
Clinical investigational studies for validation of a next-generation sequencing in vitro diagnostic...
18
Clinical investigational studies for validation of a next- generation sequencing in vitro diagnostic device for cystic fibrosis testing Expert Rev. Mol. Diagn. 14(5), 605–622 (2014) Daniel S Grosu 1 , Lynda Hague 1 , Manjula Chelliserry 1 , Kristina M Kruglyak 1 , Ross Lenta 1 , Brandy Klotzle 1 , Jonathan San 1 , Wendy M Goldstein 1 , Sharmili Moturi 1 , Patricia Devers 2 , Julie Woolworth 3 , Eric Peters 1 , Barbara Elashoff 4 , Jay Stoerker 5 , Daynna J Wolff 3 , Kenneth J Friedman 6 , W Edward Highsmith 7 , Erick Lin 1 and Frank S Ong* 1 1 Illumina, Inc., San Diego, CA 92122, USA 2 Verinata Health, Inc., an Illumina Company, Redwood City, CA 94063, USA 3 Department of Pathology and Labora- tory Medicine, Medical University of South Carolina, Charleston, SC 29425, USA 4 Myraqa, Inc., Redwood Shores, CA 94065, USA 5 Progenity, Inc., Ann Arbor, MI 49108, USA 6 Laboratory Corporation of America Ò Holding, Research Triangle Park, NC 27709, USA 7 Department of Laboratory Medicine and Pathology, Mayo Clinic College of Medicine, Rochester, MN 55905, USA *Author for correspondence: [email protected] Purpose: Clinical investigational studies were conducted to demonstrate the accuracy and reproducibility of the Illumina MiSeqDx CF System, a next-generation sequencing (NGS) in vitro diagnostic device for cystic fibrosis testing. Methods: Two NGS assays – a Clinical Sequencing Assay (Sequencing Assay) and a 139-Variant Assay (Variant Assay) – were evaluated in both an Accuracy Study and a Reproducibility Study, with comparison to bi-directional Sanger sequencing and PCR as reference methods. For each study, positive agreement (PA), negative agreement (NA), and overall agreement (OA) were evaluated. Results: In the Accuracy Study, the Sequencing Assay achieved PA of 99.7% including the polyTG/polyT region and PA of 100% excluding the region. The Variant Assay achieved PA of 100%. NA and OA were >99.99% for both Assays. In the Reproducibility Study, the Sequencing Assay achieved PA of 99.2%; NA and OA were both 99.7%. The Variant Assay achieved PA of 99.8%; NA and OA were both 99.9%. Sample pass rates were 99.7% in both studies for both assays. Conclusion: This is the first systematic evaluation of a NGS platform for broad clinical use as an in vitro diagnostic, including accuracy validation with multiple reference methods and reproducibility validation at multiple clinical sites. These NGS-based Assays had accurate and reproducible results which were comparable to or better than other methods currently in clinical use for clinical genetic testing of cystic fibrosis. KEYWORDS: clinical validation • cystic fibrosis • in vitro diagnostics • next-generation sequencing • sequencing-by-synthesis Since 1989 when patients with cystic fibrosis (CF) were found to have a deletion of three base pairs in the CF transmembrane conduc- tance regulatory gene (CFTR) [1–3], more than 1900 variants have been reported in patients affected by the disease [4]. A small percentage of these variants are known to be disease- causing [5,6], and the majority are variants of uncertain significance [7]. Furthermore, allelic heterogeneity is a challenge for translating genomic knowledge into clinical practice [8], and patients with CFTR-related diseases present with a variety of clinical phenotypes, ranging from classical CF to congenital bilat- eral absence of the vas deferens in otherwise healthy males [9–11], confounding genotype– phenotype correlations and CF prediction algorithms [12]. In 2001, the American College of Medical Genetics (ACMG) issued guidelines for population-based CF carrier screening using a panel of 25 CF-causing variants [13], with a subsequent review of the clinical evidence in 2004 resulting in an updated ACMG- recommended panel of 23 mutations [6]. More recently, the Clinical and Functional Transla- tion of CFTR Project [14] was initiated in 2008, with the goal of expanding the under- standing of disease liability of CFTR variants using a ‘phenotype-driven’ approach [15]. Phe- notypic and genomic data from 39,696 individ- uals with CF (representing >50% of CF patients worldwide) were compiled from 25 CF registries and centers and preliminary stud- ies have shown an improved screening out- come with the inclusion of more variants [5]. informahealthcare.com 10.1586/14737159.2014.916618 Ó 2014 Informa UK Ltd ISSN 1473-7159 605 Original Research Expert Review of Molecular Diagnostics Downloaded from informahealthcare.com by 175.98.98.130 on 05/22/14 For personal use only.
-
Upload
frank-s-ong-md-cpi -
Category
Documents
-
view
109 -
download
2
Transcript of Clinical investigational studies for validation of a next-generation sequencing in vitro diagnostic...

Clinical investigational studiesfor validation of a next-generation sequencingin vitro diagnostic devicefor cystic fibrosis testingExpert Rev. Mol. Diagn. 14(5), 605–622 (2014)
Daniel S Grosu1,Lynda Hague1,Manjula Chelliserry1,Kristina M Kruglyak1,Ross Lenta1,Brandy Klotzle1,Jonathan San1,Wendy M Goldstein1,Sharmili Moturi1,Patricia Devers2,Julie Woolworth3,Eric Peters1, BarbaraElashoff4, Jay Stoerker5,Daynna J Wolff3,Kenneth J Friedman6,W Edward Highsmith7,Erick Lin1 andFrank S Ong*1
1Illumina, Inc., San Diego, CA 92122,
USA2Verinata Health, Inc., an Illumina
Company, Redwood City, CA 94063,
USA3Department of Pathology and Labora-
tory Medicine, Medical University of
South Carolina, Charleston, SC 29425,
USA4Myraqa, Inc., Redwood Shores,
CA 94065, USA5Progenity, Inc., Ann Arbor, MI 49108,
USA6Laboratory Corporation of America�
Holding, Research Triangle Park,
NC 27709, USA7Department of Laboratory Medicine
and Pathology, Mayo Clinic College of
Medicine, Rochester, MN 55905, USA
*Author for correspondence:
Purpose: Clinical investigational studies were conducted to demonstrate the accuracy andreproducibility of the Illumina MiSeqDx CF System, a next-generation sequencing (NGS) in vitrodiagnostic device for cystic fibrosis testing. Methods: Two NGS assays – a Clinical SequencingAssay (Sequencing Assay) and a 139-Variant Assay (Variant Assay) – were evaluated in both anAccuracy Study and a Reproducibility Study, with comparison to bi-directional Sanger sequencingand PCR as reference methods. For each study, positive agreement (PA), negative agreement(NA), and overall agreement (OA) were evaluated. Results: In the Accuracy Study, the SequencingAssay achieved PA of 99.7% including the polyTG/polyT region and PA of 100% excluding theregion. The Variant Assay achieved PA of 100%. NA and OA were >99.99% for both Assays. Inthe Reproducibility Study, the Sequencing Assay achieved PA of 99.2%; NA and OA were both99.7%. The Variant Assay achieved PA of 99.8%; NA and OA were both 99.9%. Sample passrates were 99.7% in both studies for both assays. Conclusion: This is the first systematicevaluation of a NGS platform for broad clinical use as an in vitro diagnostic, including accuracyvalidation with multiple reference methods and reproducibility validation at multiple clinical sites.These NGS-based Assays had accurate and reproducible results which were comparable to orbetter than other methods currently in clinical use for clinical genetic testing of cystic fibrosis.
KEYWORDS: clinical validation • cystic fibrosis • in vitro diagnostics • next-generation sequencing • sequencing-by-synthesis
Since 1989 when patients with cystic fibrosis(CF) were found to have a deletion of threebase pairs in the CF transmembrane conduc-tance regulatory gene (CFTR) [1–3], more than1900 variants have been reported in patientsaffected by the disease [4]. A small percentageof these variants are known to be disease-causing [5,6], and the majority are variants ofuncertain significance [7]. Furthermore, allelicheterogeneity is a challenge for translatinggenomic knowledge into clinical practice [8],and patients with CFTR-related diseasespresent with a variety of clinical phenotypes,ranging from classical CF to congenital bilat-eral absence of the vas deferens in otherwisehealthy males [9–11], confounding genotype–phenotype correlations and CF predictionalgorithms [12].
In 2001, the American College of MedicalGenetics (ACMG) issued guidelines forpopulation-based CF carrier screening using apanel of 25 CF-causing variants [13], with asubsequent review of the clinical evidence in2004 resulting in an updated ACMG-recommended panel of 23 mutations [6]. Morerecently, the Clinical and Functional Transla-tion of CFTR Project [14] was initiated in2008, with the goal of expanding the under-standing of disease liability of CFTR variantsusing a ‘phenotype-driven’ approach [15]. Phe-notypic and genomic data from 39,696 individ-uals with CF (representing >50% of CFpatients worldwide) were compiled from 25CF registries and centers and preliminary stud-ies have shown an improved screening out-come with the inclusion of more variants [5].
informahealthcare.com 10.1586/14737159.2014.916618 � 2014 Informa UK Ltd ISSN 1473-7159 605
Original Research
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

CFTR testing may be performed for clinical diagnosis of CF inindividuals with signs or symptoms of the disease [16], newbornscreening [17–20], preconception or prenatal carrier screening [21,22],targeted treatment [23] and subject selection in clinical researchsettings [24]. For patients with suspected CF in whom the first-tier variant panel test is unable to detect both disease alleles, astep-wise reflexive testing algorithm [25], generally using Sangersequencing (Sanger) followed by Multiplex Ligation-dependentProbe Amplification, is often necessary to confirm the diagnosis.When analyzing many exons and genes simultaneously, Sangercan be time-consuming. Additionally, Sanger has reported errorrates as high as 3% [26] due to imbalanced amplification, errors inautomatic base-calling or the manual interpretation of Sangertraces. Additionally, Sanger is prone to error in regions that con-tain a repeat motif [27], and is generally unable to identify largedeletions and rearrangements that span multiple exons. Next-generation sequencing (NGS) represents a shift in approach tothe analysis of CFTR and other genetic disorders. NGS allows forrapid turn-around times and high multiplexing of samples in asingle run, while maintaining high levels of reported quality [26].Several limitations of NGS technologies have been reported, gen-erally associated with whole genome sequencing. These limita-tions include sequence-specific error motifs, difficulty inresolving long complex regions of the genome using short readsand higher base call error rates [28]. Targeted NGS approacheshave addressed many of these issues and the demand for focusedclinical applications of the technology has increased [29]. The Illu-mina MiSeqDxTM CF System (MiSeqDx CF System) is asequencing-by-synthesis [30] system that was developed as a NGS-based in vitro diagnostic (IVD) platform for comprehensivegenetic testing of suspected CF patients and carriers.
Two CF assays were developed as part of the MiSeqDx CFSystem: the MiSeqDx CF Clinical Sequencing Assay (Sequenc-ing Assay) and the MiSeqDx CF 139-Variant Assay (VariantAssay); both assays were designed for use with genomic DNA(gDNA) samples isolated from human peripheral whole bloodspecimens. The Sequencing Assay serves as a rapid turn-around,high-throughput option for patients with suspected CF whocannot be conclusively diagnosed through variant-panel testing.The Variant Assay reports on 139 variants, including those cur-rently recommended by ACMG [6] and those identified as CF-causing in the CFTR2 database (as of September 2013). Inthis report, we present the results of the accuracy and reproduc-ibility clinical investigational studies of the two CF assays.
MethodsInstitutional review board
Institutional review board (IRB) approvals for sample collec-tions were obtained from Quorum Review IRB, CopernicusGroup IRB and Mayo Clinic IRB. Approval for the AccuracyStudy was obtained from Quorum Review IRB. Approval forthe Reproducibility Study was obtained from the IRBs of Med-ical University of South Carolina and the Johns HopkinsSchool of Medicine. All samples, whether residual clinical sam-ples or cell lines, were de-identified at the collection sites.
Investigational Assays
Sequencing Assay
The Sequencing Assay was designed to interrogate 5206 uniquegenomic positions in CFTR to enable detection of all singlenucleotide variants (SNVs) and small insertions/deletions (indels)within the protein coding regions and intron/exon bound-aries, as well as two deep intronic variants (c.1679+1.6kbA>G,c.3717+12191C>T) and two large deletions (c.54-5940_273+10250del21kb, c.3964-78_4242+577del). Regions not tar-geted in the assay include other deep intronic mutations orvariants in the promoter region. This assay was designed todetect indels up to 25 bp: indels ranging from 1 to 3 bpwere tested using clinical samples, indels ranging from 4 to22 bp were tested using synthetic plasmids and indels rangingfrom 23 to 25 bp were tested in silico. All protein coding regionsin CFTR including 10 bp of flanking intronic sequence for allexons with the exception of three (exons 7, 10 and 20), 100 bp offlanking sequence at the 5´ and 3´ untranslated regions, 2 deepintronic mutations (1811+1.6kbA>G, 3489+10kbC>T) and2 large deletions (CFTRdele2,3, CFTRdele22,23) are sequenced.For exons 7 and 10, only 5 bp of flanking intronic sequence isincluded at the 5´ end of the exon to avoid proximal homopoly-meric indels. For exon 20, 30 bp of flanking intronic sequence isincluded at the 5´ end of the exon to enable detection of themutation 3272-26A>G.
Variant Assay
The Variant Assay was designed to interrogate 139 unique var-iants present at 135 unique genomic loci: 135 variants SNVs,30 small indels and 2 large deletions) that were directlyreported, including 134 CF-causing variants (per CFTR2) and1 ACMG-recommended variant (R117H), as well as 4 otherclinically relevant variants that were conditionally reported (thepolyTG/polyT region was reported when p.R117H variant wasidentified; and the variants p.I506V, p.I507V and p.F508Cwere reported when either homozygous p.F508del or homozy-gous p.I507del variants were identified). The 134 CF-causingvariants reported by the assay correspond to the 129 CF-caus-ing variant loci in the CFTR2 database (as of September2013), as the CFTR2 database includes 5 CF-causing variantloci at which the same deleterious protein level change mayarise from two distinct nucleotide changes (e.g., S466X[C>A]and S466X[C>G]). These five variant loci were listed in theCFTR2 database according to the amino acid codon (e.g.,S466X), while the Variant Assay reports each individual geno-type variant (e.g., S466X[C>A] and S466X[C>G]). The full setof 139 variants, which includes all 23 variants currently recom-mended by ACMG for carrier screening and the disease-causingvariants per the CFTR2 database, can be found in TABLE 1.
Assay overview
Library preparation was performed using Illumina TruSeqCustom Amplicon chemistry [31] and involves targeted amplifi-cation of the CFTR gene (FIGURE 1) followed by sequencing-by-synthesis [32,33] on board the MiSeqDx. Briefly, one pair
Original Research Grosu, Hague, Chelliserry et al.
606 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

Table 1. Content of Variant Assay.
Variant (common name) HGVS name Variant type
M1V c.1A>G SNV
CFTRdele2,3 c.54-5940_273+10250del21kb Del
Q39X c.115C>T SNV
E60X c.178G>T SNV
P67L c.200C>T SNV
R75X c.223C>T SNV
G85E c.254G>A SNV
394delTT c.262_263delTT Div
405+1G>A c.273+1G>A SNV
406-1G>A c.274-1G>A SNV
E92X c.274G>T SNV
E92K c.274G>A SNV
Q98X c.292C>T SNV
457TAT>G c.325_327delTATinsG Div
D110H c.328G>C SNV
R117C c.349C>T SNV
R117H c.350G>A SNV
Y122X c.366T>A SNV
574delA c.442delA Div
621+1G>T c.489+1G>T SNV
663delT c.531delT Div
G178R c.532G>A SNV
711+1G>T c.579+1G>T SNV
711+3A>G c.579+3A>G SNV
711+5G>A c.579+5G>A SNV
712-1G>T c.580-1G>T SNV
H199Y c.595C>T SNV
P205S c.613C>T SNV
L206W c.617T>G SNV
Q220X c.658C>T SNV
852del22 c.720_741delAGGGAGAATGATGATGAAGTAC Div
1078delT c.948delT Div
G330X c.988G>T SNV
R334W c.1000C>T SNV
I336K c.1007T>A SNV
T338I c.1013C>T SNV
MiSeqDx cystic fibrosis 139-Variant Assay reported variants (Bold = ACMG23; Italics = conditionally reported).†Classified in the CFTR2 database as a CF-causing variant (as of September 2013); however, Sosnay et al. classified the variant as indeterminate. The CFTR2 databaseclassification is more current and reflects the completed functional testing for this variant, which was not available at the time of the Sosnay et al. publication.Del: Large deletion variant; Div: Insertion/deletion variant; HGVS: Human genome variation society; SNV: Single nucleotide variant.
NGS for CF diagnosis Original Research
informahealthcare.com 607
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

Table 1. Content of Variant Assay (cont.).
Variant (common name) HGVS name Variant type
1154insTC c.1022_1023insTC Div
S341P c.1021T>C SNV
R347H c.1040G>A SNV
R347P c.1040G>C SNV
R352Q c.1055G>A SNV
1213delT c.1081delT Div
1248+1G>A c.1116+1G>A SNV
1259insA c.1127_1128insA Div
W401X (c.1202G>A) c.1202G>A SNV
W401X (c.1203G>A) c.1203G>A SNV
1341+1G>A c.1209+1G>A SNV
1461ins4 c.1329_1330insAGAT Div
A455E c.1364C>A SNV
1525-1G>A c.1393-1G>A SNV
S466X (C>A) c.1397C>A SNV
S466X (C>G) c.1397C>G SNV
L467P c.1400T>C SNV
1548delG† c.1418delG Div
S489X c.1466C>A SNV
S492F c.1475C>T SNV
Q493X c.1477C>T SNV
I507del c.1519_1521delATC Div
F508del c.1521_1523delCTT Div
1677delTA c.1545_1546delTA Div
V520F c.1558G>T SNV
Q525X† c.1573C>T SNV
1717-8G>A c.1585-8G>A SNV
1717-1G>A c.1585-1G>A SNV
G542X c.1624G>T SNV
S549R (c.1645A>C) c.1645A>C SNV
S549R (c.1647T>G) c.1647T>G SNV
S549N c.1646G>A SNV
G551D c.1652G>A SNV
Q552X c.1654C>T SNV
R553X c.1657C>T SNV
A559T c.1675G>A SNV
MiSeqDx cystic fibrosis 139-Variant Assay reported variants (Bold = ACMG23; Italics = conditionally reported).†Classified in the CFTR2 database as a CF-causing variant (as of September 2013); however, Sosnay et al. classified the variant as indeterminate. The CFTR2 databaseclassification is more current and reflects the completed functional testing for this variant, which was not available at the time of the Sosnay et al. publication.Del: Large deletion variant; Div: Insertion/deletion variant; HGVS: Human genome variation society; SNV: Single nucleotide variant.
Original Research Grosu, Hague, Chelliserry et al.
608 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.
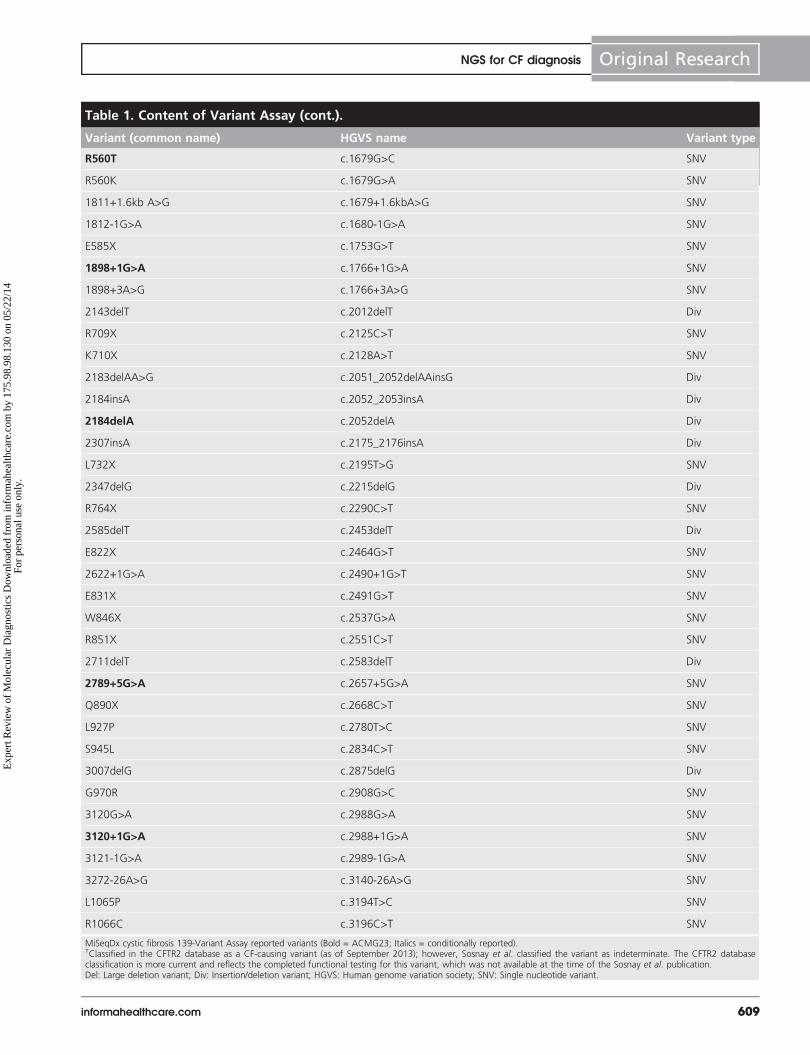
Table 1. Content of Variant Assay (cont.).
Variant (common name) HGVS name Variant type
R560T c.1679G>C SNV
R560K c.1679G>A SNV
1811+1.6kb A>G c.1679+1.6kbA>G SNV
1812-1G>A c.1680-1G>A SNV
E585X c.1753G>T SNV
1898+1G>A c.1766+1G>A SNV
1898+3A>G c.1766+3A>G SNV
2143delT c.2012delT Div
R709X c.2125C>T SNV
K710X c.2128A>T SNV
2183delAA>G c.2051_2052delAAinsG Div
2184insA c.2052_2053insA Div
2184delA c.2052delA Div
2307insA c.2175_2176insA Div
L732X c.2195T>G SNV
2347delG c.2215delG Div
R764X c.2290C>T SNV
2585delT c.2453delT Div
E822X c.2464G>T SNV
2622+1G>A c.2490+1G>T SNV
E831X c.2491G>T SNV
W846X c.2537G>A SNV
R851X c.2551C>T SNV
2711delT c.2583delT Div
2789+5G>A c.2657+5G>A SNV
Q890X c.2668C>T SNV
L927P c.2780T>C SNV
S945L c.2834C>T SNV
3007delG c.2875delG Div
G970R c.2908G>C SNV
3120G>A c.2988G>A SNV
3120+1G>A c.2988+1G>A SNV
3121-1G>A c.2989-1G>A SNV
3272-26A>G c.3140-26A>G SNV
L1065P c.3194T>C SNV
R1066C c.3196C>T SNV
MiSeqDx cystic fibrosis 139-Variant Assay reported variants (Bold = ACMG23; Italics = conditionally reported).†Classified in the CFTR2 database as a CF-causing variant (as of September 2013); however, Sosnay et al. classified the variant as indeterminate. The CFTR2 databaseclassification is more current and reflects the completed functional testing for this variant, which was not available at the time of the Sosnay et al. publication.Del: Large deletion variant; Div: Insertion/deletion variant; HGVS: Human genome variation society; SNV: Single nucleotide variant.
NGS for CF diagnosis Original Research
informahealthcare.com 609
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

of oligonucleotides is designed for each amplicon. The oligonu-cleotides were hybridized to unfragmented gDNA and appliedto a filter plate to wash away the unhybridized oligonucleotides.In the next step, a DNA polymerase extended from theupstream oligonucleotide through the targeted region, and wasligated to the 5´ end of a downstream oligonucleotide through
use of a DNA ligase. The extension-ligation products wereeluted off the filter plate and amplified using primers that addindex sequences for sample multiplexing as well as the commonadapters required for generation of clusters on the flow cell.The PCR products were purified using AMPureXP beads andnormalized using a bead-based method. Equal volumes of 8–
Table 1. Content of Variant Assay (cont.).
Variant (common name) HGVS name Variant type
R1066H c.3197G>A SNV
L1077P c.3230T>C SNV
W1089X c.3266G>A SNV
Y1092X(C>A) c.3276C>A SNV
Y1092X(C>G) c.3276C>G SNV
M1101K c.3302T>A SNV
E1104X c.3310G>T SNV
R1158X c.3472C>T SNV
R1162X c.3484C>T SNV
3659delC c.3528delC Div
S1196X c.3587C>G SNV
W1204X (c.3611G>A) c.3611G>A SNV
W1204X (c.3612G>A) c.3612G>A SNV
3791delC c.3659delC Div
3849+10kbC>T c.3717+12191C>T SNV
G1244E c.3731G>A SNV
3876delA c.3744delA Div
S1251N c.3752G>A SNV
3905insT c.3773_3774insT Div
W1282X c.3846G>A SNV
4005+1G>A c.3873+1G>A SNV
N1303K c.3909C>G SNV
4016insT c.3884_3885insT Div
Q1313X c.3937C>T SNV
4209TGTT>AA c.4077_4080delTGTTinsAA Div
CFTRdele22,23 c.3964-78_4242+577del Del
4382delA c.4251delA Div
I506V c.1516A>G SNV
I507V c.1519A>G SNV
F508C c.1523T>G SNV
PolyTG/PolyT N/A N/A
MiSeqDx cystic fibrosis 139-Variant Assay reported variants (Bold = ACMG23; Italics = conditionally reported).†Classified in the CFTR2 database as a CF-causing variant (as of September 2013); however, Sosnay et al. classified the variant as indeterminate. The CFTR2 databaseclassification is more current and reflects the completed functional testing for this variant, which was not available at the time of the Sosnay et al. publication.Del: Large deletion variant; Div: Insertion/deletion variant; HGVS: Human genome variation society; SNV: Single nucleotide variant.
Original Research Grosu, Hague, Chelliserry et al.
610 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

48 normalized libraries were combinedand diluted in hybridization buffer andloaded onto the MiSeqDx instrument.The Illumina MiSeqDx CF Systemrequires 2 � 150 bp paired sequencing tosequence the 175–225 bp amplicons inthe panel and two rounds of eight-cyclesequencing to determine the sequences ofboth indexes in each cluster.
The impacts of DNA concentrationand DNA quality were assessed for boththe Illumina MiSeqDx CF Variant Assayand Sequencing Assay. DNA input rangefrom 25 to 1250 ng produced accurateresults. For each sequencing run, 5 ml ofgDNA normalized to 50 ng/ml (total of250 ng) is recommended. It was deter-mined that DNA should be quantitatedusing a spectrophotometer and have averified A260/A280 of >1.5.
The integrated primary analysis soft-ware, real time analysis, performed imageanalysis and base calling, and assigned aquality score to each base for each cycle asthe run progressed. Secondary analysis wasperformed by MiSeq Reporter softwareand consisted of de-multiplexing individualreads to the correct sample from whichthey originated, alignment of sequences tothe reference sequence (Human GenomeReference build 19) of the targeted regions,variant calling and report generation.Alignment was performed using a bandedSmith-Waterman algorithm, and variantcalling using Starling (version 0.4.9). Atypical default value for band size duringalignment is 10; however, for the current assay, the band size wasset to 25, thus allowing indels of up to 25 bp in length to bereported. In order for a base position (variant or reference) to bereported to the user, a quantitative threshold for quality musthave been met. The results report was a text file that listed allmutations identified in the patient samples in a run, as well as anyregions that could not be called due to lack of high-quality datafor that region.
Samples for the clinical investigational studies
Accuracy study
The sample pool consisted of 355 de-identified, residual clinicalgDNA samples, and 11 gDNA samples from cell lines, for a totalof 366 unique gDNA samples. Candidate residual clinical gDNAsamples were identified from three collection sites (Progenity,Ann Arbor, MI, USA; LabCorp, Durham, NC, USA and MayoClinic, Rochester, MN, USA) and collected according to thestandard sample collection procedures at each site. Samples weresent to one central CLIA-certified laboratory (ResearchDx,
Irvine, CA, USA) for verification of eligibility (samples wererequired to have a minimum of 4 mg DNA and an A260/A280 ratio of at least 1.5). gDNA samples from cell lines werecreated according to standard procedures at ResearchDx usingcell lines (Coriell Institute for Medical Research, Camden, NJ,USA) or were directly obtained as gDNA from Coriell.
Reproducibility study
The sample pool consisted of 68 cell line-derived gDNA samplesand 8 mock blood samples (cell lines spiked into leukocyte-depleted whole blood), for a total of 76 unique samples. Sampleswere allocated into two sample panels, A and B, of 46 sampleseach. Panel A consisted of 8 mock blood samples and 38 gDNAsamples (of which 8 were present in duplicate). Panel B consistedof 46 gDNA samples (of which 8 were present in duplicate).
CFTR variants present in tested samples
For both studies, the samples were from known CF carriers orfrom patients diagnosed with CF. Samples were selected to
A
B
D
C
Index 2
Index 2
Index 1 P5
Index 1P7
P7
Customprobe 2
Customprobe 1
P5
Customprobe 2Region of interest
Customprobe 1
Figure 1. Overview of library preparation procedure for the MiSeqDx cysticfibrosis Assay. (A) Hybridization – the first step, hybridizes a pool of upstream anddownstream oligonucleotides specific to the MiSeqDx Cystic Fibrosis Clinical SequencingAssay to the sample genomic DNA. At the end of this process, a three-step wash proce-dure with a filter capable of size selection removes unbound oligonucleotides from thegenomic DNA. (B) Extension-ligation – the second step, connects the hybridized upstreamand downstream oligonucleotides. A DNA polymerase extends from the upstream oligonu-cleotides through the targeted region, followed by ligation to the 5´ end of the down-stream oligonucleotide using a DNA ligase. The result is the formation of products thatcontain the cystic fibrosis specific oligonucleotides flanked by sequences required for ampli-fication. (C) PCR amplification – the third step, amplifies the extension-ligation productsusing primers that add index sequences for sample multiplexing, as well as common adapt-ers required for cluster generation on the MiSeqDx. At the end of this process, a PCRclean-up procedure purifies the PCR products (referred to as a library). (D) Library normali-zation – the final step, normalizes the quantity of each library to ensure more equal libraryrepresentation in the final pooled library. At the end of this process, the pooled library isloaded onto the MiSeqDx for sequencing using sequencing-by-synthesis chemistry.
NGS for CF diagnosis Original Research
informahealthcare.com 611
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

include the following types of CFTR variants: SNVs, smallindels, PolyTG/PolyT variants, compound heterozygous variantsand large deletions.
Synthetic plasmid samples for rare variants
Due to the extreme rarity of some variants analyzed by the Vari-ant Assay, 52 synthetic plasmid samples were used to supplementthe clinical samples. The synthetic plasmids were designed toinclude the genomic context of the rare variants, and containedanywhere from 1 to 10 variants within the same construct. Plas-mids were linearized, diluted to gDNA equivalent copy numbersand blended with human gDNA samples of wild-type (WT)genotype at equivalent copy numbers to mimic a heterozygoussample. For 18 variants, performance was investigated using bothgDNA and synthetic DNA samples; in all instances, the resultsfor both sample types were found to be 100% concordant withbi-directional Sanger and with each other, supporting the equiva-lence of the two sample types for validation testing purposes.
Clinical investigational studies testing design
Accuracy study
Aliquots from each of the 366 gDNA samples used in theAccuracy Study were sent to PreventionGenetics (Marshfield,WI, USA) for bi-directional Sanger reference testing; PCR ref-erence testing and investigational testing by the MiSeqDx CFSystem were performed at Illumina.
Reproducibility study
Forty-eight samples were multiplexed per assay run (46 panelsamples plus 1 positive and 1 negative control). Each of thetwo sample panels was tested three-times by each of two opera-tors at each of the three clinical sites (Progenity, Johns HopkinsSchool of Medicine and Medical University of South Carolina),with one run per day over six non-consecutive days, for a totalof 276 (92 � 3) sample results per operator over the course ofthe study. Each panel was tested 18-times (3 sites � 2 opera-tors/site � 3 runs/operator). The CFTR variant profiles ofpanel samples were masked to the operators.
Reference method testing
For both the Accuracy and Reproducibility Studies, investiga-tional test results were compared with results from bi-directional Sanger and PCR assays to assess performance forboth the sequencing and the variant assays. Bi-directionalSanger was used to assess the identity of all the targeted basesin the sample pool. PCR-based assays were used to detect twolarge deletions that cannot be effectively detected with bi-directional Sanger. Investigational testing personnel weremasked to the bi-directional Sanger and PCR assay results.
Data monitoring & management
ResearchDx and Illumina completed monitoring of the testinglaboratories to ensure that all protocol requirements, US FDAregulations and investigator obligations were fulfilled. The MiS-eqDx CF System electronic reports, PCR assay result tables and
bi-directional Sanger result tables were uploaded via electronicdata transfer to a secure site via secure file transfer protocol andsubsequently imported into the clinical database by Myraqa,Inc. (Redwood Shores, CA, USA). All data issues were resolvedprior to the generation of complete datasets for analysis.
Analysis of performance
All analyses were performed independently by Myraqa, Inc.The following performance metrics were evaluated for bothstudies: sample pass rate, positive agreement (PA), negativeagreement (NA) and overall agreement (OA). The sample passrate was defined as the percentage of samples passing samplequality control metrics on the first attempt, out of the totalnumber of samples tested. PA was calculated for each variantgenotype by dividing the number of samples with agreeing var-iant calls by the total number of samples with that variant (asidentified by the reference methods). NA was calculated acrossall WT positions by dividing the number of WT positions con-cordant between the MiSeqDx CF System and the referencemethods by the total number of WT positions (as identified bythe reference methods). OA was calculated across all reportedpositions by dividing the number of positions (both variantand WT) concordant between the MiSeqDx CF System andthe reference methods by the total number of reported posi-tions (as identified by the reference methods).
ResultsClinical investigational study for accuracy
The sample pass rate in the Accuracy Study was 100% forboth assays. For the Sequencing Assay, a total of1,905,396 bases (5206 bases/sample � 366 samples) were com-pared with the reference methods; for the Variant Assay, a totalof 49,427 loci ([135 variants � 366 samples] + 17 instances ofthe listed conditionally-reported loci) were compared with thereference methods. During data analysis, it was determined thattwo pairs of samples had been switched during bi-directionalSanger reference testing. After confirmation of this switch andcorrection for data analysis, there were a total of 13 discordantcalls between the MiSeqDx CF Sequencing Assay and the refer-ence methods (all discrepancies were relative to bi-directionalSanger, as concordance with PCR was 100%) and two addi-tional calls where the MiSeqDx reported ‘No Call’ for the pol-yTG variant. A ‘No Call’ was reported by the MiSeqDx incases where the genotype quality score was insufficient to makea confident call [34]. Only 1 of the 13 discordant calls wasamong the genomic positions reported by the Variant Assay.Of the 13 discordant calls, 11 were reported within the pol-yTG/polyT region, a region often subject to manual interpreta-tion of Sanger traces and therefore associated with higher errorsin reporting. To address these polyTG/polyT discrepancies, the11 discrepant samples were combined with 15 concordant sam-ples and provided to the original Sanger investigator for ablinded re-interpretation, as a result of which, 9 of 11 discor-dant samples became concordant, and 1 of 15 concordant sam-ples became discordant. The results after re-interpretation were
Original Research Grosu, Hague, Chelliserry et al.
612 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

used for final data analysis. The forward Sanger traces andread-level view of the three outstanding discrepancies are pre-sented below (FIGURES 2–4). The reverse Sanger traces were concor-dant with the forward Sanger traces and were not includedfor (FIGURES 2–4). Although determination of polyTG/polyTregion in the clinical testing environment includes assays suchas melting curve analysis [35] or oligonucleotide ligation assay,the clinical investigational study design directly comparesNGS with bi-directional Sanger sequencing. Furthermore, theNGS data demonstrate the ability of NGS to accuratelydetermine TG/T status due to sequencing of multiple indi-vidual amplicons.
The two outstanding discordant calls that were not in the pol-yTG/polyT region were reviewed, but not sent out for re-inter-pretation. In the first case, the MiSeqDx CF System (both assays)identified a sample as homozygous for variant p.P205S, while theSanger interpretation reported the sample as heterozygous forthat variant. The forward Sanger trace (FIGURE 5B) clearly shows thevariant as being homozygous. The final analysis did not considerthis case discordant between the MiSeqDx and the referencemethods. In the second case, the MiSeqDx CF Sequencing Assayidentified a SNV, p.Q1463Q, which was not reported by Sanger(FIGURE 6). Though Sanger did not report this variant, neither couldit confidently report that position as WT. Indels are known toaffect the phase of Sanger traces, meaning that downstream callsare ambiguous. In this instance, because there were indels bothupstream and downstream of this position in question, bothdirections of Sanger reads were ambiguous due to phase. Thefinal analysis did consider this case discordant between the MiS-eqDx and the reference methods.
Based on the final analysis, the study achieved a PAof 99.7 and 100.0% for the Sequencing and VariantAssays, respectively, with NA and OA of >99.99% for bothassays TABLE 2; SUPPLEMENTARY TABLES 1 & 2 (supplementary materialcan be found online at www.informahealthcare.com/suppl/14737159.2014.916618_suppl.doc).
Comprehensive testing of all variants in the VariantAssayCombining clinical and cell line-derived samples from threestudies (Accuracy, Reproducibility and a supporting Extrac-tion study) with 52 synthetic plasmid samples allowed forthe testing of each variant reported by the Variant Assay atleast once. Comparison to the reference methods showed PAof 100% for each of the 135 loci in the Variant Assay results.For 18 variants where performance was investigated usingboth gDNA and synthetic DNA samples, the results for bothsample types were found to be 100% concordant withbi-directional Sanger and with each other in all instances,supporting the equivalence of the two sample types for vali-dation testing purposes.
Clinical investigational study for reproducibility
Across all samples and operators, the sample pass rate wasconsistently high: 99.7% for the Sequencing Assay and
99.9% for the Variant Assay. Five samples failed (due to notpassing quality control metrics as described in the section‘Methods’) the Sequencing Assay, of which two also failedthe Variant Assay. The five failed samples were five differentsamples over four operators. For the Sequencing Assay, atotal of 8,621,136 bases (5206 bases/sample � 92 samples/run � 18 runs) were compared with the reference methods;for the Variant Assay, across all operators and sites a total of223,668 loci were compared with the reference methods([135 loci/sample � 92 samples/run + 6 instances of thelisted conditionally-reported variants] � 18 runs). Duringdata review, it was determined that two samples had beenswitched during MiSeqDx testing; however, the originaluncorrected data were used in the final analysis.
After excluding the failed samples, there were a total of25 discordant calls between the MiSeqDx CF SequencingAssay and the reference methods, and an additional 13 calls,where the MiSeqDx reported ‘No Call’. Only 6 of the25 discordant calls were among the genomic loci reportedby the Variant Assay. Of the 25 discordant calls reportedbetween the Sequencing Assay and the reference methods,4 were due to the switching of two samples during Sangertesting, as described above. Of the remaining 21 discordantcalls where the MiSeqDx and reference methods eachreported a result, 18 were discordant calls of the polyTG/polyT region from one single sample. For this sample, all18 MiSeqDx runs for all operators across all sites were con-cordant with each other, but discrepant with the reportedbi-directional Sanger result. The consistency of the MiS-eqDx CF System results is strong evidence that the reportedbi-directional Sanger result was likely incorrect in this case.The remaining three discordant calls were all single cases,where 1 of the 18 MiSeqDx runs for each was discordantwith bi-directional Sanger and with the other 17 MiSeqDxruns. In the first case, the MiSeqDx identified the correctvariant (c.1408G>A) in the read-level data, but incorrectlyreported it as homozygous as opposed to heterozygous; thereported variant frequency for this run was 0.789 (the vari-ant frequency for the other runs ranged from 0.431 to0.619), making the differentiation of zygosity difficult. Inthe second case, the MiSeqDx incorrectly reported the pol-yTG variant as homozygous for (TG)10(T)7 as opposed to(TG)10(T)7/(TG)11(T)7; the variant score for this call waslower than all other replicates (51 as opposed to at least 111in the other 17 runs), indicating lower confidence in thecall, even though it passed calling thresholds. In the thirdcase, the MiSeqDx incorrectly reported a sample heterozy-gous for c.3870A>G as WT at this position; the source ofthis discrepancy was unclear, as the read-level data for thediscrepant run did not show any evidence of a heterozygousevent at this location.
Based on the final data, across all operators, sites and runs,the PA of the Sequencing Assay relative to the reference meth-ods was 99.2%. NA and OA were both 99.7%, driven by thefive sample failures. The PA of the Variant Assay relative to the
NGS for CF diagnosis Original Research
informahealthcare.com 613
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

reference methods was 99.8%. NA and OA were both 99.9%(TABLE 3; SUPPLEMENTARY TABLES 3 & 4).
Depth of coverage
With regard to sequencing depth, there was no particular cutoffthat was applied. The end-user will not be expected to usesequencing depth in making clinical decisions. The number of
samples that can be multiplexed in a given run was limited,thus ensuring that read depths did not drop due to increasedsample count. The average sequencing depth for the sequencingassay was on the order of 10,000�, and the average sequencingdepth for the Variant Assay was on the order of 1000�. Ateach locus, a consensus call was reported based on probabilitymodel that effectively considered three options: wild-type,
A
B
Figure 2. Comparison of MiSeqDx reads (A) and forward Sanger trace (B) for first PolyTG/PolyT discrepancy. The MiSeqDxreported this variant as (TG)10(T)7/(TG)12(T)5, while Sanger reported the variant as (TG)11(T)5/(TG)11(T)7. Reverse Sanger trace frombi-directional Sanger sequencing with concordant complementary data is not shown.
Original Research Grosu, Hague, Chelliserry et al.
614 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

heterozygous variant, homozygous variant. Each consensus call,regardless of whether it was variant or wild-type, was reportedwith a corresponding quality score that took into account thedistribution of base calls, base call quality, read depth, and so
on. The resultant quality score must then have passed specificthresholds in order for the consensus call to be reported. Thus,high read depth of poor quality bases would be filtered as effec-tively as overall low read depth.
A
B
Figure 3. Comparison of MiSeqDx reads (A) and forward Sanger trace (B) for second PolyTG/PolyT discrepancy. The MiSeqDxreported this variant as (TG)10(T)9/(TG)12(T)7, while Sanger reported the variant as (TG)11(T)9/(TG)12(T)7. Reverse Sanger trace frombi-directional Sanger sequencing with concordant complementary data is not shown.
NGS for CF diagnosis Original Research
informahealthcare.com 615
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

DiscussionGenetic conditions that have been challenging to diagnose dueto rare variants, or conditions that have substantial genetic het-erogeneity, have stimulated adoption of NGS in different clini-cal settings [36,37]. In CF, NGS has been utilized foridentification of rare variants and for achieving high detectionrates [38]. Trujillano et al. recently evaluated the use of a
pooled, targeted enrichment approach combined with multi-plexed sequencing-by-synthesis NGS in the molecular analysisof CFTR. The authors reported a mutation detection rate of100% and diagnostic rate of 98.9%, and demonstrated theability of NGS to detect large deletions, duplications andinversions [38]. In the Accuracy Study presented here, theMiSeqDx CF System demonstrated comparable performance,
A
B
Figure 4. Comparison of MiSeqDx reads (A) and forward Sanger trace (B) for third PolyTG/PolyT discrepancy. The MiSeqDxreported this variant as (TG)10(T)9/(TG)12(T)7, while Sanger reported the variant as (TG)11(T)9/(TG)12(T)7. Reverse Sanger trace frombi-directional Sanger sequencing with concordant complementary data is not shown.
Original Research Grosu, Hague, Chelliserry et al.
616 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

with OA ‡99.99% compared with traditional referencemethods. The high accuracy of NGS offers the promise ofdetecting even vanishingly rare variants for a large number ofgenetic diseases.
Although Sanger is well-established in clinical use, it is vul-nerable to certain analytical errors. Harismendy et al. comparedbase calls generated by three early NGS platforms with those
generated by Sanger and estimated the false-positive and false-negative rates to be 0.9 and 3.1% for NGS and Sanger, respec-tively [26]. Knowles et al. and Lemke et al. have also demon-strated the ability of NGS to identify disease-causing variantsthat were apparently missed by Sanger or other screening tech-niques [39,40]. In both the Accuracy and the ReproducibilityStudies, there were discordant results in the polyTG/polyT
A
B
Figure 5. Comparison of MiSeqDx reads (A) and forward Sanger trace (B) for zygosity discrepancy in reporting of P205Svariant. Both the Sanger trace and MiSeqDx reads unambiguously support a homozygous call for this variant; however, the Sanger resultwas initially reported as heterozygous. Reverse Sanger trace from bi-directional Sanger sequencing with concordant complementary datais not shown.
NGS for CF diagnosis Original Research
informahealthcare.com 617
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

region that were later confirmed to be the result of misinterpre-tation of Sanger traces.
The MiSeqDx Clinical Sequencing Assay and Variant Assayboth utilize a targeted approach for variant detection. TheSequencing Assay interrogates 5206 unique genomic positionsin CFTR to enable detection of all SNVs, and small indels
within the protein coding regions and intron/exon boundaries,as well as two selected deep intronic variants and two largedeletions. The Variant Assay was designed to interrogate 135variants that are directly reported and 4 variants that are condi-tionally reported. Therefore, the clinical CF NGS assays arenot intended to detect copy number variants or structural
A
B
Figure 6. Comparison of MiSeqDx reads (A) and forward Sanger trace (B) for discrepancy in reporting of Q1463Q variant.The MiSeqDx reads unambiguously support a heterozygous call for this variant; however, the Sanger traces in both directions are ambig-uous due to upstream and downstream indels in this sample. Reverse Sanger trace from bi-directional Sanger sequencing with similarambiguous sequence data is not shown.
Original Research Grosu, Hague, Chelliserry et al.
618 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

variants such as duplications, inversions or translocations, nor atotal deletion of CFTR in the heterozygous state, and are limi-tations of the assays.
The current ACMG-recommended CF carrier-screeningpanel [6], while pan-ethnic, is primarily based on mutation fre-quency in the non-Hispanic Caucasian and Ashkenazi Jewishpopulations. However, admixture of races in the US populationis increasing and in many cases individuals are not fully awareof their specific ethnic background. Rohlfs et al. reviewed acohort of over 350,000 carrier screens using a 98-variant paneland found that there was a significant difference in CFTRmutation patterns among ethnic groups [41]. Because of theincreasing difficulty in assigning a single ethnicity to individu-als, expanded CF mutation panels with variants that are specificfor different ethnicities may provide increased detection ratesfor many individuals, as recommended by Grody et al. [42].Sosnay et al. recently estimated that testing for pathogenic var-iants at 127 CFTR variant loci, with variant frequencies‡0.01% in the global CFTR2 cohort of nearly 40,000 CFpatients, would account for 95.4% of CF-causing alleles in this
cohort, leaving only 0.21% of affected individuals without atleast one pathogenic variant identified [5]. The authors alsonoted that if carrier screening was carried out for the patho-genic variants present at these 127 variant loci, the sensitivityof detecting couples at risk of having a child with CF wouldincrease to approximately 91% (from 72% with the use of thecurrent ACMG panel of 23 variants), subject to variability inallele distribution and frequency based on geography and ances-try [5]. The Variant Assay includes all variants identified aspathogenic in the CFTR2 database as of September2013 (including all 127 variant loci cited in Sosnay) and isexpected to provide enhanced clinical utility by detecting ahigher proportion of carrier couples and a higher fraction ofpathogenic variants in CF patients, thus allowing for moreinformed reproductive and treatment decisions. The Sequenc-ing Assay, by interrogating with high accuracy the vast majorityof CFTR genomic positions likely to harbor pathogenic var-iants, provides additional clinical utility in cases where CF isstrongly suspected on clinical grounds but first-tier testingcould not identify a genetic basis for the phenotype.
Table 2. Accuracy summary table.
Metric Total results No calls Discordant Percent agreement
Sequencing Assay
Positive agreement† 1547 2 3 99.68
Negative agreement 1,903,849 0 1 >99.99
Overall agreement 1,905,396 2 4 >99.99
Variant Assay
Positive agreement 379 0 0 100.00
Negative agreement 49,048 0 0 100.00
Overall agreement 49,427 0 0 100.00
Agreement results for both assays based on the Accuracy Study.†Positive agreement excluding PolyTG/PolyT calls was 100%.
Table 3. Reproducibility summary table.
Metric Total results Agreeing calls Total number (all sites) Percentagreement
Per site All sites Site 1 Site 2 Site 3 No calls Discordant
Sequencing Assay
Positive agreement† 2580 7740 2562 2553 2565 37 23 99.22
Negative agreement 2,871,132 8,613,396 2,865,930 2,855,526 2,865,932 26,006 2 99.70
Overall agreement 2,873,712 8,621,136 2,868,492 2,858,079 2,868,497 26,043 25 99.70
Variant Assay
Positive agreement 738 2214 738 733 738 3 2 99.77
Negative agreement 73,818 221,454 73,817 73,548 73,817 270 2 99.88
Overall agreement 74,556 223,668 74,555 74,281 74,555 273 4 99.88
Agreement results for both assays across all three testing sites based on the Reproducibility Study.†Positive agreement excluding PolyTG/PolyT calls was 99.61%.
NGS for CF diagnosis Original Research
informahealthcare.com 619
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

ConclusionThe clinical investigational studies presented here demonstratethat the MiSeqDx CF System is able to identify a broad spec-trum of CFTR variants with high accuracy and reproducibility.The MiSeqDx CF Assays perform well even in regions that areknown to induce Sanger errors, such as the polyTG/polyT regionand genomic positions close to indels. NGS technologies havebeen previously critiqued for limitations such as selection bias,uneven capture efficiency and variable performance in difficult-to-sequence regions including repetitive and GC-rich segmentsof CFTR that are difficult to enrich. In this report, we documentrobust performance of two NGS CF assays, wherein variants areconsistently and accurately called, even in challenging regions.The ability of NGS to simultaneously interrogate multiple typesof CFTR pathogenic variants (including large deletions) may leadto the displacement of current multi-step sequential genetic test-ing paradigm for CF. As the cost of NGS continues to drop, andperformance is shown to be comparable or superior to traditionalmethods [43], the application of NGS to many different clinicalareas [44,45] is expected to become ubiquitous.
This is the first published report of a systematic evaluationof a NGS platform for broad clinical use as an IVD, includingaccuracy validation with multiple reference methods and repro-ducibility validation at multiple clinical sites. The IlluminaMiSeqDx CF System offers enhanced clinical utility for themanagement of CF due to the analytical characteristics of NGSand the inclusion of a comprehensive set of clinically and
functionally validated CFTR variants. Furthermore, the simulta-neous FDA marketing authorization of the MiSeqDx as thefirst non-disease-specific NGS IVD platform is expected toallow any lab to test any sequence for any purpose, and repre-sents a major step forward in the application of genomic infor-mation to improve patient care [45].
Acknowledgement
The authors would like to thank the investigators for their participation in
the clinical investigational study for reproducibility. They would like to
thank G Cutting for his expert guidance. They also acknowledge L Hanson,
N Marino and P Rupprecht of Mayo Clinic College of Medicine, as well as
H Walline and T Goodman of Progenity, Inc.
Financial & competing interests disclosure
DS Grosu, L Hague, M Chelliserry, KM Kruglyak, R Lenta, B Klotzle,
J San, WM Goldstein, S Moturi, P Devers, E Lin and FS Ong are
employees of Illumina, Inc. E Peters was a former employee of Illumina
Inc. and is now an employee of Genentech, Inc. J Woolworth, B Elashoff,
J Stoerker, DJ Wolff, KJ Friedman and WE Highsmith were in receipt of
project funding from Illumina, Inc. The authors have no other relevant
affiliations or financial involvement with any organization or entity with
a financial interest in or financial conflict with the subject matter or
materials discussed in the manuscript apart from those disclosed. This
includes employment, consultancies, honoraria, stock ownership or options,
expert testimony, grants or patents received or pending or royalties.
No writing assistance was utilized in the production of this manuscript.
Key issues
• This is the first published report of a systematic evaluation of a next-generation sequencing (NGS) platform for broad clinical use as an
in vitro diagnostic, including accuracy validation with multiple reference methods and reproducibility validation at multiple clinical sites.
The clinical investigational studies presented here demonstrate that the Illumina MiSeqDx Cystic Fibrosis (CF) System is able to identify a
broad spectrum of CF transmembrane (CFTR) variants with high accuracy and reproducibility.
• The Illumina MiSeqDx CF System offers enhanced clinical utility for the management of CF due to the analytical accuracy of NGS and
the inclusion of a comprehensive set of clinically and functionally validated CFTR variants. Furthermore, the simultaneous US FDA mar-
keting authorization of the MiSeqDx as the first non-disease-specific NGS in vitro diagnostic platform is expected to allow any lab to
test any sequence by NGS for any purpose, and represents a major step forward in the application of genomic information to improve
patient care.
• The 139-Variant Assay includes all variants identified as pathogenic in the CFTR2 database as of September 2013 and is expected to
provide enhanced clinical utility by detecting a higher proportion of CF carrier couples and a higher fraction of pathogenic variants in CF
patients, thus allowing for more informed and more timely reproductive and treatment decisions.
• The Sequencing Assay, by interrogating with high accuracy the vast majority of CFTR genomic positions likely to harbor pathogenic
variants, provides additional clinical utility in cases where CF is strongly suspected on clinical grounds but first-tier testing could not iden-
tify a genetic basis for the phenotype.
• The MiSeqDx CF Assays perform well even in regions that are known to induce Sanger errors, such as the polyTG/polyT region and
genomic positions close to indels.
• The ability of NGS to simultaneously interrogate multiple types of CFTR pathogenic variants (including large deletions) may lead to the
displacement of current multi-step sequential genetic testing paradigm for CF. As the cost of NGS continues to drop, and performance
is shown to be comparable or superior to traditional methods, the application of NGS to many different clinical areas is expected to
become ubiquitous.
Original Research Grosu, Hague, Chelliserry et al.
620 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

References
1. Kerem B, Rommens JM, Buchanan JA,
et al. Identification of the cystic fibrosis
gene: genetic analysis. Science 1989;
245(4922):1073-80
2. Riordan JR, Rommens JM, Kerem B, et al.
Identification of the cystic fibrosis gene:
cloning and characterization of
complementary DNA. Science 1989;
245(4922):1066-73
3. Rommens JM, Iannuzzi MC, Kerem B,
et al. Identification of the cystic fibrosis
gene: chromosome walking and jumping.
Science 1989;245(4922):1059-65
4. Cystic fibrosis mutation database. 2014.
Available from: www.genet.sickkids.on.ca
5. Sosnay PR, Siklosi KR, Van Goor F, et al.
Defining the disease liability of variants in
the cystic fibrosis transmembrane
conductance regulator gene. Nat Genet
2013;45:1160-7
6. Watson MS, Cutting GR, Desnick RJ, et al.
Cystic fibrosis population carrier screening:
2004 revision of American College of
Medical Genetics mutation panel. Genet
Med 2004;6(5):387-91
7. Grody WW, Cutting GR, Watson MS. The
Cystic fibrosis mutation “arms race”: whenless is more. Genet Med 2007;9(11):739-44
8. Dorfman R, Nalpathamkalam T, Taylor C,
et al. Do common in silico tools predict the
clinical consequences of amino-acid
substitutions in the CFTR gene? Clin Genet
2010;77(5):464-73
9. Chillon M, Casals T, Mercier B, et al.
Mutations in the cystic fibrosis gene in
patients with congenital absence of the vas
deferens. N Engl J Med 1995;332(22):
1475-80
10. Dequeker E, Stuhrmann M, Morris MA,
et al. Best practice guidelines for molecular
genetic diagnosis of cystic fibrosis and
CFTR-related disorders – updated European
recommendations. Eur J Hum Genet 2009;
17(1):51-65
11. Groman JD, Hefferon TW, Casals T, et al.
Variation in a repeat sequence determines
whether a common variant of the cystic
fibrosis transmembrane conductance
regulator gene is pathogenic or benign. Am
J Hum Genet 2004;74(1):176-9
12. Masica DL, Sosnay PR, Cutting GR,
Karchin R. Phenotype-optimized sequence
ensembles substantially improve prediction
of disease-causing mutation in cystic
fibrosis. Hum Mutat 2012;33(8):1267-74
13. Grody WW, Cutting GR, Klinger KW,
et al. Laboratory standards and guidelines
for population-based cystic fibrosis carrier
screening. Genet Med 2001;3(2):149-54
14. Clinical and Functional Translation of
CFTR Project (CFTR2). 2014. Available
from: www.cftr2.org
15. Castellani C, team C. CFTR2: how will it
help care? Paediatr Respir Rev 2013;
14(Suppl 1):2-5
16. Farrell PM, Rosenstein BJ, White TB, et al.
Guidelines for diagnosis of cystic fibrosis in
newborns through older adults: cystic
Fibrosis Foundation consensus report.
J Pediatr 2008;153(2):S4-14
17. Massie RJ, Curnow L, Glazner J, et al.
Lessons learned from 20 years of newborn
screening for cystic fibrosis. Med J Aust
2012;196(1):67-70
18. Southern KW, Munck A, Pollitt R, et al.
A survey of newborn screening for cystic
fibrosis in Europe. J Cyst Fibros 2007;6(1):
57-65
19. Vernooij-van Langen AM, Loeber JG,
Elvers B, et al. Novel strategies in newborn
screening for cystic fibrosis: a prospective
controlled study. Thorax 2012;67(4):289-95
20. Wagener JS, Zemanick ET, Sontag MK.
Newborn screening for cystic fibrosis. Curr
Opin Pediatr 2012;24(3):329-35
21. Amos JGF, Wayne Grody,
Kristen Monaghan, et al. Technical
standards and guidelines for CFTR
mutation testing. American College of
Medical Genetics; 2002
22. Strom CM, Crossley B, Buller-Buerkle A,
et al. Cystic fibrosis testing 8 years on:
lessons learned from carrier screening and
sequencing analysis. Genet Med 2011;13(2):
166-72
23. Ramsey BW, Davies J, McElvaney NG,
et al. A CFTR potentiator in patients with
cystic fibrosis and the G551D mutation. N
Engl J Med 2011;365(18):1663-72
24. Thursfield RM, Davies JC. Cystic fibrosis:
therapies targeting specific gene defects.
Paediatr Respir Rev 2012;13(4):215-19
25. Ramos MD, Masvidal L, Gimenez J, et al.
CFTR rearrangements in Spanish cystic
fibrosis patients: first new duplication
(35kb) characterised in the Mediterranean
countries. Ann Hum Genet 2010;74(5):
463-9
26. Harismendy O, Ng PC, Strausberg RL,
et al. Evaluation of next generation
sequencing platforms for population
targeted sequencing studies. Genome Biol
2009;10(3):R32
27. Pont-Kingdon G, Gedge F,
Wooderchak-Donahue W, et al. Design and
analytical validation of clinical
DNA sequencing assays. Arch Pathol Lab
Med 2012;136(1):41-6
28. Diguistini S, Liao NY, Platt D, et al. De
novo genome sequence assembly of a
filamentous fungus using Sanger, 454 and
Illumina sequence data. Genome Biol 2009;
10(9):R94
29. Sikkema-Raddatz B, Johansson LF,
de Boer EN, et al. Targeted next-generation
sequencing can replace Sanger sequencing in
clinical diagnostics. Hum Mutat 2013;
34(7):1035-42
30. Bentley DR, Balasubramanian S,
Swerdlow HP, et al. Accurate whole human
genome sequencing using reversible
terminator chemistry. Nature 2008;
456(7218):53-9
31. Illumina. TruSeq� Custom Amplicon
Library Preparation Guide. 2012. Available
from: http://supportres.illumina.com/
documents/myillumina/b718c350-b3b2-
4234-b71a-0b832f14cda3/truseq_custom_
amplicon_libraryprep_ug_15027983_b.pdf
32. Mardis ER. Next-generation
DNA sequencing methods. Ann Rev
Genomics Hum Genet 2008;9:387-402
33. Metzker ML. Sequencing technologies - the
next generation. Nat Rev Genet. 2010;
11(1):31-46
34. Raczy C, Petrovski R, Saunders CT, et al.
Isaac: ultra-fast whole-genome secondary
analysis on Illumina sequencing platforms.
Bioinformatics 2013;29(16):2041-3
35. Costa C, Goossens M, Girodon E.
Simultaneous molecular haplotyping of both
IVS8 (TG)m and (T)n tracts in the CFTR
gene: still a challenge. Clin Chem 2006;
52(8):1621-2
36. Ng SB, Bigham AW, Buckingham KJ, et al.
Exome sequencing identifies
MLL2 mutations as a cause of Kabuki
syndrome. Nat Genet 2010;42(9):790-3
37. Saunders CJ, Miller NA, Soden SE, et al.
Rapid whole-genome sequencing for genetic
disease diagnosis in neonatal intensive care
units. Sci Transl Med 2012;4(154):
154ra135
38. Trujillano D, Ramos MD, Gonzalez J, et al.
Next generation diagnostics of cystic fibrosis
and CFTR-related disorders by targeted
multiplex high-coverage resequencing of
CFTR. J Med Genet 2013;50(7):455-62
39. Knowles MR, Leigh MW, Ostrowski LE,
et al. Exome sequencing identifies mutations
in CCDC114 as a cause of primary ciliary
dyskinesia. Am J Hum Genet 2013;92(1):
99-106
40. Lemke JR, Riesch E, Scheurenbrand T,
et al. Targeted next generation sequencing
NGS for CF diagnosis Original Research
informahealthcare.com 621
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.

as a diagnostic tool in epileptic disorders.
Epilepsia 2012;53(8):1387-98
41. Rohlfs EM, Zhou Z, Heim RA, et al.
Cystic fibrosis carrier testing in an ethnically
diverse US population. Clin Chem 2011;
57(6):841-8
42. Grody WW, Thompson BH, Gregg AR,
et al. ACMG position statement on
prenatal/preconception expanded
carrier screening. Genet Med 2013;15(6):
482-3
43. Rehm HL, Bale SJ, Bayrak-Toydemir P,
et al. ACMG clinical laboratory standards
for next-generation sequencing. Genet Med
2013;15(9):733-47
44. Ong FS, Lin JC, Das K, et al. Translational
utility of next-generation sequencing.
Genomics 2013;102(3):137-9
45. Collins FS, Hamburg MA. First
FDA authorization for next-generation
sequencer. N Engl J Med 2013;369(25):
2369-71
Original Research Grosu, Hague, Chelliserry et al.
622 Expert Rev. Mol. Diagn. 14(5), (2014)
Exp
ert R
evie
w o
f M
olec
ular
Dia
gnos
tics
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
175.
98.9
8.13
0 on
05/
22/1
4Fo
r pe
rson
al u
se o
nly.




