
Characterisation of Staphylococcus felis isolated from ... file1 Characterisation of Staphylococcus...
28
RESEARCH REPOSITORY This is the author’s final version of the work, as accepted for publication following peer review but without the publisher’s layout or pagination. The definitive version is available at: https://doi.org/10.1016/j.vetmic.2018.07.002 Worthing, K., Pang, S., Trott, D.J., Abraham, S., Coombs, G.W., Jordan, D., McIntyre, L., Davies, M.R. and Norris, J. (2018) Characterisation of Staphylococcus felis isolated from cats using whole genome sequencing. Veterinary Microbiology http://researchrepository.murdoch.edu.au/id/eprint/41409/ Copyright: © 2018 Elsevier B.V. It is posted here for your personal use. No further distribution is permitted.
Transcript of Characterisation of Staphylococcus felis isolated from ... file1 Characterisation of Staphylococcus...

RESEARCH REPOSITORY
This is the author’s final version of the work, as accepted for publication following peer review but without the publisher’s layout or pagination.
The definitive version is available at:
https://doi.org/10.1016/j.vetmic.2018.07.002
Worthing, K., Pang, S., Trott, D.J., Abraham, S., Coombs, G.W., Jordan, D., McIntyre, L., Davies, M.R. and Norris, J. (2018) Characterisation of Staphylococcus felis isolated from
cats using whole genome sequencing. Veterinary Microbiology
http://researchrepository.murdoch.edu.au/id/eprint/41409/
Copyright: © 2018 Elsevier B.V.
It is posted here for your personal use. No further distribution is permitted.

Accepted Manuscript
Title: Characterisation of Staphylococcus felis isolated fromcats using whole genome sequencing
Authors: Kate Worthing, Stanley Pang, Darren J. Trott, SamAbraham, Geoffrey W Coombs, David Jordan, LiamMcIntyre, Mark R Davies, Jacqueline Norris
PII: S0378-1135(17)31399-8DOI: https://doi.org/10.1016/j.vetmic.2018.07.002Reference: VETMIC 8009
To appear in: VETMIC
Received date: 29-11-2017Revised date: 5-7-2018Accepted date: 5-7-2018
Please cite this article as: Worthing K, Pang S, Trott DJ, Abraham S, Coombs GW,Jordan D, McIntyre L, R Davies M, Norris J, Characterisation of Staphylococcus felisisolated from cats using whole genome sequencing, Veterinary Microbiology (2018),https://doi.org/10.1016/j.vetmic.2018.07.002
This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers thatapply to the journal pertain.

1
Characterisation of Staphylococcus felis isolated from cats using whole genome
sequencing
Kate Worthing a,f, 1, Stanley Pang b,c, Darren J. Trott d, Sam Abraham b, Geoffrey W Coombs
b.c , David Jordan e, Liam McIntyre f, Mark R Davies f, and Jacqueline Norris a#
a University of Sydney, Sydney School of Veterinary Science, NSW, Australia
b Antimicrobial Resistance and Infectious Diseases Research Laboratory, School of
Veterinary Life Sciences, Murdoch University, Murdoch, Western Australia, Australia;
c PathWest Laboratory Medicine – WA, Fiona Stanley Hospital, Murdoch, Western Australia,
Australia;
d Australian Centre for Antimicrobial Resistance Ecology, School of Animal and Veterinary
Sciences, University of Adelaide, Roseworthy, South Australia, Australia;
e New South Wales Department of Primary Industries, Wollongbar, NSW, Australia
f Department of Microbiology and Immunology, at the Peter Doherty Institute for Infection
and Immunity, The University of Melbourne, Victoria, Australia
# Corresponding author: Jacqueline Norris. Sydney School of Veterinary Science, McMaster
Building B14, University of Sydney, Parramatta Rd, NSW, 2006, Australia. Phone: (+612)
9351 7095. Email: [email protected]
1. Present address: University of Melbourne, Peter Doherty Institute for Infection and Immunity, Department
of Microbiology and Immunology, VIC
ACCEPTED MANUSCRIP
T

2
Highlights
Staphylococcus felis isolates were examined using whole genome sequencing and
phenotypic tests.
Gene sequences of putative virulence factors were found in all isolates.
Ninety two percent of isolates were susceptible to all antimicrobials tested.
One isolate caused coagulation of feline plasma.
Abstract
This study used phenotypic tests and whole genome sequencing to characterise a collection of
37 clinical Staphylococcus felis isolates from cats. Samples were isolated from a range of
diseases including feline lower urinary tract disease (n = 15), otitis externa (n= 13), and ocular
disease (n= 2). Isolates were identified using MALDI-TOF MS and by BLASTn analysis of S.
felis-specific 16S rRNA, rpoB and nuc genes in whole genome sequence-based contigs.
Phenotypic antimicrobial resistance was determined using disk diffusion and broth
microdilution. Coagulase activity was assessed using feline and rabbit plasma. Genomes were
screened for putative virulence and antimicrobial resistance genes using the sequences of
known genes from other staphylococci as homologous references. Phylogenetic relationships
were inferred using single nucleotide polymorphisms. One isolate was coagulase-positive
when tested with feline plasma but all isolates were rabbit plasma coagulase-negative. No
genetic determinant of coagulase activity was identified in this isolate. A range of putative
virulence genes were found amongst isolates including genes associated with adhesion, toxin
production and immune evasion. Ninety two percent of isolates were fully susceptible to all
antimicrobials tested, which was reflected by a general absence of resistance genes. Clustering
within the phylogenetic tree suggested a multiclonal population structure; this clustering did
not correlate with disease syndrome or geographic origin of the isolate. Future studies of
ACCEPTED MANUSCRIP
T

3
veterinary staphylococci will benefit from the publicly available S. felis draft genomes that
were generated in this study.
Keywords
Staphylococcus felis, feline medicine, bacterial virulence, antimicrobial resistance,
coagulase-negative staphylococci
Introduction
The resident staphylococcal species for cats, Staphylococcus felis, was first identified as a new
coagulase-negative Staphylococcus species in 1989 (Igimi et al., 1989). The species, found on
the skin of around 25% of healthy cats (Lilenbaum et al., 1998), is the most frequently isolated
Staphylococcus species carried on the skin (Lilenbaum et al., 1998) and in the saliva of cats
(Lilenbaum et al., 1999). S. felis is coagulase-negative when tested using rabbit plasma and
produces incomplete haemolysis on sheep blood agar. Although phenotypically similar to S.
simulans and S. sciuri (Devriese et al., 1984; Igimi et al., 1989), it can be definitively
differentiated from other staphylococci by DNA-DNA hybridization. Due to its phenotypic
similarity to other coagulase-negative staphylococci (CNS), the prevalence of S. felis may have
been underestimated in early studies that did not use advanced molecular methods. While it is
now generally agreed that S. felis is a common cat commensal, its role as a feline pathogen is
more contentious (Higgins and Gottschalk, 1991; Patel et al., 2002; Litster et al., 2007; Litster
et al., 2011; Kwaszewska et al., 2015). S. felis-positive urine samples have significantly higher
pH and are more likely to contain urine crystals (Litster et al., 2007). Like many staphylococcal
species, S. felis produces urease (Igimi et al., 1989), which is the likely cause of the high pH
seen in S. felis-positive urine. As a skin commensal, S. felis could be dismissed as a contaminant
of urine samples. However, Litster et al. (2007) concluded the production of urease and the
ACCEPTED MANUSCRIP
T

4
subsequent increase in urine pH suggest S. felis is a potential feline urinary pathogen. A
subsequent study which examined a single S. felis isolate from a cat’s subcutaneous wound
identified virulence factors such as biofilm and proteolytic enzyme production, demonstrating
the organism’s potential as an opportunistic skin pathogen (Kwaszewska et al., 2015).
Overall, there is a scarcity of literature regarding the epidemiology and potential virulence
factors of S. felis, and the species population structure in cats is unknown. Furthermore, there
have been divergent reports on its antimicrobial susceptibility profile (Igimi et al., 1989; Litster
et al., 2007; Kwaszewska et al., 2015). While methicillin resistance and multidrug resistance
have been identified amongst S. felis isolates (Lilenbaum et al., 1998; Muniz et al., 2013), other
studies have reported low levels of antimicrobial resistance (Litster et al., 2007; Litster et al.,
2011). Typing methods such as multilocus sequence typing (MLST), as well as whole genome
sequencing, have already shed light on the population structure of other veterinary
Staphylococcus species (spp.) such as S. aureus (Enright et al., 2000) and S. pseudintermedius
(Solyman et al., 2013), but similar genomic epidemiology approaches have not been applied to
S. felis. Given the current scarcity of research characterising S. felis, the three aims of this study
were to: 1) investigate potential virulence factors that may aid S. felis in its colonization and
formation of opportunistic infection; 2) investigate the phenotypic and genotypic antimicrobial
profiles of a collection of clinical S. felis isolates; and 3) use whole genome sequencing to infer
the population structure of S. felis amongst Australian cats.
Methods
Collection and identification of S. felis isolates
Isolates were collected in 2013 from 22 veterinary diagnostic laboratories located in all
Australian states and mainland territories as part of the first Australian survey into
ACCEPTED MANUSCRIP
T

5
antimicrobial resistance in veterinary staphylococcal clinical isolates, as previously described
(Saputra et al., 2017). A total of 1080 coagulase-positive staphylococci (CPS) from a range of
animal species were received during the study. Although laboratories were instructed to
forward only CPS to the researchers, 172 CNS isolates were also received from eight of the 22
laboratories across Australia. A total of 74 non-consecutive CPS and CNS isolates were from
cats. For each feline isolate, the geographic postcode of the submitting veterinary clinic, the
clinical syndrome (as recorded by the submitting veterinarian) and the site of isolation were
recorded. Isolates from the urinary bladder, urinary catheters or free catch urine samples were
recorded as feline lower urinary tract isolates. Preliminary staphylococcal speciation was
determined by traditional phenotypic tests (Gram stain, catalase and coagulase testing) and
confirmed by the BD™ Bruker MALDI Biotyper™ with all isolates run in duplicate and results
with a confidence score above 1.8 deemed acceptable for speciation.
All presumptive S. felis isolates underwent whole genome sequencing as previously described
(Worthing et al., 2018a; Worthing et al., 2018b). Briefly, DNA was extracted from
staphylococcal samples using the Qiagen DNA MiniAmp kit using the manufacturer’s
instructions for Gram-positive bacteria, using a 90-minute incubation with 20mg/mL lysozyme
as the cell lysis agent (Qiagen, Valencia, USA). DNA yield and purity (A260/A280) were
measured using the Nanodrop 1000 spectrophotometer (Thermo Scientific, Delaware, USA).
DNA concentration was verified using the QuBit dsDNA kit (Thermo Scientific, Delaware,
USA). DNA library preparation was performed according to the manufacturer’s instructions
(Nextera, Illumina, San Diego, USA). Whole genome sequencing was performed using the
NextSeq 500 sequencer, according to the manufacturer’s instructions (Illumina, San Diego,
USA). De novo assembly of paired-end sequence data was undertaken using SPAdes v3.12.0
in the Nullarbor v1.4.1 bioinformatics pipeline (Seemann et al.). The mean sequencing depth
ACCEPTED MANUSCRIP
T

6
for these 28 sequences was 92.3x and mean genome size of the draft genome assemblies was
2,439,240bp with a mean of 216 contigs. The identity of presumptive S. felis isolates was
confirmed by identification of the species-specific genes, 16s ribosomal RNA (16s rRNA;
Accession number: D83364) (Takahashi et al., 1999); thermonuclease (nuc; Accession
number: AB465335.1) (Sasaki et al., 2010) and RNA polymerase subunit-B (rpoB; Accession
number: AF325878.1) (Drancourt and Raoult, 2002) in whole genome sequenced contigs.
Contigs underwent BLASTn analysis in CLC Genomics Workbench v10 (Qiagen, USA) and
had to display sequence homology and coverage length of >98% with the S. felis 16s rRNA,
nuc and rpoB reference sequences for an isolate to be deemed S. felis.
Coagulase testing
The tube coagulase test was conducted using sterile EDTA-treated rabbit plasma (Sigma
Aldrich, USA) and EDTA-treated feline plasma collected from residual blood samples sent to
the Veterinary School’s diagnostic laboratory. Four colonies were inoculated into 0.3ml plasma
and incubated at 37°C. Tubes were checked for visual clot formation after four hours, and then
incubated overnight at room temperature. Isolates were deemed coagulase-positive if an
immobile clot formed at the bottom of the tube after 4 hours incubation at 37°C or after
overnight incubation at room temperature. The slide agglutination test was used to assess the
presence of clumping factor using rabbit and feline plasma. S. aureus ATCC 29213 was used
as a positive control for all coagulase and clumping factor tests.
Antimicrobial susceptibility testing
Samples in this study were collected as part of a larger surveillance study that, amongst other
aims, sought to compare the relative diagnostic accuracy of disk diffusion and broth
microdilution in antimicrobial susceptibility testing (Abraham et al., 2015; Saputra et al., 2017;
ACCEPTED MANUSCRIP
T

7
Badger et al., 2018). Consequently, two methods of phenotypic antimicrobial susceptibility
testing (AST) were performed. Antimicrobial disk diffusion was undertaken using the direct
colony suspension method according to the Clinical and Laboratory Standards Institute (CLSI)
guidelines for bacteria isolated from animals. The diameter of the zone of inhibition was
recorded and isolates were deemed susceptible or resistant based on CLSI guidelines for
veterinary CNS, where available (CLSI, 2013a). If animal-derived data were not available,
human data from the CLSI guidelines were used. The following antimicrobials were tested by
disk diffusion: amoxicillin-clavulanic acid, cephalothin, cefoxitin, cefovecin, oxacillin,
penicillin, trimethoprim-sulfamethoxazole, ciprofloxacin, enrofloxacin, marbofloxacin,
pradofloxacin, clindamycin, gentamicin, chloramphenicol, rifampicin, azithromycin and
tetracycline. Except for the cefovecin disk (Oxoid, Basingstoke), disks were manufactured by
BD (BD BBLTM Sensi-DiskTM, Sparks). Automated AST was carried out by broth
microdilution using the Vitek 2TM system (bioMerieux, USA), according to the manufacturer’s
instructions. The following antimicrobials were tested: benzylpenicillin, oxacillin,
enrofloxacin, erythromycin, gentamicin, clindamycin, tetracycline, chloramphenicol,
rifampicin and trimethoprim sulfamethoxazole. Minimum inhibitory breakpoints for CNS were
used, as outlined in CLSI guidelines for humans (CLSI, 2013b).
Antimicrobial resistance gene screening
Genome assemblies were screened for antimicrobial resistance genes using a BLASTn
approach against a database of known resistance determinants by uploading their fasta files
onto the open-access bioinformatics website, ResFinder;
https://cge.cbs.dtu.dk//services/ResFinder/) (Zankari et al., 2012). Sequences required >90%
sequence homology and 60% target length to be deemed positive for a particular gene.
Virulence gene screening
ACCEPTED MANUSCRIP
T

8
Putative S. felis virulence genes were identified using BLASTn in CLC Genomics. A local
BLAST database of known S. aureus virulence genes was collated from the open access
repository, VirulenceFinder (https://cge.cbs.dtu.dk/services/VirulenceFinder/) (Joensen et al.,
2014). S. felis sequences with >60% sequence homology and 60% target length to the
VirulenceFinder gene sequences were then screened for homology to sequences from other
staphylococcal species using the BLASTn function of the NCBI website. The following
additional sequences that are described as staphylococcal virulence factors, but not available
from VirulenceFinder were downloaded from the NCBI website: biofilm-associated surface
protein, bap (Cucarella et al., 2001), lysozyme-resistance determinant, peptidoglycan O-
acetyltransferase, oatA (Bera et al., 2005), S. (pseud)intermedius exfoliative toxin, siet
(Terauchi et al., 2003), autolysin/adhesion, atl (Oshida et al., 1995), capsular polysaccharide
synthesis locus, cap (Sau et al., 1997), and coagulase, coa (Phonimdaeng et al., 1990),
(Accession numbers: AY220730.1, CP009046.1, AB099710.1, D17366.1, U81973.1 and
X17679.1 respectively).
Phylogenetic analysis
A phylogenetic tree was generated using a maximum-likelihood algorithm that inferred
phylogeny based on a mapped alignment of high quality single nucleotide polymorphisms
(SNPs) relative to the S. felis reference genome ATCC49168 (Accession number: CP027770.1)
(dos Santos et al., 2018). Core genome SNPs were identified using Snippy v3.2 in the Nullarbor
pipeline (Seemann et al.). SNPs associated with putative homologous recombination events
were identified and purged using Gubbins v2.3.2 (Croucher et al., 2014). Gubbins was also
used to calculate the r/m ratio of each isolate, which is a measure of nucleotide substitution
rates due to recombination compared to mutation (Vos and Didelot, 2009). The core genome
ACCEPTED MANUSCRIP
T

9
alignment with filtered SNP data was then imported into IQ-TREE v1.6.5, which was used
with a generalised time reversible plus gamma substitution model to generate a maximum
likelihood tree with 100 bootstraps (Tamura et al., 2013; Nguyen et al., 2014). Metadata was
added to the tree using the online platform, Interactive tree of life (iTOL v3, http://itol/embl/de)
(Letunic and Bork, 2016).
Results
Identification of S. felis isolates
Of the 74 isolates received from cats, 28 were CPS and 46 were CNS. Thirty-eight isolates
were initially identified as S. felis using MALDI-TOF, which accounted for 50% of the
staphylococcal isolates cultured from cats. One isolate, presumptively identified as S. felis by
MALDI-TOF, was found to be contaminated with S. aureus DNA when it underwent whole
genome sequencing; this isolate was removed from the study resulting in a final S. felis
collection of 37 isolates. S. felis submissions originated from four diagnostic laboratories, with
25 submissions from veterinary clinics in New South Wales (NSW), five in Victoria (Vic), two
in Queensland (QLD) and two from South Australia (SA). The postcode of the submitting
veterinary clinic was not recorded in three submissions. The geographic distribution of S. felis
submissions was roughly proportional to the number of coagulase-negative isolates received
over the whole study. The remaining cat isolates were identified as S. aureus (n= 18), S.
pseudintermedius (10), S. epidermidis (3), S. haemolyticus (2), and S. warneri (1). No species
could be identified for two coagulase-negative isolates, despite the isolates undergoing
MALDI-TOF in triplicate. These isolates were removed from the study. S. felis was isolated
from a range of diseases in cats including feline lower urinary tract disease (FLUTD; n= 15),
ACCEPTED MANUSCRIP
T

10
otitis externa (13), ocular disease (2), and pyoderma (1) (Table 1). For the remaining six cats,
one had acute renal failure, two had neurological signs, and for three, a clinical diagnosis was
not recorded.
As sufficient DNA purity (A260/A280 >1.8) could not be obtained from nine isolates, whole
genome sequencing could only be performed on 28 presumptive S. felis isolates. Species
identification was confirmed by demonstrating >98% homology of the S. felis-specific 16s
rRNA, rpoB and nuc genes within the 28 draft genome assemblies.
Antimicrobial resistance amongst S. felis isolates
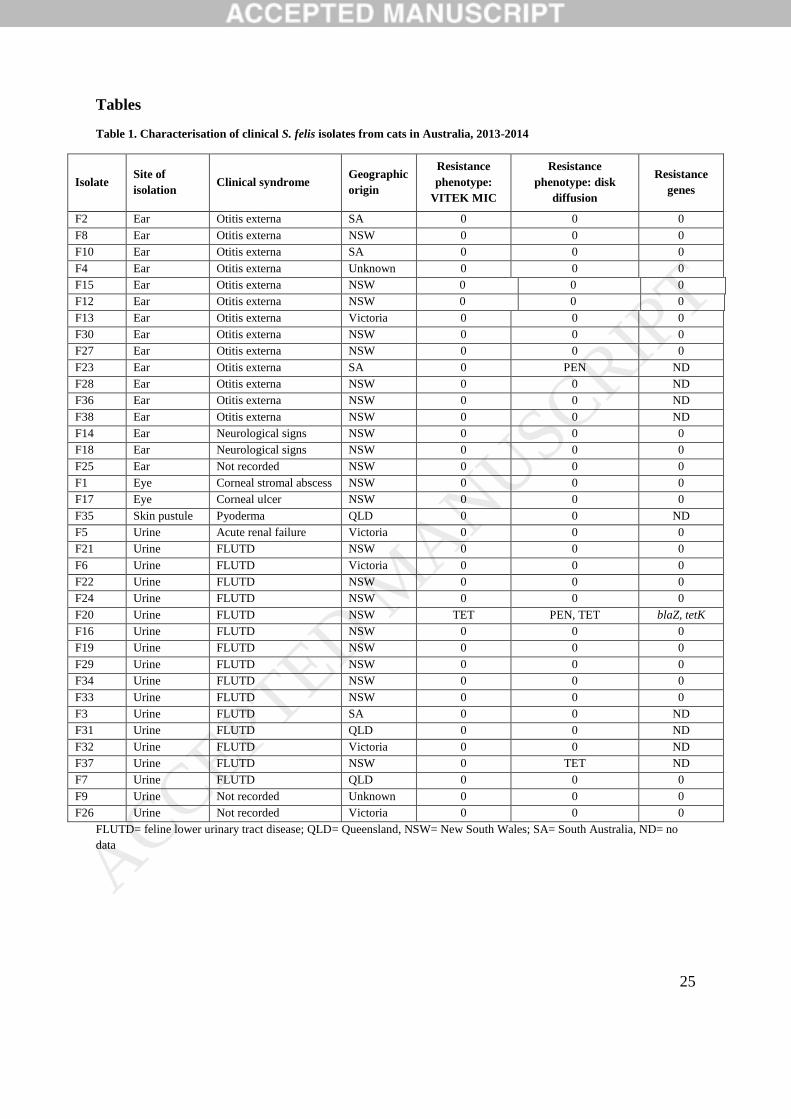
Thirty four of the 37 (92%) S. felis isolates that underwent phenotypic AST showed full
susceptibility to all antimicrobials tested (Table 1). By Vitek testing, apart from one
tetracycline-resistant isolate (MIC>1mg/L), all isolates were susceptible to all the
antimicrobials tested. Disk diffusion susceptibility testing detected additional penicillin
resistance in this same tetracycline-resistant isolate, as well as a second tetracycline-resistant
isolate (zone diameter <14mm) and additional penicillin-resistant isolate (zone diameter
<28mm). Resistance genes were rare, with blaZ and tetK found in one phenotypically
penicillin/tetracycline-resistant isolate. The remaining two penicillin-resistant and tetracycline-
resistant isolates were amongst the nine isolates for which no genomic data was available;
consequently, it is unknown whether they harboured the relevant resistance genes.
Virulence factors
One isolate (F30) was repeatedly coagulase-positive when tested using feline plasma; the
remaining isolates were coagulase-negative. F30 was coagulase-negative at four hours but
positive at 24 hours. No isolates were coagulase-positive when tested with rabbit plasma. All
isolates were negative for the clumping factor test using rabbit and feline plasma. Although all
ACCEPTED MANUSCRIP
T

11
isolates including F30 were screened for S. aureus coagulase (coa), and von Willebrand factor
binding protein (vWbp), no isolates harboured homologues of these genes associated with
phenotypic coagulase activity (Bjerketorp et al., 2004). Consequently, no genetic determinant
was identified for coagulation of feline plasma by F30. The genome size (2,463,058bp) and
G+C content (36.5%) were similar to the average of all 28 genomes examined (2,439,240bp
and 37.3%) so it did not appear that F30 had been contaminated with another organism. All
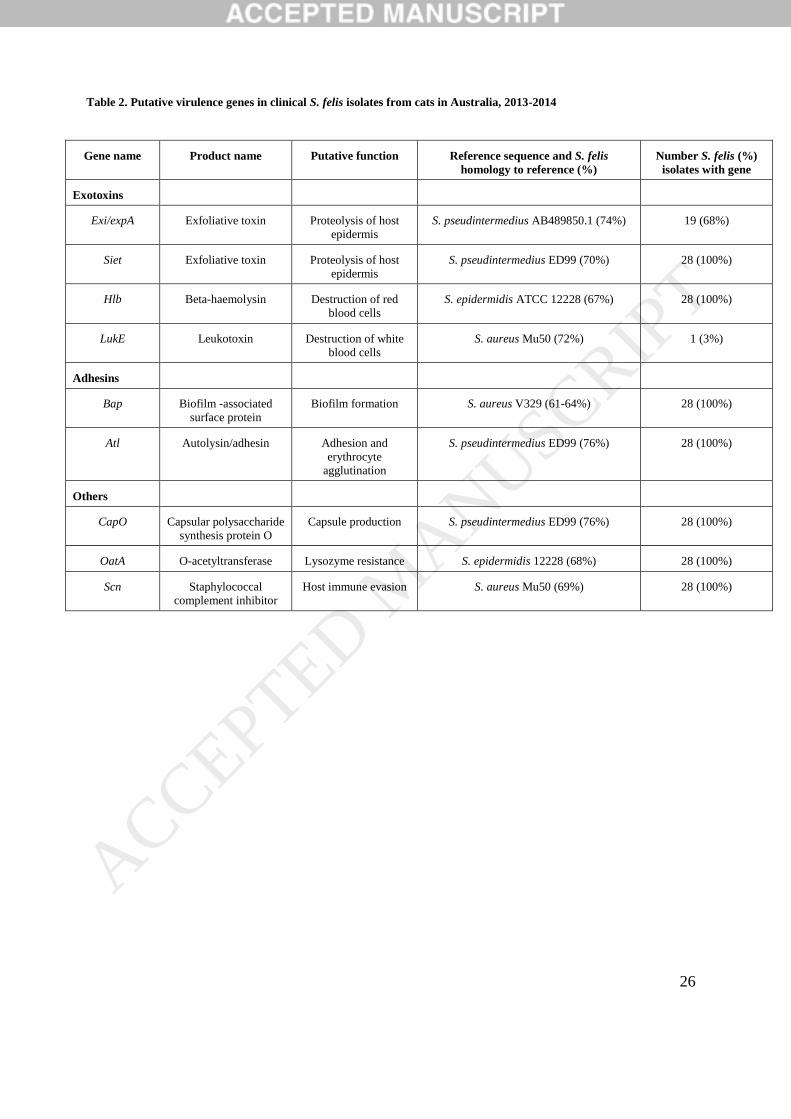
isolates harboured putative homologues of the gene sequences for β-haemolysin (hlb), biofilm-
associated protein (bap), the lysozyme-resistance determinant, O-acetyltransferase (oatA),
autolysin/adhesion (atl), exfoliative toxin (siet) and capsular polysaccharide synthesis protein,
capO (Table 2). All isolates harboured a gene with 69% homology to the S. aureus- derived
immune-evasion gene, scn. Additionally, 19/28 (68%) of isolates possessed a putative gene
with 74% sequence homology to the S. pseudintermedius exfoliative toxin gene, exi. None of
the isolates harboured homologues of the enterotoxins from the VirulenceFinder database.
Phylogenetic analysis
On average, 88.25% of the sequence reads mapped to the 2,479,423bp reference genome, S.
felis ATCC49168 (range 86.3% to 92.1%). A total of 38,705 core genome SNPs were
identified, of which 34,686 (89.6%) were identified by Gubbins as likely residing within
regions of homologous recombination. As these regions distort vertically inherited
polymorphisms, these SNPs were excluded from phylogenetic inferences. A resulting
alignment of 4,019 high quality core genome SNPs was used to assess the genetic relatedness
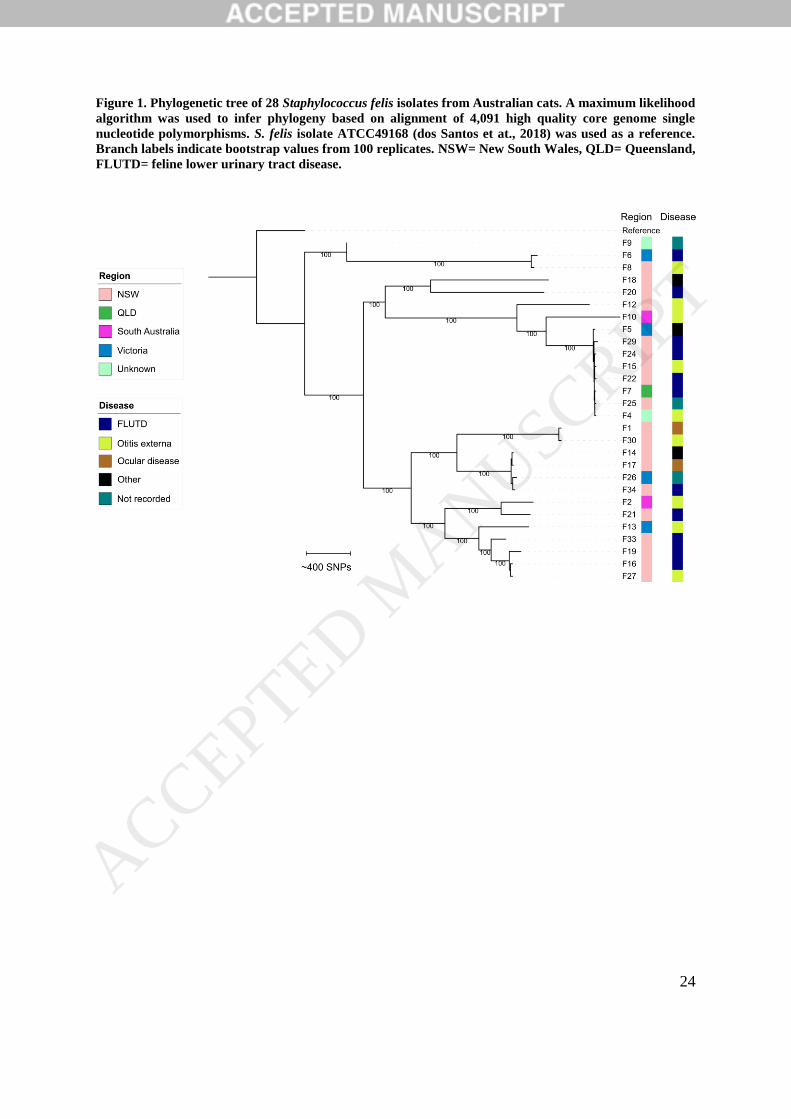
of the S. felis population. Several clusters were identified in the phylogenetic tree, suggesting
a multiclonal population structure. Isolates did not cluster according to disease syndrome or
geographic origin (Figure 1). The largest cluster, with eight closely related isolates, was
ACCEPTED MANUSCRIP
T

12
geographically diverse and included isolates from three different regions. The median r/m ratio
of S. felis isolates was 0.4 (range= 0.03 to 7.8), indicating a generally low but variable level of
recombination amongst the collection (Vos and Didelot, 2009).
Discussion
This is the first study to use whole genome sequencing to identify putative virulence factors in
S. felis and the first to report phenotypic coagulation of feline plasma by a S. felis isolate.
Despite being screened for known homologues of coagulase genes, no genetic determinant for
this phenotypic coagulation could be identified. A future study that uses long read sequence
technology on isolate F30 is now warranted to identify potentially novel determinants of
coagulation of feline plasma by F30. As the coagulase homologue was only present in one
isolate, acquisition via horizontal gene transfer seems more likely than a convergently evolved
coagulase homologue. Staphylococcal species with variable coagulase phenotypes and
genotypes have previously been reported, such as S. agnetis isolated from bovine mastitis
samples and broiler chickens (Taponen et al., 2012; Al-Rubaye et al., 2015). It is interesting
that the coagulase-positive isolate was only positive when tested with feline plasma. Thus, it
would still be deemed coagulase-negative if tested using the rabbit plasma that is traditionally
used in diagnostic laboratories. Variation in coagulation activity against different host species’
plasma has previously been observed in staphylococcal von Willebrand factor-binding protein
(vWbp), which is one of the two coagulases produced by S. aureus (Bjerketorp et al., 2004).
The presence of a feline plasma coagulase-positive isolate shows that S. felis may possess host-
specific virulence determinants towards its main host species, cats.
ACCEPTED MANUSCRIP
T

13
Staphylococcal virulence factors are generally divided into four categories: exoenzymes,
exotoxins, adhesins and others (Kuroda et al., 2001; Zhang et al., 2003). The S. felis isolates in
this study demonstrated two or more gene homologues from each category. Consistent with
previous reports, the most common infections amongst S. felis isolates were urinary tract
infections and otitis externa (Higgins and Gottschalk, 1991; Igimi et al., 1989; Litster et al.,
2011). Genomic analysis showed that most S. felis isolates harboured homologues of putative
virulence genes that have been shown to play a role in cystitis and skin infections caused by
other staphylococcal species. Specifically, 100% and 69% of isolates harboured siet and exi
homologues respectively, which are exfoliative toxin genes implicated in blister formation in
staphylococcal dermatoses (Iyori et al., 2011). Additionally, all isolates harboured an atl gene
homologue of the which, in addition to conferring host blood cell agglutination in S. aureus
isolates (Heilmann et al., 2005), is implicated in adhesion to uroepithelium by the human
urinary pathogen, S. saprophyticus (Hell et al., 1998; von Eiff et al., 2002). Our results add
further evidence that S. felis is a feline urinary pathogen, as suggested by Litster et al. (2007).
Lysozyme is a host-produced enzyme that causes bacterial cell lysis (Bera et al., 2005). Bera
et al. (2005) examined a group of staphylococci from several different species and noted
lysozyme-resistant staphylococcal species were human pathogens, while lysozyme-sensitive
species were not pathogenic in humans. The authors concluded that lysozyme resistance is a
virulence marker amongst Staphylococcus species. All S. felis isolates in our study had an oatA
gene homologue, which has been postulated to be the main determinant of lysozyme resistance
in staphylococci (Bera et al., 2005). S. felis is a commensal of feline skin and is found in saliva
(Lilenbaum et al., 1999; Lilenbaum et al., 1998), both of which produce lysozyme (Fleming,
1922), so it is logical that it would require lysozyme resistance to survive constant exposure to
host lysozyme. Although phenotypic assays of lysozyme resistance were not undertaken in our
ACCEPTED MANUSCRIP
T

14
study, the inability to obtain adequate DNA purity in nine isolates could potentially be
explained by the use of lysozyme as the cell lysis agent in our DNA extraction protocol. Most
isolates were lysed with lysozyme, which could suggest that oatA-mediated resistance was
overwhelmed by the higher concentration (20mg/mL) and longer exposure time (90 minutes)
used in our DNA extraction protocol compared to what might be expected in vivo. Lysozyme-
resistance may have hampered effective cell lysis in the remaining nine isolates. Future studies
are required to determine whether the putative virulence gene homologues identified in our
study confer phenotypic virulence in vivo.
The isolates in our study had low levels of phenotypic antimicrobial resistance and very few
harboured antimicrobial resistance genes. This is in contrast to other the staphylococcal species
collected from cats in the same surveillance study (Saputra et al., 2017; Worthing et al., 2018a)
and to other coagulase-negative species in general (Koksal et al., 2009). Our findings are in
keeping with other studies that have found low levels of antimicrobial resistance amongst
clinical S. felis isolates (Litster et al., 2007; Litster et al., 2011). Possible reasons for the absence
of antimicrobial resistance amongst clinical S. felis isolates include: a fitness cost for resistant
isolates, allowing them to be outcompeted by other commensals, or the practice of screening
for occult urinary tract infections in cats, which entails sending a sample of urine collected by
cystocentesis for culture and susceptibility as part of a diagnostic work-up, prior to empirical
therapy (Litster et al., 2009). Alternatively, via mechanisms such as restriction modification
systems, the S. felis genome could be disinclined to acquire and maintain mobile genetic
elements that confer resistance. While beyond the scope of the current study, generation of
draft genome sequences for 28 isolates will allow for future detailed examination of the
presence or absence of mobile genetic elements within the S. felis population. Disk diffusion
results appeared to show better agreement with resistance genotypes than the Vitek automated
ACCEPTED MANUSCRIP
T

15
MIC testing, but our small sample size prevented statistical confirmation of this observation.
Thus, until a robust statistical comparison of the relative diagnostic accuracy of disk diffusion
and Vitek AST can be conducted for S. felis, as has been conducted for Escherichia coli from
animals (Badger et al., 2018), we suggest diagnostic laboratories use disk diffusion over
automated testing to obtain accurate antimicrobial resistance profiles for S. felis.
Whole genome sequencing and analysis of single nucleotide polymorphisms were used to infer
relatedness amongst this group of S. felis isolates. Although our sample size is small, clustering
in the phylogenetic tree suggests that S. felis may exhibit a multiclonal population structure. A
high proportion of SNPs were flagged as residing within areas of recombination (Croucher et
al., 2014), but this proportion could be overestimated if SNPs were located within repeat
regions or mobile genetic elements rather than true regions of homologous recombination. The
median recombination to mutation ratio was low (0.4) suggesting that, similar to S. aureus,
successful strains of S. felis arise from variation attributable to point mutations rather than
recombination (Feil et al., 2003; Croucher et al., 2014; Chang et al., 2016). However, there was
a range of r/m values amongst the collection, suggesting there may be lineage-specific variation
in the rate of recombination amongst S. felis isolates. Such lineage-specific variation in r/m
ratios has also been observed in methicillin-resistant S. aureus (Chang et al., 2016). Future
studies with larger samples could examine the overall trend in r/m ratios across the species and
would also benefit from comparison of carriage and infection isolates from wild and
domesticated cats with isolates from the current study to allow a deeper understanding of the
S. felis population structure.
This study is limited by the absence of details regarding the initial culture conditions of each
isolate by the referring laboratory. We therefore cannot determine whether isolates came from
pure cultures, which would indicate that S. felis was truly the causative agent of the infections
ACCEPTED MANUSCRIP
T

16
seen in this study. There is a chance that isolates came from mixed or light growth cultures,
which would make it difficult to determine whether S. felis was the causative agent or merely
a contaminant. Future studies would benefit from recording the quality of cultures received
from infectious samples in order to prove that S. felis was the most likely causative agent of
the diseases seen. Secondly, besides phenotypic testing of the presence of coagulase, we did
not confirm the putative functions of virulence genes homologues by undertaking phenotypic
testing. Consequently, subsequent studies should compare genotype with phenotype to show
that S. felis does indeed possess the necessary array of virulence factors to be considered a true
pathogen of cats.
In conclusion, we examined a population of 37 S. felis clinical isolates from Australian cats
and found one was feline plasma coagulase-positive suggesting that host-specific coagulation
can occur in this species. Isolates mostly came from urinary tract infections and otitis, and all
isolates demonstrated sufficient putative virulence genes to suggest that they are capable of
causing disease in these body systems.
Acknowledgements
We acknowledge the assistance and support of all private, government and university
veterinary diagnostic laboratories within Australia for the provision of isolates. We gratefully
thank Dr Thomas Gottlieb, Charlotte Webster, John Huynh and the team at the Department of
Microbiology and Infectious Diseases at Concord Hospital (NSW, Australia) for their
assistance in using MALDI-TOF. We wish to acknowledge the Sydney Informatics Hub and
University of Sydney Core Research Facilities for providing subsidized access to CLC
Genomics Workbench and associated support. We thank Tanya Laird and Emily Hudson for
their assistance in processing the isolates, and Seamus O’Reilly for his ongoing support in
reviewing this manuscript.
ACCEPTED MANUSCRIP
T

17
Funding information
This work was supported by Zoetis Pty Ltd and the Australian Research Council- Linkage
Grant (grant number LP130100736).
Transparency declaration
Sam Abraham and Darren Trott have previously received funds from Zoetis Pty Ltd.
References
Abraham, S., Jordan, D., Wong, H.S., Johnson, J.R., Toleman, M.A., Wakeham, D.L.,
Gordon, D.M., Turnidge, J.D., Mollinger, J.L., Gibson, J.S., 2015. First detection of
extended-spectrum cephalosporin-and fluoroquinolone-resistant Escherichia coli in
Australian food-producing animals. J. Glob. Antimicrob. Resist. 3, 273-277.
Al-Rubaye, A.A.K., Couger, M.B., Ojha, S., Pummill, J.F., Koon, J.A., Wideman, R.F.,
Rhoads, D.D., 2015. Genome Analysis of Staphylococcus agnetis, an Agent of
Lameness in Broiler Chickens. PLoS One 10, 18.
Badger, S., Abraham, S., Saputra, S., Trott, D.J., Turnidge, J., Mitchell, T., Caraguel, C.G.B.,
Jordan, D., 2018. Relative performance of antimicrobial susceptibility assays on
clinical Escherichia coli isolates from animals. Vet. Microbiol. 214, 56-64.
Bera, A., Herbert, S., Jakob, A., Vollmer, W., Götz, F., 2005. Why are pathogenic
staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is
the major determinant for lysozyme resistance of Staphylococcus aureus. Mol.
Microbiol. 55, 778-787.
Bjerketorp, J., Jacobsson, K., Frykberg, L., 2004. The von Willebrand factor-binding protein
(vWbp) of Staphylococcus aureus is a coagulase. FEMS Microbiol. Lett. 234, 309-
314.
ACCEPTED MANUSCRIP
T

18
Chang, H.-H., Dordel, J., Donker, T., Worby, C.J., Feil, E.J., Hanage, W.P., Bentley, S.D.,
Huang, S.S., Lipsitch, M., 2016. Identifying the effect of patient sharing on between-
hospital genetic differentiation of methicillin-resistant Staphylococcus aureus.
Genome Med. 8, 18.
CLSI 2013a. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests
for Bacteria Isolated From Animals; Approved Standard—Fourth Edition. In CLSI
document VET01-A4 (Wayne, PA, USA, CLSI).
CLSI, 2013b. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Third
Informational Supplement. CLSI Document M02-A11 and M100-S23.
Croucher, N.J., Page, A.J., Connor, T.R., Delaney, A.J., Keane, J.A., Bentley, S.D., Parkhill,
J., Harris, S.R., 2014. Rapid phylogenetic analysis of large samples of recombinant
bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15-e15.
Cucarella, C., Solano, C., Valle, J., Amorena, B., Lasa, Í., Penadés, J.R., 2001. Bap, a
Staphylococcus aureus Surface Protein Involved in Biofilm Formation. J. Bacteriol.
183, 2888-2896.
Devriese, L., Nzuambe, D., Godard, C., 1984. Identification and characterization of
staphylococci isolated from cats. Vet. Microbiol. 9, 279-285.
dos Santos, A.C.M., Jie, R., Godoy, H.A., Alves, M., Pombert, J.-F., 2018. Complete
Genome Sequences of the Potential Zoonotic Pathogens Staphylococcus felis and
Staphylococcus kloosii. Genome Announce. 6, e00404-00418.
Drancourt, M., Raoult, D., 2002. rpoB gene sequence-based identification of Staphylococcus
species. J. Clin. Microbiol. 40, 1333-1338. ACCEPTED MANUSCRIP
T

19
Enright, M.C., Day, N.P.J., Davies, C.E., Peacock, S.J., Spratt, B.G., 2000. Multilocus
Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-
Susceptible Clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008-1015.
Feil, E.J., Cooper, J.E., Grundmann, H., Robinson, D.A., Enright, M.C., Berendt, T.,
Peacock, S.J., Smith, J.M., Murphy, M., Spratt, B.G., Moore, C.E., Day, N.P., 2003.
How clonal is Staphylococcus aureus? J. Bacteriol. 185, 3307-3316.
Fleming, A., 1922. On a remarkable bacteriolytic element found in tissues and secretions.
Proc. R. Soc. Lond. B. Biol. Sci. 93, 306-317.
Heilmann, C., Hartleib, J., Hussain, M.S., Peters, G., 2005. The multifunctional
Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen
and fibronectin. Infect. Immun. 73.
Hell, W., Meyer, H.G., Gatermann, S.G., 1998. Cloning of aas, a gene encoding a
Staphylococcus saprophyticus surface protein with adhesive and autolytic properties.
Mol. Microbiol. 29, 871-881.
Higgins, R., Gottschalk, M., 1991. Québec. Isolation of Staphylococcus felis from cases of
external otitis in cats. Can. Vet. J. 32, 312-313.
Igimi, Kawamura, S., Takahashi, E., Mitsuoka, T., 1989. Staphylococcus felis, a New Species
from Clinical Specimens from Cats. Int. J. Syst. Bacteriol. 39, 373-377.
Iyori, K., Futagawa-Saito, K., Hisatsune, J., Yamamoto, M., Sekiguchi, M., Ide, K., Son,
W.G., Olivry, T., Sugai, M., Fukuyasu, T., Iwasaki, T., Nishifuji, K., 2011.
Staphylococcus pseudintermedius exfoliative toxin EXI selectively digests canine
desmoglein 1 and causes subcorneal clefts in canine epidermis. Vet. Dermatol. 22,
319-326.
ACCEPTED MANUSCRIP
T

20
Joensen, K.G., Scheutz, F., Lund, O., Hasman, H., Kaas, R.S., Nielsen, E.M., Aarestrup,
F.M., 2014. Real-time whole-genome sequencing for routine typing, surveillance, and
outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501-
1510.
Koksal, F., Yasar, H., Samasti, M., 2009. Antibiotic resistance patterns of coagulase-negative
staphylococcus strains isolated from blood cultures of septicemic patients in Turkey.
Microbiol. Res. 164, 404-410.
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi,
A., Aoki, K., Nagai, Y., Lian, J., Ito, T., Kanamori, M., Matsumaru, H., Maruyama,
A., Murakami, H., Hosoyama, A., Mizutani-Ui, Y., Takahashi, N.K., Sawano, T.,
Inoue, R., Kaito, C., Sekimizu, K., Hirakawa, H., Kuhara, S., Goto, S., Yabuzaki, J.,
Kanehisa, M., Yamashita, A., Oshima, K., Furuya, K., 2001. Whole genome
sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225-1240.
Kwaszewska, A., Lisiecki, P., Szemraj, M., Szewczyk, E.M., 2015. Animal Staphylococcus
felis with the potential to infect human skin. Med. Dosw. Mikrobiol. 67, 69-78.
Letunic, I., Bork, P., 2016. Interactive tree of life (iTOL) v3: an online tool for the display
and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242-W245.
Lilenbaum, W., Esteves, A.L., Souza, G.N., 1999. Prevalence and antimicrobial susceptibility
of staphylococci isolated from saliva of clinically normal cats. Lett. Appl. Microbiol.
28, 448-452.
Lilenbaum, W., Nunes, E., Azeredo, M., 1998. Prevalence and antimicrobial susceptibility of
staphylococci isolated from the skin surface of clinically normal cats. Lett. Appl.
Microbiol. 27, 224-228.
ACCEPTED MANUSCRIP
T

21
Litster, A., Moss, S., Platell, J., Trott, D.J., 2009. Occult bacterial lower urinary tract
infections in cats—urinalysis and culture findings. Vet. Microbiol. 136, 130-134.
Litster, A., Moss, S.M., Honnery, M., Rees, B., Trott, D.J., 2007. Prevalence of bacterial
species in cats with clinical signs of lower urinary tract disease: Recognition of
Staphylococcus felis as a possible feline urinary tract pathogen. Vet. Microbiol. 121,
182-188.
Litster, A., Thompson, M., Moss, S., Trott, D., 2011. Feline bacterial urinary tract infections:
An update on an evolving clinical problem. Vet. J. 187, 18-22.
Muniz, I., Penna, B., Lilenbaum, W., 2013. Meticillin-resistant commensal staphylococci in
the oral cavity of healthy cats: a reservoir of meticillin resistance. Vet. Rec. 173, 502.
Nguyen, L.-T., Schmidt, H.A., von Haeseler, A., Minh, B.Q., 2014. IQ-TREE: a fast and
effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol.
Biol. Evol. 32, 268-274.
Oshida, T., Sugai, M., Komatsuzawa, H., Hong, Y.-M., Suginaka, H., Tomasz, A., 1995. A
Staphylococcus aureus autolysin that has an N-acetylmuramoyl-L-alanine amidase
domain and an endo-beta-N-acetylglucosaminidase domain: cloning, sequence
analysis, and characterization. Proc. Nat. Acad. Sci. 92, 285-289.
Patel, A., Lloyd, D., Howell, S., Noble, W., 2002. Investigation into the potential
pathogenicity of Staphylococcus felis in a cat. Vet. Rec. 150, 668-669.
Phonimdaeng, P., O'Reilly, M., Nowlan, P., Bramley, A.J., Foster, T.J., 1990. The coagulase
of Staphylococcus aureus 8325-4. Sequence analysis and virulence of site-specific
coagulase-deficient mutants. Mol. Microbiol. 4, 393-404.
Saputra, S., Jordan, D., Worthing, K.A., Norris, J.M., Wong, H.S., Abraham, R., Trott, D.J.,
Abraham, S., 2017. Antimicrobial resistance in coagulase-positive staphylococci
ACCEPTED MANUSCRIP
T

22
isolated from companion animals in Australia: A one year study. PLoS One 12,
e0176379.
Sasaki, T., Tsubakishita, S., Tanaka, Y., Sakusabe, A., Ohtsuka, M., Hirotaki, S., Kawakami,
T., Fukata, T., Hiramatsu, K., 2010. Multiplex-PCR method for species identification
of coagulase-positive staphylococci. J. Clin. Microbiol. 48, 765-769.
Sau, S., Bhasin, N., Wann, E.R., Lee, J.C., Foster, T.J., Lee, C.Y., 1997. The Staphylococcus
aureus allelic genetic loci for serotype 5 and 8 capsule expression contain the type-
specific genes flanked by common genes. Microbiology 143, 2395-2405.
Seemann, T., Goncalves da Silva, A., Bulach, D., Schultz, M., Kwong, J., Howden, B.
Nullarbor. San Francisco; Github.[Accessed: 04 June 2018].
Solyman, S.M., Black, C.C., Duim, B., Perreten, V., Duijkeren, E.v., Wagenaar, J.A.,
Eberlein, L.C., Sadeghi, L.N., Videla, R., Bemis, D.A., Kania, S.A., 2013. Multilocus
sequence typing for characterization of Staphylococcus pseudintermedius. J. Clin.
Microbiol. 51, 306-310.
Takahashi, T., Satoh, I., Kikuchi, N., 1999. Phylogenetic relationships of 38 taxa of the genus
Staphylococcus based on 16s rRNA gene sequence analysis. Int. J. Syst. Evol.
Microbiol. 49, 725-728.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S., 2013. MEGA6: molecular
evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725-2729.
Taponen, S., Supré, K., Piessens, V., Van Coillie, E., De Vliegher, S., Koort, J.M., 2012.
Staphylococcus agnetis sp. nov., a coagulase-variable species from bovine subclinical
and mild clinical mastitis. Int. J. Syst. Evol. Microbiol. 62, 61-65. ACCEPTED MANUSCRIP
T

23
Terauchi, R., Sato, H., Endo, Y., Aizawa, C., Maehara, N., 2003. Cloning of the gene coding
for Staphylococcus intermedius exfoliative toxin and its expression in Escherichia
coli. Vet. Microbiol. 94, 31-38.
von Eiff, C., Peters, G., Heilmann, C., 2002. Pathogenesis of infections due to coagulase
negative staphylococci. Lancet Infect. Dis. 2, 677-685.
Vos, M., Didelot, X., 2009. A comparison of homologous recombination rates in bacteria and
archaea. ISME J. 3, 199-208.
Worthing, K.A., Abraham, S., Coombs, G.W., Pang, S., Saputra, S., Jordan, D., Trott, D.J.,
Norris, J.M., 2018a. Clonal diversity and geographic distribution of methicillin-
resistant Staphylococcus pseudintermedius from Australian animals: Discovery of
novel sequence types. Vet. Microbiol. 213, 58-65.
Worthing, K.A., Abraham, S., Pang, S., Coombs, G.W., Saputra, S., Jordan, D., Wong, H.S.,
Abraham, R.J., Trott, D.J., Norris, J.M., 2018b. Molecular Characterization of
Methicillin-Resistant Staphylococcus aureus Isolated from Australian Animals and
Veterinarians. Microb. Drug Resist. 24, 203-212.
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O.,
Aarestrup, F.M., Larsen, M.V., 2012. Identification of acquired antimicrobial
resistance genes. J. Antimicrob. Chemother. 67, 2640-2644.
Zhang, Y.Q., Ren, S.X., Li, H.L., Wang, Y.X., Fu, G., Yang, J., Qin, Z.Q., Miao, Y.G.,
Wang, W.Y., Chen, R.S., 2003. Genome‐based analysis of virulence genes in a non‐
biofilm‐forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol.
49, 1577-1593.
Figure legends
ACCEPTED MANUSCRIP
T
24
Figure 1. Phylogenetic tree of 28 Staphylococcus felis isolates from Australian cats. A maximum likelihood
algorithm was used to infer phylogeny based on alignment of 4,091 high quality core genome single
nucleotide polymorphisms. S. felis isolate ATCC49168 (dos Santos et at., 2018) was used as a reference.
Branch labels indicate bootstrap values from 100 replicates. NSW= New South Wales, QLD= Queensland,
FLUTD= feline lower urinary tract disease.
ACCEPTED MANUSCRIP
T
25
Tables
Table 1. Characterisation of clinical S. felis isolates from cats in Australia, 2013-2014
Isolate Site of
isolation Clinical syndrome
Geographic
origin
Resistance
phenotype:
VITEK MIC
Resistance
phenotype: disk
diffusion
Resistance
genes
F2 Ear Otitis externa SA 0 0 0
F8 Ear Otitis externa NSW 0 0 0
F10 Ear Otitis externa SA 0 0 0
F4 Ear Otitis externa Unknown 0 0 0
F15 Ear Otitis externa NSW 0 0 0
F12 Ear Otitis externa NSW 0 0 0
F13 Ear Otitis externa Victoria 0 0 0
F30 Ear Otitis externa NSW 0 0 0
F27 Ear Otitis externa NSW 0 0 0
F23 Ear Otitis externa SA 0 PEN ND
F28 Ear Otitis externa NSW 0 0 ND
F36 Ear Otitis externa NSW 0 0 ND
F38 Ear Otitis externa NSW 0 0 ND
F14 Ear Neurological signs NSW 0 0 0
F18 Ear Neurological signs NSW 0 0 0
F25 Ear Not recorded NSW 0 0 0
F1 Eye Corneal stromal abscess NSW 0 0 0
F17 Eye Corneal ulcer NSW 0 0 0
F35 Skin pustule Pyoderma QLD 0 0 ND
F5 Urine Acute renal failure Victoria 0 0 0
F21 Urine FLUTD NSW 0 0 0
F6 Urine FLUTD Victoria 0 0 0
F22 Urine FLUTD NSW 0 0 0
F24 Urine FLUTD NSW 0 0 0
F20 Urine FLUTD NSW TET PEN, TET blaZ, tetK
F16 Urine FLUTD NSW 0 0 0
F19 Urine FLUTD NSW 0 0 0
F29 Urine FLUTD NSW 0 0 0
F34 Urine FLUTD NSW 0 0 0
F33 Urine FLUTD NSW 0 0 0
F3 Urine FLUTD SA 0 0 ND
F31 Urine FLUTD QLD 0 0 ND
F32 Urine FLUTD Victoria 0 0 ND
F37 Urine FLUTD NSW 0 TET ND
F7 Urine FLUTD QLD 0 0 0
F9 Urine Not recorded Unknown 0 0 0
F26 Urine Not recorded Victoria 0 0 0
FLUTD= feline lower urinary tract disease; QLD= Queensland, NSW= New South Wales; SA= South Australia, ND= no
data
ACCEPTED MANUSCRIP
T
26
Table 2. Putative virulence genes in clinical S. felis isolates from cats in Australia, 2013-2014
Gene name Product name Putative function Reference sequence and S. felis
homology to reference (%)
Number S. felis (%)
isolates with gene
Exotoxins
Exi/expA Exfoliative toxin Proteolysis of host
epidermis
S. pseudintermedius AB489850.1 (74%) 19 (68%)
Siet Exfoliative toxin Proteolysis of host
epidermis
S. pseudintermedius ED99 (70%) 28 (100%)
Hlb Beta-haemolysin Destruction of red
blood cells
S. epidermidis ATCC 12228 (67%) 28 (100%)
LukE Leukotoxin Destruction of white
blood cells
S. aureus Mu50 (72%) 1 (3%)
Adhesins
Bap Biofilm -associated
surface protein
Biofilm formation S. aureus V329 (61-64%) 28 (100%)
Atl Autolysin/adhesin Adhesion and
erythrocyte
agglutination
S. pseudintermedius ED99 (76%) 28 (100%)
Others
CapO Capsular polysaccharide
synthesis protein O
Capsule production S. pseudintermedius ED99 (76%) 28 (100%)
OatA O-acetyltransferase Lysozyme resistance S. epidermidis 12228 (68%) 28 (100%)
Scn Staphylococcal
complement inhibitor
Host immune evasion S. aureus Mu50 (69%) 28 (100%)
ACCEPTED MANUSCRIP
T


















