
Chris Chander & Verna Vu. Ras: family of small GTPase Ubiquitously expressed in all cell lineages &...
22
Chris Chander & Verna Vu
-
Upload
rosamund-bell -
Category
Documents
-
view
217 -
download
1
Transcript of Chris Chander & Verna Vu. Ras: family of small GTPase Ubiquitously expressed in all cell lineages &...

Chris Chander & Verna Vu

Ras: family of small GTPase
• Ubiquitously expressed in all cell lineages & organs• Cell growth, differentiation and proliferation• Mutations are prevalent in most cancer• GTP-bound is active
• Point-mutations lead to:• Interference with Ras GAP binding • Constitutively active GTP bound state
• KRAS most frequently mutated • Found at higher frequencies in pancreatic, thyroid, colon, lung, liver cancer
• Poor prognosis
• KRAS mutations are observed in 40-50% of human colorectal adenomas and carcinomas; up to 80% in relapse patients
• Ras is the prominent cancer drug targethttp://www2.le.ac.uk/

Anaphase-promoting complex/cyclosome APC/C
Proteasome
Mitotic kinase PLK1
Important Genes in Ras pathway

http://en.wikipedia.org
Challenges for cancer therapeutics:
Lack of understanding of the vulnerabilities of these cancers
Goal: inhibit cellular drug targets to selectively kill cancer cells
1. Exploiting oncogene addiction: Use inhibitors to block oncoproteins
2. Exploiting nononcogene addiction: Inhibiting proteins that are not oncoproteins but ultimately reverses oncogenic state
Vulnerabilities: aren’t obvious & can’t be predicted so genetic exploration is the most direct approach to their discovery

Genome-wide RNAi synthetic lethal screen against KRAS oncogene
Find genes whose loss of function constitutes synthetic lethality with RAS oncogene
Synthetic lethality: mutations in 2+ genes leads to cell death, but a mutation in only one of these genes does not
Found a set of proteins whose depletion selectively impaired the viability of RAS mutant cells
scbt.com

KRAS WT/G13D (Mut) screen against isogenic KRAS WT/- shRNA (WT) library
DLD-1: colorectal cancer cell line that was used• Carries G13D mutation: endogenous point mutation that exhibits addiction to
KRAS oncogene
DLD-1 Mut and WT cells were infected with 6 pools of ~13k shRNAs in triplicates
http://elledgelab.med.harvard.edu

Identify Ras synthetic lethal (RSL) candidates
RSL candidates: shRNAs that show selective depletion (dropout) in the Ras Mut but not Ras WT cells.
Analyzed relative abundance of each shRNA over time by microarray hybridization to identify antiproliferative shRNA.
shRNA were PCR-recovered and labeled with dye
Relaxed stats: 1,742 RSL shRNAs targeting 1,613 genesStringent cut off: 379 shRNAs targeting 368 genes
elledgelab.med.harvard.edu
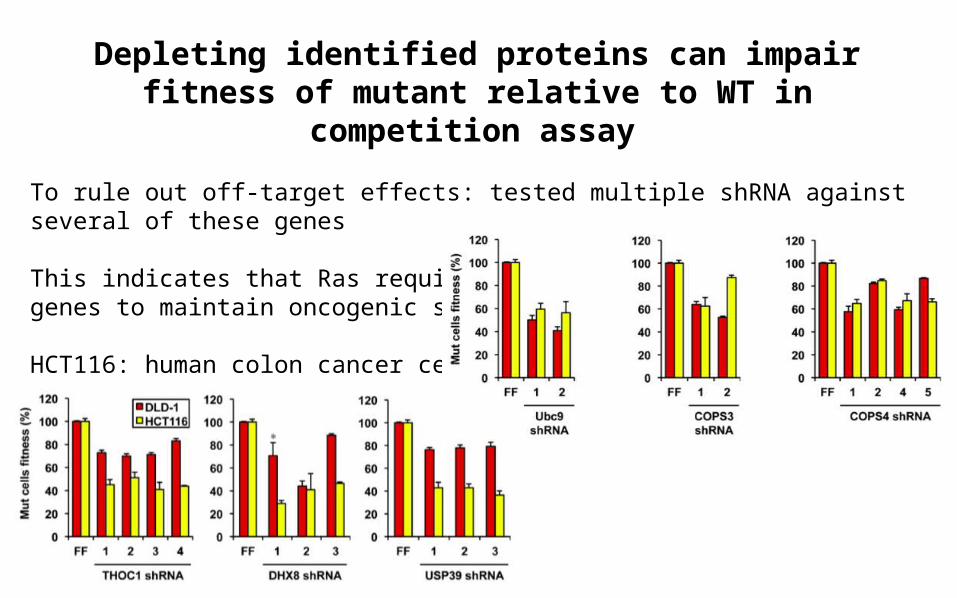
Depleting identified proteins can impair fitness of mutant relative to WT in competition assay
To rule out off-target effects: tested multiple shRNA against several of these genes
This indicates that Ras requires additional support from many genes to maintain oncogenic state
HCT116: human colon cancer cells

Many mitotic genes are identified as RSL candidates
- Ras mutants exhibit characteristics of increased mitotic stress
- When released from mitotic block higher fraction of Ras Mut exhibited lagging chromosomes and abnormal anaphases

Ras Mut are hypersensitive to mitotic stress
- Paclitaxel is a microtubule stabilizer
- Prometaphase block caused by paclitaxel in mut cells but not in WT

Ras Mutant Cells Display Hypersensitivity to PLK1 Function Inhibition
- Several shRNA to PLK1 demonstrate increased toxicity to Ras mutant cells compared to WT
- BI-2536 selectively inhibits PLK1. - Increasing concentrations of BI-2536 demonstrate a significant
decrease in mutant cell viability


Inhibition of APC/C and Proteasome Results in Ras Mutant Hypersensitivity
- APC/C targets key mitotic proteins for degradation through its activity as an E3 ubiquitin ligase
- Ras oncogene causes heightened dependence on APC/C making cells sensitive to any reduction in the activity of this complex
- CDC=subunits of APC/C complex


Inhibition of APC/C and Proteasome Results in Ras Mutant Hypersensitivity
- Proteasome activity is required for APC/C activation in addition to APC/C targeted degradation of mitotic proteins (MG132 & Bortezomib inhibit the proteasome)

NSCLC cancer cell line
- Impaired APC/C function or an enhanced requirement for APC/C function may underlie critical oncogenic stress associated with Ras mutation
- Tested sensitivity, of non- small lung cancer cell lines (NSCLC) with and without Ras mutations, to shRNA mediated knockdown of either APC1/APC4

Mice treated with PLK1 inhibitor BI-2536 demonstrate significantly subcutaneous tumor growth
In vivo BI-2536 decreases tumor growth

Human Lung Adenocarcinomas
- Patients with lung tumors having a (+) ras signature show enhanced survival in correlation with genes associated with decreasing APC/C activity
- 3 genes associated with increased survival in Ras+ signature tumor
- Simultaneous investigation of all 3 genes, expression profiles with -CDC16, -COP9, +EVI5 show near 100% survival

CDC 16 = APC/C activity (APC/C subunit)
COPS3 = APC/C activity
EVI5 = APC/C activity

Benefits & Implications:
Genome-wide RNAi synthetic lethal screen can be done by combining microarray and shRNA
• Highly parallel format makes it cost-effective & is flexible in assay design of this approach
• Signals are highly reproducible in replicate PCR
• Highly specific
• Provide additional gene targets for therapeutic exploration
• Discover larger set of oncogenic and nononcogenic targets that cancer cells rely on
• Shed new light on Ras mechanisms of action
• Potentially provide new biomarkers for patient stratification
• Future studies should look for other drugs that could be used to in target selected genes→ clinical trials

Further Reading
- Russo, M.A., Kang, K.S., Di Cristofano, A. 2013. The PLK1 inhibitor GSK461364A is effective in poorly differentiated and anaplastic thyroid carcinoma cells, independent of the nature of their driver mutations. Thyroid. 23:1284-93.
- Schlabach, M.R., Luo, J., Solimini, N.L., Hu, G., Xu, Q., Li, M.Z., Zhao, Z., Smogorzewska, A., Sowa, M.E., Ang, X.L., Westbrook, T.F., Liang, A.C., Chang, K., Hackett, J.A., Harper, J.W., Hannon, G.J., Elledge, S.J. 2008. Cancer proliferation gene discovery through functional genomics. Science. 319: 620–624.

Critiques and Limitations
- Extensive use of different pathways. Difficult to determine what each gene affected without going back through the mechanism or looking up the protein
- A few figures were difficult to interpret based on how they plotted the data


















