
Chemical Kinetics and Transition States Elementary Rate Laws k(T) Transition State Theory Catalysis.
23
Chemical Kinetics and Transition States Elementary Rate Laws k(T) Transition State Theory Catalysis
-
date post
20-Dec-2015 -
Category
Documents
-
view
219 -
download
1
Transcript of Chemical Kinetics and Transition States Elementary Rate Laws k(T) Transition State Theory Catalysis.

Chemical Kinetics and Transition States
Elementary Rate Lawsk(T)
Transition State TheoryCatalysis

I. Rate Equation for Elementary Rate Laws
• Rate =d[A]/dt = -k [A]n assuming nth order– [A] is reactant in A B (assume negligible
reverse rxn)– k is rate constant, units of 1/[concn-1-time]– [A] = [A(0)] exp (- kft)
• Now consider A ↔ B with kf rate and kr rate; i.e. there is a substantial back rxn– Then d[A]/dt = - kf[A] + kr[B] = - d[B]/dt
• Experiments rate laws, k and rxn order

Review of Elementary Rate LawsOrder Reaction Differential
Rate Law- d[A]/dt =
Integrated Rate Law[A] = f(t)
Units of k Plot Half-life
0 A P k [A] - [A]0 = -kt M/s [A] vs tSlope = - k
[A]0/2k
1 A P k[A] [A] = [A]0 e-kt 1/s ln[A] vs t
Slope = -k(ln 2)/k
2 A P k[A]2 1/[A]0 - 1/[A] = - kt (M-s)-1 1/[A] vs tSlope = k
1/k [A]0
2 A+ B P
k[A][B] kt = ([B]0 - [A]0)-1 *
ln {[A]0[B]/[B]0[A]}
(M-s)-1 Assume [A]0< [B]0
3 A P k[A]3 1/2 {[A]-2 - [A]0-2} =
kt
1/(M2-s) 3/{2k A]02}
n A P k[A]n α 1/[A]0 n-1

Equilibrium
• At equilibrium, d[A]/dt = 0 forward rate = kf[A] = kr[B] = reverse rate.
– This is the Principle of Detailed Balancing and leads to
– K =[B]eq/[A]eq = kf/kr (recall Eqn 13.23)
– Principle of Microscopic Reversibility

II. Arrhenius Eqn: k(T)
• In Ch 13, we combined the Gibbs-Helmholtz Eqn (G(T)) and the Gibbs Eqn (G = - RT ln K) – to get the van’t Hoff Eqn: d ln K/dT = ho/kT2
• Arrhenius combined the van’t Hoff eqn with K = kf/kb to get
– Differential eqn: d ln kf/dT = Ea/kT2 where Ea = forward activation energy; assume constant to integrate
– Integrated eqn: kf = A exp(-Ea/kT); as T ↑, kf ↑ if Ea > 0 (usual case shown in Fig 19.2 except see Prob 19.10)
– Therefore a plot of ln kf vs 1/T Eaand A (Fig 19.5)

Activation Energy Diagram (Fig 19.3)
• Ea= activation energy for forward rxn
• Ea‘= activation energy for reverse rxn
• ξ = rxn coordinate ho = Ea- Ea‘
• Note that Ea and Ea‘ > 0 (usual case) so ki ↑ with T for endo- and exothermic rxns
• Ex 19.1, Prob 8

III.Transition State Theory (TST)
• TS is at the top of the activation [‡] barrier between reactants and products.
• Energy landscape for chemical rxn– A + BC [A--B--C]‡ = TS AB + C
– Fig 19.7 for collinear rxn: D + H2 HD + H
• See handouts for – H + H2 H2 + H
– F + H2 HF + H

Saddle Point (Fig 19.7)
• Rxn starts in LHS valley (Morse potential) of H2 with D far away.
• D and H2 approach, potential energy ↑
• TS is at max energy along rxn coord; i.e. [H--H--D]‡ exists.
• Then H moves away and valley (another Morse potential) is HD.

Potential Energy Contour Diagram (Fig 19.8)
• The information in Fig 19.7 can be shown as a contour diagram (Fig 19.8).
• Follow rxn A + BC AB + C. Reactants are lower RH corner (energy min) and follow dotted line up to TS and then down to upper LH corner.

krsaddleshop.com

A 360 degree view
• http://my.voyager.net/~desotosaddle/saddle_pictures.htm

TST Rate Constant, k2
• A + B --k2 P overall rxn which proceeds via a TS: A + B K‡ (AB)‡ --k‡ P
• This 2-step mechanism involves an equilibrium between reactants and TS with eq. constant – K‡ = [(AB)‡]/[A][B] = [q‡/qAqB] exp (D‡/kT)
• and then the formation of products from the TS with rate constant k‡.
• d[P]/dt = k‡[(AB)‡] = k‡K‡[A][B] = k2[A][B]

TST: Reaction Coordinate
• d[P]/dt = k‡[(AB)‡] = k‡K‡[A][B] = k2[A][B]
• k2 = k‡K‡ is the connection between kinetics and stat. thermo (partition functions)
• In TST, we hypothesize a TS structure and assume that the reaction coord ξ is associated with the vibrational degree of freedom of the A—B bond that forms.

TST
• Define qξ as the partition function of this weak vibrational deg of freedom and separate it from other degs of freedom (q‡*)
• Then q‡ = q‡* qξ = product of TS q except rxn coord x q of rxn coord
• The reaction coord ξ is associated with a weak bond (small kξ and small ν ξ).

TST
• Then q‡ ≈ κkT/hνξ. • κ = transmission coefficient; 0 < κ ≤ 1.• K‡ = [q‡/qAqB] exp(D‡/kT)
= q‡*qξ/[qAqB] exp(D‡/kT)
= q‡* kT/hνξ /[qAqB] exp(D‡/kT) • k2 = k‡K‡=νξ(kT/hνξ){q‡*/[qAqB]} exp(D‡/kT)
= (kT/h) q‡*/[qAqB] exp(D‡/kT) = (kT/h) K‡*• Ex 19.2

Primary Kinetic Isotope Effect
• When an isotopic substitution is made for an atom at a reacting position (i.e. in the bond that breaks or forms in the TS), the reaction rate constant changes.
• These changes are largest for H/D/T substitutions.
• And can be calculated using eqn for k2 = (kT/h) q‡*/[qAqB] exp(D‡/kT)

Isotope Effect
• Example in text is for breaking the CH (kH) or CD (kD) bond.
• Assume that – q(CH‡)≈q(CD‡) and q(CH) ≈ q(CD)– C-H and C-D have the same force constants.
• Then C-H and C-D bond breakage depends on differences in vibration of reaction coord (ξ) or νCX or reduced mass.

Isotope Effect
• kH /kD = exp [(DCH‡ - DCD
‡ )/kT]
• = exp {-(h/2kT)[νCD - νCH ]}
• = exp {-(hνCH/2kT)[2-1/2 - 1]} since
• ν = (1/2π)√(ks/μ) ks= force const, μ = reduced mass
• Ex 19.3; see Fig 19.9
• Prob 3

Thermodynamic Properties of TS or Activated State (Arrhenius)
• K ‡* = equilibrium constant from reactants to TS without the rxn coordinate ξ.
• Define a set of thermody properties for the TS: G‡ = - kT ln K ‡* = H‡ - TS‡
– k2 = (kT/h) K‡* = (kT/h) exp(-G‡/kT)
= [(kT/h) exp(-S‡/k)] exp(-H‡/kT)
• [term] is related to Arrhenius A and H‡ is related to Ea Prob 6
• k vs T expts H‡, S‡, G‡
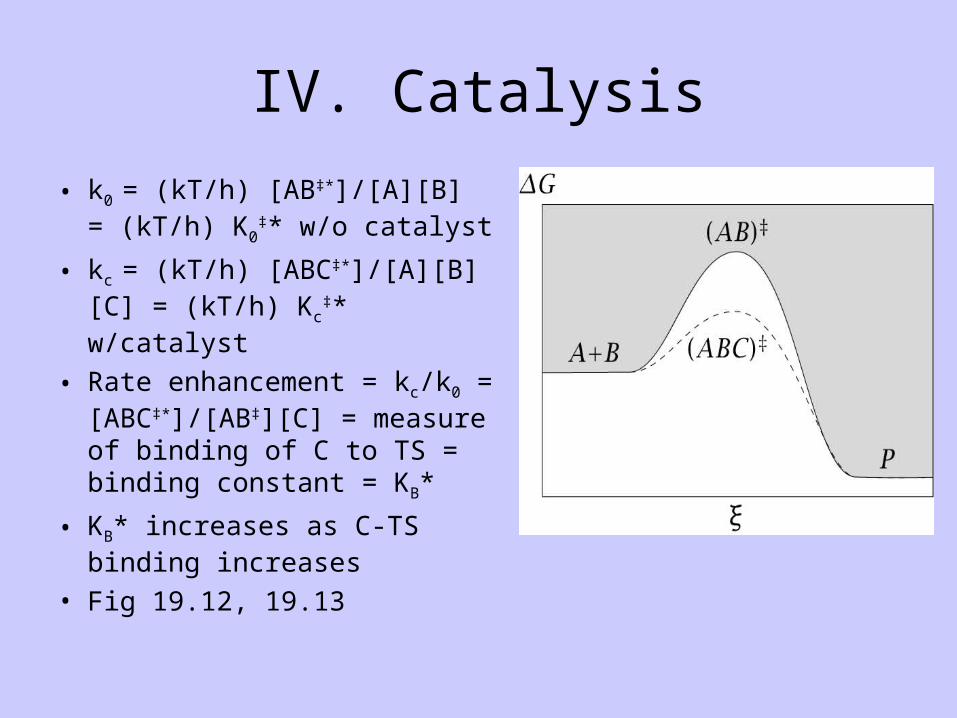
IV. Catalysis
• k0 = (kT/h) [AB‡*]/[A][B] = (kT/h) K0
‡* w/o catalyst
• kc = (kT/h) [ABC‡*]/[A][B][C] = (kT/h) Kc
‡* w/catalyst
• Rate enhancement = kc/k0 = [ABC‡*]/[AB‡][C] = measure of binding of C to TS = binding constant = KB*
• KB* increases as C-TS binding increases
• Fig 19.12, 19.13

Catalysis Mechanisms
• Catalysts stabilize ‡ relative to reactants; this lowers activation barrier.
• T 19.1: create favorable reactant orientation
• T 19.2 and Fig 19.14: reduce effect of polar solvents on dipolar transition state

Acid and Base Catalysis
• Consider a rxn R P catalyzed by H+ Then rate might be = ka [HA] [R]
• H+ is produced in AH ↔ H+ + A-
– Ka = [H+][ A-]/ [AH]
• These two rxns are coupled

Brønsted Law
• log ka = α log Ka + ca
– or log ka = - α pKa + ca
– α > 0 and ca = constant
– As Ka ↑ (stronger acid), rxn rate constant ka ↑
– Plot log ka vs pKa (Fig 19.15)
• This law proposes that presence of acid stabilizes the product.
• Omit pp 361-365


















