
Cationic copolymerization of phenyl propenyl ethers
14
JOURNAL OF POLYMER SCIENCE: Polymer Chemietry Edition VOL. 12, 613426 (1974) Cationic Copolymerization of Phenyl Propenyl Ethers KENJI YAMAMOTO and TOSHINOBU HIGASHISIURA, Department of Polymer C h m i s t y , Kyoto University, Kyoto 606, Japan synopsis Ring-substituted phenyl propenyl ethers were found to form homopolymers without any rearrangement by metal halides. Phenyl propenyl ethers were less reactive than the corresponding phenyl vinyl ethers in cationic ploymerization. In order to study the electronic effect of a substituent on the reactivity, cis-pC1, pCK1, and p-CHtO-phenyl propenyl ethers were copolymerized with phenyl propenyl ether in methylene chloride at -78°C with stannic chloride-trichloroacetic acid, and their 'H- and W-NMR spectra were measured. The reaction constant p against Hammett up wm -2.1. The cis-phenyl propenyl ethers were slightly more reactive than the corresponding trans isomers. On the other hand, an o-methyl group decreased the reactivity of phenyl propenyl ether. The low reactivity of o-methyl phenyl propenyl ether was attributed to the steric hindrance between the propagating carbocation and the monomer. INTRODUCTION Th's paper deals with the polymerization of phenyl propenyl ethers. Phenyl propenyl ethers, in spite of the large steric hindrance of their sub- stituents, were found to form the polymers wit.h cationic catalyst at low temperature. Moreover, the effect of a ring substituent to phenyl propenyl ethers on the reactivity was studied in terms of cationic copolymerization. The reactivity of alkyl propenyl ethers carrying various alkyl groups has been examined.'S2 As the difference in the alkyl group affects the monomer reactivity through both an inductive and a steric effect, it is difficult to discuss the inductive and steric effect of substituent separately. In thc present study, phenyl propenyl ethers with various substituents in the para or ortho position were copolymerized by metal halides. The introduc- tion of a substituent at tho para position is expected to be able to clarify the electronic effect of the substituent, because a para substituent does not int.erfere sterically with the reaction on the olefinic double bond. From the same point of view, the reactivity of ring-substituted phenyl vinyl ether was already investigated in cationic polymerization3 and acid-cat.alyzcd hydrolysis.' In addition, one would expect to obtain information concern- ing the steric effect in the propagation reaction of phenyl propenyl ethers by making a comparison between the reactivity of ortho- arid para-substituted phenyl propenyl ethers. Monomer reactivities were evaluated in terms of copolymerization, and thc nature of the monomers way estimated on the basis of NhlR spectra. 613 @ 1974 by John Wiley & Sons, Inc.
-
Upload
kenji-yamamoto -
Category
Documents
-
view
214 -
download
2
Transcript of Cationic copolymerization of phenyl propenyl ethers

JOURNAL OF POLYMER SCIENCE: Polymer Chemietry Edition VOL. 12, 6 1 3 4 2 6 (1974)
Cationic Copolymerization of Phenyl Propenyl Ethers
KENJI YAMAMOTO and TOSHINOBU HIGASHISIURA, Department of Polymer Chmisty, Kyoto University, Kyoto 606, Japan
synopsis Ring-substituted phenyl propenyl ethers were found to form homopolymers without
any rearrangement by metal halides. Phenyl propenyl ethers were less reactive than the corresponding phenyl vinyl ethers in cationic ploymerization. In order to study the electronic effect of a substituent on the reactivity, cis-pC1, pCK1, and p-CHtO-phenyl propenyl ethers were copolymerized with phenyl propenyl ether in methylene chloride a t -78°C with stannic chloride-trichloroacetic acid, and their 'H- and W-NMR spectra were measured. The reaction constant p against Hammett up wm -2.1. The cis-phenyl propenyl ethers were slightly more reactive than the corresponding trans isomers. On the other hand, an o-methyl group decreased the reactivity of phenyl propenyl ether. The low reactivity of o-methyl phenyl propenyl ether was attributed to the steric hindrance between the propagating carbocation and the monomer.
INTRODUCTION
Th's paper deals with the polymerization of phenyl propenyl ethers. Phenyl propenyl ethers, in spite of the large steric hindrance of their sub- stituents, were found to form the polymers wit.h cationic catalyst a t low temperature. Moreover, the effect of a ring substituent to phenyl propenyl ethers on the reactivity was studied in terms of cationic copolymerization.
The reactivity of alkyl propenyl ethers carrying various alkyl groups has been examined.'S2 As the difference in the alkyl group affects the monomer reactivity through both an inductive and a steric effect, it is difficult to discuss the inductive and steric effect of substituent separately. In thc present study, phenyl propenyl ethers with various substituents in the para or ortho position were copolymerized by metal halides. The introduc- tion of a substituent at tho para position is expected to be able to clarify the electronic effect of the substituent, because a para substituent does not int.erfere sterically with the reaction on the olefinic double bond. From the same point of view, the reactivity of ring-substituted phenyl vinyl ether was already investigated in cationic polymerization3 and acid-cat.alyzcd hydrolysis.' In addition, one would expect to obtain information concern- ing the steric effect in the propagation reaction of phenyl propenyl ethers by making a comparison between the reactivity of ortho- arid para-substituted phenyl propenyl ethers.
Monomer reactivities were evaluated in terms of copolymerization, and thc nature of the monomers way estimated on the basis of NhlR spectra.
613
@ 1974 by John Wiley & Sons, Inc.

614 YAMAMOTO A N D IIIGASHIMURA
The propagation reaction of phenyl propcnyl ethers was explained by thcsc results.
EXPERIMENTAL
Materials l'henyl propenyl et.her (PhI'E) and its ring-substituted derivatives were
prepared by the rearrangement of ally1 phcnyl ethersb which wwc syn- thmized from allyl bromide and thc corresponding Pure cis isomers were obtairicd in the rearrangement of allyl ethers by potassium tert-butoxide in dimctthyl sulfoxidc. The purities of monomws were found to be morct than 99.<5y0 by gas chromatographic analyses. Thc cis isomctrs were isomcrized by mc'rcury acetat.e7 and the rcsulting cis-tran.s mixtures were separated by fractional distillation through a spinning band column. The steric purities of the geometric isomers srparated were more than 98.0%. The geomcttric structure of 1'hPE was identificd on tht: basis of 'H-XMR coupling constants between olcfinic protons. Boiling 1)oint.s and lH-NMlt coupling constants of 1'hPE are summarized iri Tablc I. The compounds in Table I have not previously bcen reportrd except for un- subdtu ted PhI'E.
TABLE I Boiling Point and Coupling Constant of Phenyl Propenyl Ethers (PhPE:)
Substituent R
cis isomer trans isomer
Up, "Clmm Hg Jcin, IIZ Bp, 'Cjrnm Hg
1% p-Me0 o-Me0 p-h,le 0-Me
0-Cl 2,6-diMe
p-CI
72.1-72.4115 117-118/20 97-9818 82-831 15 76.577114 83-84 / 10 90/10 87-881 14
6.2. 6 . 1 6 . 1 6 . 2 6 . 1 6.0 6 . 0 6 .1
72.4--73.0; 1.5 82-8415 98-9918 83-841 15 77-78/14
90-91 I10 -
12.0. 11.8 11.6 11.9 12.1
11.6 -
Phenyl vinyl ether (PhVE) and its p-methoxy derivative were syn- thesized from the corresponding p-bromophmetholes according to t he method r e p ~ r t e d . ~ These monomers were more than 99.0% pure by gas chromatographic analyses. Solvents, catalysts, tetralin, and the phcnyl bromide used as an internal standard for gas chromatographic analysis were purificd by the usual method and distilled over calcium hydridv before use. Trichloroacetic acid (TCA) (Guaranteed Rcagent) a$ cocatalyst was used without further purification.
Procedures Polymerization was carried out. in a vessel equipped with a stopcock
In this procedure thc concentrations of water were in under dry nitrogen.

PlIENYL PROPENYL m I I E R S 615
the order of lo-' mole/l. in all the systems. The polymer obtained i n homopolymerizat.ion was precipitatcd in a large amount of methanol, washed with methanol, and dried under reduced pressure a t room tctmpera- ture. In copolymerization, polymer compositions werct determined from the amounts of residual monomers measured by gas chromatography. Monomer reactivity ratios were evaluated by the improved Fineman-Ross method.IO The intrinsic viscosity of the polymers was measured in benzene solution a t 30°C.
RESULTS
Polymerization of Pbenyl Propenyl Ethers It has bcen reported that I'hVE was polymerized with rcarrangcmerit to
o-vinylphenol by an active catioriic catalyst in a polar solvent.11.12 Therc- fore, the possible rearrangement of PhPE compounds under c:ttionic poly- merization conditions was examiricd.
When PhPE compounds were polymerized by SnCl,-TCA in methylenc chloride, gas chromatography showed no evidence. of rearrangement. Infrared and NRlR spectra of the polymers also showrd that these mono- mers polymerized without rearrangement and that the polymer was formed by the addition to the olcfinic double bond of the monomer.
In thc copolymerization of I'hVE with cis-PhPE at 0°C: by SiiClr TCA, a part of PhVE rearranged to o-vinylphenol, even in a nonpolar solvent sueh as toluene. On the other hand, PhVE did not rearrange to o-vinyl- phenol with BFa.OEt2 in methylrne chloride a t 0°C. Also, the reactivities of PhPE compounds were lower than those of alkyl propenyl ethers and their polymerization rates were very small with 13E'3.0Etz at -78°C. Therefore, we polymerized the PhPE compounds with the us(' of SnC14 as catalyst and TCA as cocatalyst a t -7S"C. Only the copolymerization of PhPE with I'hVE was carried out with BF3.0Et2 at 0°C.
TABLE I1 Properties of Polymers Obtained with SnClr-TCA in CH2C1, at. -78 '0
[C], Conversion, Softening Monomer mmole/l. T, Ivlb point,, "C -
Cis-p-MeOPhPEc 20 - 0.048 207-210 Cis-pM eOPh PE 20 -. 0.042 1 .iO- 1 .i2 Cis+-MeOPhPEd 10 66 0.038 120-121 Cis+-MeOPhPE 10 100 0.040 132-133 Cis-pMePhPE 10 100 0.041 122-123 Cis- and trans-pMePhPEe 5 25 0.045 143 Cis- and trans-o-MePhPEe 7 28 0.048 143-144
~ -
a [M]o = 10 vol-yo, polymerization time, 24 hr. b Measured at, 30°C in benzene solution.
Polymerization temperature, -30 to - 15OC. d Solvent, toluene.
Equimolar mixture of cis- and trans-isomers; polymerization time, 0.5 hr.

616 YAMAMOTO A N D IIIGASHIMURA
The viscosities and softening points of PhPE polymers are summarized in Table 11. Their softening points were relatively high but their molccular weights were not so large.
Effect of ,+Methyl Group on the Reactivity of Phenyl Vinyl Ether In order to study the effect of the @-methyl group on t-he reactivity of
PhVE, cis-PhPE was copolymerized with PhVE in methylenc chloride a t 0°C by use of BFS-OEt, as catalyst. Figure 1 shows tho copolymer com- position curve. The rcact.ivity of PhPE was found to be smaller than that. of PhVE.
Moreover, to clarify the effect of @-methyl group on the reactivity of phenyl vinyl ethers, cis-p- or cis-o-MeOI’hPE was copolymerized with p - MeOPhVE. Rearrangement of the monomers was riot observed in the copolymerization under the condition described in Figure 2. Figure 2 shows the first-order plots on the rate of the monomer consumption in the
All the polymers were whitc, amorphous powders.
0.5 1 Mi in monomer
Fig. 1. Copolymer composition curve in the copolymerization of PhVE (MI) with cis- PhPE (Mz). [MI0 = 10 VOI-O/,, BF30Eti; CHzClz; 0°C.
r - -1
Time ( m i d
Fig. 2. First-order plots of the rate of the monomer consumption in the copolymeriza- tion of equimolar mixtures of MeOPhPE and MeOPhVE: (A) cis-p-MeOPhPE- (0 ) p-MeOPhVE and (A) cis-o-MeOPhPE-(O) p-MeOPhVE. 13410 = 10 vol-o/,, [SnCI,] = 5.0 mmole/l., [TCA] = 2.6mmole/l.; CH&IZ; -78°C.

PHENYL PROPENYL ETHERS 617
copolymerization of the equimolar mixture of these monomers. In b0t.h systems, t.ho polymcrization rate of p-MeOPhVE waq found to be about twice that of cis-p- or cis-o-McOPhPE. It. is concluded that the P-methyl group decreases the reactivity of 1’hVE.
Effect of Ring Substituents on the Reactivity of Phenyl Propenyl Ether Various substituents w(m: introduced to the para or ortho position of Ph-
PE and their reactivities were compared with that of PhPE. Tablo I11 shows the copolymerization dat.a for cis-p-RleOPhPE (14,) with cis-PhPE (M2). The effect of a ring subst,itut:nt. on the rcact.ivity of PhI’E was studied on the cis isomer, because the reactivit.y of thc cis isomer was about equal to that of the corresponding trans isomer, as will be describcd lat,er. The copolymer composition curves arc shown in Figure 3, and their mono- mer reactivity ratios are summarizctd in Table IV. Except for cis-o- McPhPE, it was found that thc dcctron-donating substituent,s increase the reactivity of the monomers.
TABLE 111 Copolymerization of cis-p-MeOPhPE (MI) with cis-PhPE(M2) with SnCI,-TCA in
CHZC12 at - 7 8 ’ 0
Monomer feed, mole/l.b -_______-- ization -. . - Conver- Run M 1 MP time, min MI M2 sion, yo
Residual monomer, mole/l.c
1 0.038 0.653 8 0.022 0.528 20.5 2 0.184 0.504 2 0.173 0.487 4 . 2 3 0.310 0.332 6 0.269 0.307 10.3 4 0.369 0.290 6 0.343 0.273 6 . 7 5 0.438 0.218 6 0.415 0.215 3 . 8 6 0.569 0.076 20 0.534 0.075 5 . 6 ____ __ ~
* (MI, = 10 v01-y~; [SnCL] = 5.0 mmole/l.; [TCA] = 2.5 mmole/l. b Initial amount of monomer M I or M Z in the monomer mixture. e Amount of the residual monomers with respect to the total monomer feeds.
1 or----
MI in monomer
Fig. 3. Copolymer composition curves in the Copolymerization of cis-substituted PhPE (111) with cis-PhPE (M2): (A) o-MeOPhPE; ( A ) p-MeOPhPE; (0) p-MePhPE; (0) o-MePhPE; (0 ) p-ClPhPE; (B) o-CIPhPE.

618 YAMAMOTO AND HIGASHIMURA
The para-substituted PhPE monomers were also copolymerized with orthesubstituted ones, and the effect of substitucnt position on the re- activity of PhPE was examined. Figure 4 shows the copolymer composi- tion curves. Monomer reactivity ratios are summarized in Table IV.
TABLE I V Monomer Reactivity Ratios in the Copolymerization
Catalyzed by SnC1,-TCA in CH&& at -78°C
MI Mi TI TI
PhVE Cis-pMePhPE Cis+-MePhPE Cis-pMeOPhPE Cis-pMeOPhPE Cis-o-MeOPhPE Cis-o-MePhPE Cis-o-MeOPhPE Cis-o-ClPhPE Cis-pClPhPE
Cis-PhPE. Cis-PhPE Cis-PhPE Cis-PhPE Cis-PhPEb Cis-PhPE Cis-p.MePhPE Cis-PMeOPhPE Cis-PhPE Cis-PhPE
3.05 f 0.15 1.40 f 0.06 0.87 f 0.12 2.60 f 0.09 3.27 f 0.10 2.65 f 0.20 0.32 f 0.02 1.23 f 0.04 0.10 f 0.09 0.13 f 0.02
Polymerization temperature, 0°C ; catalyst, BFiOEh. b Solvent, toluene.
3
0.23 f 0.05 0.39 f 0.03 1.18 f 0.11 0.46 f 0.03 0.43 f 0.03 0.21 f 0.02 1.98 Z!= 0.03 0.98 f 0.05 4.77 f 0.76 4.87 Z!= 0.16
M1 in monomer
Fig. 4. Copolymer composition curves in the copolymerization of cis-o-f~ubtituted PhPE (MI) with cis-psubstituted PhPE (MI); (0) MePhPE; (0) MeOPhPE. [MI. = 10 vol-%; S ~ C ~ ~ T C A ( ~ / ~ ) ; C H I C ~ ~ ; -78°C.
- 0.2 - 0 10 20 30 40
Time (rnin.1
Fig. 5. Firstorder plots on the rate of the monomer consumption in the copolymeriza- tiop of equimolar mixture of cis-2,UiMePhPE and cis-PhPE: (0) cis-2,UiMePhPE; (0 ) cis-PhPE. [MI0 = 10 vol-%; [SnCl], = 5.0 mmole/l.; [TCA] = 2.5 mmole/l.; CHrCl,; -78°C.

PHENYL PROPENYL ETHERS 619
The reactivity of o-MeOPhPE wm found to be about equal to that of p- MeOPhPE. On the other hand, o-MePhPE was less reactive than p- MePhPE. This result is similar to that of the copolymerization of cis-o- MePhPE wit.h cis-PhPE as shown in Figure 3.
To clarify the anomalous behavior of the methyl group, an equimolar mixture of cis-2,gdiMePhPE and cis-PhPE wm copolymerized. Figure 5 shows the firsborder rate plots of monomer consumption. The polymeriza- tion rate of cis-2,gdiMePhPE was found to be about half of t.hat of cis- PhPE, in spite that the former has two electron-donating methyl groups. Also, the reactivity of cis-2,6-diMePhPE wm smaller than that of cis-o- MePhPE.
cis-PhPE was copolymerized with cis-p-MeOPhPE in methylene chloride and toluene to elucidate the effect of solvent polarity on the relative mono- mer reactivities. As shown in Table IV, the polarity of the solvent did not greatly affect relative monomer reactivities with SnCL catalyst.
Effect of Geometric Structure on Monomer Reactivity
The cis-PhPE monomers were copolymerized with the corresponding trans-PhPE monomers. Figure 6 shows the copolymer composition curves, and the corresponding monomer reactivity ratios are summarized in Table V. These results show that the cis isomers are slightly more reactive than the corresponding trans isomers.
TABLE V Monomer Reactivity Ratios in the Copolymerization of Cis- and Trans-Phenyl Propenyl
Ethers Catalyzed by SnCld-TCA in CHtCll a t -78OC
Mi Mi Tl rz
Cis-PhPE Trans-PhPE 0.93 f 0.03 0.67 f 0.02 Cis-pMePhPE Trans-pMePhPE 1 .OO f 0.05 0.82 f 0.04 Cis-o-MePhPE Trans-o-MePhPE 1.25 f 0.10 0.50 f 0.09
MI in monomer
Fig. 6. Copolymer composition curves in the copolymerization of &-(MI) and trans- (M2)PhPE monomers: (0) PhPE; (A) o-MePhPE; (0) pMePhPE. [M]o = 10 vol-%; SnCGTCA(2/1); CHiCli; -78°C.

TAB
LE V
I *H
-NM
R C
hem
ical
Shifts a
nd C
oupl
ing
Con
stan
ts in
Phe
nyl P
rope
nyl
Eth
ers
Che
mic
al s
hift,
z
Geo
met
ric
Phen
yl
R
stru
ctur
e Su
bstit
uent
R
B-CH
a H
A
HB
pr
oton
pr
oton
JH
.,-E
, JE
,,-cH
~ JH
B-C
HS
Cis
H
8.32
5.
26
3.72
2.
98
-
6.2
7.0
1.8
PO
CH
a 8.
31
5.29
3.
73
3.23
6.
44
6.1
7.0
1.8
4-
CIla
8.
30
5.30
3.
76
3.19
7.
85
6.2
7.0
1.7
4-Cl
8.
34
5.20
3.
80
3.06
-
6.0
6.9
1.8
2-O
CHa
8.28
5.
27
3.71
3.
17
6.34
6.
1 7.
0 1.
8 ZC
H3
8.30
5.
30
3.78
3.
14
7.78
6
.1
6.8
1.7
2-
c1
8.30
5.
14
3.77
3.
00
-
6.0
6.9
1.8
2,
6-C
H3
8.25
5.
53
4.19
3.
16
7.85
6.
1 6.
8 1.
8
Tra
ns
H
8.44
4.
72
3.66
2.
98
-
12.0
7.
0 1.
8 4-
OCH
a 8.
46
4.76
3.
69
3.23
6.
44
11.8
6.
9 1
.6
PCH
a 8.
47
4.74
3.
73
3.19
7.
85
11.9
7.
0 1.
6 2-
0CH
3 8.
48
4.77
3.
63
3.17
6.
35
11.6
6.
9 1
.7
ZC
H,
8.49
4.
80
3.75
3.
14
7.80
12
.1
7.0
1.6
zc
1
8.44
4.
69
3.75
3.
00
-
11.6
6.
9 1.
7
Cou
plin
g co
nsta
nt,
Hz
d

PIIENYL PROPENYL ETllERS 621
NMR Spectra of Phenyl Propenyl Ethers To determine the effect of ring substituents on t.he properties of the
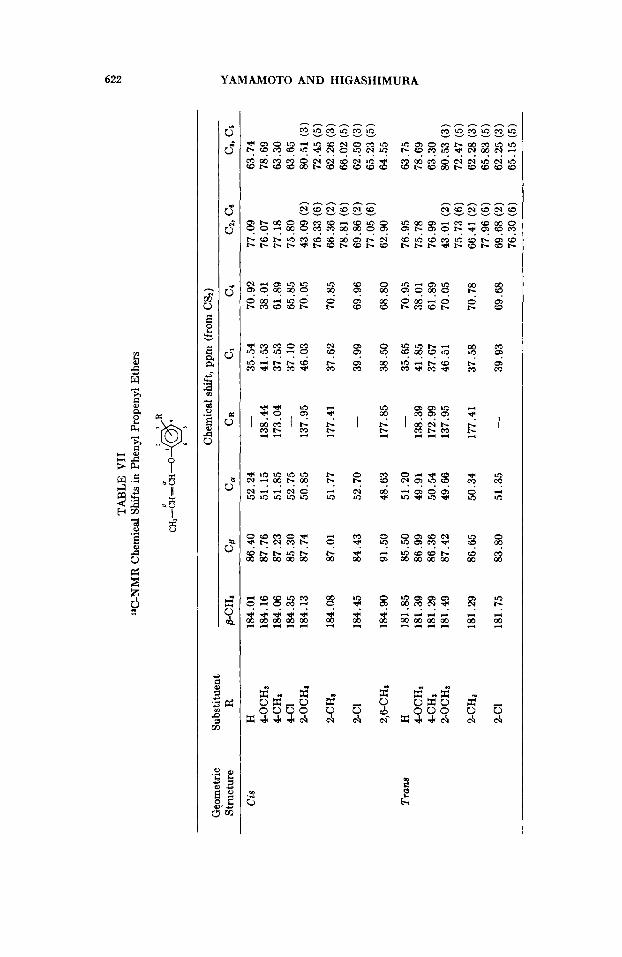
monomcr, 'H- and 13C-NhlK spectra of the monomers were mcasurctd. lH-NhlR spcxtra were measured with a Variari HA-1OOD spcctrometer, tetramcthylsilanc being used ft9 internal standard. All spectra wcrc ob- served at 100 MHz on neat liquid a t room tcmpcraturcx. The rcmlts are summarized in Table VI. Coupling constants betweon olcfiriic protons are not sensitive to ring substitution within the limits of c.xpctrimenta1 error, which coincides with tbc results for ring-substituted FhVE.I3 Also the chemical shifts of olcfinic protons did not greatly vary with the kind of the substituent, except for cis-2,6-dibIt:PhPE.
13C-XMK spect.rn were measured with a JNM-PFT-100 spectromcter (at 25.1 MHz, intcrnal standard; benzene) or with a JNM-C-GO HI, spectrometer (at 15.1 MHz, extwnal standard; benzene and methanol) at room temperature. All spect.ra were observed on neat liquid in 8-mm diameter samplc tubes. Thtt chomical shifts are summarized in Table VII. The chemical shift of 13C-NR1R of a olefinic @-carbon moves to higher field by substitution of an elcct.ron-donating group. This means that the introduction of electron-donating substitucnts increases the electron density on the olefinic @-carbon. A linear rclat,ionship held between the observed chemical shifts and Hammctt up constants of the substituents, as shown in Figures 7 and 8. The p values, dofincd by 6x - 6H = p a , arc -5.0 in cis
Fig. 7. Hammett plots of the 13C chemical shifts for the @-carbon para-subst.itut.d P h P E compounds.
of (0) cis and (0 ) trans
N :: 87
85
% Fig. 8. Harnmett plots of the 13C chemical shifts for the @-carbon of (0) c is and (0 ) tram
ortho-substituted PhPE compounds.
TAB
LE V
II
W-N
MR
Che
mic
al Shifts
in P
heny
l Pr
open
yl E
ther
s
Geo
met
ric
Subs
titue
nt
Stru
ctur
e R
Cis
H
4-
OCH
a 4-
CHa
44
1
2-O
CH
j
ZC
Hs
24
1
2,tL
CH
a
Tran
s H
WC
Hs
PCH
3 2-
OCH
a
ZC
Hi
ZC1
ul N
N
Che
mic
al s
hift,
ppm
(fr
om C
S,)
BC
Ha
CB
C,
CR
C1
c4
CZ, c
s Ca
, C5
2 E 18
4.01
86
.40
52.2
4 -
35.5
4 70
.92
77.0
9 63
.74
184.
16
87.7
6 51
.15
138.
44
41.5
3 38
.01
76.0
7 78
.69
0
184.
06
87.2
3 51
.85
173.
04
37.5
3 61
.89
77.1
8 63
.30
c3
184.
35
85.3
0 52
.75
-
37.1
0 65
.85
75.8
0 63
.65
0
184.
13
87.7
4 50
.85
137.
95
46.0
3 70
.0.5
43
.09
(2)
80.5
1 (3
) * U
76
.33
(6)
72.4
5 (5
) 70
.85
66.3
6 (2
) 62
.26
(3)
78.8
1 (6
) 66
.02
(5)
69.9
6 69
.86
(2)
62.5
0 (3
) 77
.05
(6)
65.2
3 (5
)
.* 3
184.
08
87.0
1 51
.77
177.
41
37.6
2
184.
45
84.4
3 52
.70
-
39.9
9
184.
90
91.5
0 C
18
1.85
85
.50
51.2
0 -
35.6
5 70
.95
76.9
5 63
.75
2r
181.
29
86.3
6 50
.54
172.
99
37.6
7 61
.89
76.9
9 63
.30
181 .
49
87.4
2 49
.66
137.
95
46.5
1 70
.05
43.0
1 (2
) 80
.53
(3)
75.7
3 (6
) 72
.47
(5)
181.
29
86.6
5 50
.34
177.
41
37.5
8 70
.78
66.4
1 (2
) 62
.28
(3)
77.9
6 (6
) 65
.83
(5)
181.
75
83.8
0 51
.35
-
39.9
3 69
.68
69.6
8 (2
) 62
.25
(3)
76.3
0 (6
) 65
.15
(5)
P m 5
48.6
3 17
7.85
38
.50
68.8
0 62
.90
64.5
5 E
181.
39
86.9
9 49
.91
138.
39
41.8
5 38
.01
75.7
8 78
.69
P

PHENYL PROPENYL ETHERS 623
monomers and -5.2 in trans monomers, respectively, for para-substituted PhPE. For mtho-substituted I'hl'E, in the same manner, p = -7.6 is obhined both in cis and tram monomers.
DISCUSSION
It was found that PhPE monomers formcd homopolymcrs without any rearrangement by SnCkTCA even at - 78"C, alt.hough they were less reactive than alkyl propenyl ethers. However, th r polymers were amor- phous and of low molecular weight.
PhPE monomers were less reactive than the corresponding PhVE mono- mers in cationic polymerization. A p-methyl group incrcasos the rcac- tivity of vinyl ether with a Ic!ss bulky alkoxy group. This is considervd to be due to the formation of a bridgcd carbocation as an inkrmediate in a propagation reaction." On the other hand, in a vinyl et,hcr carrying a bulky alkoxy group, the introduction of a methyl group on the olefinic &carbon decreases the rctactivity of the vinyl ether due t.o steric hindraticr between t.hese subst . i tuent .~.~~ As PhPE is sterically similar to the alkyl propenyl ether wit.h a bulky alkoxy group, the @-methyl group may decrease the reactivity of the PhVE.
The reactivity of a PhPE was found to be affected by the nature of the substituent on the phenyl group. Figure 9 shows the relationship between Hammett up and log (l/r2), that is, the relative reactivity of the ring-sub- stituted PhPE for the phenyl propenyl ether carbocation.
Before discussing the react,ivity of the monomer, i t is necessary to know the conformation of the monomer, because steric hindrance plays an impor- tant. role in the polymerization of a$-disubstituted ethylones. PhPE cannot be in the s-cis form (I) due to stcric hindrance between thc @-hy-
(I) (II) drogen or @-methyl group and the o-hydrogen of the phenyl group. Fucno et aL13 have reported that i t was difficult even for PhVE monomers to take the *cis form. In the s-trans form, the olefinic doublc: bond and the phenyl

624 YAMAMOTO AND HIGASHIMURA
1.0 ---- - -
CP
Fig. 9. Plots of log ( l h ) vs. Hammett up (from Table IV).
group are difficult to bring into the same plane because of repulsion between the olefinic a-proton and the aromatic ortho proton as shown in I1 or I11
and both groups may be twisted. Yuki et a1.l" have suggested that the vinyl ethers with bulky alkoxy groups (for example, tert-butyl vinyl ether) could assume only the gauche form. Moreover, steric hindrance between the olefinic double bond and the aromatic ring is expected to be the same, irrespective of the geometric structures and the position of the substituent on the phenyl group, as shown in I1 and 111. The 'H-NMR chemical shifts of the olefinic protons were found to be about equal between the para and the ortho isomers. This means that there is no difference between the conformations of these isomers.
The cis-fL,G-diMePhPE, howevcr, shows different features from the monosubstituted PhPE monomers. Chemical shifts for Ha and HB in 'H-NMR data and for C, in 'W-NMR data for this compound shift to higher fields than for the others. These facts suggest that the distortion between the olefinic double bond and the phenyl ring due to two o-methyl groups in cis-2,GdiMePhPE is larger than that in a monosubstituted PhPE.
It has been reported that the r-electron density is reflected in the chemi- cal shift of the 13C-NMR spectrum." The W-NMR chemical shifts of the @-carbons for monosubstituted PhPE were found to correlate linearly with

PIIENYL PROPENYL ETHERS 625
Hammett up values, and the p values, shown in Figure 7, were -5.0 in cis monomers and -5.2 in trans monomers for para-substituted PhPE mono- mers. They correspond to p = -4.84 in ring-substituted PhVE com- pounds.
The p value in the cationic copolymerization of para-substituted PhPE has been found to be about -2.1 from Figure 9. Fueno et aL3 obt.ained p = - 1.76 in the cationic copolymerization of ring-substituted PhVE com- pounds. Therefore, the ring substitutent of PhPE as well as PhVE affects the reactivity of the olefinic double bond of the monomer through the ether oxygen. It may be safe to conclude that t,he (3-methyl group does not in- fluence the resonance effect of t.he substituents through the ether oxygen atom to the olefinic double bond.
It waB presumed from 'H-NMR spectra of the monomers that the con- formations of monosubstituted PhPE monomers were the same, irrespective of the positions of the ring substituents. Nevortheless, o-MeI'hPE was less reactive than PhPE. This means that the reduction of monomer reactivity by the introduction of the emethyl group is due to steric hindrance between the propagating carbocation and the o-methyl group of the monomer, because the phenyl ring with the o-methyl group lies out of the monomer
Thus, a substituent far from the &carbon of a monomer, such as the ortho substituent, was considered to interfere sterically with the reaction. This fact supports our preceding mechanism, l 4 in which a propagating car- bocation approaches the monomer far from the (3-carbon of the monomer.
The introduction of an o-met.hoxy group did not decrease monomer reac- tivity as much as introduction of the o-methyl group. This means that the propagating carbocation interacts with the monomer, since an oxygen atom of the methoxy group is present near the olefinic double bond.
In conclusion, the (3-methyl group and the o-methyl group decreased the monomer reactivity in the cationic polymerization of PhPE. This means that the bulky substituents greatly affect monomer reactivity by twisting the phenyl ring from the monomer plane.
plane.
References
1. T. Higashimura, S. Kusudo, Y. Ohsumi, and S. Okamura, J . Polym. Sci. A-I, 6 ,
2 . T. Okuyama, T. Fueno, H. Nakatsuji, and J . Furukawa, J . Amer. Chem. Soc., 89,
3. T. Fueno, T. Okuyama, I. Matsumura, and J. Furukawa, J . Polym. Sci. A-1, 7,
4. T. Fueno, I . Matsumura, T. Okuyama, and J . Furukawa, Bull. Chem. Soc. Japan,
5. T. J . Prosser, J . Amer. Chem. Soc., 83. 1773 (1961). 6. D. S. Tarbell, Organic Readions, Vol. 11, Wiley, New York, 1944, p. 1. 7. T. Okuayama, T . Fueno, and J. Furukawa, Tetrahedron, 25, .5409 (1969). 8. P. W. Jolly, F. G. A. Stone, and K. Mackenzie, J . Chem. SOC., 1965,6416. 9. S. 11. McElvain and B. Fajardc+Pinz6n, J . Amer. Chem. Soc., 67.650 (1945).
2523 (1968) and subsequent papen.
5826 (1967) and subsequent papers.
1447 (1969).
41,818 (1968).

626 YAMAMOTO AND HIGASHIMURA
10. A. I. Ezrielev, E. L. Brokhina, and E. S. Roskin, Vyeokomol. Soedin., 11, 1670
11. C. E. Schildknecht, Vinyl and R e h d Polymers, Interscience, New York, 1952,
12. 34. Imoto and J. Takenaka, Kobumhi Kagaku, 13,268 (1956). 13. T. Fueno, 0. Kajimoto, K. Izawa, and >I. Masago, BuU. Chem. Soc. Japan, 46,
14. T. Higashimura, T. Masuda, S. Okamura, and T. Yonezawa, J. Polym. Sci. A-I, 7,
15. A. Mizote, T. Tanaka, T. Higwhimura, and S. Okamura, J . Polym. Sci. A, 3,2567
16. H. Yuki, I(. Hetada, and M. Takeshita, J. Polym. Sci. A-I, 7,667 (1969). 17. hl. Karplus and J. A. Pople, J. Chem. Phys., 38,2803 (1963). 18. 0. Kajimoto, M. Kobayashi, and T. Fueno, Bull. Chem. Soe. Japan, 46, 1422
(1969).
p. 625.
1418 (1973).
3129 (1969).
(1965).
(1973).
Received November 7,1973 Revised December 14,1973


















