the
appropriate doses of pancreatic
enzyme replacement therapy (PERT)
is essential. Recommendations for
PERT follow previous guidelines to
ensure adequate digestion and avoid
risk of fibrosing colonopathy.135
Evaluation of PERT dose and
adherence should occur at each visit
(Supplemental Table 4). Table 1:
Recommendation 45.
Behavior
To meet growth goals, families of
preschoolers with CF and health care
professionals should establish individ-
ualized energy-intake goals and
routinely monitor progress.10, 136–139
Table 1: Recommendation 46.
In multiple studies, families
consistently reported challenges with
mealtime energy-intake goals due to
demanding mealtime behaviors,
leading to significant stress.8, 9, 124–127
These mealtime behavioral
challenges can predict calorie
intake and weight gain.140 Regular
assessment for mealtime behavior
challenges should be performed
and proactive behavioral assistance
should be provided when needed
Table 5.10, 136, 137, 141–143 Table 1:
Recommendation 47.
CF health care professionals with
appropriate training should provide
behavioral therapy for preschoolers
with CF at nutritional risk, exhibiting
challenging mealtime behaviors, or
not meeting energy-intake goals.
Behavioral and nutrition treatment
can lead to improvements in
attaining energy-intake
goals.10, 136–140, 143, 144 Table 1:
Recommendation 48.
Gastrointestinal
Conditions associated with abdominal
pain may contribute to malabsorption,
14
TABLE 4 General Calorie Guidelines for Age and Gender: The Recommendations Are a Consolidation
of 3 References118–120
Age, y Boys Girls
2–3 1290–1430 kcal/d 1200–1250 kcal/d
100–105 kcal/kg/d 95–100 kcal/kg/d
3–4 1430–1520 kcal/d 1285–1290 kcal/d
95–100 kcal/kg/d 85–90 kcal/kg/d
4–5 1520–1655 kcal/d 1285–1505 kcal/d
95–100 kcal/kg/d 85–90 kcal/kg/d
5–6 1655–1780 kcal/d 1505–1605 kcal/d
85–95 kcal/kg/d 80–85 kcal/kg/d
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016 15
FIGURE 2ANutrition algorithm: Tier One Initial Evalutaion.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
impaired quality of life, reluctance to
perform airway clearance therapies,
and loss of appetite in children with
CF. In a large retrospective study of
children with CF, abdominal pain
frequency in children <6 years
of age was no different from the
general population.145 Abdominal
pain should not be attributed to
pancreatic enzyme dosing.145 If
pain persists after assessment for
common causes of abdominal pain in
CF, or “red flag” symptoms, listed in
Supplemental Table 5, are present,
refer to pediatric gastroenterology.
16
FIGURE 2BNutrition algorithm: Tier Two Consultation and In-depth Diagnostic Evaluation. (continued)
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
Studies of preschoolers with CF have
shown that constipation, 146 distal
intestinal obstruction syndrome, 147
gastroesophageal reflux disease, 148
small bowel overgrowth, 149 and
celiac disease150 can occur. Providing
parents with a questionnaire directed
at gastrointestinal symptoms can
assist in detecting disease.151, 152 CF
health care professionals unfamiliar
with the diagnosis and management
of these conditions should refer the
child to a pediatric gastroenterologist.
Table 1: Recommendations 49–50.
Children with CF who are
pancreatic sufficient (PS) do
not necessarily have normal
17
FIGURE 2CNutrition algorithm: Tier Three Consideration of G-Tube. (continued)
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al 18
FIGURE 2DNutrition algorithm: BMI and Weight for Age.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
pancreatic function.153 Over time,
they may develop PI. Diarrhea is
an unreliable indicator of PI. Poor
weight gain or growth may be a late
indicator of PI, and micronutrient
deficiency may be present. Fecal
elastase, 72-hour stool for fecal
fat, and cholecystokinin/secretin-
stimulated pancreatic function
testing may be used to diagnose PI.
Fecal elastase is the simplest, most
available screening test. Table 1:
Recommendation 51.
Children with PS and CFTR mutations
associated with milder disease are at
risk for acute pancreatitis, which may
lead to episodes of acute, recurrent,
or chronic pancreatitis.154 Specific
CFTR mutations cannot reliably
predict which children will get
pancreatitis.155 Acute pancreatitis is
a potentially life-threatening disease
associated with severe pain, nausea,
and vomiting. Although infrequent
in preschoolers, providers should be
aware that pancreatitis can occur in
preschoolers who are PS. Table 1:
Recommendation 52.
Vitamin B12 is absorbed exclusively
in the terminal ileum. Individuals
with resection of the terminal ileum
will become vitamin B12 deficient,
which may take 1 to 3 years to
develop.156, 157 These individuals
should be screened by using serum
B12 levels or urinary methylmalonic
acid. Providers should not wait for
the development of hematologic
signs to screen for deficiency. Table
1: Recommendation 53.
UNANSWERED QUESTIONS
With few clinical trials evaluating
monitoring, therapeutics, and
nutritional care, determining
care pathways for preschoolers
with CF is challenging. Additional
research on monitoring for lung
disease in preschoolers to advance
early detection and potentially
improve outcomes is needed. MRI
and other imaging modalities that
avoid radiation exposure may
hold promise for evaluation of
early CF lung disease. MBW, after
further validation, may become
incorporated into clinical care.
Therapeutic trials evaluating efficacy
of chronic respiratory medications,
including dornase alfa and HS, are
important; increasing treatment
complexity with additional therapies
must be weighed against the
potential of preventing irreversible
damage. CFTR modulators
are a potentially life-changing
therapeutic strategy for CF. The
recent approval of ivacaftor for
preschoolers will hopefully allow
for early disease prevention. As
modulators and correctors are
developed for other mutations,
it is important to recognize the
substantial benefits for the youngest
patients. Gaps in nutritional,
behavioral, and gastrointestinal
studies in this age group remain.
Additional research on timing of
nutritional interventions and of the
conversion from PS to PI is needed.
Determining the prevalence of key
gastrointestinal disorders is needed
to assist providers with testing and
diagnostic decisions.
CONCLUSIONS
The care of the preschool-aged child
with CF includes complex, time-
consuming treatment regimens
and overcoming behavioral
challenges common in this age
group to maintain lung health and
optimize growth. We hope that these
guidelines will help CF care teams
and families make informed decisions
regarding care of the 2- to 5-year-old
children with CF.
ACKNOWLEDGMENTS
The authors acknowledge Elaine
Skopelja, Indiana University, for
her assistance with the literature
review.
19
ABBREVIATIONS
ACT: airway clearance therapy
BAL: bronchoalveolar lavage
CF: cystic fibrosis
CFTR: cystic fibrosis
transmembrane
conductance regulator
CT: computed tomography
FEV1: forced expiratory volume
in 1 second
HS: hypertonic saline
LCI: lung clearance index
MBW: multiple-breath washout
OP: oropharyngeal
PCP: primary care provider
PERT: pancreatic enzyme
replacement therapy
PI: pancreatic insufficiency
PICO: population, intervention,
comparison, outcome
PS: pancreatic sufficiency
TABLE 5 Behavioral Assessment and Management of Mealtimes
Assessment of behavior at mealtimes
It is important to assess for a regular schedule of meal and snack times, for mealtime duration, and
for child behaviors and family stress related to diffi culties in meeting energy-intake goals and a
balanced variety of foods.
Setting energy-intake goals
For families of preschoolers to succeed with meeting growth goals, it is important that energy-intake
goals individualized to the child are set and then monitored in terms of progress.
Inclusion of behavioral expertise when evaluating concerns about growth
Whenever concerns with growth and/or family stress related to mealtimes (and with meeting energy-
intake goals) are present, it is critical to consider a multidisciplinary approach to understanding
the problem, developing a treatment plan, and assessing the success (or need to further modify)
the plan. Inclusion of expertise in child behavior management and knowledge of the evidence-based
behavioral and nutrition treatment is strongly recommended whenever possible.
Recommending behavioral and nutrition treatment to improve energy intake and growth
A treatment plan focused on increasing energy intake and improving growth in preschoolers with
CF will likely be more successful with inclusion of evidence-based interventions that are part of
behavioral and nutrition treatment.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
REFERENCES
1. Borowitz D, Robinson KA, Rosenfeld
M, et al; Cystic Fibrosis Foundation.
Cystic Fibrosis Foundation evidence-
based guidelines for management of
infants with cystic fi brosis. J Pediatr.
2009;155(suppl 6):S73–S93
2. Mogayzel PJ Jr, Naureckas ET, Robinson
KA, et al; Pulmonary Clinical Practice
Guidelines Committee. Cystic fi brosis
pulmonary guidelines. Chronic
medications for maintenance of lung
health. Am J Respir Crit Care Med.
2013;187(7):680–689
3. Stallings VA, Stark LJ, Robinson KA,
Feranchak AP, Quinton H; Clinical
Practice Guidelines on Growth and
Nutrition Subcommittee; Ad Hoc
Working Group. Evidence-based
practice recommendations for
nutrition-related management of
children and adults with cystic fi brosis
and pancreatic insuffi ciency: results of
a systematic review. J Am Diet Assoc.
2008;108(5):832–839
4. Sly PD, Gangell CL, Chen L, et al;
AREST CF Investigators. Risk factors
for bronchiectasis in children
with cystic fi brosis. N Engl J Med.
2013;368(21):1963–1970
5. Aurora P, Bush A, Gustafsson P, et al;
London Cystic Fibrosis Collaboration.
Multiple-breath washout as a marker
of lung disease in preschool children
with cystic fi brosis. Am J Respir Crit
Care Med. 2005;171(3):249–256
6. Kerby GS, Rosenfeld M, Ren CL, et al.
Lung function distinguishes preschool
children with CF from healthy controls
in a multi-center setting. Pediatr
Pulmonol. 2012;47(6):597–605
7. Brumback LC, Davis SD, Kerby GS,
et al. Lung function from infancy to
preschool in a cohort of children
with cystic fi brosis. Eur Respir J.
2013;41(1):60–66
8. Spieth LE, Stark LJ, Mitchell MJ, et al.
Observational assessment of family
functioning at mealtime in preschool
children with cystic fi brosis. J Pediatr
Psychol. 2001;26(4):215–224
9. Mitchell MJ, Powers SW, Byars
KC, Dickstein S, Stark LJ. Family
functioning in young children with
cystic fi brosis: observations of
interactions at mealtime. J Dev Behav
Pediatr. 2004;25(5):335–346
10. Powers SW, Jones JS, Ferguson KS,
Piazza-Waggoner C, Daines C, Acton JD.
Randomized clinical trial of behavioral
and nutrition treatment to improve
energy intake and growth in toddlers
and preschoolers with cystic fi brosis.
Pediatrics. 2005;116(6):1442–1450
11. Yen EH, Quinton H, Borowitz D. Better
nutritional status in early childhood
is associated with improved clinical
outcomes and survival in patients
with cystic fi brosis. J Pediatr.
2013;162(3):530–535.e1
12. Robinson KA, Saldanha IJ, McKoy
NA. Development of a framework
to identify research gaps from
systematic reviews. J Clin Epidemiol.
2011;64(12):1325–1330
13. Simon GR, Baker C, Barden III GA,
et al; Committee on Practice and
Ambulatory Medicine; Bright Futures
Periodicity Schedule Workgroup.
2014 recommendations for pediatric
preventive health care. Pediatrics.
2014;133(3):568–570
14. Committee on Infectious Diseases.
Recommendations for prevention and
control of infl uenza in children, 2013–
2014. Pediatrics. 2013;132(4). Available
at: www. pediatrics. org/ cgi/ content/
full/ 132/ 4/ e1089
15. Committee on Infectious Diseases,
American Academy of Pediatrics.
Recommended childhood and
adolescent immunization schedule—
United States, 2014. Pediatrics.
2014;133(2):357–363
16. Committee on Environmental Health;
Committee on Substance Abuse;
Committee on Adolescence; Committee
on Native American Child. From the
American Academy of Pediatrics:
20
review and assisted with manuscript preparation and revision; Drs Brady, Schwarzenberg, and Rosenfeld reviewed selected articles, and each compiled and
edited contributions for major sections of the manuscript and drafted sections of the manuscript; Drs Cannon, Condren, Guill, Guillerman, Powers, Tompkins, and
Zemanick and Ms Clark, Ms Leone, Ms Maguiness, and Ms Monchil reviewed selected articles and contributed written sections to the manuscript; and all authors
reviewed and approved the fi nal manuscript as submitted
DOI: 10.1542/peds.2015-1784
Accepted for publication Dec 29, 2015
Address correspondence to Thomas Lahiri, MD, Director, Pediatric Pulmonology, University of Vermont Children’s Hospital, Smith 571, 111 Colchester Ave,
Burlington, VT 05401. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2016 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE: The authors have indicated they have no fi nancial relationships relevant to this article to disclose.
FUNDING: Funding was provided by the Cystic Fibrosis Foundation.
POTENTIAL CONFLICT OF INTEREST: Dr Cannon's institution has received funding from Vertex Pharmaceuticals, Gilead Sciences, Insmed, and Novartis
Pharmaceuticals for running the studies supported by Cystic Fibrosis Therapeutics funds. Dr Cannon is an inventor on a patent for an antimicrobial licensed to
Akron Research Commercialization Corporation, DBA Nebusil. Dr Guillerman has consulted for PTC Therapeutics, Inc, received funding from the Cystic Fibrosis
Foundation Therapeutics, Inc, and compensation from Vertex Pharmaceuticals. Dr Rosenfeld has received grant funding from Vertex Pharmaceuticals. Dr Davis
has served as a board member for Vertex Pharmaceuticals and served as an unpaid consultant for Eli Lilly. The other authors have indicated they have no
potential confl icts of interest to disclose.
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
Policy statement—Tobacco use:
a pediatric disease. Pediatrics.
2009;124(5):1474–1487
17. Uyan ZS, Ozdemir N, Ersu R, et
al. Factors that correlate with
sleep oxygenation in children with
cystic fi brosis. Pediatr Pulmonol.
2007;42(8):716–722
18. van der Giessen LJ, Gosselink R,
Hop WC, Tiddens HA. Recombinant
human DNase nebulisation in children
with cystic fi brosis: before bedtime
or after waking up? Eur Respir J.
2007;30(4):763–768
19. de Castro-Silva C, de Bruin VM,
Cavalcante AG, Bittencourt LR, de
Bruin PF. Nocturnal hypoxia and sleep
disturbances in cystic fi brosis. Pediatr
Pulmonol. 2009;44(11):1143–1150
20. Bakker EM, van der Meijden JC,
Nieuwhof EM, Hop WC, Tiddens
HA. Determining presence of lung
disease in young children with cystic
fi brosis: lung clearance index, oxygen
saturation and cough frequency. J Cyst
Fibros. 2012;11(3):223–230
21. Aurora P, Stocks J, Oliver C, et al;
London Cystic Fibrosis Collaboration.
Quality control for spirometry in
preschool children with and without
lung disease. Am J Respir Crit Care
Med. 2004;169(10):1152–1159
22. Marostica PJ, Weist AD, Eigen H, et al.
Spirometry in 3- to 6-year-old children
with cystic fi brosis. Am J Respir Crit
Care Med. 2002;166(1):67–71
23. Vilozni D, Bentur L, Efrati O, et al.
Spirometry in early childhood in
cystic fi brosis patients. Chest.
2007;131(2):356–361
24. Latzin P, Fehling M, Bauernfeind A,
Reinhardt D, Kappler M, Griese M.
Effi cacy and safety of intravenous
meropenem and tobramycin
versus ceftazidime and tobramycin
in cystic fi brosis. J Cyst Fibros.
2008;7(2):142–146
25. Muramatu LH, Stirbulov R, Forte WC.
Pulmonary function parameters and
use of bronchodilators in patients
with cystic fi brosis. J Bras Pneumol.
2013;39(1):48–55
26. Marson FA, Bertuzzo CS, Ribeiro AF,
Ribeiro JD. Polymorphisms in ADRB2
gene can modulate the response
to bronchodilators and the severity
of cystic fi brosis. BMC Pulm Med.
2012;12:50
27. Davies PL, Doull IJ, Child F. The
interrupter technique to assess
airway responsiveness in children
with cystic fi brosis. Pediatr Pulmonol.
2007;42(1):23–28
28. Aurora P, Stanojevic S, Wade
A, et al; London Cystic Fibrosis
Collaboration. Lung clearance index
at 4 years predicts subsequent
lung function in children with cystic
fi brosis. Am J Respir Crit Care Med.
2011;183(6):752–758
29. Rosenfeld M, Farrell PM, Kloster M, et
al. Association of lung function, chest
radiographs and clinical features in
infants with cystic fi brosis. Eur Respir
J. 2013;42(6):1545–1552
30. Terheggen-Lagro SW, Arets HG, van der
Laag J, van der Ent CK. Radiological
and functional changes over 3 years in
young children with cystic fi brosis. Eur
Respir J. 2007;30(2):279–285
31. Li Z, Kosorok MR, Farrell PM, et al.
Longitudinal development of mucoid
Pseudomonas aeruginosa infection
and lung disease progression in
children with cystic fi brosis. JAMA.
2005;293(5):581–588
32. Kosorok MR, Zeng L, West SE, et
al. Acceleration of lung disease in
children with cystic fi brosis after
Pseudomonas aeruginosa acquisition.
Pediatr Pulmonol. 2001;32(4):277–287
33. Sanders DB, Li Z, Rock MJ, Brody AS,
Farrell PM. The sensitivity of lung
disease surrogates in detecting chest
CT abnormalities in children with
cystic fi brosis. Pediatr Pulmonol.
2012;47(6):567–573
34. Sanders DB, Li Z, Brody AS, Farrell PM.
Chest computed tomography scores
of severity are associated with future
lung disease progression in children
with cystic fi brosis. Am J Respir Crit
Care Med. 2011;184(7):816–821
35. Brasfi eld D, Hicks G, Soong S, Tiller
RE. The chest roentgenogram in
cystic fi brosis: a new scoring system.
Pediatrics. 1979;63(1):24–29
36. Chrispin AR, Norman AP. The
systematic evaluation of the chest
radiograph in cystic fi brosis. Pediatr
Radiol. 1974;2(2):101–105
37. Koscik RE, Kosorok MR, Farrell PM,
et al. Wisconsin cystic fi brosis chest
radiograph scoring system: validation
and standardization for application to
longitudinal studies. Pediatr Pulmonol.
2000;29(6):457–467
38. Sly PD, Brennan S, Gangell C, et
al; Australian Respiratory Early
Surveillance Team for Cystic Fibrosis
(AREST-CF). Lung disease at diagnosis
in infants with cystic fi brosis detected
by newborn screening. Am J Respir
Crit Care Med. 2009;180(2):146–152
39. Stick SM, Brennan S, Murray C, et
al Bronchiectasis in infants and
preschool children diagnosed
with cystic fi brosis after newborn
screening. J Pediatr. 2009;155(5):623–
628.e1
40. de Jong PA, Nakano Y, Lequin MH,
et al. Progressive damage on high
resolution computed tomography
despite stable lung function in cystic
fi brosis. Eur Respir J. 2004;23(1):93–97
41. Gustafsson PM, De Jong PA, Tiddens
HA, Lindblad A. Multiple-breath inert
gas washout and spirometry versus
structural lung disease in cystic
fi brosis. Thorax. 2008;63(2):129–134
42. Mott LS, Park J, Murray CP, et al; AREST
CF. Progression of early structural lung
disease in young children with cystic
fi brosis assessed using CT. Thorax.
2012;67(6):509–516
43. Davis SD, Fordham LA, Brody AS, et
al. Computed tomography refl ects
lower airway infl ammation and
tracks changes in early cystic
fi brosis. Am J Respir Crit Care Med.
2007;175(9):943–950
44. Kuo W, Ciet P, Tiddens HA, Zhang
W, Guillerman RP, van Straten M.
Monitoring cystic fi brosis lung disease
by computed tomography. Radiation
risk in perspective. Am J Respir Crit
Care Med. 2014;189(11):1328–1336
45. Thia LP, Calder A, Stocks J, et al;
London Cystic Fibrosis Collaboration. Is
chest CT useful in newborn screened
infants with cystic fi brosis at 1 year of
age? Thorax. 2014;69(4):320–327
46. de Jong PA, Ottink MD, Robben SG, et
al. Pulmonary disease assessment
21 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
in cystic fi brosis: comparison of CT
scoring systems and value of bronchial
and arterial dimension measurements.
Radiology. 2004;231(2):434–439
47. Owens CM, Aurora P, Stanojevic S, et al;
London Cystic Fibrosis Collaboration.
Lung Clearance Index and HRCT are
complementary markers of lung
abnormalities in young children with
CF. Thorax. 2011;66(6):481–488
48. Yankaskas JR, Marshall BC, Sufi an B,
Simon RH, Rodman D. Cystic fi brosis
adult care: consensus conference
report. Chest. 2004;125(suppl
1):1S–39S
49. Douglas TA, Brennan S, Berry L, et al;
members of AREST CF and ACFBAL
Trial. Value of serology in predicting
Pseudomonas aeruginosa infection
in young children with cystic fi brosis.
Thorax. 2010;65(11):985–990
50. Wainwright CE, Vidmar S, Armstrong
DS, et al; ACFBAL Study Investigators.
Effect of bronchoalveolar lavage-
directed therapy on Pseudomonas
aeruginosa infection and structural
lung injury in children with cystic
fi brosis: a randomized trial. JAMA.
2011;306(2):163–171
51. Jain K, Wainwright C, Smyth AR.
Bronchoscopy-guided antimicrobial
therapy for cystic fi brosis. Cochrane
Database Syst Rev. 2013;(12):CD009530
52. De Boeck K, Alifi er M, Vandeputte S.
Sputum induction in young cystic
fi brosis patients. Eur Respir J.
2000;16(1):91–94
53. Mussaffi H, Fireman EM, Mei-Zahav
M, Prais D, Blau H. Induced sputum
in the very young: a new key to
infection and infl ammation. Chest.
2008;133(1):176–182
54. Al-Saleh S, Dell SD, Grasemann H, et al
Sputum induction in routine clinical
care of children with cystic fi brosis. J
Pediatr. 2010;157(6):1006–1011.e1
55. Byrnes CA, Vidmar S, Cheney JL,
et al; ACFBAL Study Investigators.
Prospective evaluation of respiratory
exacerbations in children with
cystic fi brosis from newborn
screening to 5 years of age. Thorax.
2013;68(7):643–651
56. VanDevanter DR, Elkin EP, Pasta
DJ, et al Changing thresholds and
incidence of antibiotic treatment
of cystic fi brosis pulmonary
exacerbations, 1995–2005. J Cyst Fibro.
2013;12(4):332–337
57. Wagener JS, Rasouliyan L, VanDevanter
DR, et al; Investigators and
Coordinators of the Epidemiologic
Study of Cystic Fibrosis. Oral, inhaled,
and intravenous antibiotic choice for
treating pulmonary exacerbations
in cystic fi brosis. Pediatr Pulmonol.
2013;48(7):666–673
58. Bilton D, Canny G, Conway S, et al
Pulmonary exacerbation: towards
a defi nition for use in clinical trials.
Report from the EuroCareCF Working
Group on outcome parameters
in clinical trials. J Cyst Fibros.
2011;10(suppl 2):S79–81
59. Regelmann WE, Schechter MS,
Wagener JS, et al; Investigators of
the Epidemiologic Study of Cystic
Fibrosis. Pulmonary exacerbations in
cystic fi brosis: young children with
characteristic signs and symptoms.
Pediatr Pulmonol. 2013;48(7):649–657
60. Rabin HR, Butler SM, Wohl ME, et
al; Epidemiologic Study of Cystic
Fibrosis. Pulmonary exacerbations
in cystic fi brosis. Pediatr Pulmonol.
2004;37(5):400–406
61. Pittman JE, Johnson RC, Davis SD.
Improvement in pulmonary function
following antibiotics in infants with
cystic fi brosis. Pediatr Pulmonol.
2012;47(5):441–446
62. Briggs EC, Nguyen T, Wall MA,
MacDonald KD. Oral antimicrobial use
in outpatient cystic fi brosis pulmonary
exacerbation management: a single-
center experience. Clin Respir J.
2012;6(1):56–64
63. Padman R, McColley SA, Miller DP, et
al; Investigators and Coordinators
of the Epidemiologic Study of Cystic
Fibrosis. Infant care patterns at
epidemiologic study of cystic fi brosis
sites that achieve superior childhood
lung function. Pediatrics. 2007;119(3).
Available at: www. pediatrics. org/ cgi/
content/ full/ 119/ 3/ e531
64. van Ewijk BE, van der Zalm MM, Wolfs
TF, van der Ent CK. Viral respiratory
infections in cystic fi brosis. J Cyst
Fibros. 2005;4(suppl 2):31–36
65. Olesen HV, Nielsen LP, Schiotz PO.
Viral and atypical bacterial infections
in the outpatient pediatric cystic
fi brosis clinic. Pediatr Pulmonol.
2006;41(12):1197–1204
66. Wat D, Gelder C, Hibbitts S, et al
The role of respiratory viruses
in cystic fi brosis. J Cyst Fibros.
2008;7(4):320–328
67. Mogayzel PJ Jr, Naureckas ET, Robinson
KA, et al; Cystic Fibrosis Foundation
Pulmonary Clinical Practice Guidelines
Committee. Cystic Fibrosis Foundation
pulmonary guideline. Pharmacologic
approaches to prevention and
eradication of initial Pseudomonas
aeruginosa infection. Ann Am Thorac
Soc. 2014;11(10):1640–1650
68. Flume PA, Robinson KA, O’Sullivan
BP, et al; Clinical Practice Guidelines
for Pulmonary Therapies Committee.
Cystic fi brosis pulmonary guidelines:
airway clearance therapies. Respir
Care. 2009;54(4):522–537
69. Subbarao P, Balkovec S, Solomon
M, Ratjen F. Pilot study of safety and
tolerability of inhaled hypertonic saline
in infants with cystic fi brosis. Pediatr
Pulmonol. 2007;42(5):471–476
70. Dellon EP, Donaldson SH, Johnson R,
Davis SD. Safety and tolerability of
inhaled hypertonic saline in young
children with cystic fi brosis. Pediatr
Pulmonol. 2008;43(11):1100–1106
71. Rosenfeld M, Davis S, Brumback L, et
al. Inhaled hypertonic saline in infants
and toddlers with cystic fi brosis:
short-term tolerability, adherence,
and safety. Pediatr Pulmonol.
2011;46(7):666–671
72. Elkins MR, Robinson M, Rose BR,
et al; National Hypertonic Saline
in Cystic Fibrosis (NHSCF) Study
Group. A controlled trial of long-term
inhaled hypertonic saline in patients
with cystic fi brosis. N Engl J Med.
2006;354(3):229–240
73. Rosenfeld M, Ratjen F, Brumback L, et
al; ISIS Study Group. Inhaled hypertonic
saline in infants and children younger
than 6 years with cystic fi brosis: the
ISIS randomized controlled trial. JAMA.
2012;307(21):2269–2277
74. Subbarao P, Stanojevic S, Brown M,
et al. Lung clearance index as an
outcome measure for clinical trials in
young children with cystic fi brosis. A
pilot study using inhaled hypertonic
22 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
saline. Am J Respir Crit Care Med.
2013;188(4):456–460
75. Quan JM, Tiddens HA, Sy JP, et al;
Pulmozyme Early Intervention Trial
Study Group. A two-year randomized,
placebo-controlled trial of dornase alfa
in young patients with cystic fi brosis
with mild lung function abnormalities.
J Pediatr. 2001;139(6):813–820
76. McPhail GL, Acton JD, Fenchel MC,
Amin RS, Seid M. Improvements in
lung function outcomes in children
with cystic fi brosis are associated
with better nutrition, fewer chronic
Pseudomonas aeruginosa infections,
and dornase alfa use. J Pediatr.
2008;153(6):752–757
77. Jones AP, Wallis C. Dornase alfa for
cystic fi brosis. Cochrane Database
Syst Rev. 2010;(3):CD001127
78. Furuya ME, Lezana-Fernández JL,
Vargas MH, Hernández-Sierra JF,
Ramírez-Figueroa JL. Effi cacy of human
recombinant DNase in pediatric
patients with cystic fi brosis. Arch Med
Res. 2001;32(1):30–34
79. Shah PL, Conway S, Scott SF, et al A
case-controlled study with dornase
alfa to evaluate impact on disease
progression over a 4-year period.
Respiration. 2001;68(2):160–164
80. Hodson ME, McKenzie S, Harms HK, et
al; Investigators of the Epidemiologic
Registry of Cystic Fibrosis. Dornase
alfa in the treatment of cystic
fi brosis in Europe: a report from
the Epidemiologic Registry of
Cystic Fibrosis. Pediatr Pulmonol.
2003;36(5):427–432
81. Konstan MW, Wagener JS, Pasta DJ,
et al; Scientifi c Advisory Group and
Investigators and Coordinators of
Epidemiologic Study of Cystic Fibrosis.
Clinical use of dornase alpha is
associated with a slower rate of FEV1
decline in cystic fi brosis. Pediatr
Pulmonol. 2011;46(6):545–553
82. Amin R, Subbarao P, Lou W, et
al. The effect of dornase alfa on
ventilation inhomogeneity in patients
with cystic fi brosis. Eur Respir J.
2011;37(4):806–812
83. Robinson TE, Goris ML, Zhu HJ, et al.
Dornase alfa reduces air trapping in
children with mild cystic fi brosis lung
disease: a quantitative analysis. Chest.
2005;128(4):2327–2335
84. Nasr SZ, Kuhns LR, Brown RW, Hurwitz
ME, Sanders GM, Strouse PJ. Use
of computerized tomography and
chest x-rays in evaluating effi cacy of
aerosolized recombinant human DNase
in cystic fi brosis patients younger
than age 5 years: a preliminary study.
Pediatr Pulmonol. 2001;31(5):377–382
85. Rozov T, de Oliveira VZ, Santana MA, et
al; Pulmozyme Study Group. Dornase
alfa improves the health-related
quality of life among Brazilian patients
with cystic fi brosis—a one-year
prospective study. Pediatr Pulmonol.
2010;45(9):874–882
86. Wagener JS, Rock MJ, McCubbin MM,
Hamilton SD, Johnson CA, Ahrens RC;
Pulmozyme Pediatric Bronchoscopy
Study Group. Aerosol delivery and
safety of recombinant human
deoxyribonuclease in young children
with cystic fi brosis: a bronchoscopic
study. J Pediatr. 1998;133(4):486–491
87. McKenzie SG, Chowdhury S, Strandvik
B, Hodson ME; Investigators of the
Epidemiologic Registry of Cystic
Fibrosis. Dornase alfa is well tolerated:
data from the epidemiologic registry
of cystic fi brosis. Pediatr Pulmonol.
2007;42(10):928–937
88. Ratjen F, Saiman L, Mayer-Hamblett
N, et al. Effect of azithromycin on
systemic markers of infl ammation in
patients with cystic fi brosis uninfected
with Pseudomonas aeruginosa. Chest.
2012;142(5):1259–1266
89. Saiman L, Mayer-Hamblett N, Anstead
M, et al; AZ0004 Macrolide Study
Team. Open-label, follow-on study of
azithromycin in pediatric patients
with CF uninfected with Pseudomonas
aeruginosa. Pediatr Pulmonol.
2012;47(7):641–648
90. Renna M, Schaffner C, Brown K, et al.
Azithromycin blocks autophagy and
may predispose cystic fi brosis patients
to mycobacterial infection. J Clin
Invest. 2011;121(9):3554–3563
91. Levy I, Grisaru-Soen G, Lerner-Geva
L, et al. Multicenter cross-sectional
study of nontuberculous mycobacterial
infections among cystic fi brosis
patients, Israel. Emerg Infect Dis.
2008;14(3):378–384
92. Binder AM, Adjemian J, Olivier
KN, Prevots DR. Epidemiology of
nontuberculous mycobacterial
infections and associated chronic
macrolide use among persons with
cystic fi brosis. Am J Respir Crit Care
Med. 2013;188(7):807–812
93. Wainwright CE, Quittner AL, Geller
DE, et al Aztreonam for inhalation
solution (AZLI) in patients with cystic
fi brosis, mild lung impairment,
and P. aeruginosa. J Cyst Fibros.
2011;10(4):234–242
94. Oermann CM, Retsch-Bogart GZ,
Quittner AL, et al. An 18-month study
of the safety and effi cacy of repeated
courses of inhaled aztreonam lysine
in cystic fi brosis. Pediatr Pulmonol.
2010;45(11):1121–1134
95. Retsch-Bogart GZ, Quittner AL, Gibson
RL, et al. Effi cacy and safety of
inhaled aztreonam lysine for airway
Pseudomonas in cystic fi brosis. Chest.
2009;135(5):1223–1232
96. McCoy KS, Quittner AL, Oermann
CM, Gibson RL, Retsch-Bogart
GZ, Montgomery AB. Inhaled
aztreonam lysine for chronic airway
Pseudomonas aeruginosa in cystic
fi brosis. Am J Respir Crit Care Med.
2008;178(9):921–928
97. Treggiari MM, Retsch-Bogart G, Mayer-
Hamblett N, et al; Early Pseudomonas
Infection Control (EPIC) Investigators.
Comparative effi cacy and safety
of 4 randomized regimens to treat
early Pseudomonas aeruginosa
infection in children with cystic
fi brosis. Arch Pediatr Adolesc Med.
2011;165(9):847–856
98. Gibson RL, Emerson J, McNamara S,
et al; Cystic Fibrosis Therapeutics
Development Network Study Group.
Signifi cant microbiological effect of
inhaled tobramycin in young children
with cystic fi brosis. Am J Respir Crit
Care Med. 2003;167(6):841–849
99. Taccetti G, Bianchini E, Cariani L, et
al; Italian Group for P aeruginosa
Eradication in Cystic Fibrosis. Early
antibiotic treatment for Pseudomonas
aeruginosa eradication in patients
with cystic fi brosis: a randomised
multicentre study comparing
two different protocols. Thorax.
2012;67(10):853–859
23 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
100. Hennig S, McKay K, Vidmar S, et
al Safety of inhaled (Tobi(R)) and
intravenous tobramycin in young
children with cystic fi brosis. J Cyst
Fibros. 2014;13(4):428–434
101. Nguyen TT, Thia LP, Hoo AF, et al; London
Cystic Fibrosis Collaboration (LCFC).
Evolution of lung function during the
fi rst year of life in newborn screened
cystic fi brosis infants. Thorax.
2014;69(10):910–917
102. Stutman HR, Lieberman JM, Nussbaum
E, Marks MI. Antibiotic prophylaxis in
infants and young children with cystic
fi brosis: a randomized controlled trial.
J Pediatr. 2002;140(3):299–305
103. Smyth AR, Walters S. Prophylactic anti-
staphylococcal antibiotics for cystic
fi brosis. Cochrane Database Syst Rev.
2012;(12):CD001912
104. Ratjen F, Comes G, Paul K, Posselt HG,
Wagner TO, Harms K; German Board
of the European Registry for Cystic
Fibrosis (ERCF). Effect of continuous
antistaphylococcal therapy on the rate
of P. aeruginosa acquisition in patients
with cystic fi brosis. Pediatr Pulmonol.
2001;31(1):13–16
105. Chatfi eld S, Owen G, Ryley HC, et al
Neonatal screening for cystic fi brosis
in Wales and the West Midlands:
clinical assessment after fi ve years of
screening. Arch Dis Child. 1991;66(spec
no. 1):29–33
106. Weaver LT, Green MR, Nicholson K, et
al. Prognosis in cystic fi brosis treated
with continuous fl ucloxacillin from
the neonatal period. Arch Dis Child.
1994;70(2):84–89
107. Lo DK, Hurley MN, Muhlebach MS,
Smyth AR. Interventions for the
eradication of methicillin-resistant
Staphylococcus aureus (MRSA) in
people with cystic fi brosis. Cochrane
Database Syst Rev. 2013;2:CD009650
108. Solís A, Brown D, Hughes J, Van Saene
HK, Heaf DP. Methicillin-resistant
Staphylococcus aureus in children
with cystic fi brosis: an eradication
protocol. Pediatr Pulmonol.
2003;36(3):189–195
109. Macfarlane M, Leavy A, McCaughan
J, Fair R, Reid AJ. Successful
decolonization of methicillin-resistant
Staphylococcus aureus in paediatric
patients with cystic fi brosis (CF) using
a three-step protocol. J Hosp Infect.
2007;65(3):231–236
110. Dalboge CS, Pressler T, Hoiby N, Nielsen
KG, Johansen HK. A cohort study of
the Copenhagen CF Centre eradication
strategy against Staphylococcus
aureus in patients with CF. J Cyst
Fibros. 2013;12(1):42–48
111. Davies J, Sheridan H, Bell N, et al.
Assessment of clinical response
to ivacaftor with lung clearance
index in cystic fi brosis patients
with a G551D-CFTR mutation and
preserved spirometry: a randomised
controlled trial. Lancet Respir Med.
2013;1(8):630–638
112. Ramsey BW, Davies J, McElvaney NG,
et al; VX08–770–102 Study Group. A
CFTR potentiator in patients with cystic
fi brosis and the G551D mutation. N
Engl J Med. 2011;365(18):1663–1672
113. Accurso FJ, Rowe SM, Clancy JP, et al.
Effect of VX-770 in persons with cystic
fi brosis and the G551D-CFTR mutation.
N Engl J Med. 2010;363(21):1991–2003
114. Davies CA, Robertson S, Green Y,
Rosenfeld M. 200. An open-label study
of the safety, pharmacokinetics, and
pharmacodynamics of ivacaftor in
patients aged 2 to 5 years with CF and
a CFTR gating mutation: the KIWI study.
Pediatr Pulmonol. 2014;(suppl 38):286
115. Rosenfeld M, Robertson S, Green
Y, Cooke J, Lawal A, Davies JC.
WS01.5 An open-label study of the
safety, pharmacokinetics, and
pharmacodynamics of ivacaftor in
patients aged 2 to 5 years with cystic
fi brosis and a CFTR gating mutation:
the KIWI study. J Cyst Fibros. 2015;14:S2
116. Centers for Disease Control and
Prevention. Data table of weight-for-
age charts. August 2001. Available at:
www. cdc. gov/ growthcharts/ html_
charts/ wtage. htm. Accessed July 28,
2014
117. AAP Committee on Nutrition. Kleinman
RE, Greer FR, eds. Pediatric Nutrition.
7th ed. Elk Grove Village, IL: American
Academy of Pediatrics; 2014
118. McCammon RW. Human Growth and
Development. Springfi eld, IL.: Thomas;
1970
119. US Department of Agriculture. Offi ce
of Disease Prevention and Health
Promotion. Dietary guidelines for
Americans 2010. Available at: www.
dietaryguidelines . gov. Accessed July
28, 2014
120. Dietary Reference Intakes for Energy.
Carbohydrate, Fiber, Fat, Fatty Acids,
Cholesterol, Protein, and Amino Acids
(Macronutrients). Washington D.C.: The
National Academies Press; 2005
121. Driscoll KA, Modi AC, Filigno SS, et
al. Quality of life in children with CF:
psychometrics and relations with
stress and mealtime behaviors.
Pediatr Pulmonol. 2015;50(6):560–567
122. Groleau V, Schall JI, Dougherty KA, et
al Effect of a dietary intervention on
growth and energy expenditure in
children with cystic fi brosis. J Cyst
Fibros. 2014;13(5):572–578
123. Poustie VJ, Russell JE, Watling RM,
Ashby D, Smyth RL; CALICO Trial
Collaborative Group. Oral protein
energy supplements for children with
cystic fi brosis: CALICO multicentre
randomised controlled trial. BMJ.
2006;332(7542):632–636
124. Piazza-Waggoner C, Driscoll KA,
Gilman DK, Powers SW. A comparison
using parent report and direct
observation of mealtime behaviors
in young children with cystic
fi brosis: Implications for practical
and empirically based behavioral
assessment in routine clinical care.
Child Health Care. 2008;37(1):38–48
125. Powers SW, Mitchell MJ, Patton
SR, et al Mealtime behaviors in
families of infants and toddlers
with cystic fi brosis. J Cyst Fibros.
2005;4(3):175–182
126. Ward C, Massie J, Glazner J, et al.
Problem behaviours and parenting in
preschool children with cystic fi brosis.
Arch Dis Child. 2009;94(5):341–347
127. Sheehan J, Massie J, Hay M, et al.
The natural history and predictors
of persistent problem behaviours
in cystic fi brosis: a multicentre,
prospective study. Arch Dis Child.
2012;97(7):625–631
128. Rosenfeld M, Casey S, Pepe M, Ramsey
BW. Nutritional effects of long-term
gastrostomy feedings in children
with cystic fi brosis. J Am Diet Assoc.
1999;99(2):191–194
129. Best C, Brearley A, Gaillard P, et
al. A pre-post retrospective study
24 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume 137 , number 4 , April 2016
of patients with cystic fi brosis
and gastrostomy tubes. J Pediatr
Gastroenterol Nutr. 2011;53(4):453–458
130. Efrati O, Mei-Zahav M, Rivlin J, et al.
Long term nutritional rehabilitation by
gastrostomy in Israeli patients with
cystic fi brosis: clinical outcome in
advanced pulmonary disease. J Pediatr
Gastroenterol Nutr. 2006;42(2):222–228
131. Bradley GM, Carson KA, Leonard
AR, Mogayzel PJ Jr, Oliva-Hemker
M. Nutritional outcomes following
gastrostomy in children with
cystic fi brosis. Pediatr Pulmonol.
2012;47(8):743–748
132. Tangpricha V, Kelly A, Stephenson A, et
al; Cystic Fibrosis Foundation Vitamin
D Evidence-Based Review Committee.
An update on the screening, diagnosis,
management, and treatment of
vitamin D defi ciency in individuals
with cystic fi brosis: evidence-based
recommendations from the Cystic
Fibrosis Foundation. J Clin Endocrinol
Metab. 2012;97(4):1082–1093
133. Brodlie M, Orchard WA, Reeks GA,
et al. Vitamin D in children with
cystic fi brosis. Arch Dis Child.
2012;97(11):982–984
134. Scurati-Manzoni E, Fossali EF, Agostoni
C, et al. Electrolyte abnormalities in
cystic fi brosis: systematic review
of the literature. Pediatr Nephrol.
2014;29(6):1015–1023
135. Borowitz DS, Grand RJ, Durie PR;
Consensus Committee. Use of
pancreatic enzyme supplements for
patients with cystic fi brosis in the
context of fi brosing colonopathy. J
Pediatr. 1995;127(5):681–684
136. Powers SW, Piazza-Waggoner C, Jones
JS, Ferguson KS, Daines C, Acton JD.
Examining clinical trial results with
single-subject analysis: an example
involving behavioral and nutrition
treatment for young children with
cystic fi brosis. J Pediatr Psychol.
2006;31(6):574–581
137. Stark LJ, Quittner AL, Powers SW,
et al. Randomized clinical trial of
behavioral intervention and nutrition
education to improve caloric intake
and weight in children with cystic
fi brosis. Arch Pediatr Adolesc Med.
2009;163(10):915–921
138. Stark LJ, Opipari-Arrigan L, Quittner
AL, Bean J, Powers SW. The effects of
an intensive behavior and nutrition
intervention compared to standard of
care on weight outcomes in CF. Pediatr
Pulmonol. 2011;46(1):31–35
139. Powers SW, Stark LJ, Chamberlin
LA, et al. Behavioral and nutritional
treatment for preschool-aged
children with cystic fi brosis: a
randomized clinical trial. JAMA Pediatr.
2015;169(5):e150636
140. Opipari-Arrigan L, Powers SW, Quittner
AL, Stark LJ. Mealtime problems
predict outcome in clinical trial to
improve nutrition in children with CF.
Pediatr Pulmonol. 2010;45(1):78–82
141. McDonald CM, Haberman D, Brown N.
Self-effi cacy: empowering parents of
children with cystic fi brosis. J Cyst
Fibros. 2013;12(5):538–543
142. McClellan CB, Cohen LL, Moffett K.
Time out based discipline strategy for
children’s non-compliance with cystic
fi brosis treatment. Disabil Rehabil.
2009;31(4):327–336
143. Hourigan SE, Helms SW, Christon
LM, Southam-Gerow MA. Improving
nutrition in toddlers with cystic
fi brosis: feasibility of a behavioral
parent-training intervention. Clin Pract
Pediatr Psychol. 2013;1(3):235–249
144. Children’s Hospital Medical Center
Cincinnati. National Institute of
Diabetes and Digestive and Kidney
Diseases. ClinicalTrials.gov [Internet].
Bethesda (MD): National Library of
Medicine (US). 2000–2014 Aug 20. A
Multi-Site Randomized Clinical Trial of
Behavioral and Nutrition Treatment
Designed to Help Preschoolers With
Cystic Fibrosis Optimize Growth.
NLM Identifi er: NCT00241969.
Available at: https:// clinicaltrials.
gov/ ct2/ show/ NCT00241969? term=
Behavioral+and+Nu trition+Treatment
+for+Preschoolers +with+Cystic+Fibr
osis& rank= 1. Accessed January 22,
2015
145. Baker SS, Borowitz D, Duffy L,
Fitzpatrick L, Gyamfi J, Baker RD.
Pancreatic enzyme therapy and clinical
outcomes in patients with cystic
fi brosis. J Pediatr. 2005;146(2):189–193
146. Tabbers MM, DiLorenzo C, Berger MY,
et al; European Society for Pediatric
Gastroenterology, Hepatology, and
Nutrition; North American Society
for Pediatric Gastroenterology.
Evaluation and treatment of functional
constipation in infants and children:
evidence-based recommendations
from ESPGHAN and NASPGHAN.
J Pediatr Gastroenterol Nutr.
2014;58(2):258–274
147. Houwen RH, van der Doef HP, Sermet I,
et al; ESPGHAN Cystic Fibrosis Working
Group. Defi ning DIOS and constipation
in cystic fi brosis with a multicentre
study on the incidence, characteristics,
and treatment of DIOS. J Pediatr
Gastroenterol Nutr. 2010;50(1):38–42
148. Vandenplas Y, Rudolph CD, Di Lorenzo
C, et al; North American Society
for Pediatric Gastroenterology
Hepatology and Nutrition;
European Society for Pediatric
Gastroenterology Hepatology and
Nutrition. Pediatric gastroesophageal
refl ux clinical practice guidelines:
joint recommendations of the North
American Society for Pediatric
Gastroenterology, Hepatology,
and Nutrition (NASPGHAN) and the
European Society for Pediatric
Gastroenterology, Hepatology, and
Nutrition (ESPGHAN). J Pediatr
Gastroenterol Nutr. 2009;49(4):498–547
149. Lisowska A, Wójtowicz J, Walkowiak J.
Small intestine bacterial overgrowth
is frequent in cystic fi brosis: combined
hydrogen and methane measurements
are required for its detection. Acta
Biochim Pol. 2009;56(4):631–634
150. Hill ID, Dirks MH, Liptak GS, et al;
North American Society for Pediatric
Gastroenterology, Hepatology and
Nutrition. Guideline for the diagnosis
and treatment of celiac disease in
children: recommendations of the
North American Society for Pediatric
Gastroenterology, Hepatology and
Nutrition. J Pediatr Gastroenterol Nutr.
2005;40(1):1–19
151. Agréus L, Svärdsudd K, Nyrén O, Tibblin
G. Reproducibility and validity of a
postal questionnaire. The abdominal
symptom study. Scand J Prim Health
Care. 1993;11(4):252–262
152. Brunner HI, Johnson AL, Barron AC,
et al. Gastrointestinal symptoms and
their association with health-related
quality of life of children with juvenile
rheumatoid arthritis: validation
25 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

LAHIRI et al
of a gastrointestinal symptom
questionnaire. J Clin Rheumatol.
2005;11(4):194–204
153. Wilschanski M, Novak I. The cystic
fi brosis of exocrine pancreas.
Cold Spring Harb Perspect Med.
2013;3(5):a009746
154. Ooi CY, Dorfman R, Cipolli M, et al.
Type of CFTR mutation determines
risk of pancreatitis in patients with
cystic fi brosis. Gastroenterology.
2011;140(1):153–161
155. Terlizzi V, Tosco A, Tomaiuolo R, et
al Prediction of acute pancreatitis
risk based on PIP score in children
with cystic fi brosis. J Cyst Fibros.
2014;13(5):579–584
156. Simpson RM, Lloyd DJ, Gvozdanovic D,
Russell G. Vitamin B12 defi ciency in
cystic fi brosis. Acta Paediatr Scand.
1985;74(5):794–796
157. Garcia-Naveiro RUJ. Maldigestion and
malabsorption. In: Wyllie R, Hyams JS,
Kay M, eds. Pediatric Gastrointestinal
and Liver Disease, 4th ed. Philadelphia,
PA: Elsevier Saunders; 2011:337–349
26 by guest on October 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2015-1784 originally published online March 23, 2016; 2016;137;Pediatrics
Schwarzenberg, Connie L. Tompkins, Edith T. Zemanick and Stephanie D. DavisKaren Maguiness, Lisa Monchil, Scott W. Powers, Margaret Rosenfeld, Sarah JaneMichelle E. Condren, Margaret F. Guill, R. Paul Guillerman, Christina G. Leone,
Thomas Lahiri, Sarah E. Hempstead, Cynthia Brady, Carolyn L. Cannon, Kelli Clark,Preschoolers With Cystic Fibrosis
Clinical Practice Guidelines From the Cystic Fibrosis Foundation for
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/137/4/e20151784including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/137/4/e20151784#BIBLThis article cites 130 articles, 29 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/pulmonology_subPulmonologyhttp://www.aappublications.org/cgi/collection/nutrition_subNutritionfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on October 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2015-1784 originally published online March 23, 2016; 2016;137;Pediatrics
Schwarzenberg, Connie L. Tompkins, Edith T. Zemanick and Stephanie D. DavisKaren Maguiness, Lisa Monchil, Scott W. Powers, Margaret Rosenfeld, Sarah JaneMichelle E. Condren, Margaret F. Guill, R. Paul Guillerman, Christina G. Leone,
Thomas Lahiri, Sarah E. Hempstead, Cynthia Brady, Carolyn L. Cannon, Kelli Clark,Preschoolers With Cystic Fibrosis
Clinical Practice Guidelines From the Cystic Fibrosis Foundation for
http://pediatrics.aappublications.org/content/137/4/e20151784located on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://pediatrics.aappublications.org/content/suppl/2016/03/23/peds.2015-1784.DCSupplementalData Supplement at:
1073-0397. ISSN:60007. Copyright © 2016 by the American Academy of Pediatrics. All rights reserved. Print
the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on October 12, 2018www.aappublications.org/newsDownloaded from