
Clinical Evaluation in the EU for Medical Devices: Understanding the Changes in MEDDEV 2.7.1 Rev 4...
48
Understanding the changes in MEDDEV 2.7.1 rev 4 and Their Impact Presented by: Keith Morel, PhD VP Regulatory Compliance
-
Upload
greenlightguru -
Category
Health & Medicine
-
view
1.582 -
download
0
Transcript of Clinical Evaluation in the EU for Medical Devices: Understanding the Changes in MEDDEV 2.7.1 Rev 4...

Understanding the changes in MEDDEV 2.7.1 rev 4 and Their Impact
Presented by: Keith Morel, PhDVP Regulatory Compliance

greenlight.guru
• Quality Management Software Exclusively for Medical Device Companies
• Single Source of Truth• Manage design controls, risk and
quality in a single, easy-‐to-‐use solution
• Customers & Partners All Over the Globe: • Represented on 5 continents
• www.greenlight.guru
Jed Johnson PhD, CTO
Nanofiber Solutions
“greenlight.guru has been instrumental for us moving so
quickly through the ISO certification and I would highly
recommend it.”

Qserve Group
• Largest European Medical device consulting firm
• A global reach: • Offices in Amsterdam, San
Francisco, Boston and Nanjing• One-‐stop-‐shopping • All medical device RA/QA/CA
needs• Pragmatic project approach
• www.qservegroup.com
• Strong International team• Technical, regulatory, quality and
clinical competences• Ex EU Notified Body, FDA and
CFDA staff• Dr Gert Bos is a partner (RAPS
board member; key NB rep. in MDR/IVDR negotiations)
• Medical Doctor in management • WOFE in China• Approvedas a CRO• Native speakers in 8 languages

Contents
• Structure of the new Document• The process of developing the new MEDDEV • Who, how, timing?
• New expectations around Clinical Data & Evaluation in the EU Regulations surrounding MEDDEV 2.7.1 rev 4 (i.e. the context)• New requirements in MEDDEV 2.7.1 rev 4 (i.e. the content)• Summary and Conclusions

Structure of the Document: rev 3 vs rev 4
Rev 3 Rev 4
46 pages 64 pages10 sections 12 sections
6 Appendices 12 Appendices

Structure of the Document: rev 3 vs rev 4

Structure of the Document: rev 3 vs rev 4
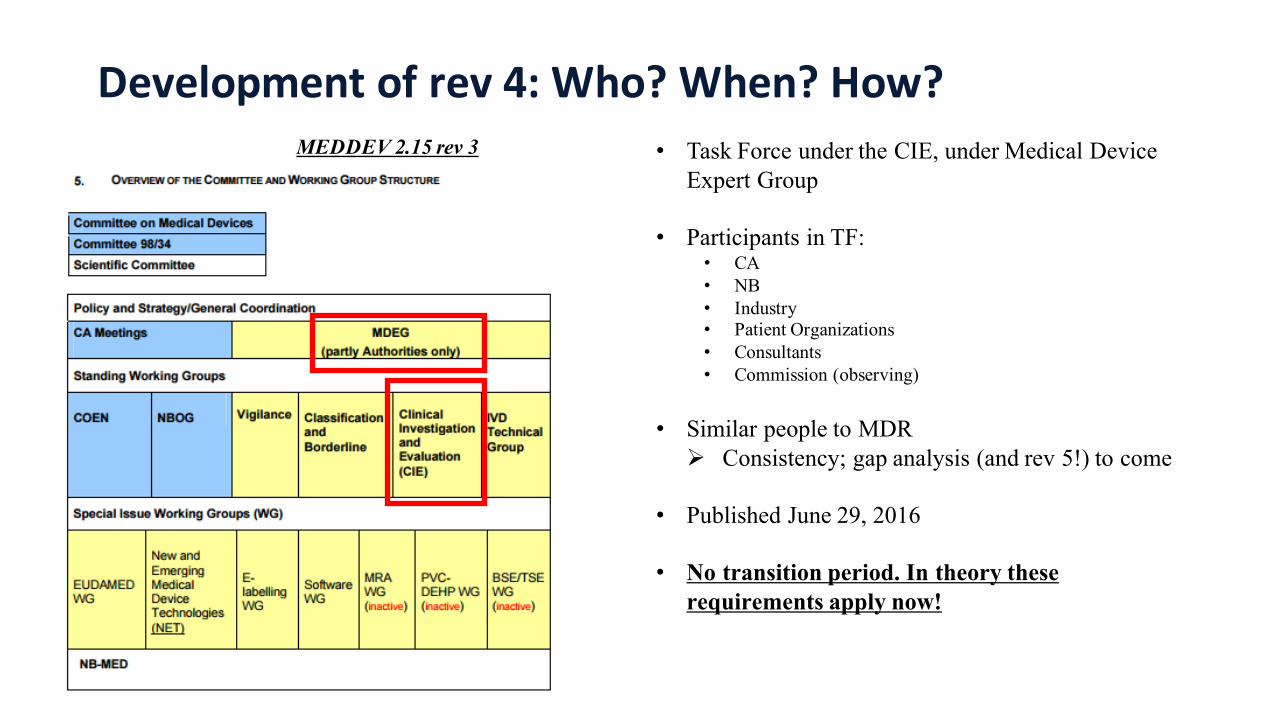
Development of rev 4: Who? When? How?MEDDEV 2.15 rev 3 • Task Force under the CIE, under Medical Device
Expert Group
• Participants in TF:• CA• NB• Industry• Patient Organizations• Consultants• Commission (observing)
• Similar people to MDRØ Consistency; gap analysis (and rev 5!) to come
• Published June 29, 2016
• No transition period. In theory these requirements apply now!

Increasing Requirements in Recent Years
Joint audits
UAV
CoC audits
COEN - MS
EUDAMED
Electronic submission
UDI
MDR / IVDR
EU oversight
Clinical trials
Scrutiny procedure
and MORE….
2010 2013 ~2020
MEDDEV 2.7.1 rev 4
Mid 2016

New expectations surrounding rev 4
1. Joint NB Audits• Involvement of clinical experts in technical file reviews to be improved• Appropriateness of Clinical expertise/competence of clinical expert for device under review• NB QMS procedures for ensuring appropriateness/validity of clinical experts opinion• Clinical competence of NB staff• Depth of review of clinical investigations

New expectations surrounding rev 4
1. Joint NB Audits (continued)• Processes for assessment CER needs improvement• Review of PMS and PMCF plans• Thoroughness of assessment of CER, e.g.
‒ all relevant clinical practice considered; ‒ all hazardous situations considered?; ‒ not following structure/guidance in MEDDEV 2.7.1; ‒ intended use supported by data?

New expectations surrounding rev 4
2. New Regulations for Medical Devices and IVDs in Europe• See recent recorded webinar by Dr Gert Bos• http://bit.ly/2a9naRI
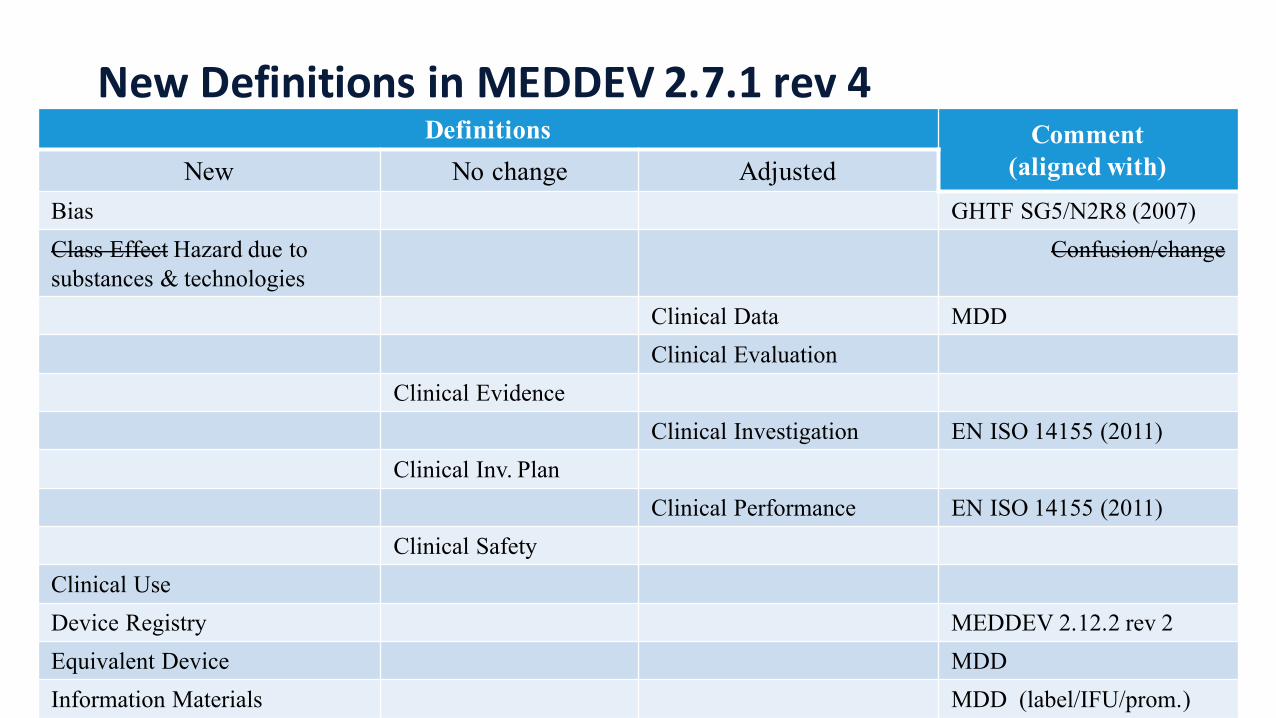
New Definitions in MEDDEV 2.7.1 rev 4Definitions Comment
(aligned with)New No change AdjustedBias GHTF SG5/N2R8 (2007)Class Effect Hazard due to substances & technologies
Confusion/change
Clinical Data MDDClinical Evaluation
Clinical EvidenceClinical Investigation EN ISO 14155 (2011)
Clinical Inv. PlanClinical Performance EN ISO 14155 (2011)
Clinical SafetyClinical UseDevice Registry MEDDEV 2.12.2 rev 2Equivalent Device MDDInformation Materials MDD (label/IFU/prom.)
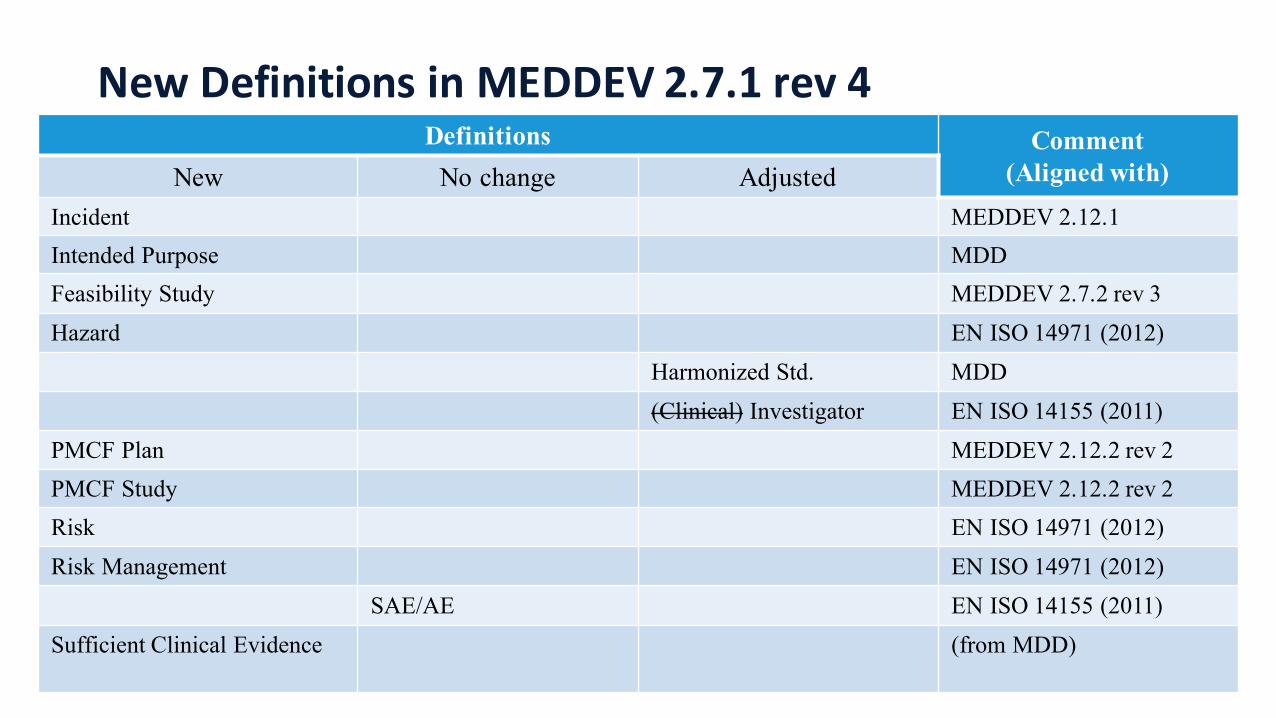
New Definitions in MEDDEV 2.7.1 rev 4Definitions Comment
(Aligned with)New No change AdjustedIncident MEDDEV 2.12.1Intended Purpose MDDFeasibility Study MEDDEV 2.7.2 rev 3Hazard EN ISO 14971 (2012)
Harmonized Std. MDD(Clinical) Investigator EN ISO 14155 (2011)
PMCF Plan MEDDEV 2.12.2 rev 2PMCF Study MEDDEV 2.12.2 rev 2Risk EN ISO 14971 (2012)Risk Management EN ISO 14971 (2012)
SAE/AE EN ISO 14155 (2011)Sufficient Clinical Evidence (from MDD)

Important New/Adjusted Definitions in rev 4
• PMCF Plan• the documented, proactive, organised methods and procedures set up by the manufacturer to collect clinical data based on the use of a CE-‐marked device corresponding to a particular design dossier or on the use of a group of medical devices belonging to the same subcategory or generic device group as defined in Directive 93/42/EEC. The objective is to confirm clinical performance and safety throughout the expected lifetime of the medical device, the acceptability of identified risks and to detect emerging risks on the basis of factual evidence

Important New/Adjusted Definitions in rev 4
• PMCF Study• A study carried out following the CE marking of a device and intended to answer specific questions relating to clinical safety or performance (i.e. residual risks) of a device when used in accordance with its approved labelling.
• Sufficient Clinical Evidence• clinical data of an amount and quality to guarantee the scientific validity of the conclusions (adapted from section 2.3.1 of Annex X of MDD)

Clinical Evaluation – what is it? (6.1)
1.Clinical evaluation is a methodologically sound ongoing procedure to collect and analyse clinical data pertaining to a medical device and to assess whether there is Sufficient Clinical Evidence to confirm compliance with relevant essential requirements for safety and performance when using the device according to the manufacturer’s Instructions for Use
2.Very clear in rev 4 CER needed for ALL classes of medical device (class I and up!)
3.Core issues are the proper determination of the benefit/risk profile in the intended target groups and medical indications, and demonstration of acceptability of that profile based on the state of the art in the medical fields concerned

Clinical Evaluation – focus for evaluators (6.1)# Issue Focus for evaluators1 Intended use - ER 1, 3 Is this supported by Sufficient Clinical Evidence (SCE)?2 Clinical Performance & Benefits - ER 1, 3, 6 Are these supported by Sufficient Clinical Evidence (SCE)?3 Risk avoidance/risk mitigations in
“Information Material” ER 1, 3, 6Are these supported by Sufficient Clinical Evidence (SCE)?
4 Usability for intended users (e.g. professionals, lay-persons, disabled persons) – ER 1
Is this supported by Sufficient Clinical Evidence (SCE)?
5 Suitability of “Information Material” for intended users – ER 1
Is this supported by Sufficient Clinical Evidence (SCE)?
6 Sufficient instructions for intended users (e.g. pregnant women, paediatrics) – ER 1
Consistent with the clinical data?
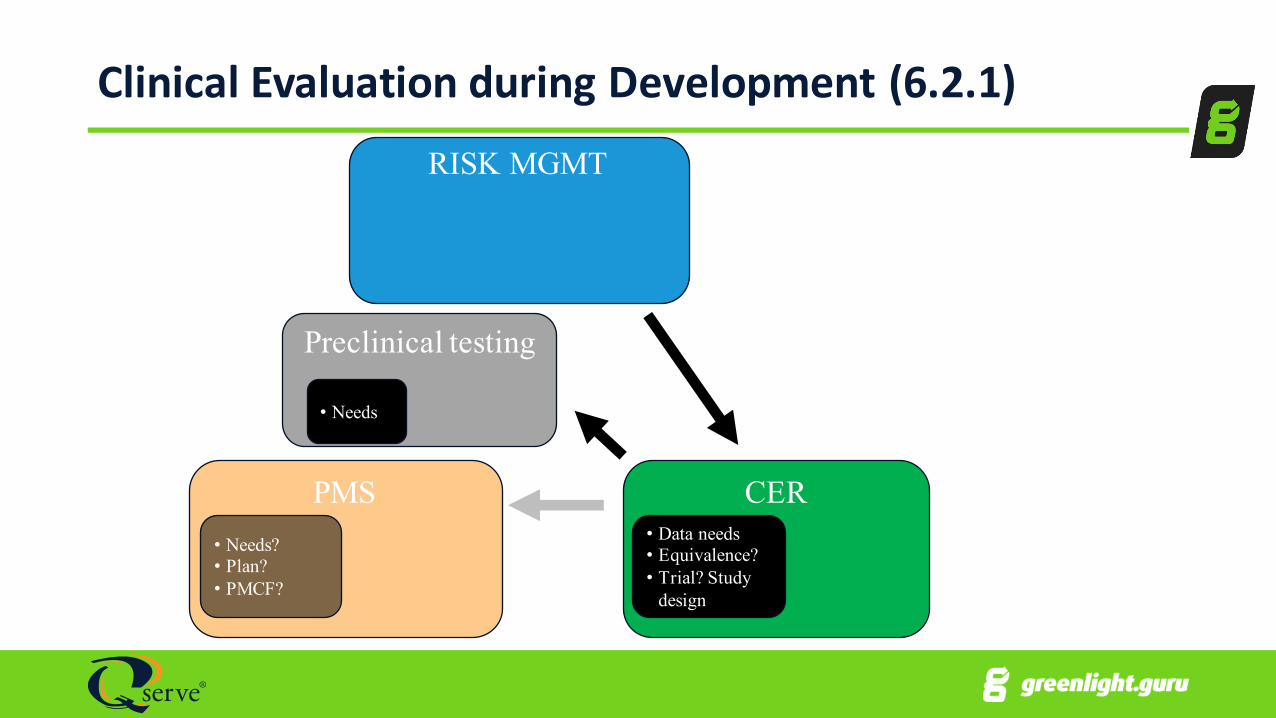
Clinical Evaluation during Development (6.2.1)RISK MGMT
PMS CER• Data needs• Equivalence?• Trial? Study
design
• Needs?• Plan?• PMCF?
Preclinical testing
• Needs

Clinical Evaluation for Initial CE Marking (6.2.2)RISK MGMT
CERPMS
INFORMATION MATERIALS
• Sufficient Clinical Evidence to meet ERs?• Significance of any clinical
risks after risk control
• Needs (subtle, long terms effects)• Plan• PMCF?
• IFU appropriate for intended users?
• All clinical risks in RMR?• Risks controls ID and verified?• Risk-Benefit justified?
Preclinical testing
• All risk controls in IFU? Validated?• All warnings etc in RMR?
• Usability demonstrated?
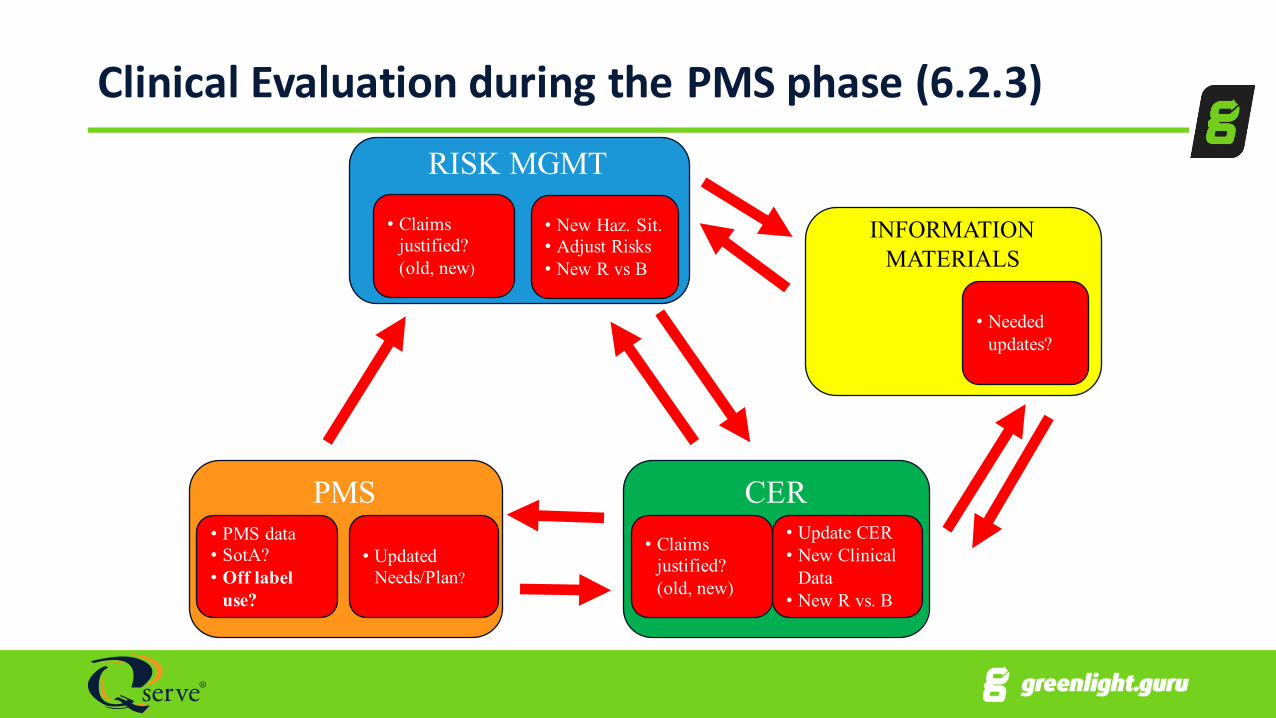
Clinical Evaluation during the PMS phase (6.2.3)
INFORMATION MATERIALS
• Needed updates?
PMS
• Updated Needs/Plan?
• PMS data• SotA?• Off label
use?
RISK MGMT
• New Haz. Sit.• Adjust Risks• New R vs B
• Claims justified? (old, new)
CER• Update CER• New Clinical
Data• New R vs. B
• Claims justified? (old, new)

Clinical Evaluation during the PMS phase (6.2.3)
1.Manufacturer must DEFINE and JUSTIFY frequency of updating the CER, based on:-‐i. Device has significant risks? (invasiveness, high risk patient population,
severity of disease … see MEDDEV 2.12.2 rev 2)ii. Device is well established? (innovation, design changes, number of devices
used, are there risks/uncertainties in long term & has this period been covered? …)
2.Update CER when:i. New PMS info with potential to change current evaluation ii. If no such information is received, at least:
a. AT LEAST Annually if “significant risk”/not well establishedb. Every 2-‐5 years if no “significant risk”/IS well established; MUST JUSTIFY(coordinate with NB for certificate renewals)

How is Clinical Evaluation Performed? (6.3)
Scope
Section 7App. A3
ID data
Section 8App. A4-A5
Appraisal
Section 9App. A6
Analysis
Section 10App A7-A8
Finalize CER
Section 11App. A9-A10

Who performs Clinical Evaluation? (6.4)
• Suitably qualified individual or a team• The manufacturer should take the following aspects into consideration:• requirements for evaluators in line with the nature of the device under evaluation and its clinical performance and risks.• The manufacturer should be able to justify the choice of the evaluators (qualifications; documented experience*), and to present a declaration of interest* (financial interests, conflicts of interest – see A11) for each.
‒ *Reviewed by NB. ‒ *“Independence” does not appear. ‒ * Not explicit how they are to assess these and what they are to do

Who performs Clinical Evaluation? (6.4)
• As a general principle, the evaluators should possess knowledge of the following:1. the device technology and its application;2. research methodology (including clinical investigation design and
biostatistics);3. diagnosis and management of the conditions intended to be managed or
diagnosed by the device, knowledge of alternative treatments, treatment standards and technology
4. information management (e.g. scientific background or librarianship qualification; experience with relevant databases)
5. regulatory requirements; and6. medical writing (e.g. post-‐graduate experience in a relevant science or in
medicine; training and experience in medical writing);

Who performs Clinical Evaluation? (6.4)
• The evaluators should have at least the following training and experience in the relevant field:1. a higher degree and 5 years of documented professional experience; or2. 10 years of documented professional experience if a higher degree is not a
prerequisite for a given task.
• There may be circumstances where the level of evaluator expertise may be less or different, this should be documented and justified

Scoping (7.0)
• Define scope, based on the ER which need to be addressed (e.g. 1, 3, 6 MDD)
• Nice table in rev 4 highlighting the focus for the CER in the pre-‐ and post-‐CE Marking situation (see previous slides)‒ Off-‐label use is included in the post-‐CE phase now (cf. rev 3)
• Annex 7 gives lots of addition guidance on how to analyse clinical data to show meet the ERs (7 pages!)
• Won’t address all of this, but a few key points …

Scoping (7.0/Annex 7) – Key Points
• ER 1, Safety• Risk Mgmt -‐> all hazards covered by harmonized stds?
‒ e.g. 60601-‐1-‐1 electrical hazards – hence don’t need clinical data‒ e.g. Usability – 62366 – doesn’t give design specifics, therefore may need clinical data to show risk of use errors, risks due to ergonomic issues are reduced AFAP
• ER 1, Acceptable R-‐B ratio• ER 1, Clinical risk • ER 3, Performance

Scoping (7.0/Annex 7) – Key Points
• ER 6, Undesirable side effects• Must be acceptable when weighed against the benefits of using the device• Need clinical data on the:
‒ Nature, severity & frequency of undesirable side effects‒ Must have sufficient sample size (N) to guarantee the scientific validity of the conclusion (OTHERWISE ER 6 NOT MET!)
‒ Consider the state of the art (alternative therapies, baseline devices ..)
• Make sure studies are big enough!(Binomial Distribution) 1 2 3 4 5 6
Chance of observing at least 1 event (%) 80 80 80 90 90 90Actual probability of SAE (%) 10 5 1 10 5 1Number of subjects needed 15 32 161 22 45 230

Identification of Data (8.0, 8.1)
• Various sources identified1.Data generated & held by Manufacturer
a. Clinical Investigation data (pre-‐market)b. Animal/Performance datac. PMS data
a. PMS/trend reportsb. Vigilance reportsc. Complaintsd. FSCAe. Compassionate use/Humanitarian Use

Identification of Data (8.2)
2.Data not held by the manufacturer – Data from Literature • Literature searching identifies potential sources of data for establishing:• Current knowledge/State of the Art
‒ Alternative therapies, benchmark devices, medical condition, patient population‒ Potential clinical hazards‒ Justify criteria for equivalence‒ Justify validity of any surrogate end-‐points
• Data for the device in question

Key points in rev 4 re Literature Searching (8.2)
1.Search strategy should be thorough & objectivea. PICOb. Cochrane Handbook for Systematic Reviewsc. PRIMSAd. MOOSE
2.Use Multiple search strategies to get all information
3.Thorough search of all journals • MEDLINE/PUBMED good starting point• May need to include EMBASE to ensure adequate coverage of EU journals
4.Need a protocol for literature review (as in rev 3)
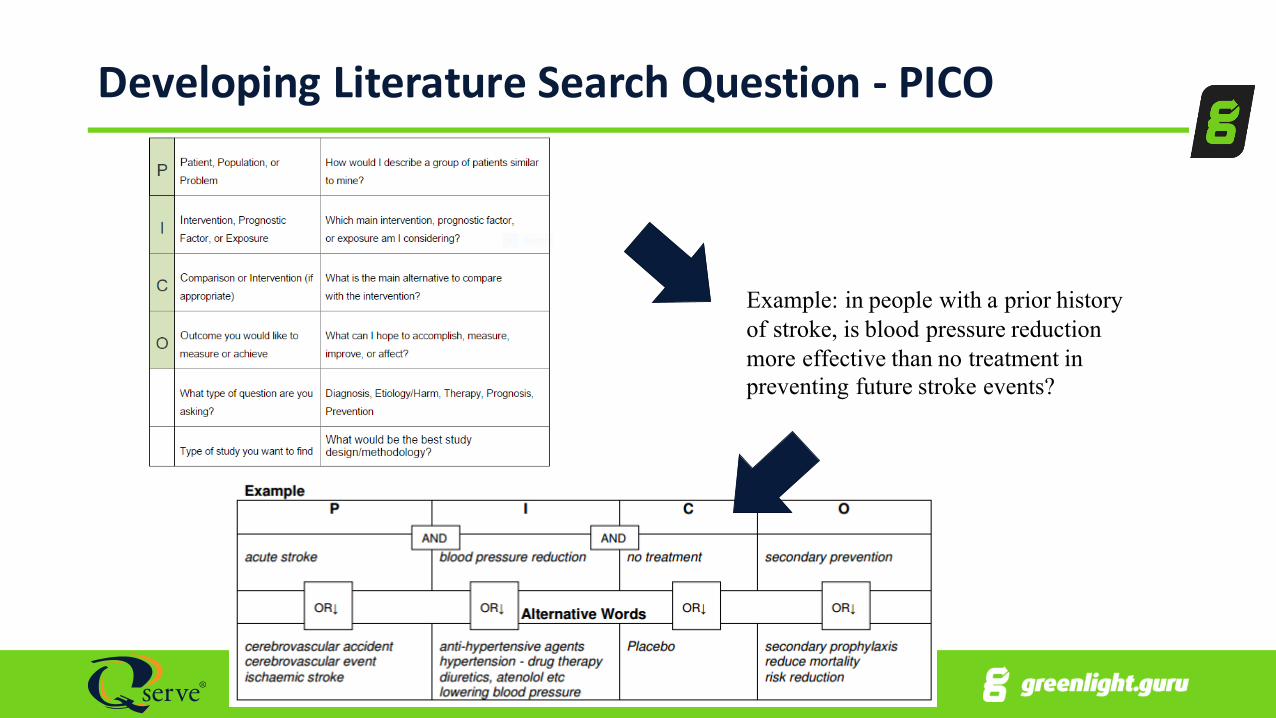
Developing Literature Search Question -‐ PICO
Example: in people with a prior history of stroke, is blood pressure reduction more effective than no treatment in preventing future stroke events?

Appraisal of Clinical Data (9)
• Purpose is to estimate the value of the contribution of each piece of clinical data to the assessment of clinical safety & performance• Uncertainty about the value of the data comes from:
1. Methodological quality of the data2. Relevance of the data to the intended use
• Many ways to do this (e.g. see next 3 slides)• Need an Appraisal Plan (part of CER) detailing:
1. Criteria for determining the quality and scientific validity of each piece of data2. Criteria for relevance to the evaluation3. Criteria for how to weight the contribution of each piece of data

Contribution of each data set -‐ Appraisal Criteria -‐ 1Data Suitability
• Appropriate device (D)• Appropriate device application (use) (A)• Appropriate patient group (P)• Acceptable report/data collection (R)
Data Contribution
• Data source type (T)• Outcome measures (O)• Follow-‐up (F)• Statistical significance (S)• Clinical significance (C)
Each “Letter” or “Category” receives a 1, 2, or 3
Then weight the data1 = actual device (or equivalent) as intended (on label), etc.2 = actual device (or equivalent) off label in same anatomy, etc.3 = competitor devices/state-of-art, different anatomy, etc.
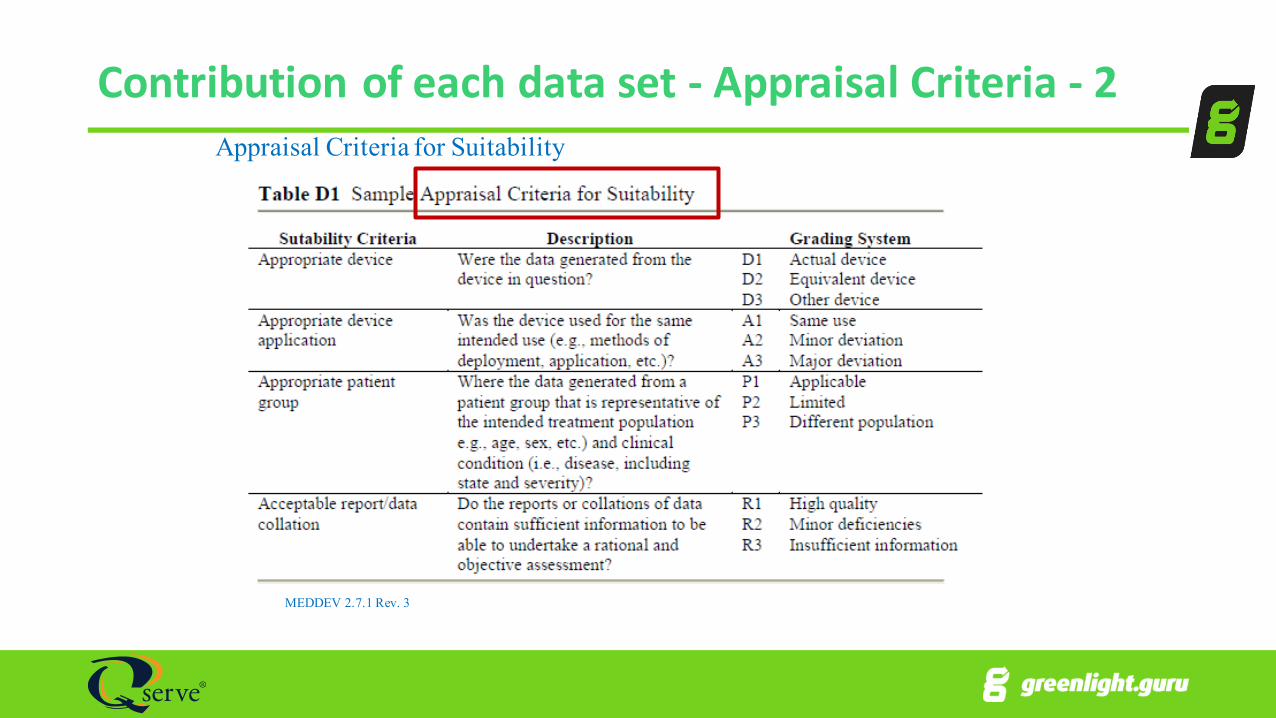
Contribution of each data set -‐ Appraisal Criteria -‐ 2
MEDDEV 2.7.1 Rev. 3
Appraisal Criteria for Suitability

Contribution of each data set -‐ Appraisal Criteria -‐ 3
MEDDEV 2.7.1 Rev. 3
Appraisal Criteria for Data Contribution

Appraisal of Clinical Data (9)
• Lots of advice on how to assess the quality and scientific validity of the data now given (section 9.3)
• How to determine the relevance of the data to the clinical evaluation (9.3.2)• Pivotal Data must come from:
1. The device under evaluation2. An equivalent device (see next slide)
• Weighting • Use pre-‐defined criteria • RCT noted as highest weight (increasing emphasis on RCT)

Equivalence (A1)
• Lots of details given. Some highlights1.Can only be based on a Single device (but …)
2.Same general criteria given (Clinical, Technical, Biological) with a few additions/clarifications
3.All 3 of these criteria must be fulfilled for a device to be “equivalent”4.Any differences must be clearly identified, disclosed, evaluated and explanation for why the differences are not expected to significantly affect the performance and safety of the device are to be given

Equivalence (A1)
4.“If measurements are possible, clinically relevant specifications and properties should be measured both in the device under evaluation and the device presumed to be equivalent, and presented in comparative tabulations”• i.e. must OWN (or have complete access to) Technical File/DD!
5.The manufacturer is expected to: include the supporting non-‐clinical information (e.g. pre-‐clinical study reports) in the technical documentation of the device• i.e. must OWN (or have complete access to) Technical File/DD!
6.The only clinical data that are considered as relevant are the data obtained when the equivalent device: Is a medical device that conforms to the requirements of the medical device directives. • i.e. if non-‐CE Marked (e.g. 510k, CFDA approval ..) MAY be able to use it, but must justify – same device intended use, patient population, medical condition … etc; also issues of:
‒ Any differences in patient population, and‒ clinical practice in local geography cf. Europe

Analysis (10)
• Goal of Analysis = to determine if the appraised data sets (taking into account the pivotal data, other own data, and the literature review report) available for a medical device collectively demonstrate compliance with each of the Essential Requirements in relation to its intended use‒ (See previous slides)
• Evaluators needs to assess:• Compliance to the ERs based on the clinical data and analysis• Determine any gaps and if a clinical investigation is needed to fill those gaps
ID pivotal datasets
Look for consistency across such
datasets

When Should Additional Clinical Investigations be conducted (A2)?
Considerations Additional Clinical Investigation?
Implant (Annex X, 1.1a MDD)
Yes/(most likely)
High risk, class III (Annex X, 1.1a MDD)
Novel technologies
New clinical use for existing technology
Any gaps in data to identify benefits, risks, claims, side effects? (class I, IIa, Ib)

When Should Additional Clinical Investigations be conducted (A2)?• If gap analysis reveals any gaps in data are sufficient to verify that device meets ERs• Special attention should be paid to:• new design features, including new materials,• new intended uses (including new medical indications), • new claims, • new types of users (e.g. lay persons),• seriousness of direct and/or indirect risks, • contact with mucosal membranes or invasiveness,• …..

When Should Additional Clinical Investigations be conducted (A2)?• Special attention should be paid to (continued):• ………• increasing duration of use or numbers of re-‐applications, • incorporation of medicinal substances, • use of animal tissues (other than in contact with intact skin), • issues raised when medical alternatives with lower risks or more extensive benefits to patients are available or have become newly available,• whether the data of concern are amenable to evaluation through a clinical investigation

In summary
1. Rev 4 is a complete rewrite of the MEDDEV2. It is clearer, more prescriptive3. It is much harder to demonstrate equivalence now -‐ essentially
only if you own the technical file for the devices to be used4. Clinical Investigations will need to be conducted more often5. The requirements for the expertise of the evaluators has
increased, and the requirements are clearer

In summary
6. The focus for CER at various stages is clearer now7. Greater expectations are being put on the NB now (hence more
in-‐depth reviews of CERs)• The clinical expertise requirement for staff has increased.• It is expected they medical doctors/professionals on staff. • The QMS procedures for assessment of CER have increased.• Clinical experts will now have to be part of the on-‐site audit team where CER are assessed on site (e.g. renewals, class IIa and IIb initial assessments). • A CEAR will have to be written now, and provided to Expert Panel for review

In summary
8. It is clearer when PMCF is needed now (more often, in general)9. The links between CER-‐Risk-‐PMS and CER-‐IFU are emphasized
(maybe stricter reviews by NB?)10. The scoping of the CER is clearer now. Which ERs are to be address
by the CER (1,3,6 for MDD) are now much more explicit, as is the assessment required to be conducted and recorded in the CER for each of these.
11. Assessment of Usability more explicitly part CER now12. Include EMBASE now (and criteria for selection of adequate
databases)13. MEDDEV 2.7.1 rev 4 was published June 29, 2016.

Thank you for your attention
What questions can we answer?
Keith Morel, PhDVP Regulatory Compliancek e i t h . m o r e l @ q s e r v e g ro u p . c o m+ 1 9 1 6 6 0 6 4 8 2 0 ( c e l l )






![Policy Clarification and Premarket Notification [510(k ...fdagov-meddev-gen/... · Policy Clarification and Premarket Notification [510(k)] Submissions for Ultrasonic Diathermy Devices](https://static.fdocuments.net/doc/165x107/5afcba557f8b9a864d8c84d7/policy-clarification-and-premarket-notification-510k-fdagov-meddev-genpolicy.jpg)











