
CHAPTER- I BIOACTIVE METABOLITES OF MARINE...
68
1 CHAPTER- I BIOACTIVE METABOLITES OF MARINE ORGANISMS
Transcript of CHAPTER- I BIOACTIVE METABOLITES OF MARINE...

1
CHAPTER- I
BIOACTIVE METABOLITES OF MARINE ORGANISMS

1
BIOACTIVE METABOLITES OF MARINE ORGANISMS
Terrestrial plants have been used to treat human diseases since the ancient ages.
Studies of the secondary metabolites of terrestrial plants were begun in the 1800’s1and
have proven to be rich sources of natural drugs that are used for the treatment of fatal
diseases such as cancer (taxol) and microbial infections (penicillin). However, due to
resurgence of pathogenic microorganisms and parasites that have developed resistance to
traditional chemotherapies, natural products chemists are increasingly turning to new
sources in the search for biologically active compounds, and specifically to marine
realm.2,3
Life on earth arose from the ocean, and living marine resources continue to
provide essential ecosystem services on which all life depends. Only in recent years has
the extent to which the Ocean is a host to a vast diversity of species and ecosystems been
recognized.
Only in the last 30 years have the seas begun to yield the secrets of the deep ocean
floor. For example, the deep ocean is home to communities of organisms whose
productivity is based on chemosynthesis instead of photosynthesis, the latter being the
process by which most plant life on the earth and in the sea converts sunlight into usable
biological energy. Other whole new ecosystems have been discovered in the ocean in
recent years, and the vast majority of species remain to be discovered.
The seas and oceans are estimated to be 141 million square miles in area and
cover three-fourth’s of the earth’s surface and harbours over 500 million species such as
algae, soft corals, sponges, gorgonians etc. in about thirty phyla 4 (Fig. I-01).

1

2
It takes a lot of energy to produce a natural product, so why do the organisms
bother? The answer is that most of the organisms are sessile as adults, meaning that they
grow attached to something else, perhaps a rock or another animal. As larvae, most of the
invertebrates can get around but once they enter their juvenile phase, they select a home,
settle there and metamorphose into adulthood if they choose any advantageous spot. Now
as they are sessile they have to keep other animals from moving into their space and
should protect themselves from temperature ranges from below freezing temperatures in
Antarctic waters to about 350 0C in deep hydrothermal vents and pressure range (1-1000
atm) which can turn a man into a jelly fish. !
This extensive variability has equipped many marine organisms with the
appropriate mechanisms to survive in a hostile milieu in terms of extreme temperatures,
changes in salinity and pressure, as well as overcoming the effects of mutation, bacteria
and viral pathogens. Marine organisms especially those which are sedentary in nature
have evolved biochemical and physiological mechanisms that include the production of
bioactive compounds for such purposes as communication, protection against predation,
infection and competition. Because of the physical and chemical conditions in the marine
environment almost every class of marine organism exhibits a variety of molecules with
unique structural features.
Difference in physical and chemical environment on the land and seas naturally
causes substantial variation in the nature of metabolites and their biogenesis. Halogens
and isocyanide groups appear frequently as substituents in the metabolites of algae or
sponges or coelenterates; yet these functionalities are very rarely observed as products of

3
terrestrial biochemistry. The metabolites of marine origin often differ in absolute
configuration (or stereochemistry) from their terrestrial counter parts. It is, however,
pertinent to note that the marine environment provides different biosynthetic conditions
to those found on land. 5 By the buffering action of sodium carbonate and bicarbonate
the pH of sea water is always maintained at 8.2-8.5. Sea water contains up to 40% salt
and has an osmotic pressure 15-25 atm.6 In order to cope with such an environment the
marine organisms may have specialized cell structures, especially the composition of the
membrane.
The composition of marine secondary metabolites, while in part, identical to those
from terrestrial plants and animals, is also significantly modified by other factors such as
the aquatic medium, partial availability of sun light up to certain depth and easy mobility
of the nutrients both organic and inorganic, besides the absence of human predation. The
halide rich sea water environment, consisting of Cl-, 19,000 mg/liter; Br-, 65 mg/liter and
I-/IO3-, 5 X10-4 mg/liter, has readily allowed marine organisms to incorporate bromine,
chlorine, and iodine in that order into covalent organic structures. Furthermore, these
elements are not only incorporated into diverse structural types, but they appear to play
essential role in terpene biosynthesis. The utilization of halogens in terrestrial secondary
metabolism is, by comparison, a rare process observed in only a few microorganisms.7
The halogenation process attains a major significance in the sea.
The organic compounds obtained by solvent extraction of a marine organism are
considered to be the secondary metabolites of that organism. Secondary metabolites are
derived from the primary metabolites (amino acids, proteins, fats and polysaccharides) of
the organism. Primary metabolites are needed for the basic cell metabolic processes but

4
the secondary metabolites are not needed in the cell metabolism. The exact function of
the secondary metabolites is not clear. Probably they are the chemicals produced by the
organism in response to environment stresses and also to serve as defense agents against
predators.8 The sea is a potential source of bioactive compounds. The bioactive
compounds could be compounds, which are directly useful in human medicine or as
allelochemics,9 i.e. as reagents by which organisms of one species affect the growth,
health, behaviour or population biology of another species (excluding substances used
only as food by the second species). Examples of the later type are found abundantly in
marine natural products. It is not surprising because to survive in the competitive marine
environment, the marine organism has to produce agents to meet different requirements,
for example, to drive away its predators, to maintain and expand its living space and for
its successful reproduction. As a result of these requirements several bioactive
compounds are possible from the numerous kinds of predator-prey combinations
occurring in the marine environment.
Metabolites of marine organisms are often believed to play an active role in
chemical defense.10,11 The metabolite which is responsible for the chemical defense
mechanism may be present during the active growth of the marine organisms or of
reproduction of the organism alone (seasonal variation), in which case a role as feeding
deterrents or as protectors of new tissue or offspring may be implied, where a more
general defense role is inferred the chemical is likely to be present at all times. Changes
in metabolic composition in sponges in response to environmental changes have been
detected.12 In many cases the compounds that are produced in artificial environments
(such as under laboratory cultures) are identical to those produced in situ. 13 In total

5
contrast, seemingly identical colonies of organisms such as sponges or algae or soft
corals that have been collected in the same location may sometimes have different
chemical composition.
Due to technical barriers there has been a lack of extensive marine folk medicine
in the western world. A Chinese pharmacopoeia recommends seaweed based recipes for a
number of disorders such as pain, abscesses, menstrual disorders and cancer. Seaweed
remedies were also used by the San blas Indians in Panama.14 The relationship between
marine sponges and medicines goes back to Alexandrian Physicians and was described
by Roman historian Plinius. Physicians used sponges that were saturated with iodine to
stimulate coagulation of the blood. Sponges were soaked with pure wine and put on the
left part of the chest in case of heartaches and soaked in urine to treat bites of poisonous
animals. They were used against all kinds of wounds, bone fractures, dropsy, stomach
aches, infectious diseases, etc. At least since the 18th century, Russian, Ukrainian and
Polish physicians have used a fresh water sponge called Badiaga for the treatment of
patients.
The dry powder of this sponge is rubbed on the chest or back of patients with lung
diseases or on the sore places in cases of foot and leg aches such as rheumatism.15
Oficjalski (1937) discovered that Badiaga is not really one sponge, but mixtures of
several freshwater sponges that differ depending on the region. In Poland it consisted of
powder of “Euspongilla lacustris, Ephydatia fluviatilis and Meyenia muelleri,” while the
Russian Badiaga was a mixture of “Euspongilla lacustris, Ephydatia fluviatilis, Spongilla
fragilis and Carterius stepanowi.” He suggested that the high iodine concentration in all
sponge species gives rise to the wholesome effect of Badiaga. At present stodal, syrup

6
containing roasted ‘Spongia officinalis’ is used for homeopathic treatment of dry and
asthmatic cough in the western world.16
The deep knowledge about nerve transmission has been learnt using squid and its
giant nerve axons and the mesenteries of vision have been unraveled using the eyes of
horse shoe crabs, sharks and skates. The surf clam is proving an excellent model for the
cell cycle and its regulation while the sea urchin is a model for understanding the
molecular basis of cellular reproduction and development.17
By the early 1950s, researchers began to view the oceans as a new and untouched
source of potentially useful compounds. Several research groups around the world have
joined hands in marine chemical research and the author has made an attempt to present
their contributions in a nutshell in the following pages. The activities of these metabolites
are immense and mainly depend on the ingenuity of man (Chart I-01).
In recent years, marine natural product bioprospecting has yielded a considerable
number of drug candidates. Most of these molecules are still in preclinical or early
clinical development (Table I-02) but some are already on the market, such as
cephalosporins, cytarabine (Ara-C), vidarabine (Ara-A), ecteinascidin 743 (ET 743) and
ziconotide.
The unusual nucleosides spongouridine and spongothymidine, isolated from
marine sponge Tethya crypta in 1950s, served as lead structures for the development of
commercially viable anti-viral drug Ara-A and the anticancer drug, Ara-C, while
Cephalosporin C, isolated from marine fungus Cephalosporium acremonium in 1940s,
led to the development of the cephalosporin antibiotics. But serious attempts to tap the
vast potential of marine organisms as sources of bioactive metabolites were started only

7
in late 1960s by the interdisciplinary marine research groups at the Scripps Institute of
Oceanography in California, the University of Hawaii and Bedford Institute of
Oceanography, Canada.
The discovery of sizeable quantities of prostaglandins, which had just been
discovered as important mediators involved in inflammatory diseases, fever and pain,
isolated from the gorgonian Plexuara homomalla by Weinheimer and Spraggins18 was
usually considered as the “take-off point” of any serious search for drugs from the sea.
In the early 1970’s researchers at the University of Oklahoma screened about 2000
extracts of diverse marine species and found that nine percent contained compounds with
anti-tumor activity. Inspired by these results and other screening studies, Hoffmann-
LaRoche of Switzerland entered the field in a big way by establishing the Roche
Research Institute of Marine Pharmacology near Sydney, Australia. Even after six years
of intensive research, not a single drug could be developed and understandably the
company closed its venture in 1980. During the 1990’s, workers at several laboratories,
including those of Dr.Richard Moore (University of Hawaii), Dr.William Gerwick
(Oregon State University) and colleagues, had begun to screen extracts of cyanobacteria
(blue-green algae) for various biological activities using predominantly mechanism and
enzyme based assays.
However, search for pharmaceuticals from marine resources continued both in
United States and Japan. Major drug makers, Upjohn, Novartis, Johnson and Johnson,
Knoll, Eli Lilly and others, had started supporting marine natural product research in
University Laboratories worldwide. Pharma Mar, a Spanish company dedicated wholly
to development of anticancer drugs from marine organisms was started in 1986. It has a

8
collection of about 40,000 marine organisms, which has yielded about 150 anticancer
compounds, of which 14 are preclinical candidates, five are in late-stage evaluation and
four are in clinical development. Aplidine, a cyclodepsipeptide from a tunicate has been
approved in Europe for acute lymphocyte leukemia and has already received marketing
authorization. Mera Pharmaceuticals exploring microalgae and Cerylid (Victoria,
Australia) with a good collection of marine invertebrates are the other companies actively
pursuing the development of drugs from marine natural products.
It is now understood that the search for drugs from the ocean resources is rather
slow and arduous task and many laboratories around the world have continued programs
to achieve the goal. Beginning the studies on a humble number of compounds from
marine sources, copious literature on marine natural products and their utility has
emerged. In an article by Grant and Mackie “Drugs from the Sea-Fact or Fantasy?” it is
concluded that marine organisms are potential resources of bioactive compounds.19 The
chemical and biological perspectives have been reviewed by experts in the field of
marine natural products in “Marine Natural Products: Chemical and Biological
Perspectives” edited during 1978-83 by Professor P.J.Scheuer of the University of
Hawaii.20 In 1987, he initiated a new series of volumes “Bioorganic Marine Chemistry.”21
Halstead’s22 three volume series “Poisonous and Venomous Marine Animals of the
World” covers the history, biology, morphology, toxicology, pharmacology and
chemistry of all known poisonous and venomous marine organisms. Marderosian 23 in his
inspiring review article “Marine Pharmaceuticals” alerts the pharmaceutical community
about the enormous potential of the sea as a storehouse of new and different
pharmaceuticals of many types. Kaul and Daftari 24 in their article “Marine

9
Pharmacology: Bioactive Molecules from the Sea” focused on pharmacological activities
of pure compounds isolated from marine organisms. Garson 25 reviewed the biosynthesis
of marine natural products in an excellent report “Biosynthetic studies on Marine Natural
Products”. Riguera 26 detailed interesting bioactive compounds from marine organisms
and Kornprobst et al. 27 reviewed sulfated compounds from marine organisms. Bakus 28
expounded the chemical ecology of marine organisms in an informative and inspiring
report “Chemical Ecology of Marine Organisms: an overview”. Colwell 29 illustrated the
immense potential of biotechnology to the development of marine sciences in an
inspiring article “Biotechnology in Marine Sciences”. Burja et al.30 reviewed various
compounds isolated from marine cyanobacteria and their biological activities in an article
“Marine Cyanobacteria- a prolific source of natural products”. Biotechnological
applications of cultivation of marine sponges were discussed by Osinga et al.31 in an
article ‘Cultivation of marine sponges for metabolite production: applications for
biotechnology?’ Forty-two articles on various aspects of marine biotechnology were
featured in a special issue of the Journal of Biotechnology.32 Berman et al.33 reviewed
‘Marine microorganisms as a source of new natural products’ while Pietra 34 discussed
prospects of secondary metabolites from marine microorganisms in the article ‘Secondary
metabolites from marine microorganisms-bacteria, protozoa, algae and fungi-
achievements and prospects’.
The individual reviews in the edition of “Chemical Reviews: Marine Natural
products Chemistry”35 which include chemical studies of marine bacteria, the
biosynthesis of marine natural products, bioactive peptides and amino acid derived
metabolites from the marine organisms, marine toxins and alkaloids etc. Kelecom 36

10
reviewed ‘Chemistry of marine natural products: yesterday, today and tomorrow’ while
Scheuer37 presented an introductory educational article ‘Products of chemistry -
Exploring the Ocean-Stating the case for the Chemistry’. A book entitled Drugs from the
Sea consisted of a number of reviews running the whole gamut of marine drug discovery
38-47, like ‘Marine microorganisms and drug discovery: current status and future
potential’, ‘Microalgae as a drug source’, ‘Search for biologically active substances from
marine sponges’, ‘Cytotoxic substances from opisthobranch molluscs’, ‘-Conotoxin
MVIIIA: from marine snail venom to analgesic drug’, KRN-7000 as a new type of
antitumour and immunostimulatory drug’, ‘Zoanthamines, antiosteoporotic alkaloids’,
‘Symbiotic bacteria in sponges: sources of bioactive substances’, and ‘The halichondrins:
chemistry, biology, supply and delivery’. Cimino and Ghiselin 48 gave an evolutionary
narrative of marine natural products chemistry which included a taxonomic survey while
Capon 49 highlighted the molecular diversity seen in the results obtained by the
University of Melbourne’s marine natural products group in an article ‘Marine
bioprospecting – trawling for treasure and pleasure’. A database, MarinLit, dedicated to
marine literature is maintained by the marine chemistry group at University of
Canterbury, New Zealand.50 The task of tracking and cataloging the steady stream of
fascinating new structures has been undertaken up to 2002 by D. John Faulkner51 and
later by research group at University of Canterbury52 in periodical reviews, “Marine
Natural Products”. In addition, numerous books53-55, symposia proceedings56-58,
reviews59, specialist and non-specialist articles 60-67 all devoted to marine natural products
have been published. In the year 2003 the members of the Marine Drugs and Food

11
Institute at Ocean University of China in Quindao, China launched a journal “Marine
Drugs” dedicated to marine chemistry and pharmacology. 68
Schwartsmann69 discussed semisynthetic and biocatalytic methods for
enhancement of structural diversity and bioactivity along with latest developments of
underwater breathing apparatus in an article ‘Enhancement of marine natural product
structural diversity and bioactivity through semisynthesis and biocatalysis’. Newman et
al. 70 summarized preclinical and clinical trial data for a range of marine natural products
in an article ‘Marine Natural Products and Related Compounds in Clinical and Advanced
Preclinical Trials’. There has been a steady and continuous flow of research papers on
marine natural products in almost all scientific journals. Marine organisms biosynthesize
compounds as simple as iodoform and as complex as maitotoxin.71,72 The number of
compounds that have been isolated from various marine organisms has virtually soared
and now exceeds 17,00043,73 with hundreds of new compounds still being discovered
every year.25,26,49-53 From 1969-2008 approximately 400 patents on bioactive marine
natural products were issued. The marine derived products currently on the market are
listed in Table I-01
Although the number of clinically used drugs developed from marine sources is
less, a number of promising compounds that have been identified are either already at
advanced stages of clinical trials (Table I-02) or have been selected as promising
candidates for extended preclinical evaluation with the combined efforts of natural
product chemists and pharmacologists. The stronghold of marine natural products that
entered clinical trials is in the area of cancer treatment. A brief account of these is given
below.

12
Table I-01 Marine derived drugs in market
Product Application Original source Method of production
Ara-A Antiviral drug Marine sponge Microbial fermentation of analog
Ara-C Anticancer drug Marine sponge Chemical synthesis of analog
Okadaic acid Molecular probe: phosphatase inhibitor Dinoflagellate Cell culture
Manoalide Molecular probe: phospholipase A2 inhibitor
Marine sponge, Luffartella vartabilis
Wild harvest of sponge
Vent TM DNA polymerase
Polymerase chain reaction enzyme
Deep sea hydrothermal vent bacterium
Recombinant protein
Fomulaid (Market Bio-seiencess, Columbia, MD)
Fatty acids used as additive in infant formula nutritional supplement
Marine microalgae Cell culture
Acquorin Bioluminescent calcium indicator
Bio luminescent jellyfish, acquora victora
Recombinant protein
Green Fluoresecent Protein (GFP)
Reporter gene Bio luminescent jellyfish, acquora victora
Recombinant protein
Phycoerythrin Conjugated antibodies used in ELISAs and flow cytometry
Red algae Cell culture
Resilience Marine extract additive in skin creams
Caribbean gorgonian, Pseudopterogorgta elisabethea
Wild harvest of gorgonian

13
Trabectedin (Yondelis, ecteinasicidin-743, ET-743): The marine alkaloid ecteinascidin
743 (ET-743) is by far the most advanced anticancer compound which belongs to the
tetrahydroisoquinoline alkaloid class. It is produced from the ascidian Ecteinascidia
turbinata. This is approved by the EMEA in September 2007 for the treatment74,75 of
advanced soft tissue sarcoma. ET-743 is in late stages of phase III clinical trials for the
treatment of ovarian cancer (with Johnson & Johnson in the US) and other ongoing Phase
II trials include paediatric sarcomas, breast and prostate cancers. The alkaloid was shown
to be a minor-groove alkylator of DNA and disrupts cell cycle causing cell proliferation
inhibition and to cause inhibition of MDR1 gene transcription47 the latter being
responsible for the well-known phenomenon of multi-drug resistance (MDR), which
causes tumors to become insensitive to anticancer drugs and is a severe obstacle for
chemotherapy. ET-743 furthermore elicits non-p53-mediated apoptosis in tumor cells.
One of the predominant toxicities exhibited by ET-743 in preclinical studies was
hepatotoxicity, particularly in the female rat, and similar effects had been seen in human
patients but could be controlled by dose-reduction. However, in recent publication,
Donald et al.76 demonstrated that pretreatment with high-dose dexamethasone gave
complete protection against hepatotoxicity in this animal. Thus such a treatment in
humans may well be a method of controlling this Et-743 –related toxic side effect. This is
produced commercially by semi-synthesis from the eubacterium derived
cyanosafracin B.77
Didemnin B: Didemnins provoked interest way back in the 1980s due to their
pronounced anti-tumour activity because of their interference with the protein synthesis.58

14
Its further development as anticancer drug was recently abandoned, after Phase II trials,
due to its hepatotoxic side effects.
Aplidine: Dihydrodidemnin B (Aplidine) appears to be less toxic and even 5 to 6 times
more effective than didemnin B with a broad spectrum activity both in vitro and in vivo
against various types of cancer diseases such as colorectal, lymphoma, thyroid and renal
cancers. 59 It further shows anti-angiogenic activity in experimental models. Phase II
clinical trials are underway in Europe for renal and colon carcinomas. European
Commission has approved aplidine for acute lymphoblastic leukaemia and other trials
covering renal, head and neck, and medullary thyroid are ongoing, simultaneously.60
Phase II clinical trials are ongoing by Pharma Mar.78-80
Kahalalide F belongs to the family of dehydroaminobutyric acid containing cyclic
peptides isolated from the Hawaiin mollusk, Elysia rufences. It has shown antitumor
activity probably by interfering with lysosome function in prostate, colorectal and lung
cancer cell lines.60,61 It is currently in phase II trials for prostate cancer and other solid
tumours.81-84
Bryostatins are macrocyclic lactones isolated from the marine bryozoan Bugula naritina.
Bryostatin 1 is one of the most abundant and best-studied compounds of this series and is
found to have anticancer and immunostimulating activities. Bryostatin 1 binds to protein
kinase C with high affinity, which may be the mechanistic basis for both observed
anticancer and immunostimulatory activities. Since the publication of the first structure
by Pettit in 1982, these molecules have been the target of many synthetic groups but only

15
three of the 20 reported bryostatins have been synthesized. It is presently in Phase I/II
clinical trials.85-89
Halichondrin B: (eribulin (E-7389, NSC-707389) is one of the series of compounds
originally isolated and reported by Umera et al. in 1985 from the Japanese sponge
Halichondria okadai. It is a tubulin interactive agent, affecting tubulin depolymerization
at a site close to, but distinct from the Vinca site. It is currently in Phase II/III clinical
trials.90-96
Dolastatin-15: (Synthadotin, ILX-651) This is an orally active third generation dolastatin
derivative that was licensed by Iiex from BASF Pharma. It is currently in phase II clinical
trials by Genzyme.97-99
The other areas in which considerable development has been made are pain and
inflammation. It is in this area where conotoxins have gained their importance.
Conotoxins are a group of peptides produced by shells, which have a variety of biological
actions.
-Conotoxin MVIIA: (SNX-III) This 25-residue peptide with three interlocking
cystinyl bridges was originally isolated by Olivera. The pain-killing marine natural
product has successfully completed Phase III clinical trials for two applications: to
alleviate pain associated with malignant diseases (Cancer and AIDS) and as an analgesic
for nonmalignant neuropathic pain.42
Ziconotide: (Prialt) This is a semisynthetic analogue of conotoxin with a 25 amino acid
peptide exhibiting three disulfide bonds. It is an N-type calcium channel -conotoxin
MVIIA. This compound is launched by Elan in both US and Europe in 2005 for the

16
treatment of patients suffering from chronic pain.100 In March 2006 Eisai obtained the
rights to market ziconitide in Europe. The compound proved to be 1,000 times more
active than morphine in animal models of nociceptic pain. The activity of ziconotide is
due to the blockage of N-type calcium channels and thereby inhibits the release of
neurotransmitter. In contrast to morphine, it does not induce the development of
tolerance, constipation or respiratory depression. Following the discovery of this Conus
peptide in the late 1970s, no efforts were spared to develop more potent synthetic
analogues. US FDA approved ziconotide for hard-to-treat pain associated with cancer,
AIDS and neuropathies.
Xen-2174: This conotoxin is undergoing clinical evaluation, as all the other conotoxins
have been halted or discontinued. Xen-2174 a 13-aminoacid peptide with two cysteine
bridges isolated from Conus marmoreus has been found to inhibit the norepinephrine
transporter (NET), a known CNS drug target that is inhibited by the antidepressant
desipramine.101-103 Currently Xen-2174 is in Phase I/II trials for the treatment of cancer
pain.104 Other conotoxins which have been halted or discontinued are contulakin G
(CGX-1160) and conotoxin – G (CGX-1007).
Methopterosin (OAS 1000): The extracts of Caribbean gorgonian Pseudopterogorgia
elizabethae showed anti-inflammatory activity and are nowadays used as an ingredient
for cosmetic skin care products.105 The activity is due to the diterpene glycosides-
pseudopterosins-which inhibit phospholipase A2. Methopterosin (OAS-1000), a
derivative of pseudopterosin, is currently under Phase I trials.

17
IPL 576: A synthetic analog of the steroid contignasterol isolated from the sponge
Petrosia contignata,106-108 it is in phase II trials as a leukocyte-suppressing anti-
inflammatory drug for the treatment of asthma.
GTS 21: 3-(2,4-dimethoxy benzylidene)-Anabaseine, is a selective 7-nicotinic
acetylcholine receptor partial agonist in clinical development at Taiho to treat
Alzheimer’s disease and schizophrenia.109
Trodusquemine (MSI-1436): This is a sulfated aminosterol from the dog fish shark
Squalus acanthias along with the closely related squalamine and other steroids.110
Squalamine has been shown to inhibit endothelial cell proliferation but clinical trials in
cystic fibrosis and oncology were halted in July 2007. In contrast to squalamine,
trodusquemine suppresses mammalian appetite through inhibition of protein tyrosine
phosphatase 1B.111-113

18
N
N
N
N
NH2
Ara - A
OHO
HOOH
N
S
O
HOOCCH(CH2)3CN H
COOHCH2OCO CH 3
NH2
O
Cephalosporin C
HN
N
OR
O
Spongouri d ine R = MeSpongot hym idine R = H
Ma it o t o x in
O O
O O
O
O
OH
O
O
O
OOH
OH
O O
O O
OO
OOH
HO
O
OOH
O
OO
O
O
O
OO
O
O
O
OO
OHOSO3Na
OH
OHHO OH
HO
OH OH
OHNaO3SOHO OH
OH
OHOH
HOOH OH OH
OH
OH
HO
OH
O O
NH
OH
O
O
O NH
O
NNMe
OO
N
ONMe
O
NR
O
OMe
H
Didem nin B R = CH(OH)CH3Dehydrodidem n in B (Apl id ine)
R = COCH3
NNMe
O S
NH
OOH
OMeHIO
OAc
OO
MeO
HO
H
Ec t einasc idin 743
K RN 7000
O
OH
OH
OHO
OH
(CH2)13CH3
NH
(CH2)24CH3O
OH
OH
HOO
HOOH
ThrGlySerCysArg
SerGly
Cys Cys Asp TyrMet
LeuArgSerCys
Lys
Ala Gly
LysGlyLysCysH2N
Lys
CysH2N
Ziconotide

19
5-MeHex
Val-5
NH
NH
N
O
NHO N
O
H2N
NH
O
O
HNO
NHN
OO
N
O
NHO
O
HONH
NH
O
O
H
H
H
L-OrnD-allolle-2
Thr-1
D-allolle-1
Val-2
L-PheThr-2
Val-3
L-Pro
Z-Dhb
Val-1
Val-4
Kahalalide F
Methopterosin
Me
OOH
Me
HMe
O
OHHO
OH
RSqualamine H
Trodusquemine
NNNH OH
OSO3-
R
H2N NH
NN
OMe
OMe
E
.2 HCl
GTS-21
O
O
OO O
O
O
OOO
HH
HH
HH
OH
H
H H H
H
OH
H
H HH
H
O
O HO
OHOOH OH
HH
Halichondrin B
Discodermolide
OO H
OH
OH OCONH2
HO
OH

20
A major issue that has to be considered when a marine chemical entity is selected
for clinical development is sustainable and industrially feasible supply of the material.
A crucial step is the incorporation of a sustainable supply, which has delayed the
development of these agents. Marine organisms for drug discovery research have been
collected using various methods such as scuba diving, submersibles, dredging and
trawling. Submersibles (Fig. I-02) enable scientists, to access unusual habitats, such as
vent communities and deep-sea benthic habitats.
Fig. 1-02: Submersible
However, the metabolites occur in trace amounts in the organism and a steady source of
supply from wild harvest can’t provide enough of the target compound for the
development studies.
The concentrations of many highly active compounds in marine invertebrates are
often minute, sometimes accounting for less than 10-6 % of the wet weight. ET-743 is the
best example to illustrate this problem. For example, in order to obtain approximately 1 g
of the promising anti-cancer agent ET-743, close to 1 metric tonne of the tunicate

21
Ecteinascidia turbinata has to be harvested and extracted. Scientists at PharmaMar
performed an elegant semisyntheses from the marine Pseudomonas fluorescens
metabolite cyanosafracin B that provided cGMP grade ET-743 from a 21-step synthetic
process on a scale large enough to provide enough material for clinical trials. This was
feasible despite a low overall yield of 1.4% because the starting material could be
obtained on a large scale by fermentation.46
Various approaches to overcome this problem are
1. Chemical synthesis
2. Controlled harvesting
3. Mari-culture: Favoring the growth of the organism in its natural milieu.
4. Aquaculture: Culture of the organism under artificial conditions.
5. Genetic intervention (Cloning)
6. Semi synthesis: Use of a parent/related compound as the starting point
followed by a short / industrially effective synthetic process.
Marine invertebrates are laden with bacterial symbionts, often in high density.
Because many marine natural products structurally resemble bacterial compounds, it has
long been proposed that the marine chemicals are produced by bacterial symbionts.
Based on the symbiotic hypothesis of marine natural products synthesis, two basic
strategies can be used to supply for development. First, bacteria can be cultured from
these organisms followed by detection of the compound in the culture. Second, genes for
the biosynthesis of important molecules can be cloned and heterologously expressed.
Recently Hildebrand et al. 116 reported the cloning and partial sequencing of the putative
bryostatin biosynthetic gene Bry A which contains the required functionality to make

22
most of the important ‘recognition domain’ portion of the bryostatin, potentially enabling
production of this portion via genetic engineering. In combination with chemical
synthesis or further genetics this could provide the first example of a symbiotic natural
product supplied by the genetic approach.
Both the sea and land-based aquaculture methods can be considered as an
alternative to harvesting wild specimens. Bugula neritina, the source of bryostatins and
Ecteinasicidia turbinata, the source of ecteinascidin-743 have been produced under
controlled conditions by Cal Bio Marine Technologies 117 (Carlsbad, CA) (Fig. I-03 and
Fig. I-04)
Fig. I-03: Aquacultured growth plate of Bugula neritina
Fig. I-04: Aquacultured colony of Ecteinascidia turbinata

23
Table I-02: Potential Therapeutic Compounds isolated from Marine Organisms
Source Compound Disease area Status References Tunicata
Ecteinascidia turbinata Ecteinascidin 743 Cancer Phase III
74,75,77
Aplidium albicans Dehydrodidemnin B (Aplidine)
Cancer Phase II 78-80
Bryozoa Bugula neritina Bryostatin 1 Cancer Phase II 85-89 Elysia rubefscens Kahalalide F Cancer Phase I 80-83 Molluscans Conus magnus Ziconotide Chronic pain Phase III 93
Dolabella auricularia Dolastatin 10 Cancer
Development discontinued
by NIH
114
Dolabella auricularia Dolastatin 15 Cancer
Phase II under preparation
97-99
Porifera (sponges) Halichondria okadai Halichondrin B Cancer Phase II / III 90-96
Hemiasterella minor Hemiasterlin (E 7974)
Cancer Phase I 115
Discodermia dissoluta Discodermolide Cancer
Discontinued due to toxicity
114
Agelas mauritiamus KRN 7000 Cancer
Discontinued due to
cytotoxin antagonism
114
Pseudopterogorgia elizabethae Methopterosin
Wound healing/
Inflammation Phase I
105
Petrosia contignata IPL576902 (11) Inflammation/
Asthma Phase II
106-108
Nemertia Amphiponus lactifloreus GTS-21
Alzheimer’s/ schizophrenia
Phase I 109
Others
Spisula polynmya Spisulosine Cancer Discontinued
due to lack of efficacy
114

24
Squalus acanthias Trodusquemine Antihyperlipi
demic, Antidiabetic
110-113
Squalus acanthias Squalamine Cancer Trials
discontinued 114
Conceptually, it is clear that the marine ecosystem offers a huge potential in the
naturally based pharmacopoeia of this century. However, an unfavorable balance between
discovery and the very small number of candidates incorporated for clinical evaluation
exists. So it appears that a better and more pragmatic approach is urgently needed in
order to translate innovative discoveries into active clinical therapeutics.

25
Present research work of the author
India is surrounded on three sides by oceanic waters especially in near tropical or
tropical zones thereby harbouring innumerable genera and species of marine plants and
animals. A few research groups in India have been engaged in these efforts on marine
metabolites with assistance from government funding agencies. One of the prominent
groups under the leadership of Prof. Ch. Bheemasankara Rao and Prof. D. Venkata Rao
has been pursuing research on bioactive metabolites from sea organisms of the Indian
Ocean for the past two decades. The author who is part of this investigative group had
gone through extensive literature in this area and is much fascinated to put his efforts in
the investigation. After a preliminary screening the following organisms were chosen for
extensive chemical investigations
Dendrilla nigra Dendy (Sponge)
Hyatella cribriformis Hyatt (Sponge)
Synoicum indicum (Ascidian)
The author isolated 3,4 - diaryl pyrrole alkaloids, phenolic compounds,
sesterterpene with interesting scalarane skeleton, sphingolipids, polyaromatic brominated
phenolic compounds. During the study of 3,4-diaryl pyrrole alkaloids, the author has
come across extremely interesting data on this group of compounds from marine
organisms and preferred to incorporate a detailed account on this class of compounds in
the following pages.

26
3, 4-DIARYL PYRROLE ALKALOIDS
Marine ascidians or tunicates and sponges are prominent producers of metabolites
derived from amino acids. The amino acid DOPA (2-amino, 3- (3’, 4’– dihydroxy
phenyl) propionic acid) in particular, appears to play an important role in the metabolism
of these marine invertebrates, serving as the apparent precursor of several alkaloidal
metabolites isolated from this source. DOPA metabolic products range from peptides to
polycyclic alkaloids. Examples of peptide products are the tetrapeptides, halocyamines
and the tripeptides known as tunichromes.
Alkaloids apparently derived via DOPA metabolism include the lamellarins,
ningalins, polycitrins, lukianols, purpurone, storniamides, dictyodendrins, all of them
having a central pyrrole ring usually with 3,4- diaryl substitutions.
O
N
R1R1R1
R1
R1
O
Lamellarins R1=H or Me R2=H,OMe or OH R3=H or OH
R2
R1
R3
N COOMe
HO OH
OMe
RO
Lamellarins O and P R=H or OH
N
BrHO
Br
Br OR
Br
Polycitrins R=H or Me
OO
OH
N
HO
R
OH
Lukianols R=H or I
O
OH
O
NHN
HOR2
HN
OHR3
OHHOHO
OH OHR1
OH
O O
Storniamides A R1=OH R2=R3=H B R1=R3=OH R2=H C R1=H R2=R3=OH D R1= R2=R3=OH
NH
OO
O O
HO OH HOOH
Ningalin A

27
N
HO OH HO OH
Purpurone
O O
HO
HO
HO
OH
OH
N
HO
Dictyodendrin AOH
NH
OSO3Na
OH
OH
MeOOC
DOPA – derived metabolites are isolated from many ascidians which accumulate
massive quantities of ionic vanadium and iron from seawater and suggested to participate
in this process. In didemnid ascidians vanadium accumulation is negligible, but iron
accumulation is a prominent feature generating in vivo iron concentrations up to 107
times higher than that found in seawater. While an understanding of this process appears
incomplete, the metal binding phenomenon has been partially explained by complexation
with O-catechol functionalities. The mechanism involves chelation of metals such as
Fe3+ and V5+ by dopa residues.
Lamellarins:
Lamellarins are first isolated from the prosobranch mollusk Lamellaria sp. in
1985.118 They were later obtained from the ascidians Didemnum chartaceum 137
(Seychelles ascidian) Didemnum sp. 150,168 (a Great Barrier Reef ascidian) and the sponge
Dendrilla cactos.161,162 Sulfated lamellarins 169 were also isolated from an unidentified
ascidian collected from the Arabian Sea.
Lamellarins fall into three structural groups depending on whether the central
pyrrole ring is fused (lamellarins A-N) or unfused (lamellarins O-R) to adjacent aromatic
rings and on the presence between atoms 5 and 6 of the quinoline moiety of either a
single(lamellarins I-L) or a double bond (lamellarins B, D, M,N).

28
The first four lamellarin alkaloids, 118 lamellarins A (1), B (2), C (3), and D (4)
were reported from the mollusk Lamellaria species collected near Koror, Palau. The
structure of lamellarin A was determined by an X-ray crystallographic study and the
structures B-D were assigned by interpretation of spectral data. Lamellarin A exists as a
1:1 mixture of two geometrical isomers due to restricted rotation about C1-C11 bond. At
concentration of 19μg/ml, lamellarin D caused 78% inhibition of cell division in the
fertilized sea urchin egg assay while lamellarin C caused 15% inhibition and lamellarins
A and B were inactive.
The first total synthesis of lamellarins D and H was accomplished by using N-
ylide mediated pyrrole ring formation and subsequent lactonization. 119 In 2002, Ishibashi
et al. 120 synthesized ten derivatives of lamellarin D and evaluated them for cytotoxicity
against a Hela cell line in an effort to examine their structure activity relationship. It
appeared that the hydroxyl groups at positions C-8 and C-20 of lamellarin-D were
important structural requirements for cytotoxic activity, while the hydroxyl group at C-14
and the two methoxy groups at C-13 and C-21were not necessary for the activity.
Tardy et al. 121 exploited the lamellarin D pharmacophore for the development of
topoisomerase I-targeted anticancer agents. Lamellarin D is a potent poison of human
topoisomerase I endowed with remarkable cytotoxic activities against tumor cells and
potentially stabilizes topoisomerase1-DNA covalent complexes so as to promote the
formation of DNA single strand breaks. The 5-6 double bond is an essential element for
the poisoning of topoisomerase I and the antiproliferative activity. The three phenolic OH
groups at positions 8, 14 and 20 are important structural elements but they can be

29
substituted without the loss of activity and the cationic proline and valine derivatives at
these positions are selected for preclinical development.
In 2005, an efficient and highly convergent total synthetic route to lamellarin D
was achieved in 8 steps. 122
Vanhuyse et al. 123 reported that lamellarin D is a potent apoptic agent and its
cytotoxic action is fully maintained in multidrug resistant cells compared to the sensitive
parental cell line. The multidrug resistance transporter proteins P-glycoprotein, multidrug
resistance protein and lung resistance protein have been associated with treatment failure.
They are plasma membrane transporters responsible, at least in part, for the resistance of
tumor cells to chemically and/or functionally unrelated drugs. It is therefore important to
identify new anticancer agents that are not sensitive to these export pumps. Lamellarin D
is found to be equally toxic to murine leukemia P388 cells and its camptothecin resistant
cells P388CPT5 expressing a functional P glycoprotein. These results are interesting and
tend to reinforce the potential of this marine alkaloid as a lead compound to design novel
topoisomerase 1 targeted anticancer agents.
Iwao et al. reported the synthesis of Lamellarin D, L and N.124 A library of open
lactone analogues of lamellarin D was prepared from the scaffold of methyl 5,6-dihydro-
pyrrolo [2,1-a] isoquinoline-3-carboxylate by introducing various aryl groups through
sequential and regioselective bromination followed by Pd(o)-catalyzed Suzuki cross
coupling chemistry. The SAR study concluded that more than 75% of the open chain
lamellarin D analogues tested showed cytotoxicity in a low micromolar GI50 range. 125

30
Iwao et al. 126 reported an efficient route for the synthesis of 1-dearyllamellarin D
using directed lithiation, Suzuki-Miyaura coupling and palladium–catalyzed direct
arylation as the key reactions.
Albericio and Alvarez reported the design and synthesis of poly (ethylene glycol)
derivatives of lamellarin D with the aim of modulating their physicochemical properties
and improving the biological activity. Mono-, di-, and tri-PEG conjugates with improved
solubility were obtained from corresponding partially protected phenolic derivatives of
lamellarin D. Conjugates were evaluated for their cytotoxicity. 127
Alvarez et al. 128 reported the design and synthesis of Lamellarin D conjugates
with a nuclear localization signal peptide and a poly (ethylene glycol)-based dendrimer.
Conjugates were obtained from the corresponding protected lamellarin D. Conjugates
were found to be more cytotoxic than the parent compound.
Hu et al. 129 reported their work on developing 1,2-diphenyl-5,6-dihydropyrrolo
[2,1-a] isoquinolines as hybrids of combretastatin A4 and lamellarin D with the aim of
retaining the cytotoxicity and antimitotic activities of the parent compound. Ruchirawat
et al. prepared Lamellarin D analogues containing a lactam rather than a lactone. Their
work was the first report of a SAR studies done for a change at this position. 130
In late 2010, Li et al. 131 reported a new, concise synthesis of lamellarin D and
lamellarin H in seven steps.
Recent advances in the pharmacological development of Lamellarin D include
the identification of new cellular targets of this compound and new insights in to its
mechanism of action. The maleate-aspartate shuttle was recently identified as a new

31
target for Lamellarin D by proton NMR- based metabolomic. 132 Certain protein kinases
relevant to cancer have recently been reported as targets of Lamellarin D, Lamellarin N
and other lamellarins. 133 Lamellarin D exerts its anti-tumor activity through
complementary pathways: a nuclear route via Topoisomerase I (Topo I) inhibition 134,135
and mitochondrial targeting by induction of mitochondrial permeability transition (MPT).
136
In 1988 Lindquist and Fenical 137 reported four new lamellarin class alkaloids,
lamellarins E (5), F (6), G (7) and H (8) from the marine ascidian Didemnum chartaceum
of the Indian Ocean. The structure of lamellarin E was determined by spectroscopic and
X-ray crystallographic methods. The structures of lamellarins F-H were elucidated by
interpretation of NMR spectral data, which relied heavily upon JC-H and 2-3JC-H correlation
experiments.
Prosobranch mollusks of the family Lamellariidae have been described as specific
predators of colonial ascidians. 138-140 These mollusks preying on ascidians appear to have
followed a similar evolutionary pattern as the numerous opisthobranchs preying on
chemically rich sponges, bryozoans, coelenterates and algae. The report of lamellarins E-
H from D.chartaceum indicates that Lamellaria species most likely acquired lamellarins
A-D from a similar ascidian food source.
In 1997, Steglich’s group described a biomimetic synthesis of lamellarin G
trimethyl ether. 141 In 2001, Ruchirawat et al. reported the efficient synthesis of lamellarin
G trimethyl ether. The synthesis involved formation of the core pyrrole-[2, 1-α]
isoquinoline followed by formation of the lactone ring. 142 In 2003, Iwao et al. reported

32
the synthesis of lamellarin G trimethyl ether starting from a symmetric 3,4-dihydroxy
pyrrole bis-triflate derivative. 143
Handy et al. reported a modular synthesis of the lamellarin G trimethyl ether that
was based on application of three iterative sequential and regioselective halogenation /
Suzuki cross coupling events. 144 In 2006, lamellarin G was synthesized by the key step
of formation of 3,4-diarylpyrrole-2,5-dicarboxylic acids from arylpyruvic acids and 2-
arylethyl amines. 145
Opatz and Liermann reported a convergent synthesis of lamellarin U and
lamellarin G trimethyl ether from a readily available deprotonated α-aminonitrile as an
AB ring building block. 146 Gupton et al. reported formal synthesis of Lamellarin G
trimethyl ether and ningalin B via formation of the polysubstituted pyrrole derivatives,
from a vinylogous iminium salt derivative. 147
In 2005, lamellarin H and its derivatives were synthesized by using the fabricated
intermediate, 2,4,5-trimethoxy-α-chloroacetophenone as key starting material for this
synthesis. 148 In 2006, the same group reported the synthesis of the second intermediate of
lamellarin H, 1-(3,4-dimethoxy phenyl)-8,9-dimethoxy-2-(2,4,5-trimethoxyphenyl)-
pyrrole-[2,1-α]-isoquinoline. 149
An Australian Didemnum species has yielded six new alkaloids 150 lamellarins I
(9), J (10), K (11), L (12), M (13) and N (14) together with lamellarins A to D that were
previously obtained from the prosobranch mollusk Lamellaria species. These structures
were deduced using chemical and spectroscopic methods. Lamellarin K and L exhibit
immunomodulatory effects. 151

33
Lamellarin K has been synthesised using an intramolecular (3+2) cycloaddition
reaction to form the central pyrrole ring. 152 In 2001 lamellarin I and K were synthesized
based on a [3+2] cycloaddition. 153 In 2006, Steglich et al. developed biomimetic
synthesis of lamellarin K in four steps. 154
Lamellarin L has been synthesized by using biometric strategy. 155 In 2000,
Steglich et al. developed biomimetic total synthesis of the non symmetrical lamellarin L
in five steps. 156 Lamellarin L and U were generated via a total solid phase synthesis.
157,158 In 2004, Ruchirawat et al. obtained lamellarins K and L in three steps. 159 In 2006,
Iwao et al. synthesized lamellarin L. 160
Lamellarins O (15) and P (16) were isolated from the sponge Dendrilla cactos
that was obtained by dredging off South Australia. 161 The next two lamellarins Q (17)
and R (18) were isolated from a recollection of the Dendrilla cactos from New South
Wales. 162
Lamellarins O and Q were synthesized using Stille, Suzuki, and Negishi cross-
coupling reactions as the key steps. 163 Boger et al reported the total synthesis of
lamellarin O, utilizing a common heterocyclic azadiene Diels- Alder reaction. 164 In 2004,
Albericio’s group described an efficient solid-phase strategy for synthesis of lamellarins
O and Q using Merrifield resin and N-protected methyl 3, 4-dibromopyrrole-2-
carboxylate as a scaffold. 165
The total syntheses of lamellarins O, P, Q and R have been achieved by using
cross-coupling and the directed lithiation as key reactions. 166

34
In 2007, Liu et al. reported 167 four new lamellarin-like phenolic pyrroles,
neolamellarin A (19), neolamellarin B (20), 5-hydroxy neolamellarin B (21) and 7-
hydroxy neolamellarin A (22) from the marine sponge Dendrilla nigra which was
collected from shallow water in Saipan. These structures closely resemble the structure of
the known Dendrilla cactos compound lamellarin O (15). Compound (22) inhibited
hypoxia induced HIF-1 activation in T47D cells. Hypoxia induction of vascular
endothelial growth factor (VEGF), a potent angiogenic factor and HIF-1 target gene, was
also inhibited by (22) at the secreted protein level.
Lamellarin S (23) was isolated from a south – eastern Australia tunicate of the
genus Didemnum. 168
An unidentified ascidian from the Arabian Sea (Trivandrum Coast of India)
yielded nine new alkaloids of the lamellarin class T (24), U (25), V (26), W (27), X (28)
and Y (29) together with lamellarin N. Lamellarins T, U, V and Y were obtained as 20-
sulfated derivatives. 169 This is the first report of lamellarin sulfates.
In 2006, Banwell et al. reported the synthesis of lamellarin T hybrids and were
evaluated for anti-mitotic and cytotoxic properties. The key steps include selective
lithium - for halogen exchange, Negishi, and Suzuki-Miyaura cross-coupling reaction. 170
Davis et al. 171 reported 20-sulfated derivatives of lamellarins B, C and L, the 8-
sulfated derivative of lamellarin G and also the lamellarin Z (30). Lamellarin G 8- sulfate
is the first example of this class of compounds sulfated at C-8 position, while lamellarin
Z is the first example of dimethoxylated lamellarin. An aberration in the integration of
signals in the 1H NMR spectra of the 20-sulfated derivatives of lamellarins B, C and L

35
led to NMR relaxation studies.T1 ( relaxation time) values were calculated for all protons
in the sulfated lamellarins and their corresponding non sulfated derivatives. Interestingly
the protons ortho to the sulfate group have T1 values up to five times larger than the
corresponding protons in their non sulfated derivatives. These are due to the isolation of
these protons which have minimal relaxation pathways.
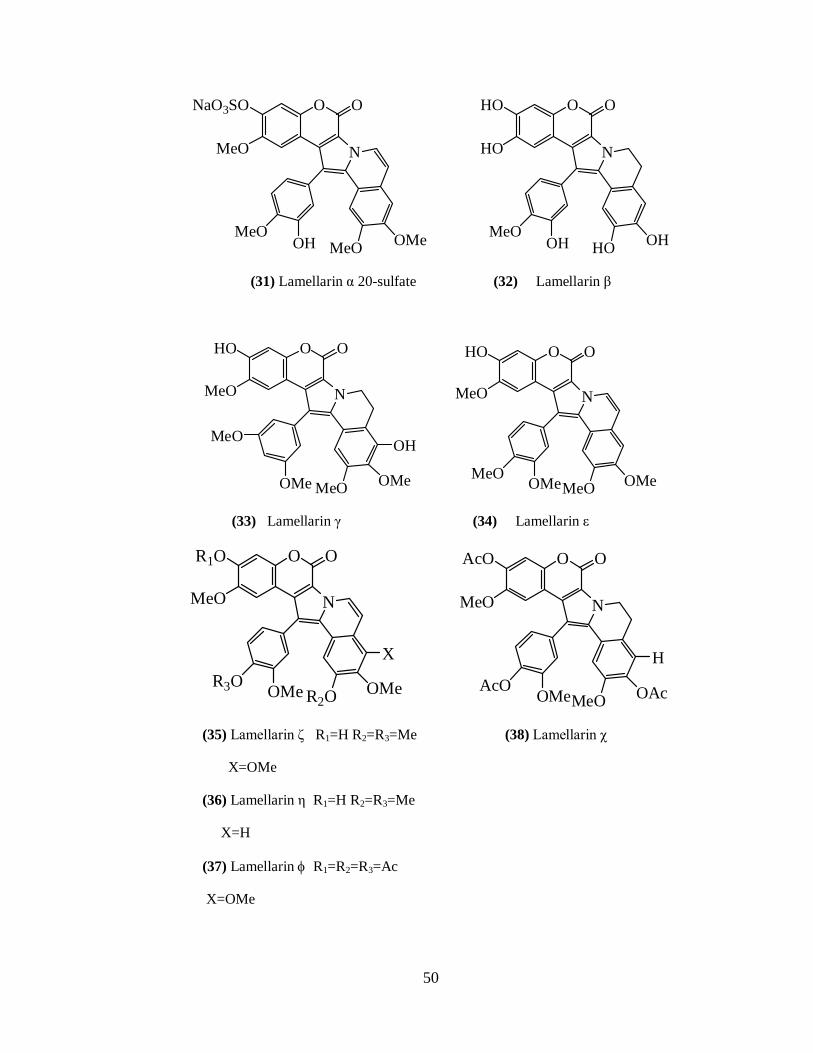
Rami Reddy et al. 172 reported lamellarin α 20-sulfate (31) from an unidentified
ascidian collected from the Arabian Sea near Trivandrum, India. They also screened
lamellarins for HIV-1 integrase inhibitory activity and found them showing selective
inhibition. Of all the lamellarins, lamellarin α 20-sulfate significantly inhibits integrase
protein in vitro and HIV viral replication in cultured cells. Lamellarin α 20-sulfate acts in
a part of the viral life cycle consistent with inhibition of integration.
These findings provide a new class of compounds for potential development of
clinically useful HIV integrase inhibitors. Embedded within the lamellarins is a coumarin
moiety. Coumarins have been found to be active as integrase inhibitors. Sulfated
compounds have also been found to be inhibitory in some cases.
In 2006, the first total synthesis of lamellarin α 20-sulphate was developed in 14
steps. 173
Ham and Kang 174 reported lamellarin β (32) from a purple unidentified
Didemnum species collected in the Indian Ocean. This alkaloid showed cytotoxicity
against human promyelocytic leukemia HL-60 with an IC50 of 4.8 μg/ml.
Lamellarins γ (33), and ε (34) along with eight known lamellarin alkaloids M,
K, K-diacetate, K – triacetate, U, I, C–diacetate and X – triacetate were isolated from the

36
Indian ascidian Didemnum obscurum. 175 The structures were established using standard
spectroscopic techniques. Lamellarins are cytotoxic and showed good antitumor activity.
Many antioxidant compounds possess anticancer properties. Hence the lamellarin
compounds were screened for anti oxidant activity. The absence of 8-OH resulted in the
decreased potency of anti oxidant activity and 14 -OH is not necessary for the activity.
Reddy et al. 176 reported lamellarin - (35), lamellarin-(36), lamellarin - (37),
lamellarin - (38) along with seven known lamellarins, lamellarin K, I, J, K–triacetate
lamellarin L-triacetate, F, T-diacetate and they also screened for cytotoxic activity against
colorectal cancer cells (COLO - 205). Lamellarin - and , L–triacetate and F have
shown excellent activity against test cancer cell lines.
To establish a more comprehensive structure activity relationship relatively large
quantities of lamellarins are required. However, because the natural sources of
lamellarins, some species of ascidians, sponges, and mollusks, provide these compounds
in only minute quantities, total synthesis is a vital alternative in providing these
compounds for detailed biological evaluations. Ploypradith et al. 177 reported four
polymer-supported reagents which are utilized in multi-step synthesis of lamellarins.
1. Amberlyst A-26Br3- 2. Polymer bound pyridine hydrobromide perbromide
(PVPHP), 3.Amberlyst A-26 NaCO3- 4. Amberlyst – 15
Marco et al. 178 compared the lamellarin activities with camptothecin. Eukaryotic
topoisomerase is the target for the anticancer drug camptothecin. They have built
molecular models of the ternary complexes formed between the DNA Top1 ensemble and
lamellarin D or camptothecin fully intercalated in to the duplex DNA and studied by

37
means of nanosecond molecular dynamics simulations in aqueous solutions. Results
showed that 20-OH and 8-OH of lamellarin D participated in hydrogen-bonding
interactions with the side chains of Glu 356 and Asn 722. It was also found that
lamellarin D stabilizes Top1 cleavage at CG sites in addition to TG sites observed for
camptothecin. They also confirmed the deleterious effect of replacing the 20-OH in
lamellarin D with hydrogen using a set of thermodynamic integration free energy
simulations.
Ruchirawat et al. synthesized 28 natural (lamellarins C,E,F,G,I,J,K,L,T,U,Yand χ)
and unnatural lamellarins with either a saturated or an unsaturated D ring. 179
Thipnate et al. studied lamellarins using receptor independent (R1) 4D
quantitative SAR (QSAR models). 180,181 They obtained valuable 3D pharmacophore
information from a set of 25 structurally complex lamellarins screened insilico against
human hormone dependent T47D breast cancer cells. Overall, they identified formation
of an intermolecular hydrogen bond and the hydrophobic interactions of substituents at
C-10, C-11 and C-12, as the most important features for cytotoxicity against the cancer
cells. They also suggested that hydrophobic substitutions at C-3’ and C-4’ could enhance
cytotoxicity.
Finally, lamellarins not only have interesting structural features, but also exhibit a
wide array of significant biological activities, including cell division inhibition,
cytotoxicity, HIV-1integrase inhibition, antioxidant activity and immunomodulatory
activity and they definitely provide a new class of compounds for potential development
of clinically useful agents.

38
Further studies can contribute to a better understanding of the mechanism of
lamellarins and their analogues and will be beneficial for the ongoing pharmacological
optimization of this class of compounds.
Pyrrole-Derived Alkaloids Related to Lamellarins:
Ningalins:
Four DOPA – derived alkaloids Ningalins A (39), B (40), C (41) and D (42) three
of which possess new carbon skeletons were isolated from a western Australian ascidian
of the genus Didemnum. 182 The structures were elucidated by interpretation of overall
spectral data and by 2DNMR correlation methods. Ningalins A-D are composed of C18,
C25, C32 and C40 condensed aromatic systems with the unifying theme that all are derived
via the condensation DOPA.
Ningalin A is synthesized using Diels-Alder strategy. 183 Ningalin B is synthesized
and shown to be a multi-drug resistant reversal agent. 184 In 2002, Bullington et al.
reported the synthesis of ningalin B. 185 A formal synthesis of ningalin B was reported in
2003. 186 Gupton et al. reported the application of vinylogous iminium salt derivatives to
a convenient and efficient synthesis of ningalin B hexamethylether. 187 Steglich et al.
reported a biomimetic synthesis of ningalin B. 188
Peschko et al. reported the synthesis of the ningalin C. 189 In 2005, Boger et al.
reported the synthesis of ningalin D in nine steps. 190
Ningalins possess remarkable multidrug resistance (MDR) modifier activity. Key
analogue derivatives of ningalins were examined resulting in the discovery of a potent

39
MDR reversal agent that hypersensitizes P-glycoprotein resistant tumor cell lines to front
line conventional therapeutic agents. 191
Ningalin derivatives have been evaluated for their properties of potent reversal of
MDR and use in drug combinations against human colon carcinoma xenograft in nude
mice. 192
Storniamides:
Storniamides A (43), B (44), C (45), and D (46) were isolated from a sponge
Cliona species collected near San Antonio Oeste, Rio Negro, and Argentina. 193 They
showed antibiotic activity against Gram-positive bacteria (Staphylococcus aureus,
Bacillus subtilis and Micrococcus luteus) at 50 μg/disk. Five tyrosine units take part in
the building of the structural frame work of the storniamide, while subsequent
hydroxylation in the aromatic rings gives rise to different compounds.
Boger et al. reported the total synthesis of permethyl storniamide A. 183 In 2003 a
formal synthesis of permethyl storniamide A was reported, which was based on a highly
efficient route to 3,4-diarylpyrrole marine alkaloids. 186
In 2002 Furstuner et al. 194 made an assessment of the DNA cleaving properties of
the pyrrole alkaloid derivatives permethyl storniamides, lamellarins, ningalins etc. They
concluded that an increase in the number of peripheral methylations of such pyrrole
alkaloids causes a sharp decrease in their antitumoral activity.

40
Polycitrins:
Polycitone A (47) and polycitrins A (49) and B (50) were isolated from the
marine ascidian Polycitor species. 195 The penta-O-methyl derivative of polycitone A
was found to inhibit the growth of SV40 transformed fibroblast cells in a concentration of
10 ug/ml. Polycitone A inhibited the RDDP and DDDP activities of HIV-1 RT. 196
Polycitone B (48) was isolated from the ascidian Polycitor africanus from
Madagascar. 197
In 1995, Steglich et al. described the biomimetic total synthesis of polycitrin A. 198
The same group reported shorter synthetic route to polycitrin A in three steps. 199 Beccali
et al. reported the synthesis of Polycitrin B. 200 In 2002, Steglich’s group first synthesized
both Polycitones A and B. 201 In 2006, Gupton’s group demonstrated a new and efficient
relay strategy to synthesize Polycitones A and B. 202
Purpurone:
Purpurone (51) was isolated from the marine sponge Iotrochota species. 203 Its
structure was established mainly on the basis of NMR spectroscopic data. Purpurone is
having ATP-citrate lyase inhibitory activity. ATP-citrate lyase inhibition is anticipated to
reduce the production of acetyl CoA and can affect both lipogenesis and
cholesterogenesis, and can be a strategic target for hypercholesterolemia therapy.
Purpurone is synthesized by Peschko and Steglich 204 in 2000. Jia et al. 205
reported the direct synthesis of polysubstituted pyrroles from readily available aldehydes
and amines and their application to the total synthesis of purpurone.

41
Fifteen new purpurone related alkaloids named baculiferins A-O (52-66), were
isolated from the Chinese marine sponge Iotrochota baculifera, 206 together with the
known alkaloids purpurone (51) and ningalin A(39). Most of the new compounds contain
one to three O-sulfate units. Their structures were determined by extensive spectroscopic
analysis including 1H and 13C NMR (COSY, HMQC, HMBC) and ESIMS data.
Baculiferins were found to possess anti-HIV-1 activity.
Lukianols:
Lukianols A (67) and B (68) were isolated from an unidentified encrusting
tunicate 207 from Palmyra Atoll and characterised by spectroscopy. They were found to be
cytotoxic.
Lukianol A was synthesized using Diels Alder strategy. 183 Lukianol A was
convergently synthesized using Stille, Suzuki, or Negishi cross-coupling reactions as the
key step. 208 In 1999 Boger’s group reported lukianol A by utilizing a common
heterocyclic azadiene Diels-Alder reaction. 209 In 2000, Wong’s group reported the
formal total synthesis of lukianol A. 210 In 2006, Steglich et al. synthesized lukianol A in
which the lukianol skeleton is assembled in a single step. 211 In 2007, the same group
reported a short synthesis of lukianol A. 212
Dictyodendrins:
Dictyodendrins A (69), B (70), C (71), D (72) and E (73) were isolated from the
Japanese sponge Dictyodendrilla verongiformis. 213 They were found to possess
telomerase inhibitory activity.

42
Furstner et al. reported flexible total syntheses of the telomerase inhibitors
dictyodendrin B, C and E. 214
Sato et al. 215 reported three potent aldose reductase inhibitors (74, 75, 76) isolated
from a Japanese marine sponge, Dictyodendrilla species and were characterized by
chemical and physical evidence and X-Ray crystallographic analysis.
Aldose reductase catalyzes reduction of aldoses such as glucose and galactose, to
the corresponding polyols, such as sorbitol and galactol respectively. Intracellular
accumulation of the polyols may result in diabetic complications. Aldose reductase
inhibitors may therefore provide an effective means for the prevention and treatment of
such diseases.
Some other miscellaneous 3,4-diaryl pyrrole alkaloids are didemnimides,
granulatimides, rigidins, staurosporine aglycones, arcyriaflavins etc.
Didemnimides:
The alkaloids didemnimides A (77), B (78), C (79) and D (80) which inhibit
predation by fish, were isolated from the ascidian Didemnum conchyliatum, which was
collected from the sea grass blades in mangrove habitats in Bahamas. 216
Didemnimides A & B were synthesized by Hughes et al. 217 Didemnimide C was
synthesized in four steps by Steglich et al. 218 Piers et al also reported the synthesis of
didemnimide C in three steps. 219
Berlinck et al reported didemnimide E (81) from Didemnum granulatum. 220

43
Granulatimides:
Didemnum granulatum from Brazil contained the G 2 cell cycle checkpoint
inhibitors, granulatimide (82) and isogranulatimide 220 (83) the structures of which were
confirmed by synthesis. Isogranulatimide was isolated also from Didemnum
conchyliatum, from Bahamas. 221 Didemnum granulatum also yielded a novel alkaloid 6-
bromo granulatimide 222 (84).
Rigidins:
Rigidin A (85), a phosphodiesterase inhibitor, was isolated from the ascidian
Eudistoma rigida 223 and was synthesized by Sakamoto et al. 224 An additional synthesis of
rigidin was also reported. 225
Rigidins B (86), C (87), and D (88) were isolated from an Okinawan collection of
Cystodytes sp. 226 and these were found to be mildly cytotoxic towards L1210 murine
leukemia cell line.
Magedov et al. reported a four-step synthesis of alkaloids rigidins A, B, C and
D.227
Rigidin E (89) was isolated from a Papua New Guinea collection of a Eudistoma
species.228
Halitulin:
Halitulin (90) was isolated from the South African sponge Haliclona
tulearensis.229 Its structure is elucidated mainly on the basis of spectroscopic data as well
as chemical modifications. Halitulin was found to be cytotoxic against several tumor

44
cells, P-388 murine leukemia, A-549 human lung carcinoma, HT-29 human colon
carcinoma and MEL-28 human melanoma in concentrations of 12-25 ng/ml. It was
synthesized by Heinrich et al.230
Staurosporins and arcyriaflavins:
11-hydroxy staurosporine (91) was first isolated from the ascidian Eudistoma
species from Pohnpei231 and was found to be a cytotoxic protein kinase C inhibitor.
A West African Eudistoma species yielded arcyriaflavin A (92) and staurosporine
aglycone which was responsible for the strong cytotoxicity and protein kinase C activity
of crude extracts.232
The prosobranch mollusk Coriocella nigra contained an additional cytotoxic
staurosporin analogue 4-N-demethyl 11-hydroxy staurosporine (93) 233. Another
staurosporin derivative, 3’-demethoxy 3, 3’-dihydroxystaurosporine (94) along with 4-N-
demethyl 11-hydroxy staurosporine was isolated from Eudistoma toealensis and its
predatory flatworm Pseudoceros species. 234 Both the ascidian Eudistoma toealensis and
its predatory flatworm Pseudoceros species collected in Chuuk and Micronesia yielded
three new staurosporine235 derivatives (95, 96, and 97).
Laatch et al. 236 reported a new staurosporine, N-carboxamido-staurosporine (98)
isolated from the culture broth of the marine-derived Streptomyces sp. QD518 from the
Jiaozhou Bay of Quindao, China. The structures were determined by spectroscopic
methods and by comparison of the NMR data with those of structurally related known
natural products, which were isolated from the same strain.

45
Two new indolocarbazole alkaloids, 7-oxo-3, 8, 9-trihydroxystaurosporine (99)
and 7-oxo-8, 9-dihydroxy-4’-N-demethyl staurosporine (100), were isolated from the
samples of the marine ascidian Cystodytes solitus collected in Tanzania. Their structures
were determined by a combination of spectroscopic techniques. Both compounds
displayed strong cytotoxicity against three human cell lines. 237
3, 4-diaryl pyrrole alkaloids from marine organisms showed varied biological
activities and these are summarized in Table 1-03.
Table I-03. Biological activities of 3,4 diaryl pyrrole alkaloids from marine organisms.
Compound Activity Compound No.
Reference
lamellarin D Cytotoxic,
multi drug resistance reversal agent
Topoisomerase1 inhibitor Mitochondrial permeability-
transition inhibitor
4 [120,127,128]
[123]
[121,132,133] [136]
lamellarin K Immunomodulatory effects 11 [151]
lamellarin L 7-hydroxyneolamellarin A
Immunomodulatory effects Hypoxia-induciblefactor-1 inhibitor
12 22
[151] [166]
lamellarin α 20-sulfate HIV integrase inhibition 31 [172]
lamellarin β Cytotoxic 32 [174] lamellarin ζ Cytotoxic 35 [176]
lamellarin χ Cytotoxic 38 [176] lamellarin L- triacetate Cytotoxic 12a [176]
lamellarin F Cytotoxic 6 [176] ningalin B Multi drug resistance reversal 40 [184]

46
agent
storniamides A-D Antibiotic 43-46 [193] polycitone A Cell growth inhibitor
HIV-1RT inhibitor 47 [195]
[196] Purpurone
Baculiferins (C,E-H,K-N)
ATP-citrate lyase inhibitor
HIV-1 inhibitor 51
(54,56-59, 62-65)
[203]
[206]
lukianols A,B Cytotoxic 67,68 [207] dictyodendrins A-E Telomerase inhibitor 69-73 [213]
aldose-reductase inhibitors aldose-reductase inhibitors 74-76 [215] Granulatimide G2 cell cycle check point
inhibitor 82 [220]
Isogranulatimide G2 cell cycle check point inhibitor
83 [220]
rigidin A Phosphodiesterase inhibitor 85 [223]
rigidins B, C, D Cytotoxic 86-88 [226] Halitulin Cytotoxic 90 [229]
11-hydroxy staurosporine 7-oxo-3,8,9-trihydroxy
staurosporine
7-oxo-8,9-dihydroxy-4’-N-demethyl staurosporine
Cytotoxic protein kinase C inhibitor
Cytotoxic
Cytotoxic
91
(99)
(100)
[231]
[237]
[237]

47
LAMELLARINS
O
NX
HO
MeO
HO
MeOMeO OMe
OMe
O
(1) Lamellarin A X = OH(3) Lamellarin C X = H
O
N
HO
MeO
HOMeO OR
X
O
OMe(2) Lamellarin B R=Me X = OMe(4) Lamellarin D R=X = H
O
N
R1O
R2O
MeOMeO OR4
R5
O
OR3
(5) Lamellarin E R1=R3=H R2= R4=Me R5= OH (6) Lamellarin F R1= H R2=R3=R4=Me R5= OH(7) Lamellarin G R1= Me R2=R3=R4=R5=H Lamellarin G 8-sulfate R4=SO3
O
N
HO
HO
HOHO OH
O
OH
(8) Lamellarin H
O
N
HO
MeO
R2OMeO OR3
X
O
R1O
(09) Lamellarin I R1=R2=R3= Me X=OMe (10) Lamellarin J R1=R2= Me R3 =X=H (11) Lamellarin K R1=R3= Me R2=H X=OH (12) Lamellarin L R1=Me R2=R3=X=H
O
N
AcO
MeO
HOMeO OAc
O
AcO
(12a) Lamellarin L triacetate

48
O
N
SO3-O
MeO
HOMeO OH
O
MeO
(12b) 20 sulfate of Lamellarin L
O
N
HO
MeO
R2OMeO OR3
X
O
R1O
(13) Lamellarin M R1=R3= Me R2= H X=OH (14) Lamellarin N R2= Me R1= R3 =X=H
N COOMe
HO OH
OMe
OX
(15)Lamellarin O X=H(16)Lamellarin P X=OH (18) Lamellarin R
N COOMe
HO OH
OH
NH
COOMe
HO OH
(17)Lamellarin Q
O
N
HO
HO
HOMeO OH
O
OH
(23) Lamellarin S

49
NR O
HO OH
OH(19)Neolamellarin A R=H(22)7-Hydroxyneolamellarin A R=OH
NO
HO OH
OH(20) Neolamellarin B R=H(21) 5-Hydroxyneolamellarin B R=OH
R O
O
N
R1O
MeO
MeOR2O OMe
O
OH
(25) Lamellarin U R1=H R2=Me 20-Sulfate of lamellarin U R1=SO3Na(29) Lamellarin Y R1=SO3Na R2=H
O
N
HO
MeO
MeOMeO OR
O
OH
(27) Lamellarin W R=Me X=OMe (28) Lamellarin X R=Me X=OH
X
O
N
RO
MeO
MeOMeO OMe
O
OH(24) Lamellarin T R=X=H (26) Lamellarin V R=H X=OH 20-Sulfate of lamellarin T R=SO3Na20-Sulfate of lamellarin V R=SO3Na
X
OMe
O
N
MeO
HO
HOMeO OH
O
OH
(30) Lamellarin Z
50
O
N
NaO3SO
MeO
MeOMeO OMe
O
OH
O
N
HO
HO
MeOHO OH
O
OH
(31) Lamellarin α 20-sulfate (32) Lamellarin β
O
N
HO
MeO
MeO OMe
O
OMe
MeO OH
O
N
HO
MeO
MeO OMe
O
OMeMeO
(33) Lamellarin γ (34) Lamellarin ε
O
N
R1O
MeO
R2O OMe
O
OMeR3OX
O
N
AcO
MeO
MeO OAc
O
OMeAcOH
(35) Lamellarin ζ R1=H R2=R3=Me (38) Lamellarin χ
X=OMe
(36) Lamellarin η R1=H R2=R3=Me
X=H
(37) Lamellarin R1=R2=R3=Ac
X=OMe

51
NH
OO
O O
HO OH HOOH
(39) Ningalin A
NO
HO OH HO OH
HO OH(40) Ningalin B
N
HO OH HO OH
HO OH(41) Ningalin C
O
OHOH
O
N
HO OH
HO OH
(42) Ningalin D
OO
HO
HO
NHN
HOR2
HN
OHR3
OHHO
HO
OH OH
R1
OH
O O
Storniamides A R1=OH R2=R3=H(43) B R1=R3=OH R2=H(44) C R1=H R2=R3=OH(45) D R1= R2=R3=OH(46)

52
N
BrHO
Br
BrOH
Br
OH
O O
(47) Polycitone A
Br
OH
Br
BrHO
Br
NH
BrHO
Br
BrOH
Br
O O
(48) Polycitone B
BrOH
Br
BrHO
Br
N
BrHO
Br
BrOR
Br
(49) Polycitrin A R=H(50) Polycitrin B R=Me
OO
OH
N
HO OH HO OH
(51) Purpurone
O O
HO
HO
HO
OH
OH
N
HO OR1 R2O OH (52) Baculiferin A R1=SO3H R2=R3=R4=H(53) Baculiferin B R3=SO3H R1=R2=R4=H(54) Baculiferin C R3=R4=SO3 H R1=R2=H(55) Baculiferin D R1=R2=SO3H R3=R4=H(56) Baculiferin E R1=R3=SO3H R2=R4=H(57) Baculiferin F R1=R4=SO3H R2=R3=H(58) Baculiferin G R1=R3=R4=SO3H R2=H(59) Baculiferin H R1=R2=R3=SO3H R4=H
O O
R3O
HO
HO
OH
OR4

53
N O
HO OHOH
(60) Baculiferin I R1=SO3H R2=R3=H(61) Baculiferin J R1=H R2=SO3H R3=H(62) Baculiferin K R1=R2=34=H
O
HO
HO
OR2
R3
OR1
N
HO OR HOOH
(63) Baculiferin L R=H(64) Baculiferin M R=SO3H
O O
HO
HO OH
OHCOOH
N O
HO OHOH
(65) Baculiferin N
O
HO
HO
OH
COOH
NH
OO
HO OR HOOH
(66) Baculiferin O R=SO3H
O O
N
HO
X
OH
(67) Lukianol A X=H(68) Lukianol B X=I
O
OH
O
N
HO
(69) Dictyodendrin AOH
NH
OSO3Na
OH
OH
MeOOC
N
HO
(70) Dictyodendrin BOH
NH
OSO3Na
OH
OH
O

54
N
HO
(71) Dictyodendrin C R=H(72) Dictyodendrin D R=SO3Na
OR
NH
OSO3Na
O
OH
ON
HO
(73) Dictyodendrin EOH
NH
OSO3Na
O
OHOH
N
HO
(74) Aldose reductase inhibitor 1. M=Na(75) Aldose reductase inhibitor 2. M=H
OH
NH
OSO3M
O
OH
O
OH
HO
N
HO
(76) Aldose reductase inhibitor 3OH
NH
OSO3H
O
OHOH
MeOOC
NH
NNHN
R1
R2
O O
(77)Didemnimide A R1=R2=H (78) B R1=Br R2=H (79) C R1=H R2=Me (80) D R1=Br R2=Me (81) E R1=Me R2=H
NH
O O
(82) Granulatimide
HNN
HN
NH
O O
(83) Isogranulatimide
N
HNN
NH
O O
(84) 6-Bromogranulatimide
HNN
HN
Br

55
NH
N
NH
OHR2
O
O
O
R3
R1 OH
(85) Rigidins A R1=R2=R3=H (86) B R1=R3=H R2=OMe (87) C R1=OMe R2=R3=H (88) D R1=R2=OMe R3=H (89) E R1=R2=H R3=Me
N
HO OH
(90) Halitulin
N N
HO OH
N
NH
O
(91) 11-Hydroxystaurosporine
NNHO O
MeONHMe
NH
O
(92) Arcyriaflavin A
HN
HN
O
NH
O
(93)4'-N-demethyl11-hydroxystaurosporine
NNHO O
MeONH2

56
NH
O
(94) 3'-demethoxy-3,3'-dihydroxystaurosporine
NN O
HONHMe
OH
NH
O
(95) R1=H R2=R3=R4=Me R5=OH(96) R1=R3= R4=H R2=Me R5=OH(97) R1= R2 =R3= R4 = R5= H
NN O
R2ON
R5
R1
R4 R3
NH
O
(98) N-Carboxamido-staurosporine
NN O
NH3C CONH2
H3CO
NH
O
(99) 7-oxo-3,8,9-trihydroxystaurosporine (R1=OH, R2=Me )(100) 7-oxo-8,9-dihydroxy- 4'-N-demethylstaurosporine (R1=R2=H )
NN O
NHR2H3CO
O
OHHO R1

57
REFERENCES:
1. E. S. Heimer, J. B. Someone, Chemical Ecology,1970, Academic Press, NewYork.
2. S. Frormann, G. Jas, Business Briefing: Future Drug Discover, 2002, 84.
3. G. M. Cragg, D. J. Newman, Expert Opinion on Investigational Drugs, 2000, 9, 2783.
4. D. J. D. Vries and M. Hall, Drug Der. Res., 1994, 33, 161.
5. J.T. Baker, Natural Products and drug development, Proceedings of the Alfred Benzon Symposium 20, ed. P.Krosgaard-larsen, S.B.Christensen, and K.H.Munksgaard, 1984, 145.
6. J. P. Riley and R. Chester, Introduction to Marine Chemistry, Academic Press, New York, 1971.
7. J. F. Sidu and J. F. D. Bernadis, Lioydia., 1973, 36, 107.
8. D. H. Williams, M. J. Stone, P. R. Hawk. and S. K. Rahman, J. Nat. Prod., 1989, 52, 1189.
9. R. H. Whittaker and P. P. Feeney, Science, 1971, 171, 757.
10. G. Cimino and S.D. Rosa, Tetrahedron, 1985, 41, 1093.
11. J. E. Thompson, R. Pwalker, S. J. Wratten and D. J. Faulkner, Tetrahedron, 1982, 38, 1865.
12. J. E. Thompson, P. T. Murthy, P. R. Bergquist and E. A. Evans, Bio. Chem. Syst. Ecol., 1987, 15, 595.
13. B. Howard, A. M. Nonomura, and W. Fenical, Bio. Chem. Syst. Ecol., 1980, 8, 329.
14. J. Jimeno, G. F. Cloth, J. M. Fsousa-fero, P. Scheuer and K. Rinehart, New Marine derived anticancer Therapeutics-A Journey from sea to clinical trials. Mar. Drugs., 2004, 1, 14.
15. S. Detmer, M. C. R. Franssen, R. Osinga, T. Johannes and H. R. Wijffels, Marine sponges as pharmacy, Marine Biotechnology., 2005, 7, 142.
16. J. Sweldlow, Nature and Medicine; National Geographic Mil Eds. Paris, 2000.
17. J. Rajeev kumar and Xu Zi-rong, Biomedical compounds from the marine organisms: a review, Mar. Drugs., 2004, 2, 123.
18. A. J. Weinheimer and R. L. Spraggins, The occurrence of two new prostaglandin derivatives (15-epi-PGA2 and its acetate, methyl ester) in the gorgonian Plexuara homonella. Chemistry of Coelenterates. XV. Tetrahedron Lett., 1974, 15, 5185.
19. P. T. Grant and A. M. Mackie, Drugs from the Sea – Fact or Fantasy, Nature, 1977, 267, 786.
20. Marine Natural Products: Chemical and Biological Perspectives: Ed. P.J.Scheuer, Academic Press, New York, Vol. I-V, 1978-1983.
21. Bioorganic Marine Chemistry: Ed. P.J. Scheuer, Springer-verlag, Berlin, Vol. I- VI, 1987-1992.
22. Poisonous and Venomous Marine animals of the World: Ed. B.W. Halstead, U.S. Govt. Printing Office, Washington D.C, Vol.I-III, 1965, 1967, 1970.
23. A. D. Marderosian, Marine Pharmaceuticals, J. Pharm. Sci., 1969, 58, 1.
24. P. N. Kaul. And P. Daftari, marine Pharmacology: Bioactive molecules from the Sea, Ann. Rev. Pharmacol. Toxicol., 1986, 26, 117.

58
25. M. J. Garson, Biosynthetic studies on Marine Natural Products, Nat. Prod. Rep., 1989, 6, 143.
26. R. Riguera, Interesting bioactive compounds from marine organisms, J. Mar. Biotechnol., 1997, 5, 187.
27. J. M. Kornprobust, C. Sallenave, G. Barnathan, sulfated compounds from marine organisms, Comp. Biochem. Physiol., Sect.B, 1998, 119, 1.
28. G. J. Bakus, N. M. Target and B. Schulte, Chemical Ecology of Marine Organisms: an overview, J. Chem. Ecol., 1986, 12, 951.
29. J. Colwell, Biotechnology in Marine Sciences, Science, 1983, 222, 19.
30. A. M. Burja, B. Banaigs, E. Abou-Mansour, J. G. Burgess and P. C. Wright, Tetrahedron, 2001, 57, 9347.
31. R. Osinga, J. Tramper and R. H. Wijffels, Cultivation of marine sponges for metabolite production: applications for biotechnology? Trends Biotechnol., 1998, 16, 130.
32. M. H. G. Munro, J. W. Blunt, E. J. Dumdei, S. J. H. Hickford, R. E. Lill, S. Li, C. N. Battershill, A. R. Duckworth, J. Biotechnol., 1999, 70,15.
33. V.S.Berman, M.Greenstein and W.M.Maise, Marine microorganisms as a source of new natural products, Adv .Appl. Microbiol., 1997, 43, 57.
34. F. Pietra, Secondary metabolites from marine microorganisms-bacteria, protozoa, algae and fungi-achievements and prospects, Nat. Prod. Rep., 1997, 14, 453.
35. Various authors, Marine Natural Products Chemistry, Chem. Rev., 1993, 93, 1671.
36. A. Kelecom, An. Acad. Bras. Cienc., 1999, 71, 249.
37. P.J. Scheuer, J. Chem. Educ., 1999, 76, 1075.
38. P. R. Jensen and W. Fenical, Drugs from the Sea, ed. N. Fusetani. Karger, Basel (Switzerland), 2000, pp. 6.
39. Y. Shimizu, Microalgae as a drug source, in Drugs from the Sea, ed. N. Fusetani, Karger, Basel (Switzerland), 2000, pp. 30.
40. M. Kobayashi, Search for biologically active substances from marine sponges, in Drugs from the Sea, ed. N. Fusetani, Karger, Basel (Switzerland), 2000, pp. 46.
41. K. Yamada, M. Ojika, H. Kigoshi and K. Suenaga, Cytotoxic substances from opisthobranch molluscs, in Drugs from the Sea, ed. N. Fusetani, Karger, Basel (Switzerland), 2000, pp. 59.
42. B. M. Olivera, -Conotoxin MVIIIA: from marine snail venom to analgesic drug, in Drugs from the Sea, ed. Fusetani, N. Karger, Basel (Switzerland), 2000, pp. 74.
43. T. Natori, K. Motoki, T. Higa and Y. Koezuka, KRN7000 as a new type of antitumour and immune-stimulatory drug, in Drugs from the Sea, ed. N. Fusetani, Karger, Basel (Switzerland), 2000, pp 86.
44. M. Kuramoto, K. Yamaguchi, T. Tsuji and D. Uemura, Zoanthamines, antiosteoporotic alkaloids, in Drugs from the Sea, ed. Fusetani, N., Karger, Basel (Switzerland), 2000, pp. 98.
45. D. J. Faulkner, M. K. Harper, M. G. Haygood, C. E. Salomon and E. W. Schimdt, Symbiotic bacteria in sponges: sources of bioactive substances, in Drugs from the Sea, ed. N.Fusetani, Karger, Basel (Switzerland), 2000, pp. 107.
46. I. Manzanares, C. Cuevas, R. Garcia-Nieto, E. Marco, F. Gago, Curr. Med. Chem.-Anti-Cancer Agents, 2001, 1, 257.

59
47. J. B. Hart, R. E. Lill, S. J. H. Hickford, J. W. Blunt and M. H. G. Munro, The halichondrins: chemistry, biology, supply and delivery, in Drugs from the Sea, ed. N.Fusetani, Karger, Basel (Switzerland), 2000, pp.134.
48. G. Cimino and M. T. Giselin, Marine natural products chemistry as an evolutionary narrative, Mar. Chem. Ecol., 2001, 115.
49. R. J. Capon, Marine bioprospecting-trawling for treasure and pleasure, Eur. J. Org. Chem., 2001, 4, 633.
50. Marin Lit database, Department of Chemistry, University of Canterbury: http://www.chem.canterbury.ac.nz/marinlit/marinlit.html.
51. D. J. Faulkner, Nat. Prod. Rep., 1984, 1, 251; ibid, 1984, 1, 551; ibid, 1986, 3, 1; ibid, 1987, 4, 539; ibid, 1988, 5, 613; ibid, 1990, 7, 269; ibid, 1991, 8, 97; ibid, 1992, 9, 323; ibid, 1993, 10, 497; ibid, 1994, 11, 355; ibid, 1995, 12, 223, ibid, 1996, 13, 75; ibid, 1997, 14, 259; ibid, 1998, 15, 113, ibid, 1999, 16, 155; ibid, 2000, 17, 7; ibid, 2001, 18, 1; ibid, 2002, 19, 1.
52. H. E. Belarbi, A. C. Gomez, Y. Chisti, F. G. Camacho and E. M. Grima, Biotechnology Advances., 2003, 21, 585.
53. Marine Natural Products Chemistry, Ed. D. J. Faulkner and W. H. Fenical, Plenum Press, New York, 1977.
54. Bioactive Compounds from Sea: Ed. J. H. Harold, E. L. Charles and D. Marcel, INC, New York, 1974.
55. Proceedings of the NATO conference on Marine Natural Products: Ed. D. J. Faulkner and W. H. Fenical, Plenum Press, New York, 1977, 87, 165, 239.
56. Bioactive Compounds from Marine Organisms: Ed. M. F. Thomson, R. Nagabhushanam and R. Sarojini, Oxford & IBH Publishing Co. Pvt. Ltd., New Delhi, 1991.
57. K. Scotlandi, S. Perdichizzi, M. C. Manara , M. Serra, S. Benini, V. Cerisano, R. Strammiello, M. Mercuri, G. Reverter-Branchat, G. Faircloth, M. D’Incalci, P. Picci, Clin. Cancer Res., 2002, 8, 389.
58. D. J. Faulkner and R. J. Andersen, Natural Chemistry of the Marine Environment: The Sea: Ed., Goldberg, E.D, Wiley, New York, 1974, 5, 679.
59. D. J. Faulkner, Tetrahedron, 1977, 33, 1421.
60. J. Jimeno, G. Faircloth, J. M. F. Sousa-Faro, P. Scheuer and K. Rinehart, Marine Drugs, 2004, 2, 1607.
61. D. J. Faulkner, Antibiotics from the Marine Organisms: Top. Antibiot. Chem., 1978, 2, 9.
62. D. J. Faulkner, Biomedical uses for Natural Marine Chemicals: Oceanus, 1992, 35, 29.
63. L. Minale, Medicine from the Sea: Managing the Ocean-Resources, Research, Law: Ed.Richardson, G. J, Lomond Publications, 1985, 87.
64. P. N. Kaul, Pure Appl.Chem., 1982, 54, 1963.
65. G. D. Ruggieri, Science, 1976, 194, 491.
66. F. Pietra., Gazz. Chim. Italia, 1989, 115, 443.
67. M. T. Hamann, Enhancing Marine natural Product Structural Diversity and Bioactivity through Semisynthesis and Biocatalysis, Curr. Pharm. Design., 2003, 9, 879.
68. www.mdpi.net/marinedrugs.
69. G. Schwartsmann, A. B. D.Rocha, R. G. S. Berlinck, and J. Jimeno, Lancet Oncol., 2001, 2, 221.

60
70. D. J. Newman and M. G. Cragg, J. Nat. Prod., 2004, 67, 1216.
71. T. Nanomura, M. Sasaki, N. Matsumori, M. Miata, K. Tachibana and T. Yasumoto, Angew. Chem, Int. Ed. Engl., 1996, 35, 1675.
72. J. B. Hart, R. E. Lill,S. J. H. Hickford, J. W. Blunt and M. H. G. Munro, The halichondrins: chemistry, biology, supply, delivery, in Drugs from the Sea, ed. W. Fusetani, Karger, Basel (Switzerland), 2000, 134.
73. G. Cimino and M. T. Giselin., Mar. Chem. Ecol., 2001, 15.
74. N. J. Carter and S. J. Keam, Drugs, 2007, 67, 2257.
75. F. Grosso, R. L. Jones, G. D. Demetri, I. R. Judson, J. Y. Blay, A. L. Cesne, R. Sanfillppo, P. Casieri, P. Collini, P. Dileo, C. Spreafico, S. Stacchiotti, E. Tamborini, C. Tercero C. J. Jimeno, M. D. Incalci, A. Gronchi., J. A. Fletcer, S. Pilotti and P. G. Casal, Lance Oncol., 2007, 8, 595.
76. S. Donald, R.D. Verschoyle, P. Greaves, T.W. Gant, T. Colombo, M. Zaffaroni, R. Frapolli, M. Zucchetti, M. D Incater, D. Meco, R. Riccardi, L. Lopez-Lazaro, J. Jimeno, A. Gescher, J. Cancer Res., 2003, 63, 5902.
77. R.Cuevas, M. J. M. P. Mrtin, J.L. Chicharro, C. R. Fernandez, M. Flores, A. Francesch, P. Gallego, M. Zarzuelo, F. D. L. Calle, J. Garcia, C. Polanco, I. Rodriguez and I. Manzanares, Org.Lett., 2002, 2, 2545.
78. K. L. Rinehart, J. B. Gloer, J. C. Cook, S. A. Mizak and T. A. Scohill , J. Am. Chem. Soc., 1981, 103, 1857.
79. C.L. Touneau, E. Raymond and S. Faivre, Curr. Pharm. Des., 2007, 324, 3427.
80. M. J. M. Alonso, L. S. Gonzalez, N. Zarich, T. Martinez, E. Alvarez, J. M. Rojas and A. Munoz, J. Pharmacol. Exp.Ther., 2008, 324, 1093.
81. M.T. Hamann and P. J. Scheuer, J. Am. Chem. Soc., 1993, 115, 5825; A. M. Lopez, J. C. Jimenez, M. Royo, E. Giralt and F. Albericio, J. Am. Chem. Soc., 2001, 123, 11398; I. Bonnard, I. Manzanares and K. L. Rinehart, J. Nat. Prod., 2003, 66, 1466.
82. Y. Suarez, L. Gonzalez, A. Cuadrado, M. Berciano, M. Lafarga and A. Munoz, Mol. Cancer Ther., 2003, 2, 863.
83. J. Jimeno, M. Aracil and J. C. Tercero, J. Transl. Med., 2006, 4, 3.
84. A. M. Piggott and P. Karuso, Chem. BioChem., 2008, 9, 524.
85. G. R. Pettit, C. L. Herald, D. L. Doubek, D. L. Herald, E. Arnold and J. Clardy, J. Am. Chem. Soc., 1982 104, 6846; G. R. Pettit, D. L. Herald, F. Gao, D. Sengupta and C. L. Herald, J. Org. Chem., 1991, 56, 1337; D. E. Schaufelberger, M. P. Koleck, J. A. Beutler, A. M. Vatakis, B. Alvarado, P. Andrews, L. V. Marzo, G. M. Musch, J. Roach, J. T. Ross, W. B. Lebherz, M. P. Reeves, R. M. Eberwein, L. L. Rodgers, R. P. Testerman, K. M. Snader and S. Forenza, J. Nat. Prod., 1991, 54, 1265.
86. S. Sudek, N. B. Lopanik, L. E.Waggoner, M. Hilderbrand, C. Anderson, H. Liu, A. Patel, D. H. Sherman and M. G. Haygood, J. Nat. Prod., 2007, 70, 67; K. H. Sharp, S. K. Davidson and M. G. Haygood, ISME J, 2007, 1, 693.
87. K. J. Hale, M. G. Hummersone, S. Manaviazar and M. Frigerio, Nat. Prod. Rep., 2002, 19, 413.
88. S. Banerjee, Z. Wang, M. Mohammad, F. H. Sarkar and R. M. Mohammad, J. Nat. Prod., 2008, 71, 492.
89. M. K. Sun and D. L. Alkon, CNS Drug Rev., 2006, 12, 1.
90. Y. Hirata, D. Uemura, Pure Appl.Chem., 1986, 58, 701.

61
91. D. Uemura, K. Takahashi, T. Yamamoto, C. Katayama, J. Tanaka, Y. Okumura and Y. Hirata, J. Am. Chem. Soc., 1985, 107, 4796; Y. Hirata and D. Uemura , Pure Appl. Chem., 1986, 58 , 701.
92. T. D. Aicher, K. R. Buszek, F. G. Fang, C. J. Forsyth, S. H. Jung, Y. Kishi, M. C. Matelich, P. M. Scola, D. M. Spero and S. K. Yoon , J. Am. Chem. Soc., 1992, 114, 3162.
93. M. J. Towle, K. A. Salvato, J. Budrow, B. F. Wels, G. Kuznetsov, K. K. Aalfs, S. Welsh,W. Zheng, B. M. Seletsky, M. H. Palme, G. J. Habgood, L. A. Singer, L. V. D. Pietro, Y. Wang, J. J. Chen, D. A. Quincy, A. Davis, K. Yashimatsu, Y. Kishi, M. J. Yu and B.A. Littlefield, Cancer Res., 2001, 61, 1013.
94. M. Seletesky, Y. Wang, L. D. Hawkins, M. H. Palme, G. J. Habgood, L.V. D. Pietro, M. J. Towle, K. A. Salvato, B. F. Wels, K. K. Aalfs, Y. Kishi, B. A. Littlefield and M. J. Yu, Bioorg. Med. Chem. Lett., 2004, 14 , 5551.
95. G. Kuznetsov, M. J.Towle, H. Cheng, T. Kawamura, K. T. Dyke, D. Liu,Y. Kishi, M. J. Yu and B. A. Littlefield, Cancer Res., 2004, 64, 5760.
96. S. Newman, Curr. Opin. Invest. Drugs., 2007, 8, 1057.
97. R. Bai, S. J. Friedman, G. R. Pettit and E. Hamel, Biochem. Pharmacol., 1992, 43, 2637; G. R. Pettit, Y. Kamano, C. L. Herald, Y. Fujii, H. Kizu, M. R. Boyd, F. E. Boettner, D. L. Doubek, J. M. Schmidt, J. C. Chapuis and C. Michael, Tetrahedron, 1993, 49, 9151.
98. A. Ray, T. Okouneva, T. Manna, H. P. Miller, S. Schmid, L. Arthaud, R. Luduena, M. A. Jordan and C. Michael, Tetrahedron, 2007, 67, 3767.
99. K. K. Rasila and C.Verschraegen, Curr. Opin. Invest. Drugs., 2005, 6, 631.
100. M.S. Wallace, Expert Rev. Neurother., 2006, 6, 1423.
101. I. A. Sharpe, J. G .Loughnan, M. L. Thomas, L. Adams, D. A. Atkins, A. Palant, E. Craik, J. Adams, D. J. P. F. Alewood and R. J. Lewis, Nat. Neurosci., 2002, 4, 902.
102. E. S. Lovelace, C. J. Armishaw, M. L. Colgrave, E. Wahistrom, P. F. Alewwood, N. L. Daly and D. J. Craik, J. Med. Chem., 2006, 49, 6561.
103. F. A. Paczkowski, I. A. Sharpe, Dutertre and R.J. Lewis, J. Biol. Chem., 2007, 282, 17837.
104. Xenome: Further information available at http://www.xenome.com
105. B. C. M. Potts and D. J. Faulkner, J. Nat. Prod., 1992, 55, 1701.
106. D. L. Burgoyne, R. J. Andersen and T. M. Allen, J. Org. Chem., 1992, 57, 525.
107. F. R. Coulson and S. R. O’Donnell, Inflamm. Res., 2000, 49, 123.
108. Y. Shen and D. L. Burgoyne, J. Org. Chem., 2002, 67, 3908.
109. H. Kitawaga T. Takenouchi, R. Azuma, K. A. Wesnes, W. G. Kramer, D. E. Clody, and A. L. Burnett, Neuropsychopharm., 2003, 28, 542.
110. M. N. Rao, A. E. Shinnar, L. A. Noecker, T. L. Chao, B. Feibush, B. Snyder,I. Sharkansky, A. Sarkahian, X. Zhang, S. R. Jones, W. A. Kinney and M. Zasloff, J. Nat. Prod., 2000, 63, 631; K. S. Moore, S.Wehrli, H. Roder, M. Rogers, J. N. Forrest Jr, D. McCrimmon and M. Zasloff, Proc. Natl. Acad. Sci. U.S.A., 1993, 90, 1354.
111. R. S. Ahima, H. R. Patel, N.Takahashi, Y.Q.S. M.Hileman and M. A. Zasloff, Diabetes, 2002, 51, 2099.

62
112. M. N. Chernova, D. H. Vandorpe, J. S. Clark, J. I. Williams, M. A. Zasloff, L. Jiang, S. Lalper, Am. J. Physiol: Regul. Integr. Comp. Physiol, 2005, 289, R1644.
113. M. McLane, K. A. Lantz, A. G. Emeigh Hart, A.V. Albright, H. L.Hung and H. R. Wolfe, WO Pat .Appl, 2007, 124086.
114. M. S. Butler, Nat. Prod. Rep., 2008, 25, 475.
115. S. Campagna and M. Mihaela, WO Pat. Appl. 2006, 121857.
116. M. Hildebrand, Chem. Biol., 2004, 11, 1543.
117. A. Shirley, I. Pomponi, The bioprocess-technological potential of the sea J. Biotech., 1999, 70, 5.
118. R. J. Anderson, D. J. Faulkner, C. H. He, G. D. V. Duyne and J. Clardy, J.Am.Chem.Soc., 1985,107, 5492.
119. F. Ishibashi, Y. Miyazaki and M. Iwao, Tetrahedron, 1997, 53, 5951.
120. F. Ishibashi, S.Tanabe, T. Oda and M. Iwao, J. Nat. Prod., 2002, 65, 500.
121. C. Tardy, M. Facompre, W. Laine, B. Balderyrou, D. G. Gravalos, A. Francesch, C. Mateo, A. Pastor, J. A. Jimenez, I. Manzanares, C. Cuevas and C. Bailly, Bioorganic & Medicinal Chemistry , 2004, 12, 1697.
122. D. Pla, A. Marchal, C. A. Olsen, F. Albericio, and M. Alvarez, J. Org. Chem., 2005, 70, 8231.
123. M. Vanhuyse, J. Kluza, C. Tardy, G. Otero, C. Cuevas, C. Bailly and A. Lansiaux, Cancer letters, 2005, 221, 165-175.
124. N. Fujikawa, T. Ohta, T. Yamaguchi, T. Fukuda, F. Ishibashi, and M. Iwao, Tetrahedron, 2006, 62, 594.
125. D. Pla, A. Marchal, C. A. Olsen, A. Francesch, C. Cuevas, F. Albericio, M. Alvarez, J. Org. Chem., 2006, 49, 3257.
126. T. Ohta, T. Fukuda, F. Ishibashi, and M. Iwao, J. Org. Chem., 2009, 74, 8143.
127. D. Pla, A. Francesch, P. Calvo, C. Cuevas, R. Aligue, F. Albericio and M. Alvarez, Bioconjugate Chem., 2009, 20, 1100.
128. D. Pla, M. Marti, J. F.Sinfren, D. Pulido, A. Francesch, P. Calvo, C. Cuevas, M. Royo, R. Aligue, F. Albericio and M. Alvarez, Bioconjugate Chem., 2009, 20, 1112.
129. L. Shen, X. Yang, B. Yang, Q. He, and Y. Hu, Eur.J.Med.Chem., 2010, 45, 11.
130. S.V. Boonya, N. Yotapan, C. Woo, C. J. Burns, S. Ruchirawat and N. Thasana, Chem.-Asian J., 2010, 5,2113.
131. Q. Li, J. Jiang, A. Fan, Y. Cui and Y. Jia, Org. Lett., 2011, 13,312.
132. M. B. Robert, S. Lim, C. Barthomeuf and D. Morvan, Biochem.Pharmacol., 2010, 80, 1170.
133. D. Baunbaek, N.Trinkler, Y. Ferandin, O. Lozach, P. Ploypradith, S.Ruchirawat, F. Ishibashi, M. Iwao and L. Meijer, Mar.Drugs, 2008, 6, 514.
134. C. Ballot, J. R. Kluza, A. Martoriati, U. Nyman, P. Formstecher, B. Joseph, C. Bailly and P. Marchetti, Mol. Cancer Ther., 2009, 8, 3307.
135. D. Pla, A. Sischka, F. Albericio, M. Alvarez, X. B. Fernandez and D. Anselmetti, Small, 2009, 5, 1269.
136. C. Ballot, J. Kluza, S. Lancel, A. Martoriati, S. Hassoun, L. Mortier, J. C. Vienne, G. Briand, P. Formstecher, C. Bailly, R. Neviere and P. Marchetti, Apoptosis, 2010, 15, 769.
137. N. Lindquist and W. Fenical, J. Org. Chem., 1988, 53, 4570.
138. D. W. Behrens, Veliger, 1980, 22, 323.
139. E. H. Morris, D. P. Abbott and E. D. Haderlie, Eds. Intertidal Invertebrates of California; Stanford, (1980).

63
140. G. Lambert, Veliger, 1980, 22, 340.
141. A. Heim, A. Terpin, W. Steglich, Angew. Chem. Int. Ed. Engl., 1997, 36, 155.
142. S. Ruchirawat, T. Mutarapat, Tetrahedron Lett., 2001, 42,1205.
143. M. Iwao, T. Takeuchi, N. Fujikawa, T. Fukuda, F. Ishibashi, Tetrahedron Lett., 2003, 44, 4443.
144. S. T. Handy, Y. N. Zhang, H. Bregman, J. Org. Chem., 2004, 69, 2362.
145. C. Peschko, C.Winklhofer, A. Terpin, W. Steglich, Synthesis, 2006,3048.
146. J. C. Liermann and T. Opatz, J.Org.Chem., 2008, 73, 4526.
147. J.T.Gupton, B. C.Giglio, J. E. Eaton, E. A. Rieck, K. L. Smith, M. J. Keongh, P. J. Barelli, L. T. Firich, J. E. Hempel, T. M. Smith, and R. P. F. Kanters, Tetrahedron, 2009, 65, 4283.
148. Y. C. You, G. Yang, A. L. Wang, D. P. Li, Curr.Appl.Phys., 2005, 5, 535.
149. Y.C.You, A. L.Wang, D. P. Li, G. Yang, Biomed. Mater, 2006, 1, L7.
150. A. R. Carroll, B. F. Bowden and J. C. Coll, Aust. J. Chem., 1993, 46, 481.
151. Isolation, activity and synthesis in progress in Heterocyclic chemistry; G.W.Gribble, J.A.Joule, Eds Elsevier; New York (2004).
152. M. Banwell, B. Flynn and D. Hockless, Chem.Commun., 1997, 2259.
153. M. Diaz, E .Guitian, E. Castedo, Synlett., 2001,1164.
154. C. Peschko, C. Winklhofer, A. Terpin, W. Steglich, Synthesis, 2006, 3048.
155. C. Peschko, C.Winklhofer and W. Steglish, Chem. Eur. J., 2000, 6, 1147.
156. C. Peschko, C.Winklhofer, W.Steglich, Chem. Eur. J., 2000, 6, 1147.
157. P. Cironi, I. Manzanares, F. Albericio, M. Alvarez, Org. Lett., 2003, 5, 2959.
158. D. Fernandez, A. Ahaidar, G. Danelon, P. Cironi, M. Marfil, O. Perez, C. Cuevas, F. Albericio, J. A. Joule, M. Alvarez, Monatsh. Chem., 2004, 135, 615.
159. P. Ploypradith, C. Mahidol, P. Sahakitpichan, S. Wongbundit, S. Ruchirawat, Angew.Chem.,Int.Ed., 2004, 43, 866.
160. N. Fujikawa, T. Ohta, T. Yamaguchi, T. Fukuda, F. Ishibashi, M. Iwao, Tetrahedron, 2006, 62, 594.
161. S. Urban, M. S. Butler and R. J. Capon, Aust. J. Chem., 1994, 47, 1919.
162. S. Urban, L. Hobbs, J. N. A. Hooper and R. J. Capon, J. Org. Chem., 1995, 48, 1491.
163. M. G. Banwell, B. L. Flynn, E. Hamel and D. C. R. Hockless, Chem. Commun.,1997, 207.
164. D. L. Boger, C.W. Boyce, M. A. Labroli, C. A. Sehon, Q. Jin, J. Am. Chem. Soc., 1999, 54,121.
165. M. Marfil, F.Albericio, M. Alvarez, Tetrahedron, 2004, 60, 8659.
166. T. Fukuda, E.I.Sudo, K. Shimokawa and M. Iwao, Tetrahedron, 2008, 64, 328.
167. R. Liu, Y. D. Zhou and D. G. Nagle, J. Nat. Prod., 2007, 70, 1741.
168. S. Urban and R. J. Capon, Aust. J. Chem., 1996, 49, 711.

64
169. M. V. Rami Reddy, D. J. Faulkner, Y.Venkateswarlu and M. Rama Rao, Tetrahedron, 1997, 53, 3457.
170. M. G. Banwell, E. Hamel, D. C. R. Hockless, P. V. Pinard, A. C.Willis, D. J.Wong, Bioorg. Med. Chem., 2006, 14, 4627.
171. R. A. Davis, A. R. Carroll, G. K. Pierens and R. J. Quinn, J. Nat. Prod., 1999, 62, 419.
172. M. V. Rami Reddy, D. J. Faulkner, Y. Venkateswarlu, M. Rama Rao, D. Rhodes, M. S. T. Hansen, K. Rubins and F. D. Bushman, J. Med. Chem., 1999, 42, 1901.
173. T. Yamaguchi, T. Fukuda, F. Ishibashi, M. Iwao, Tetrahedron Lett., 2006, 47, 3755.
174. J. Ham and H. Kang, Bull. Korean Chem. Soc., 2002, 23, 163.
175. P. Krishnaiah, V. L. N.Reddy, G. Venkataramana, K. Ravinder, M. Srinivasulu and T. V. Raju, J. Nat. Prod., 2004, 67, 1168.
176. S. M. Reddy, M. Srinivasulu, N. Satyanaryana, K. Kondapi and Y.Venkateswarlu, Tetrahedron, 2005, 16, 9242.
177. P. Ploypradith, R. K. Kagan and S. Ruchirawat, J. Org. Chem., 2005, 70, 5119.
178. E. Marco, W. Laine and C. Tardy, J. Med. Chem., 2005, 48, 3796.
179. P. Ploypradith, T. Petchmance, P. Sahakitpichan, N. D. Litvinas, S. Ruchirawat, J. Org. Chem., 2006, 71, 9440.
180. P. Thipnate, J. Liu, S. Hannongbua, and A. J. Hopfinger, J. Chem. Inf. Model., 2009, 49, 2312.
181. P. Thipnate, M. Chittchang, N. Thasana, P. Saparpakorn, P. Ploypradith, and S. Hannongbua, Monatsh .Chem., 2011, 142, 97.
182. H. Kang and W. Fenical, J. Org. Chem., 1997, 62, 3254.
183. D. L. Boger, C. W. Boyce, M. A. Labroli, C. A. Sehon and Q. Jin, J. Am. Chem. Soc., 1999, 121, 54.
184. D. L. Boger, D. R. Soenen, C. W. Boyce, M. P. Hedrick and Q. Jin, J. Org. Chem., 2000, 65, 2479.
185. J. L. Bullington, R. R. Wolff, P. F. Jackson, J. Org. Chem., 2002, 67, 9439.
186. M. Iwao, T. Takeuchi, N. Fujikawa, T. Fukuda, F. Ishibashi, Tetrahedron Lett., 2003, 44, 4443.
187. J. T. Gupton, S. C. Clough, R. B. Millar, J. R. Lukens, C. A. Henry, R. P. F. Kanters, J. A. Sikorski, Tetrahedron, 2003, 59, 207.
188. C. Peschko, C. Wiklhofer, A. Terpin, W. Steglich, Synthesis, 2006,3048.
189. C. Peschko and W. Steglich, Tetrahedron Lett., 2000, 41, 9477.
190. A. Hamasaki, J. M. Zimpleman, I. Hwang, D. L. Boger, J. Am. Chem. Soc., 2005, 127, 10767.
191. D. R. Soensen, J. Hwang, M. P. Hedrick and D. L. Boger, Bioorg. Med. Chem. Lett., 2003, 13, 1777.
192.
T. C. Chou, Y. B.Guan, D. R. Soenen, S. J. Danishefsky, D. L. Boger, Cancer Chemother.Pharmacol., 2005, 56, 379.
193. J. A. Palermo, M. Florencia and A. M. Seldes, Tetrahedron, 1996, 52, 8, 2727.
194. A. Furstner, H. Krause and O. R. Thiel, Tetrahedron, 2002, 58, 6373.
195. A. Rudi, I. G. Berg, Z. Stein, F. Frolow, Y. Benayahu, M. Schleyer and Y. Kashman, J. Org. Chem., 1994, 59, 999.

65
196. S. Loya, A. Rudi, Y. Kashman, A. Hizi, Biochem. J., 1999, 344, 85.
197. A. Rudi, T. Evan, M. Aknin and Y. Kashman, J. Nat. Prod., 2000, 63, 832.
198. A.Terpin, K. Polborn, W. Steglich, Tetrahedron, 1995, 51, 9941.
199. C. Winklhofer, A.Terpin, C. Peschko, W. Steglich, Synthesis, 2006, 3043.
200. E. M. Beccalli, F. Clerici and A. Marchesini, Tetrahedron, 2000, 56, 2699.
201. A.T. Kreipl, C. Reid, W. Steglich, Org. Lett., 2002, 4, 3287.
202. J. T. Gupton, R. B. Miller, K. E. Krumpe, S. C. Clough, E. J. Banner, R. P. F. Kanters, K. X. Du, K. M. Keertikar, N. E. Lauerman, J. M. Solano, B. R. Adams, D. W. Callahan, B. A. Little, A. B. Scharf, J. A. Sikorski, Tetrahedron, 2005, 61, 1845.
203. G. W. Chan, T. Francis, R. Thureen, P. H. Offen, N. J. Pierce, J. W. Westley and Jonson, J.Org. Chem., 1993, 58, 2544.
204. C. Peschko and W. Steglich, Tetrahedron Lett., 2000, 41, 9477.
205. Q. Li, A. Fan, Z. Lu, Y. Cui, W. Lin and Y. Jia, Org. Lett., 2010, 12, 4066.
206. G. Fan, Z. Li, S. Shen, Y. Zeng, Y. Yang, M. Xu, T. Bruhn, H. Bruhn, J. Morschhauser, G. Bringmann, W. Lin, Bioorg. Med. Chem., 2010, 18, 5466.
207. W. Y. Yoshida, K. K. Lee, A. R. Carroll and P. J. Scheuer, Helv. Chim. Acta, 1992,75, 1721.
208. M. G. Banwell, B. L. Flynn, E. Hamel, D. C. R. Hockless, Chem. Commun., 1997, 207.
209. D. l. Boger, C. W. Boyce, M. A. Labroli, C. A. Sehon, Q. Jin, J. Am. Chem. Soc., 1999, 121, 54.
210. J. H. Liu, Q. C. Yang, T. C. W. Mark, H. N. C. Wong, J. Org. Chem., 2000, 65, 3585.
211. C. Peschko, C. Winklhofer, A. Terpin, W. Steglich, Synthesis, 2006, 3048.
212. C. Hinze, A. Kreipl, A. Terpin, W. Steglich, Synthesis, 2007, 608.
213. K. Warbi, S. Matsunaga, R. W. M. Van Soest and N. Fusetani, J.Org. Chem., 2003, 68, 2765.
214. A. Furstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2006, 128, 8087.
215. A. Sato, T. Morishita, T. Shiraki, S. Yoshioka, H. Horikoshi, H. Kuwano, H. Hanzawa and T. Hata, J. Org. Chem., 1993, 58, 7632.
216. H. C. Vervoort, S.E. -gross Richards, W. Fenical, A. Y. Lee and J. Clardy, J. Org. Chem., 1997, 62,1486.
217. T.V. Hughes and M. P. Cava, Tetrahedron Lett., 1998, 39, 9629.
218. A. Terpin, C. Winklhofer, S. Schumann, W. Steglich, Tetrahedron, 1998, 54, 1745.
219. E. Piers, R. Britton, R. J. Andersen, J. Org. Chem., 2000, 65, 530.
220. R. G. S. Berlink, R. Britton, E. Piers, L. Lim, M. Roberge, R. M. D. Rocha and Andersen, J. Org. Chem., 1998, 63, 9850.
221. H. C. Vervoort, W. Fenical and P. A. Keifer, J. Nat. Prod., 1999, 62, 389.

66
222. R. Britton, J. H. H. L. D. Oliveira, R. G. S. Berlink and R. J. Andersen, J. Nat. Prod., 2001, 64, 254.
223. J. Kobayashi, J. Cheng, Y. Kikuchi, M. Ishibashi, S. Yamamura, Y. Ohizumi, T. Ohta and S. Nozoe, Tetrahedron Lett., 1990, 31, 4617.
224. T. Sakamoto, Y. Kondo, S. Sato and H. Yamanaka, Tetrahedron Lett., 1994, 35, 2919.
225. T. Sakamoto, Y. Kondo, S. Sato and H. Yamanaka, J. Chem. Soc., Perkin Trans, 1996, 1,459.
226. M. Tsuda, K. Nozawa, K. Shimbo and J. Kobayashi, J. Nat. Prod., 2003,66, 292.
227. L. V. Frolova, N. M. Evdokimov, K. Hayden, I. Malik, S. Rogelj, A. Kornienko and I. V. Magedov, Org. Lett., 2011, 13, 1118.
228. R. A. Davis, L.V. Christensen, A.D. Richardson, R.M. da Rocha and C. M. Ireland, Mar. Drugs, 2003, 27.
229. Y. Kashman, G. K. Goldshlager, M. D. G. Gravalos and M. Schleyer, Tetrahedron lett., 1999, 40, 997.
230. M. R. Heinrich, W. Steglich, M. G. Banwell and Y. Kashman, Tetrahedron, 2003, 59, 9239.
231. R. B. Kennel and P. J. Scheuer, J. Org. Chem., 1992, 57, 6327.
232. P. A. Horton, R. E. Longley, O. J. M. Cornell and L. M. Ballas, Experientia, 1994, 50, 843.
233. C. L. Cantrell, A. Groweiss, K. R. Gustafson and M. R. Boyd, Nat. Prod. Lett., 1999, 14, 39.
234.
P. Schupp, C. Eder, P. Proksch, V. Wray, B. Schneider, M. Herderich and V. Paul, J. Nat. Prod., 1999, 62, 959.
235. P. Schupp, P. Proksch and V.Wray, J. Nat. Prod., 2002, 65, 295.
236. S. J. Wu, S. Fotso, F. Li, S. Quin, G. Kelter, H. H. Fiebig and H. Laatsch, J. Antibiot., 2006, 59, 33.
237. F. Reyes, R. Fernandez, A. Rodriguez, S. Bueno, D. C. Equilior, A. Francesch and C. Cuevas, J. Nat. Prod., 2008, 71, 1046.


















