
Chirasil-Dex-TFA: A new polysiloxane-bonded and immobilizable cyclodextrin stationary phase for...
10
Chirasil-Dex-TFA: A New Polysiloxane-Bonded and Immobilizable Cyclodextrin Stationary Phase for Enantiomer Separation by GC Martin Jung and Volker Schurig' lnstitut fur Organische Chernie der Universitat, Auf der Morgenstelle 18, 7400 Tubingen, FRG Key Words: Enantioselective GC Polysiloxane-anchored cyclodextrin stationary phase Chirasil-Dex-TFA Immobilized 2,6-di-O-alkyl-3-O-acyl-cyclodextrin Summary Heptakis(2,6-di-O-methyl-3-O-trifluoroacetyl)-~-cyclodextrin has been chemically bonded to a polydimethylsiloxane via an octamethylenespacer and subsequently immobilizedon to the surface of fused silica capillaries, the first time a cyclodextrin previously observed with Chirasil-Val [ 191 and a Pirkle- type chiral polysiloxane [20] Permethylated cyclodextrin can thus be used as chiral selector in capillary supercritical fluid chromatography also [21,22] and even in capillary electrophoresis [23,24] derivative with mixed substituentshas been immobilizedin this way. The enantioselectivity of the new chiral stationary phase has been investigated as a function of the cyclodextrin content. It was found to be higher than that reported for heptakis (2,6-di-0-methyl-3-O-trifluoroacetyl)-~-cyclodextrin or hepta- kis(2,6-di-0-methyl-3-O-acetyl)-~-cyclodextrin dissolved in polysiloxane (e.9. OV-1701). The degree of immobilizationwas over 90 %.The temperature stability is, however, inferior to that of Chirasil-Dex (a permethylated cyclodextrin): the maximum operating temperature is approximately 200 "C. 1 Introduction In addition to peralkylated cyclodextrins, e.g. permethylated fi-cyclodextrin dissolved in OV-1701 [ 1 ,Zj or undiluted perpentyl- ated cyclodextrin 131, 2,6-dialkyl-3-acylcyclodextrins [2,4-111 have been employed as versatile chiral selectors for the gas chromatographic separation of the enantiomers of a large number of racemates. Because acyl groups in the 3- positions are capable of additional polar interactions, e.g. hydrogen bonding or dipole ~ dipole interactions, their presence often facilitates the analysis of more polar analytes, e.g. lactones, cyclic ethers, diols, amines, or amino alcohols. Cyclodextrins with acyl groups in the 6- positions were, however, found to be not useful because peracylated cyclodextrins generate very broad peaks [9] and 2,3-dipentyl- 6-acyl cyclodextrins show hardly any enantioselectivity [ 121 In this study, heptakis(2,6-di-O-methyl-3-O-trifluoroacetyl)-fi- cyclodextrin (previously investigated as a solution in OV-1701 121) has been chemically bonded to a polysiloxane backbone via an octamethylene spacer. The preparation of Chirasil-Dex-TFA is similar to that of Chirasil-Dex [13,141 (in which permethylated fi-cyclodextrin is attached to a polydimethylsiloxane containing Si-H groups by hexachloroplatinic acid (HzPtCl&catalyzed hydrosilylation [15,16]), Chirasil-Val-Nova 1171, and Chirasil- Metal [18]. The resulting chiral polysiloxanes can be thermally immobilized on to fused silica or glass surfaces, as has also been Following a study involving more than 100 racemates, we recently described the advantages of Chirasil-Dex over the original dissolved system (i.e. permethylated 0-cyclodextrin in OV-1701) [25].An important practical advantage lies in the possibility of rinsing the column in the event of contamination or during on-column injection. In another study we have systematically investigated different types of Chirasil-Dex stationary phase with regard to immobilization properties, long-term column stability, polarity, and the effect of cyclodextrin concentration (261. Simi- larly, we report here on the properties of Chirasil-Dex-TFA (Figure 1). In this work, the immobilization of a cyclodextrin derivative with mixed substituents on to the capillary surface has been achieved for the first time. r -I (TFA - 01, (OCH31, Figure 1 Structure of Chirasil-Dex-TFA. 0 1993 Dr. Alfred Huethig Publishers Journal of High Resolution Chromatography 289
-
Upload
martin-jung -
Category
Documents
-
view
217 -
download
3
Transcript of Chirasil-Dex-TFA: A new polysiloxane-bonded and immobilizable cyclodextrin stationary phase for...

Chirasil-Dex-TFA: A New Polysiloxane-Bonded and Immobilizable Cyclodextrin Stationary Phase for Enantiomer Separation by GC Martin Jung and Volker Schurig' lnstitut fur Organische Chernie der Universitat, Auf der Morgenstelle 18, 7400 Tubingen, FRG
Key Words: Enantioselective GC Polysiloxane-anchored cyclodextrin stationary phase Chirasil-Dex-TFA Immobilized 2,6-di-O-alkyl-3-O-acyl-cyclodextrin
Summary Heptakis(2,6-di-O-methyl-3-O-trifluoroacetyl)-~-cyclodextrin has been chemically bonded to a polydimethylsiloxane via an octamethylene spacer and subsequently immobilized on to the surface of fused silica capillaries, the first time a cyclodextrin
previously observed with C hirasil-Val [ 191 and a Pirkle- type chiral polysiloxane [20] Permethylated cyclodextrin can thus be used a s chiral selector in capillary supercritical fluid chromatography also [21,22] and even in capillary electrophoresis [23,24]
derivative with mixed substituents has been immobilized in this way. The enantioselectivity of the new chiral stationary phase has been investigated as a function of the cyclodextrin content. It was found to be higher than that reported for heptakis (2,6-di-0-methyl-3-O-trifluoroacetyl)-~-cyclodextrin or hepta- kis(2,6-di-0-methyl-3-O-acetyl)-~-cyclodextrin dissolved in polysiloxane (e.9. OV-1701). The degree of immobilization was over 90 %.The temperature stability is, however, inferior to that of Chirasil-Dex (a permethylated cyclodextrin): the maximum operating temperature is approximately 200 "C.
1 Introduction In addition to peralkylated cyclodextrins, e. g. permethylated fi-cyclodextrin dissolved in OV-1701 [ 1 ,Zj or undiluted perpentyl- ated cyclodextrin 131, 2,6-dialkyl-3-acylcyclodextrins [2,4-111 have been employed as versatile chiral selectors for the gas chromatographic separation of the enantiomers of a large number of racemates. Because acyl groups in the 3- positions are capable of additional polar interactions, e .g . hydrogen bonding or dipole ~
dipole interactions, their presence often facilitates the analysis of more polar analytes, e.g. lactones, cyclic ethers, diols, amines, or amino alcohols. Cyclodextrins with acyl groups in the 6- positions were, however, found to be not useful because peracylated cyclodextrins generate very broad peaks [9] and 2,3-dipentyl- 6-acyl cyclodextrins show hardly any enantioselectivity [ 121
In this study, heptakis(2,6-di-O-methyl-3-O-trifluoroacetyl)-fi- cyclodextrin (previously investigated as a solution in OV-1701 121) has been chemically bonded to a polysiloxane backbone via an octamethylene spacer. The preparation of Chirasil-Dex-TFA is similar to that of Chirasil-Dex [13,141 (in which permethylated fi-cyclodextrin is attached to a polydimethylsiloxane containing Si-H groups by hexachloroplatinic acid (HzPtCl&catalyzed hydrosilylation [15,16]), Chirasil-Val-Nova 1171, and Chirasil- Metal [18]. The resulting chiral polysiloxanes can be thermally immobilized on to fused silica or glass surfaces, as has also been
Following a study involving more than 100 racemates, we recently described the advantages of Chirasil-Dex over the original dissolved system (i.e. permethylated 0-cyclodextrin in OV-1701) [25]. An important practical advantage lies in the possibility of rinsing the column in the event of contamination or during on-column injection. In another study we have systematically investigated different types of Chirasil-Dex stationary phase with regard to immobilization properties, long-term column stability, polarity, and the effect of cyclodextrin concentration (261. Simi- larly, we report here on the properties of Chirasil-Dex-TFA (Figure 1). In this work, the immobilization of a cyclodextrin derivative with mixed substituents on to the capillary surface has been achieved for the first time.
r -I
(TFA - 01, (OCH31,
Figure 1
Structure of Chirasil-Dex-TFA.
0 1993 Dr. Alfred Huethig Publishers Journal of High Resolution Chromatography 289

Enantiomer Separations on Chirasil-Dex-TFA
2 Experimental
2.1 Synthesis
2.2.1 Synthesis of Monokis-6-O-(oct-7-enyl)-~-cyclodextrin The mono-octenylated cyclodextrin was prepared and purified as described previously [26]
2.1.2 Synthesis of Mon okis(2 - 0- me thy1 - 6- 0 - (Oct - 7- en yl ))hexa -
The 2,6- dimethylation of the mono-octenylated cyclodextrin was performed analogously to that of underivatized cyclodextrins [27], After the described precipitation from chloroform - n-hexane, the crude product was purified by column chromatography (5 x 20 cm column filled with 32-63 pm silica gel; eluent, toluene - ethanol, 4: 1 v/v; TLC, Rf = 0.41, detection with a mixture of phenol and conc. sulfuric acid followed by heating). Thus, 3.8 g octenylated P-cyclodextrin were converted into 3.3 g crude dimethylated product which was divided into two halves for column chroma- tography. The final yield was 2.65 g of the purified product as a white powder.
'H-NMR (ppm, 250 MHz, deuterochloroform (CDC13)): 3.39 (sing- let, OMe6). 3.62 (singlet, OMeZ), 4.89-4.95 (multiplets, low intensity, olefinic CHz), 4.95 (doublet, anomeric H), 5.05 (doublet, OH3), 5.72-5.82 (multiplet, low intensity, olefinic CH).
13C-NMR (ppm, 60 MHz, CDC13): 58.8 (OMe6), 60.1 (OMe2), 70.1 (C5), 70.7(C6), 73.0(C3), 81.9(C2), 83.3(C4), 101.1 (Cl), 114.1 (C8 of octenyl group), 139.0 (C7 of octenyl group).
kis(2,6-di-O-methyl)-/3-cyclodextrin
2.1.3 Synthesis of Monokis(2-0-methyl-3-0-trifluoroacetyl-6- 0-(oct-7-enyl))hexakis(2,6-di-0-methyl-3-0-trifluoroace- tyl)-P-cyclodextrin
The 2,6-dimethylated monokis-6-0-(oct-7-enyl)-~-cyclodextrin (2.55 g) was trifluoroacetylated as previously described 12,281 for the non-octenylated analog. The sodium trifluoroacetate was removed by filtration of the dichloromethane (CH2C12) solution. Recrystallization of the crude oil from dichloromethane - n- hexane and drying in vacuo yielded 1.9 g of a white or slightly brownish solid (TLC, Rf = 0.62, toluene - ethanol, 4 : l v/v; [a];) +88.8 (CH2C12, c = 0.8)). The purity was checked by ion-spray mass spectroscopy I291 (Figure 2).
2106
lo?
.- f a- A 751 5 6 7 I1 TFAgroups
2009 I I1 di-octenylated
2202 "
1800 2000 2200 mlz
Figure 2
Ion-spray mass spectrum of monokis(2-O-methyl-3-O-trifluoroace- tyl-6-0 - (oct-7-enyl)) hexakis(2,6-di-O-methyl-3-O-trifluoroacetyl)- p-cyclodextrin (M = 2099.3; in the presence of LICI). The spectrum shows the peaks belonging to (M + Li)+ and (at low intensity) to other adducts.
IH-NMR (ppm, 250 MHz, CDC13): 3.33 and 3.39 (singlets, OMe6 and OMeZ), 4.89 and 4.93 (multiplets, low intensity, olefinic CHz), 5.01 (doublet, anomeric H), 5.72-5.82 (multiplet, low intensity, olefinic CH).
13C-NMR(ppm, 60MHz, CDC13): 25.2,28.7,29.2, and33.6(octenyl group), 59.1 (OMe6), 59.8 (OMeZ), 70.2 (C5), 70.8 and 71.0 (C6), 79.6 (C4), 98.6 (Cl), 112.2 and 116.8 (CF), 114.2 (C8 of octenyl group), 138.9 (C7 of octenyl group), 156.9 (CO).
2.1.4 Synthesis of Chirasil-Dex-TFA
The hydrosilylation of 300 mg of the above cyclodextrin derivative and 1200 mg dimethylpolysiloxane (MW = 3000) containing 10 % silane (Si-H) groups was performed analogously to that recently described in detail for Chirasil-Dex [26]. Much care has to be taken when washing with methanol since part of the product (contain- ing a high percentage of bonded cyclodextrin) is readily soluble in methanol. Yield: 0.9 g of a highly viscous to waxy product,
NMR (in addition to the cyclodextrin signals mentioned above - the olefinic signals have disappeared): 'H-NMR, -0.15 to +0.25 (SiMe groups) and 13C-NMR, -3 to +4 (SiMe groups).
Analogous methods of preparation were used for the other types of Chirasil-Dex-TFA described.
[a]!& +15.4 ( C = 0.8 (CHZC12)).
2.2 Solutes
Most solutes were commercial samples. Others were obtained from Prof. Berson, Yale University, New Haven, USA (solute D), Prof. De Meijere, Hamburg, FRG (solute O), and Dr Jauch [33] (solute S). Spiroketals and dioxolanes were prepared as reported in the literature 134,351. Determination of natural enantiomeric composition and synthesis of the hazelnut aroma compound filbertone (solute E) were performed as described elsewhere [36,37].
2.3 Open-Tubular Columns
Fused silica tubing (25 m x 0 25 mm; MicroQuartz, Munich, FRG) was heated at 260 "C for 2 h with a low flow of hydrogen (inlet pressure 0.1-0.2 bar). Static coating with 0.4 % solutions of Chirasil-Dex-TFA in diethyl ether was used to coat the columns, without further deactivation, with ca 0.25 pm films. At the Van Deemter optimum flow rate column efficiencies were > 3000 effective theoretical plates per meter for n-tridecane at 110 "C; for most chiral solutes at 0.4 bar inlet pressure the column efficien- cies were 1000-3000 effective plates per meter. Much care should be taken not to expose the columns to air while hot or during storage.
2.4 Immobilization
When immobilized stationary phases were used, immobilization was accomplished thermally (20 h at 180-190 "C, or 175 "C for carboxy-functionalized polysiloxanes) with a very slow flow of hydrogen, as described previously [30-321.
2.5 Instrumentation
Gas chromatography was performed with Carlo-Erba (Fi- sons/Carlo-Erba. Milan, Italy) Fractovap 2150 and 2350, VEGA, and MEGA gas chromatographs equipped with FID and suitable for operation with fused silica open tubular columns. Hydrogen (99.999 %) was used as carrier gas without further purification; 10
290 VOL. 16, MAY 1993 Journal of High Resolution Chromatography

Enantiomer Separations on Chirasil-Dex-TFA
m x 0 25 mm i d capillary columns were operated at an inlet pressure of 0 4 bar, the split ratio was 1 100 Retention times and peak widths were determined using a Shimadzu C-R3A integra- tor Net retention times were measured relative to the methane peak
3 Results and Discussion
3.1 Synthesis
Although the synthesis of monokis(2- 0-methyl-3- O-trifluoroace- tyl-6- O-(oct-7-enyl))hexakis(2,6-di-O-methyl-3- O-trifluoroace- ty1)-P-cyclodextrin (or cyclomaltoheptaose, cf [27] for nomen- clature) is accomplished in three steps, each of which may yield a mixture of products, a reasonable amount of a derivative of satisfactory quality could be obtained
The purity of the derivative was checked by ion-spray mass spectrometry (291 (Figure 2) A small amount of lithium chloride (LiC1) was added in order to suppress the formation of cluster ions other than (M + Li)' Although a daughter spectrum of the 2106 a m u peak (tandem mass spectrometry) did not show any of the adjacent peaks (thus demonstrating that the 2009 a m u peak
Table 1
does not arise from the former by fragmentation), larger concen- trations of lithium chloride caused a partial loss of trifluoroacetyl (TFA) groups during analysis The piesence of the 2106 a.m.u. peak a s the largest peak is further evidence that the octenyl group is not attached in the 3-position (otherwise only 6 TFA groups could be accommodated), in addition, 'H- and l3C-NMR experi- ments (including spin-echo experiments) clearly showed that the octenyl groups are attached in one of the 6-positions (301.
3.2 Comparison with Cyclodextrin Derivatives Dissolved in
Chromatographic data obtained from a number of selected solutes are listed in Table 1 and some typical chromatograms are shown in Figure 3. Where available, the separation factors, a , for the previously investigated dissolved system [2,7,38] (1.e hepta- kis(2,6-di-0-methyl-3-O-TFA)-~-cyclodextrin dissolved in OV- 1701 (9.1-12.3 % by weight, molality 0.05-0.07)) are also given. Significant improvements are observed throughout. This is partly because of the higher cyclodextrin concentration used here (cf. below) and partly because of the higher degree of trifluoroacety- lation (the derivative used previously contained only 3 to 6 TFA groups, a s was subsequently shown by ion-spray mass spectros-
Polysiloxane
Capacity factors, k, for the separation of the enantiomers of selected racemic solutes on Chirasil-Dex-TFA, and separation factors, a, for separation of the same enantiomers using both Chirasil-Dex-TFA (B; and (in brackets) the dissolved system (D).
Solute Temp. ha' ab) I"C1 BC' (Did'
~ ~~
A 3,3,5-Trimethylcyclohexanone
A ~ ~~
80 106 3 2 513 130 4 2 1612 (1 21) 155 1 0 1329
B trans-2,6-Dimethylcyclohexanone 110 7 6 1.203 (1 06) \\."
C 2-Methylcyclohexanone 70 109.1 1.076 (1.03)
D Bicyclo(3.1 .O]hex-l-en-3-one
b 120 20.4 1.430 (1.14) 155 2.1 1.274
E E-5-methylhept-2-en-40ne (filbertone) 80 27 4 1476 100 6 3 1357 110 4 2 1 3 1 4
F Pulegone 110 15.4 1.177 (1 05) 115 9 8 1.148
100 19.7 1.131 (1 04) 115 6.9 1 0 9 7
G Isomenthol
H Neomenthol
P O H
100 17.9 1.109 (1.03)
Journal of High Resolution Chromatography VOL. 16, MAY 1993 291

Enantiomer Separations on Chirasil-Dex-TFA
I trans-2,3-Dimethyl-l,4-dioxaspiro[4.4]nonane 100 5.2 1.259 (1.05) 110 3.3 1.184
K 2-Methyl- 1,5-dioxaspiro[ 5.51undecane 100 10.2 1.098 (1.03) 110 6.9 1.070
a) 100 4.6 1.161 (1.04) b) 100 6.1 1.224 (1.07)
70 21.4 1.653 (1.16)
L 2-Ethyl-1,6-dioxaspiro[4.4lnonane (chalcogran; 2 pairs of enantiomers)e)
M trans-2,5-Dimethoxytetrahydrofuran H3C0 Q’‘.. OCH, 90 4.4 1.475
~ _ _ _ _ _ _ _
N trans-2,5-Diethoxytetrahydrofuran 70 22 1 1862 H , C , O ~ ’ OCz& 90 49 1497
130 37.9 1.141 (1.03) 150 9.5 1.101
a) 80 24.4 1.208 (1.03) b) 80 25.3 1.078 (1.02)
P 2-(Isobut-l-enyl)-4-methyltetrahydropyran (rose oxide: a, trans; b, cis)
Q 2-Ethyl-6-methyl-5,6-dihydro-y-pyrone 140 14.4 1.120 (1.07)
~ ~
R y-Decalactone ~~~
170 12.2 1.163 (1.05)
160 13.2 1.292 (1 17) 180 3.6 1.236
S 2-0xabicyclo[3.3.0loct-7-en-3-one
T Phenyloxirane Ph A 110 50 1037 (= 1)
U 2-Methyl-3-phenyloxirane (a, trans; b, cis)
Ph
a) 110 4.9 1.044 b) 95 12.7 1.219 (1.03)
110 5.2 1.172 125 2.6 1.122
V 2-Methyl-2-phenyloxirane 110 4.2 1.031 (= 1) Ph$
W 2,2-Dimethyl-3-phenyloxirane Ph*
110 5.0 1.209 (1.05)
X 2,3-Dimethyl-3-phenyloxirane (cis and trans)e) a) 110 4.4 1.173 P h A b) 110 6.5 1.101
Y 2,2,3-Trimethyl-3-phenyloxirane P h f i
110 5.6 1.216
a) second enantiomer; t 2 % b) * 0.002 c, bonded phase (Chirasil-Dex-TFA; [a];5 = +15.4)
d, dissolved system as described previously (impure 2,6-dimethyl-3-trifluoroacetyl-~-cyclodextrin in OV-1701, 12.5 % by weight) order of elution not known
292 VOL. 16, MAY 1993 Journal of High Resolution Chromatography

Enantiomer Separations on Chirasil-Dex-TFA
Figure 3
A
I t?
rl
w 0 05 1 mm
- Y
14
( i i )
a = 2.51
7 I 0 10 2b 30 min
I I i 0 1 2 min
trans n
d 1 I I - 0 2 4 min 0 1 2 mm 0 1 min
Journal of High Resolution Chromatography
b 1 min
VOL. 16, MAY 1993 293

Enantiomer Separations on Chirasil-Dex-TFA
Figure 3 contin'd
I I 0 1 min
(xi)
1 I I I I
0 05 1 15 min
X
W
11
12
I 1 I I I I o 1 2 3 4 Smin
I L L '
c
ClO
I
1
2 n
3
i c I I I I I I 0 1 2 3 4 5 min
Figure 3
Separations achieved on a 10 m x 0.25 mm i.d. fused silica capillary column coated with immobilized Chirasil-Dex-TFA (compound iden- tification letters are as used in Table 1): (i) the enantiomers of racemic 3,3,5-trimethylcyclohexanone (A) and racemic bicy- clo[3.l.0]hex-l-en-3-one (D) at 155 "C; (ii) the enantiomers of racemic 3,3,5-trimethylcyclohexanone (A) at 80 "C; (iii) the enan- tiomers of racemic trans-2,3-dimethyl-l,4-dioxaspiro[4.4]nonane (I) and 2-methyl-1,5-dioxaspiro[5.5]undecane (K) at 110 "C; (iv) the enantiomers of racemic 2-ethyl-6-methyl-5,6-dihydro-y-pyrone (0) at 140°C; (v) the isomers and enantiomers of chalcogran (L) at 100 "C; (vi) the isomers and enantiomers of 2,5-dimethoxytetrahy- drofuran (M) at 90°C; (vii) the enantiomers of racemic 4,7,11- trioxapentacyclo[6.3.0.02~6.03~10.05~g]undecane (0) at 150 "C; (viii) the enantiomers of racemic 2-oxabicyclo[3.3.0]oct-7-en-3-one (S) at 180 "C; (ix) the enantiomers of racemic y-lactones (y-hexalactone to y-dodecalactone), column temperature programmed from 160 "C (held for 1 min) to 190 "C at 10 Vmin; (x) the isomers and enantiomers of racemic methyl-substituted styrene oxides (phenyloxiranes; U and V), column temperature programmed from 85 "C (held for 2 min) at 4 "/min; (xi) the isomers and enantiomers of racemic methyl- substituted styrene oxides (phenyloxiranes; Wand X) at 111 "C.
copy [29] and TLC) The large enantioselectivity of this stationary phase towards many chiral solutes enables very rapid baseline separations of the enantiomers on 10 m columns, separations which are sometimes complete in only a few seconds (e g Figure 3(1))
For some solutes which seem to interact strongly with the cyclodextrin (e g solutes A, M. N, and 0) a remarkable behavior IS observed if the analysis temperature is reduced to well below that required for baseline separation of the enantiomer, separa-
294 VOL. 16, MAY 1993 Journal of High Resolution Chromatography

Enantiomer Separations on Chirasil-Dex-TFA
tion factors, a, reach very high values, but this is accompanied by significant peak broadening not observed for other analytes at those temperatures. An example is shown in Figure 3(ii).
A decomposition phenomenon was observed for some a,p unsa- turated ketones such as filbertone (Figure 4) on a column containing a particularly high level of residual platinum (used as
120 "C
0 2 4 6 Bmin
Figure 4
Separation of the enantiomers of racemic E-5-methylhept-2-en- 4-one (filbertone; E) on a 10 m x 0.25 mm i.d. fused silica capillary column column coated with immobilized Chirasil-Dex-TFA rich in residual platinum which is leading to solute decomposition (inlet pressure: 0.4 bar hydrogen).
the catalyst for hydrosilylation). On other columns less contami- nated with platinum, however, the extent of decomposition was negligible.
The separation factors observed were also larger than those reported for 10 % heptakis (2,6-di-O-methy1-3-0-acetyl)-P- cyclodextrin in OV-1701 191 (e.9. a = 1.035 for y-decalactone at 170 "C, a = 1.093 for trans-2,6-dimethylcyclohexanone at 100 "C) or 16 % heptakis(Z16-di- 0-methyl-3- 0-heptafluorobutyry1)-P-
cyclodextrin in OV-1701 (391 (e.g. a = 1.07 for y-decalactone at 150 "C, a = 1.06 for trans-2,6-dimethylcyclohexanone at 110 "C, a = 1.14for trans-2.5-dimethoxytetrahydrofuranat 70 "C, a = 1.03 for 2-methylcyclohexanone at 85 "C). Heptakis(2,6-di-O-methyl- 3-O-acetyl)-~-cyclodextrin in OV-1701 has been found to be one of the most useful cyclodextrin stationary phases in GC, being capable of separating the enantiomers of a third of the chiral solutes tested, many of which could not be resolved on the other phases investigated 191.
Whereas the choice of acyl groups seems to have only a small influence on enantioselectivity, the use of the pentyl group rather than methyl, e.g. in heptakis(2,6-di-O-pentyl-3-O-TFA)-P- cyclodextrin (8,401, leads to a deterioration in a , considering that these cyclodextrin derivatives are usually not diluted with poly- siloxanes (e.g. a = 1.01 for styrene oxide at 80 "C, a = 1.18 for trans-2,5-dimethoxytetrahydrofuran at 70 "C, a = 1.03 for 2- methylcyclohexanone at 80 "C). It should, however, be noted that whereas a- or P-cyclodextrin derivatives usually seem to be most useful 19,411, for 2,6-di-0-pentyl-3-0-acylcyclodextrins the y ring size yields chiral selectors with much wider applicability [6,8], Thus, 80 % of 150 racemates which were separated on 2,6- dipentyl-3-TFA-cyclodextrins could be separated on the y deriv- ative (81.
3.3 Dependence of the Separation Factor, a, on the Concen-
In previous studies of both permethylated P-cyclodextrin dis- solved in OV-1701 (421 and Chirasil-Dex 1261, we have plotted a against percentage (or molality) of cyclodextrin for many chiral solutes. It was demonstrated that increasing the concentration of permethyl-P-cyclodextrin beyond ca 30 % by weight did not result in further improvement in enantiomer resolution in a given time 1261. A mathematical equation has been derived which, assuming a knowledge of the strength of the complex between the chiral solute and the cyclodextrin, correctly predicts the dependence of a on the cyclodextrin molality [42].
In this work we used Chirasil-Dex-TFA with an [a]:5 value of +15.4 (CH2C12, c = 0.8), corresponding to 16.5 % heptakis(2,6-
tration of Cyclodextrin
Table 2
Separation factors, a=), for selected solutes on Chirasil-Dex-TFA containing different percentages of cyclodextrin.
Solute Temp. CD weight percentageb) and la]& 1 "Cl 5.5 13.2 16.5 (27)c)
+5.1 + 12.3 + 15.4 (+ 25)
A B E P G I K M N R W
3,3,5-Trimethylcyclohexanone trans-2,6-Dimethylcyclohexanone Filbertone Pulegone Isomenthol trans-2,3-Dimethyl-l,4-dioxa spiro(4.4lnonane 2-Methyl- 1,5-dioxaspiro[5.51undecane trans-2,5-Dimethoxytetrahydrofuran trans-2,5-Diethoxytetrahydrofuran y-Decalactone 3,3-Dirnethyl-Z-phenyloxirane
130 110 110 110 100 100 100 90 90
170 110
1.263 1.115 1.107 1.059 1.053 1.088 1.038 1.156 1.120 1.076 1.080
1.543 1.195 1.274 1.145 1.115 1.215 1.082 1.415 1.392 1.156 1.183
1.612 1.203 1.314 1.177 1.131 1.259 1.098 1.475 1.497 1.163 1.209
(1.61) (1 21) (1.23) (1.13) (1.11) (1.23) (1.11) (1.42) (1.48) (1.14) (1.25)
a) * 0.002 b)onlv one of the carbon atoms of the octamethvlene spacer is counted with the cyclodextrin, i.e. the weight percentages refer to 2.6-dimethyl- 3-t~ifluoroacetyl-~-cyclodextrin rather than to the octenylated analog
c, inaccurate data because of broad peaks
Journal of High Resolution Chromatography VOL. 16, MAY 1993 295
Enantiomer Separations on Chirasil-Dex-TFA
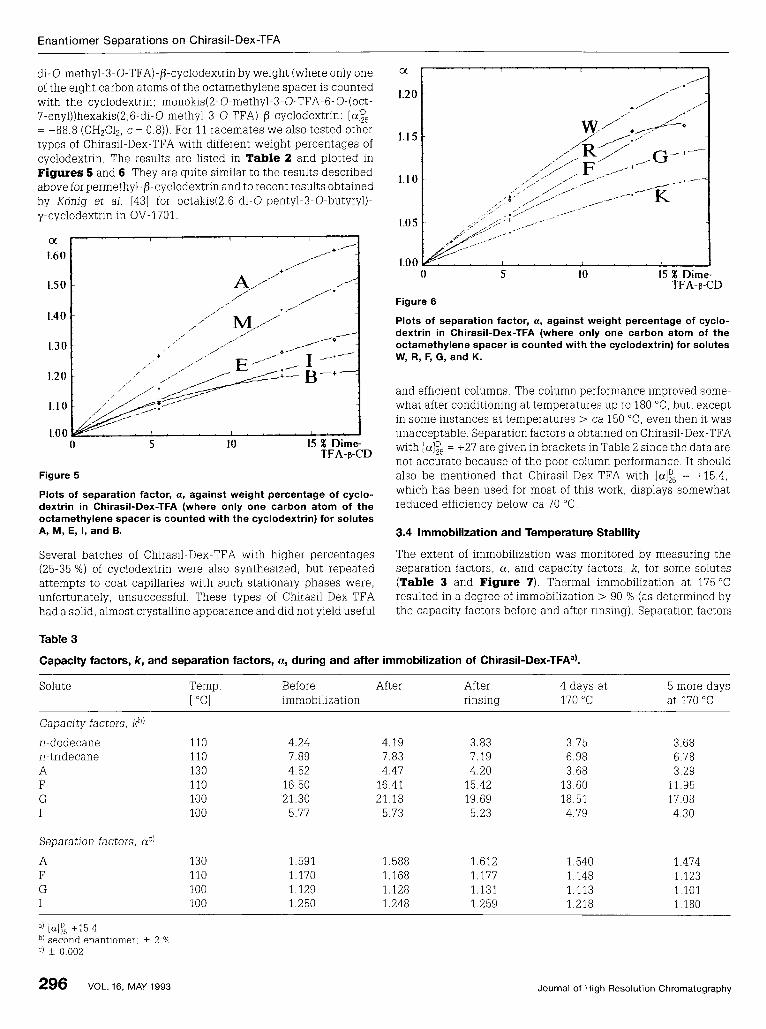
di-O-methyl-3-O-TFA)-/?-cyclodextrin by weight (where only one of the eight carbon atoms of the octamethylene spacer is counted with the cyclodextrin; monokis( 2- 0-methyl-3-0-TFA-6- 0-( oct- 7-enyl))hexakis(2,6-di-0~methyl-3-O-TFA)-/?~cyclodextrin: = +88.8 (CH~CIZ, c = 0.8)). For 11 racemates we also tested other types of Chirasil-Dex-TFA with different weight percentages of cyclodextrin. The results are listed in Table 2 and plotted in Figures 5 and 6 . They are quite similar to the results described above for permethyl-b-cyclodextrin and to recent results obtained by Konig et al. [43] for octakis(2,6-di-O-pentyl-3-O-butyryl)- y-cyclodextrin in OV-1701
, / / - - 0 - 130 -
0 5 10 15 % Dirne- TFA-B-CD
Figure 5
Plots of separation factor, a, against weight percentage of cyclo- dextrin in Chirasil-Dex-TFA (where only one carbon atom of the octamethylene spacer is counted with the cyclodextrin) for solutes A, M, E, I, and B.
Several batches of Chirasil-Dex-TFA with higher percentages (25-35 %) of cyclodextrin were also synthesized, but repeated attempts to coat capillaries with such stationary phases were, unfortunately, unsuccessful. These types of Chirasil-Dex-TFA had a solid, almost crystalline appearance and did not yield useful
1.20
1.1 5
1.10
1.05
1.00 0 5 10 15 % Dirne-
TFA-B-CD Figure 6
Plots of separation factor, a, against weight percentage of cyclo- dextrin in Chirasil-Dex-TFA (where only one carbon atom of the octamethylene spacer is counted with the cyclodextrin) for solutes W, R, F, G, and K.
and efficient columns The column performance improved some- what after conditioning a t temperatures up to 180 "C, but, except in some instances at temperatures > ca 150 "C even then it was unacceptable Separation factors a obtained on Chirasil-Dex-TFA with [a]F5 = +27 are given in brackets in Table 2 since the data are not accurate because of the poor column performance It should also be mentioned that Chirasil Dex TFA with [a]F5 = +15 4, which has been used for most of this work, displays somewhat reduced efficiency below ca 70 "C
3.4 Immobilization and Temperature Stability
The extent of immobilization was monitored by measuring the separation factors, a , and capacity factors, k, for some solutes (Table 3 and Figure 7) Thermal immobilization at 175°C resulted in a degree of immobilization > 90 % (as determined by the capacity factors before and after rinsing) Separation factors
Table 3
Capacity factors, k, and separation factors, a, during and after immobilization of Chirasil-Dex-TFAa).
Solute Temp. Before After After 4 days at 5 more days [ "CI immobilization r i n s i n g 170 "C at 170 "C
Capacity factors, kb)
n-dodecane n-tridecane A F G I
separation factors, ac)
A F G I
110 110 130 110 100 100
130 110 100 100
4.24 7.89 4.52
16.50 21.30
5.77
1.591 1.170 1.129 1.250
4.19 7.83 4.47
16.41 21.18
5.73
1.588 1.168 1.128 1.248
3.83 7.19 4.20
15.42 19.69 5.23
1.612 1.177 1.131 1259
3.75 6.98 3.68
13.60 18.51 4.79
1.540 1.148 1.113 1.218
3.68 6.78 3 29
11 95 17.08 4.30
1.474 1123 1101 1180
a) [ a ] & +15.4 h, second enantiomer. k 2 % C) f 0.002
296 VOL. 16, MAY 1993 Journal of High Resolution Chromatography

Enantiomer Separations on Chirasil-Dex-TFA
k 20
15
10
5 -
'-
-
-
-
I
I
0 ; I e ore a ter I a ter ' ifmrnobilizatiofn 1 nLsing 2 ~d7rT 5 ; ~ i k ~
rinsing Figure 7
Capacity factors k, of solutes G, F, A, I, n-dodecane, and n-tridecane during and after immobilization of Chirasil Dex TFA (data from Table 3).
improved slightly during immobilization and efficiency (Nefr) did not change noticeably.
After immobilization, the long-term column stability was tested at 170 "C over several days. The temperature stability of Chirasil- Dex-TFA is, unfortunately, not as good as that of Chirasil-Dex containing permethylated cyclodextrin. Whereas for the latter a values remained unchanged and k values decreased only slightly even after several days at 220 "C, here we observed a slow decrease in both k and a values for chiral solutes even after a few days at 170 "C. Column efficiency, however, remained unchanged, and the capacity factors, k, of n-alkanes decreased much less than those of the chiral solutes. Keeping the column at 190 "C for one night had an effect very similar to that which resulted from keeping it at 170 "C for 9 days. One night at 200 "C resulted in a reduction in a from 1.57 to 1.29 for solute A. The capacity factor, k , of solute A decreased by 50 %, whereas those of the n-alkanes decreased by only 9 %. Again, efficiency remained almost unchanged. It has to be emphasized, however, that using the columns up to approximately 200 "C under programmed conditions or for short periods does not result in measurable reductions in a or k .
These data demonstrate that the slow deterioration is caused not by further cross-linking within the stationary phase nor by column bleeding, but rather by a gradual decomposition of the chiral selector, e.g. by hydrolysis of TFA groups. Temperature stability may be improved by the use of acetyl rather than TFA substi- tuents. Thus Konig et al. [44] observed no loss of column performance for 2,6-di-0-pentyl-3-0-acetylcyclodextrin station- ary phases after prolonged operation at 220 "C, although no data (k, etc.) are given. Armstrong et a]., on the other hand, reported that 2,6-di-0-pentyl-3-0-TFA-cyclodextrins form stable films only up to 180 "C; at 200 "C droplets formed and efficiency deteriorated [81.
4 Conclusion Below 190 "C, Chirasil-Dex-TFA is a versatile new chiral station- ary phase, separating enantiomers with high separation factors and resolution. We have demonstrated that our procedure of chemically binding cyclodextrins to a polysiloxane, followed by immobilization on to the capillary surface, can also be applied to cyclodextrin derivatives with mixed substituents.
Acknowledgments
This work has been supported by Deutsche Forschungsgemeinschaft and by Fonds der Chemischen Industrie. We wish to thank Dr J. Metzger for recording the ion-spray mass spectra.
References
V. Schurig and H.-P. Nowotny. in: A. Zlatkis (Ed.), Proc. Advances in Chromatography, Berlin, 1987; V. Schurig and H.-P. Nowotny. J Chromatogr. 441 (1988) 155.
H:P. Nowotny, D. Schmalzing, D. Wistuba. and V. Schurig, HRC 12 (1989) 383.
W.A. Konig, S. Lutz, P. Mischnick-Lubbecke, B Brassat, and G. Wenz, J. Chromatogr 447 (1988) 193.
W.A. Konig, S Lutz, G. Wenz. and E. von der Bey, HRC & CC 11 (1988) 506.
W.A. Konig, S, Lutz, C. Colberg, N. Schmidt, G Wenz, E. von der Bey, A. Mosandl, 6. Gunther, and A. Kustermann, HRC & CC 11 (1988) 621.
W.A. Konig. R. Krebber, and P. Mischnick, HRC 12 (1989) 732
V. Schurig, M. Jung, D. Schmalzing, M. Schleimer, C. Duvekot, J.C. Buyten. J.A. Peene, and P. Mussche, HRC 13 (1990) 470.
W.Y. Li. L. Jin, and D.W. Armstrong, J. Chromatogr. 509 (1990) 303.
W. Keim, A. Kohnes, W. Meltzow, and H. Romer, HRC 14 (1991) 507.
C. Bicchi. G. Artuffo, A. D'Amato, G. Pellegrino, A. Galli, and M. Galli. HRC 14 (1991) 701.
G. Stoev, J. Chromatogr. 589 (1992) 257.
W.A. Konig. D. Icheln, and I. Hardt, HRC 14 (1991) 694.
P. Fischer, R. Aichholz, U. Bolz, M. Juza. and S. Krimmer, Angew. Chem. 102 (1990) 439; Angew. Chem. Int. Ed. Engl. 29 (1990) 427.
V. Schurig, D. Schmalzing, U. Muhleck. M. Jung, M. Schleimer, P. Mussche, C. Duvekot, and J.C. Buyten, HRC 13 (1990) 713.
J .L . Speier, J.A. Webster, and G.H. Barnes, J. Am. Chem. SOC. 79 (1956) 974.
J.S. Bradshaw, S.K. Aggarwal, C.A. Rouse, B. J. Tarbet, K.E. Markides, and M.L. Lee, J. Chromatogr. 405 (1987) 169.
M. Walser, Doctoral Thesis, University of Tubingen (1987).
V. Schurig, D. Schmalzing, and M. Schleimer, Angew. Chem 103 (1991) 994; Angew. Chem. Int. Ed. Engl. 30 (1991) 987.
H. Frank, HRC & CC 11 (1988) 787.
F.J. Ruffing, J.A. Lux, W. Roeder. and G. Schomburg. Chromatogra- phia 26 (1988) 19.
V. Schurig, Z. Juvancz, G. Nicholson, and D. Schmalzing. HRC 14 (1991) 58.
D. Schmalzing, G.J Nicholson, M. Jung, and V. Schung. J Microcol. Sep. 4 (1992) 23.
S. Mayer and V. Schurig, HRC 15 (1992) 129.
S. Mayer and V. Schurig, J . Liquid Chromatogr., 16 (1993) 915
D. Schmalzing, M. Jung, S. Mayer, J. Rickert, and V. Schurig. HRC 15 (1992) 723.
M. Jung and V. Schurig, J. Microcol. Sep. 5 (1993) 11.
K. Takeo. Carbohydr. Res. 200 (1990) 481.
F. Cramer, G Mackensen, and K Sensse. Chem. Ber. 102 (1969) 494.
J. Metzger, M. Jung, D. Schmalzing, E. Bayer, and V. Schurig. Carbohydr. Res. 222 (1991) 23.
Journal of High Resolution Chromatography VOL. 16, MAY 1993 297

Enantiomer Separations on Chirasil-Dex-TFA
1301 D. Schrnalzing. Doctoral Thesis, University of Tubingen (1991).
[31] G. Lai, G. Nicholson, U. Miihleck, and E. Bayer, J. Chromatogr. 540
[32] G. Lai, U. Mdhleck. G.J. Nicholson, J. Schmid. and E. Bayer,
[33] J. Jauch, Diploma Thesis, university of Tubingen (1988).
1341 R Weber, K. Hintzer. and V. Schurig, Naturwissenschaften 67 (1980)
1351 V. Schurig and D . Wistuba, Tetrahedr Lett. 25 (1984) 747.
1361 J. Jauch, D. Schrnalzing, V. Schurig, R. Emberger, R. Hopp, M. Kopsel, W. Silberzahn, and P. Werkhoff, Angew. Chem. 101 (1989) 1039; Angew. Chem. Int. Ed. Engl. 28 (1989) 977.
1371 V. Schurig, J. Jauch, D . Schmalzing, M. Jung, W. Bretschneider, R. Hopp, and P. Werkhoff, Z. Lebensm. Unters. Forsch. 191 (1989) 28.
1381 D Schrnalzing. unpublished results.
(1991) 217.
Chromalographia 32 (1991) 241.
453.
1391 V. Schurig and M. Jung, in D. Stevenson and I.D. Wilson (Eds), Recent Advances in Chiral Separations, Proc. Internat. Symp. on Chiral Separations, Guildford, Surrey, UK, 12-15 September, 1989, Plenum Press, New York, USA (1990) 117-134; M. Jung, Diploma Thesis, University of Tubingen, FRG (1989).
[40] H. Jin and D . W. Arrnstrong, Sepu 9 (1991) 148.
[41] H.-P. Nowotny, Doctoral Thesis, University of Tubingen, FRG (1990).
1421 M. Jung, D. Schmalzing. and V. Schurig, J. Chromatogr. 552 (1991) 43.
1431 I . Hardt and W.A. Konig, J. Microcol. Sep. 5 (1993) 35.
[44] W.A. Konig, R. Krebber, and G. Wenz, HRC 12 (1989) 641.
Ms received: December 23, 1992 Accepted: March 26, 1993
298 VOL. 16, MAY 1993 Journal of High Resolution Chromatography


















