![Case Report T-cell lymphoblastic lymphoma with elevated ...ijcem.com/files/ijcem0060907.pdfpleural effusion (PE) [3]. The results can closely mimic tuberculous pleural effusion (TPE)](https://static.fdocuments.net/doc/165x107/5e498f0048356572d95aac84/case-report-t-cell-lymphoblastic-lymphoma-with-elevated-ijcemcomfiles-pleural.jpg)
Casitas B lymphoma mutations in childhood acute lymphoblastic leukemia
7
GENES, CHROMOSOMES & CANCER 51:250–256 (2012) Casitas B Lymphoma Mutations in Childhood Acute Lymphoblastic Leukemia Lindsay Nicholson, Thomas Knight, Elizabeth Matheson, Lynne Minto, Marian Case, Maryna Sanichar, Simon Bomken, Josef Vormoor, Andy Hall, and Julie Irving Paediatric Oncology and Haematology Group, Northern Institute for Cancer Research, Newcastle University,Newcastle uponTyne,UK Casitas B-lineage lymphoma (CBL) proteins are RING finger ubiquitin E3 ligases that attenuate the signaling of receptor tyrosine kinases and are mutated in a number of myeloid disorders. In this study, mutational screening of the linker-RING domains of CBL and CBLB was performed by denaturing high performance liquid chromatography in a cohort of diagnostic (n ¼ 180) or relapse (n ¼ 46) samples from children with acute lymphoblastic leukemia. Somatic mutations were identified in three children, giving an overall incidence of 1.7% and involved small deletions affecting the intron/exon boundaries of exon 8, leading to skipping of exon 8 and abolishing E3 ligase function. Mutated primary samples were associated with constitutive activation of the RAS pathway and sensitivity to MEK inhibitors was shown. Thus, mutation of CBL is an alternative route to activate the RAS pathway and may identify children who are candidates for MEK inhibitor clinical trials. V V C 2011 Wiley Periodicals, Inc. INTRODUCTION Casitas B-lineage lymphoma (CBL) proteins are a highly conserved family of RING finger ubiquitin E3 ligases that serve to attenuate the signaling of a variety of receptor tyrosine kinases (RTK). Recently, somatic mutations of CBL have been identified in a range of myeloid disorders including acute myeloid leukemia (AML), chronic myelo-monocytic leukemia (CMML), ju- venile myelo-monocytic leukemia (JMML), and myeloproliferative syndrome (Caligiuri et al., 2007; Sargin et al., 2007; Dunbar et al., 2008; Grand et al., 2009; Loh et al., 2009; Makishima et al., 2009; Reindl et al., 2009; Kales et al., 2010; Muramatsu et al., 2010; Ogawa et al., 2010). Mutations principally affect the linker-RING do- main of the CBL protein and are often associated with acquired uniparental disomy (aUPD) at the CBL gene locus at 11q23.3. Functional studies in vitro and in vivo show that CBL mutations are transforming and induce a myeloproliferative dis- ease which can progress to leukemia and are asso- ciated with enhanced RTK signaling, manifested down numerous key signaling pathways including the RAS, PKB and JAK-STAT pathways (Bandi et al., 2009; Sanada et al., 2009; Rathinam et al., 2010). In addition, somatic mutations of another family member, CBLB, have also been reported (Caligiuri et al., 2007; Makishima et al., 2009). In JMML and CMML, CBL mutations were found to be mutually exclusive with respect to RAS- pathway activating mutations such as NRAS, KRAS, and PTPN11, suggesting that CBL may play an important a role in deregulating this key survival pathway (Loh et al., 2009; Sanada et al., 2009; Muramatsu et al., 2010). Acute lymphoblastic leukemia (ALL) is the most common cancer in children and is character- ized by a clonal expansion of poorly differenti- ated lymphoid precursors within the bone marrow. While cure rates are high, novel thera- pies are needed for those children that relapse. ALL is a heterogeneous disease, with a vast array of genomic aberrations and we recently showed that mutations in genes impacting on the RAS pathway, such as NRAS, KRAS, PTPN11, and FLT3 represent one of the most common genetic abnormalities, being found in a third of patients (Case et al., 2008). Primary leukemic blasts with mutations were associated with constitutive acti- vation of the RAS pathway and differential sensi- tivity to MEK inhibitors was shown in patients with the highest levels of RAS pathway activa- tion, suggesting that drugs such as MEK Additional Supporting Information may be found in the online version of this article. Supported by: Leukaemia and Lymphoma Research. *Correspondence to: Julie Irving, Northern Institute for Cancer Research, Paul O’Gorman Building, Framlington Place, Newcastle upon Tyne, Tyne and Wear, UK, NE2 4HH. E-mail: j.a.e.irving@ ncl.ac.uk Received 3 June 2011; Accepted 6 October 2011 DOI 10.1002/gcc.20949 Published online 10 November 2011 in Wiley Online Library (wileyonlinelibrary.com). V V C 2011 Wiley Periodicals, Inc.
-
Upload
lindsay-nicholson -
Category
Documents
-
view
217 -
download
5
Transcript of Casitas B lymphoma mutations in childhood acute lymphoblastic leukemia

GENES, CHROMOSOMES & CANCER 51:250–256 (2012)
Casitas B Lymphoma Mutations in Childhood AcuteLymphoblastic Leukemia
Lindsay Nicholson, Thomas Knight, Elizabeth Matheson, Lynne Minto, Marian Case, Maryna Sanichar,
Simon Bomken, Josef Vormoor, Andy Hall, and Julie Irving
Paediatric Oncologyand Haematology Group,Northern Institute for Cancer Research,Newcastle University,Newcastle uponTyne,UK
Casitas B-lineage lymphoma (CBL) proteins are RING finger ubiquitin E3 ligases that attenuate the signaling of receptor
tyrosine kinases and are mutated in a number of myeloid disorders. In this study, mutational screening of the linker-RING
domains of CBL and CBLB was performed by denaturing high performance liquid chromatography in a cohort of diagnostic
(n ¼ 180) or relapse (n ¼ 46) samples from children with acute lymphoblastic leukemia. Somatic mutations were identified
in three children, giving an overall incidence of 1.7% and involved small deletions affecting the intron/exon boundaries
of exon 8, leading to skipping of exon 8 and abolishing E3 ligase function. Mutated primary samples were associated
with constitutive activation of the RAS pathway and sensitivity to MEK inhibitors was shown. Thus, mutation of CBL is an
alternative route to activate the RAS pathway and may identify children who are candidates for MEK inhibitor clinical
trials. VVC 2011 Wiley Periodicals, Inc.
INTRODUCTION
Casitas B-lineage lymphoma (CBL) proteins
are a highly conserved family of RING finger
ubiquitin E3 ligases that serve to attenuate the
signaling of a variety of receptor tyrosine kinases
(RTK). Recently, somatic mutations of CBL have
been identified in a range of myeloid disorders
including acute myeloid leukemia (AML),
chronic myelo-monocytic leukemia (CMML), ju-
venile myelo-monocytic leukemia (JMML), and
myeloproliferative syndrome (Caligiuri et al.,
2007; Sargin et al., 2007; Dunbar et al., 2008;
Grand et al., 2009; Loh et al., 2009; Makishima
et al., 2009; Reindl et al., 2009; Kales et al., 2010;
Muramatsu et al., 2010; Ogawa et al., 2010).
Mutations principally affect the linker-RING do-
main of the CBL protein and are often associated
with acquired uniparental disomy (aUPD) at the
CBL gene locus at 11q23.3. Functional studies in
vitro and in vivo show that CBL mutations are
transforming and induce a myeloproliferative dis-
ease which can progress to leukemia and are asso-
ciated with enhanced RTK signaling, manifested
down numerous key signaling pathways including
the RAS, PKB and JAK-STAT pathways (Bandi
et al., 2009; Sanada et al., 2009; Rathinam et al.,
2010). In addition, somatic mutations of another
family member, CBLB, have also been reported
(Caligiuri et al., 2007; Makishima et al., 2009). In
JMML and CMML, CBL mutations were found
to be mutually exclusive with respect to RAS-
pathway activating mutations such as NRAS,
KRAS, and PTPN11, suggesting that CBL may
play an important a role in deregulating this key
survival pathway (Loh et al., 2009; Sanada et al.,
2009; Muramatsu et al., 2010).
Acute lymphoblastic leukemia (ALL) is the
most common cancer in children and is character-
ized by a clonal expansion of poorly differenti-
ated lymphoid precursors within the bone
marrow. While cure rates are high, novel thera-
pies are needed for those children that relapse.
ALL is a heterogeneous disease, with a vast array
of genomic aberrations and we recently showed
that mutations in genes impacting on the RAS
pathway, such as NRAS, KRAS, PTPN11, and
FLT3 represent one of the most common genetic
abnormalities, being found in a third of patients
(Case et al., 2008). Primary leukemic blasts with
mutations were associated with constitutive acti-
vation of the RAS pathway and differential sensi-
tivity to MEK inhibitors was shown in patients
with the highest levels of RAS pathway activa-
tion, suggesting that drugs such as MEK
Additional Supporting Information may be found in the onlineversion of this article.
Supported by: Leukaemia and Lymphoma Research.
*Correspondence to: Julie Irving, Northern Institute for CancerResearch, Paul O’Gorman Building, Framlington Place, Newcastleupon Tyne, Tyne and Wear, UK, NE2 4HH. E-mail: [email protected]
Received 3 June 2011; Accepted 6 October 2011
DOI 10.1002/gcc.20949
Published online 10 November 2011 inWiley Online Library (wileyonlinelibrary.com).
VVC 2011 Wiley Periodicals, Inc.
inhibitors which have already entered clinical trial
for adult solid tumors, may be a useful, novel
therapeutic approach in ALL (Case et al., 2008;
Bennouna et al., 2011). However, we also found
evidence for constitutive activation of the RAS
pathway in the absence of known activating
mutations and thus we hypothesized that CBLmutations were a likely alternative route to dysre-
gulate the RAS and possibly other key survival
pathways. Thus in this study, we mutationally
screened the linker-RING domains of CBL and
CBLB in a large cohort of ALL patients and
report the first identification of CBL mutations in
this disease.
MATERIALS AND METHODS
Patients
Bone marrow or peripheral blood samples were
obtained from children (less than 17 years) who
presented with ALL within the northern region
of England between 1994 and 2011. The study
had ethical approval (reference 2002/11 and 07/
H906). All diagnosis, remission, and relapse
events were pathologically confirmed. The major-
ity of this cohort were previously screened for
mutations in FLT3, PTPN11, NRAS, KRAS, andBRAF and were reported in Case et al. (2008).
Cell Lines and Cytotoxicity Assays
Cell lines used in the study were cultured in
RPMI 1640 medium containing L-glutamine, sup-
plemented with 10% fetal bovine serum, at 37�C,in an atmosphere of 5% CO2 and were routinely
tested for mycoplasma contamination using
MycoAlertVR
(Lonza, Basel, Switzerland). In vitro
cytotoxicity assays were performed using the
MTS assay (Promega, Southampton, UK) as pre-
viously described (Nicholson et al., 2010), which
assesses the capacity of metabolically active cells
to reduce formazan. Briefly, cell lines or patient
cells were plated out in triplicate at a seeding
density of 2 � 104 or 2 � 105 cells per well,
respectively, into 96-well plates and dosed with
either the FLT3 inhibitor PKC-412 (Sigma-
Aldrich, Dorset, UK), the MEK inhibitor U0126,
(Calbiochem, Nottingham, UK) or a control vehi-
cle (0.01% v/v DMSO). After 96-hr incubation,
the MTS reagent was added and the resulting ab-
sorbances were averaged and expressed as a per-
centage of the control vehicle. Survival curves
were plotted using GraphPad Prism software ver-
sion 4.0.
Mutational Screening and Sequencing/Cloning
Exons 8 and 9 (CBL) and exon 9 (CBLB), con-stituting the linker-RING domain, were screened
for mutations using denaturing high performance
liquid chromatography (dHPLC) on a Transge-
nomic WAVE machine using genomic DNA or in
some cases whole genome amplified DNA.
Amplicons were analyzed before and after spiking
with known wild type product to allow detection
of homozygous mutations. Primer sequences and
dHPLC conditions are shown in Supporting In-
formation Table 1. Direct sequencing and PCR
product cloning were performed using standard
techniques.
RT-PCR
Total RNA was extracted using a Qiagen
RNeasy kit and cDNA synthesized using a High
Capacity cDNA Reverse Transcription Kit
(Applied Biosystems). PCR was carried out using
primers described elsewhere (Reindl et al., 2009)
and the products visualized by electrophoresis
using a 2% agarose gel and GelRed Nucleic Acid
Stain (Biotium, Hayward, USA).
Western Blotting
Whole cell lysates were prepared using cell
lysis buffer supplied by Cell Signalling Technol-
ogy (NEB, UK) according to the manufacturers’
instructions. Protein was fractionated using 12%
SDS-polyacrylamide gels (BioRad, Hercules, CA)
and transferred onto PVDF membrane (BioRad).
Membranes were probed with antibodies directed
TABLE 1. Clinical Characteristics of CBL Mutant Positive Cases
Patient Age Sex WCC x 109/l FAB Immunophenotype Cytogenetics Day 28 MRD
L825 14 F >100 L1 CD10� B precursor ALL Normala 1.30%L914 7 F 9.6 L1 CD10þ B precursor ALL High hyperdiploid <0.01%L919 2 M 1.5 L1 CD10þ B precursor ALL Pseudodiploid Not done
aIn 16 metaphases.
F, female; M, male; WCC, white cell count; FAB, French American British morphology classification; MRD, minimal residual disease.
CBL MUTATIONS AND CHILDHOOD ALL 251
Genes, Chromosomes & Cancer DOI 10.1002/gcc
to p-STAT5, FLT-3, p-AKT, AKT (all purchased
from Cell Signaling Technology, Beverly, MA),
p-ERK and ERK (Santa Cruz Biotechnology,
Santa Cruz, CA) as previously described (Case
et al., 2008). All immunoblots were probed with
an antibody directed towards b-actin (Calbio-
chem, UK), which served as a loading control.
Secondary antibodies used were horseradish
peroxidise conjugates of either anti-rabbit or
anti-mouse immunoglobulins (Dako, Denmark).
Blots were visualized by ECL-Plus detection
(Amersham, UK) and exposed to Kodak Medical
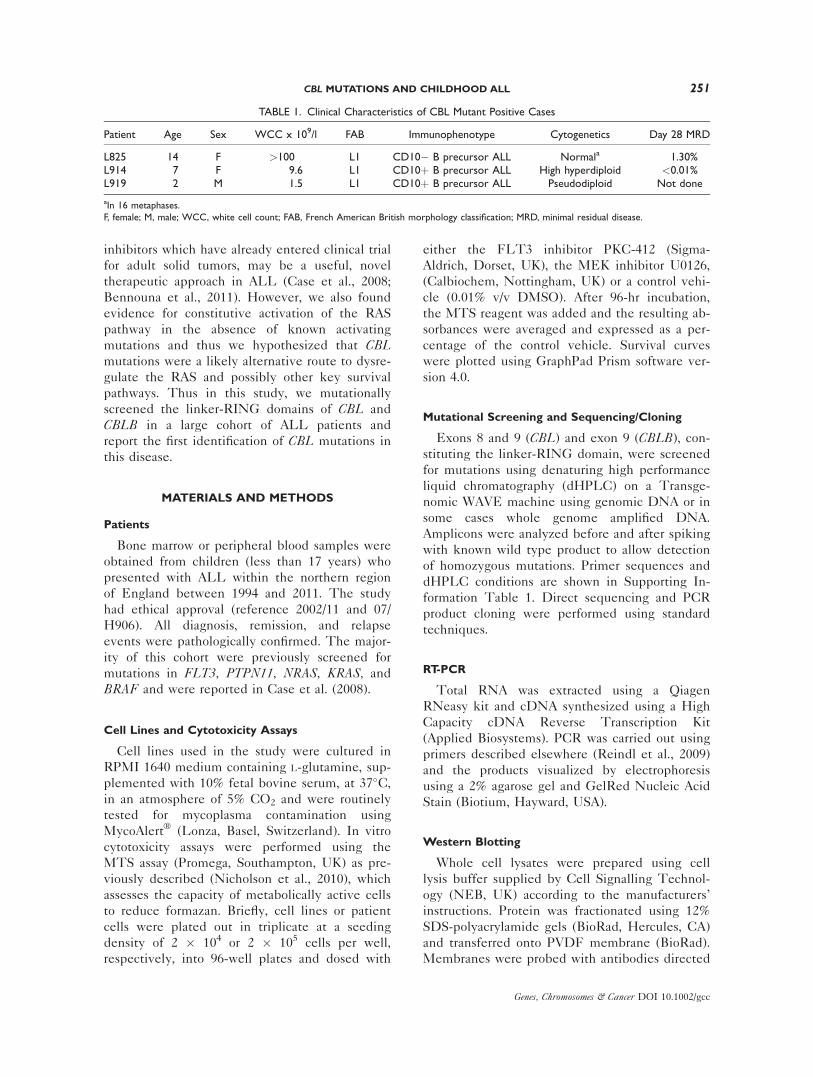
Figure 1. Detection and characterization of CBL mutations in primary ALL. A: dHPLC chromato-grams of CBL run under non-denaturing conditions, showing aberrant peaks with reduced retention timein samples L825, L914 and L919, compared with the wild type control. B–D: Sequence chromatogramsfor CBL mutations in patients L825 (B), L914 (C) and L919 (D). E: Schematic of CBL mutations in ALL.
252 NICHOLSON ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc
X-ray film (GRI, UK). Cell lines which harbored
the relevant mutations or were wild-type served
as positive and negative controls, respectively,
and were ran alongside patient L825.
Flow Minimal Residual Disease
This was performed using the method
described in Irving et al. (2009). Briefly, samples
were collected into Acid Citrate Dextrose-A
tubes and red cell lysed using a standard ammo-
nium chloride procedure. At diagnosis, eight
antibody combinations were assessed with a
common CD34/CD19/CD10 spine and a mini-
mum of 50,000 events acquired on a FACSCali-
bur (Becton Dickinson, Oxford, UK).
Combinations, in which the leukemic blasts fell
into empty spaces, distinct from regions housing
normal B cell progenitors, were identified as leu-
kemia-associated immunophenotypes (LAIPs).
At least 2 LAIPS were then tracked in follow up
Day 28 samples and a minimum of 500,000
events acquired. Samples were considered posi-
tive if the number of leukemic cells identified
with one or more LAIP combinations was equal
to or greater than 0.01%.
RESULTS AND DISCUSSION
Mutational screening of CBL (exons 8 and 9)
and CBLB (exon 9) was performed by dHPLC in
a cohort of 180 diagnostic and 46 relapse ALL
samples. Heteroduplexes, indicative of a DNA
alteration, were identified in four samples, three
in CBL (patient numbers, L825, L914, and L919)
(Fig. 1A) and 1 in CBLB (L526). Direct sequenc-
ing of CBLB amplicons from patient L526
showed a T>C transition, P387P, which was also
present in the matched constitutive genomic
DNA and is a rare synonymous SNP (rs9657925).
For CBL, cloning and sequencing of amplicons
identified, from patient L825 a 4bp insertion/37
bp deletion at the exon 8/intron 8-9 boundary in
2 from 9 clones (22%; 95%CI, 5–56%); from
L914, a 6 bp insertion/21 bp deletion in 4 from
20 clones (20%; 95% CI, 8–42%) and from L919,
a 3 bp insertion/187 bp deletion in 5 from 9
clones (56%; 95% CI, 27–81%), the latter two
mutations affecting the intron 7-8/exon 8 bound-
ary (Figs. 1B–1E). DHPLC analyses of constitu-
tive DNA from these patients confirmed that
these deletions were acquired somatic mutations.
In addition, the mutational status of hot spot
exons of FLT3, NRAS, KRAS, and PTPN11 was
assessed and found to be wild type for patient
L825, while L914 also had a FLT3 I836 deletion.
Unfortunately, there was insufficient material to
perform additional mutational screening in L919.
All three samples had a high percentage of leuke-
mic cells (>90%) and unlike L914, CBL muta-
tions in L825 and L919 were present in the
majority of blasts, presumably heterozygously,
although without LOH analyses at the CBL
locus, we cannot rule out that the mutation exists
in a subpopulation of cells in a homozygous state,
concomitant with 11q aUPD.
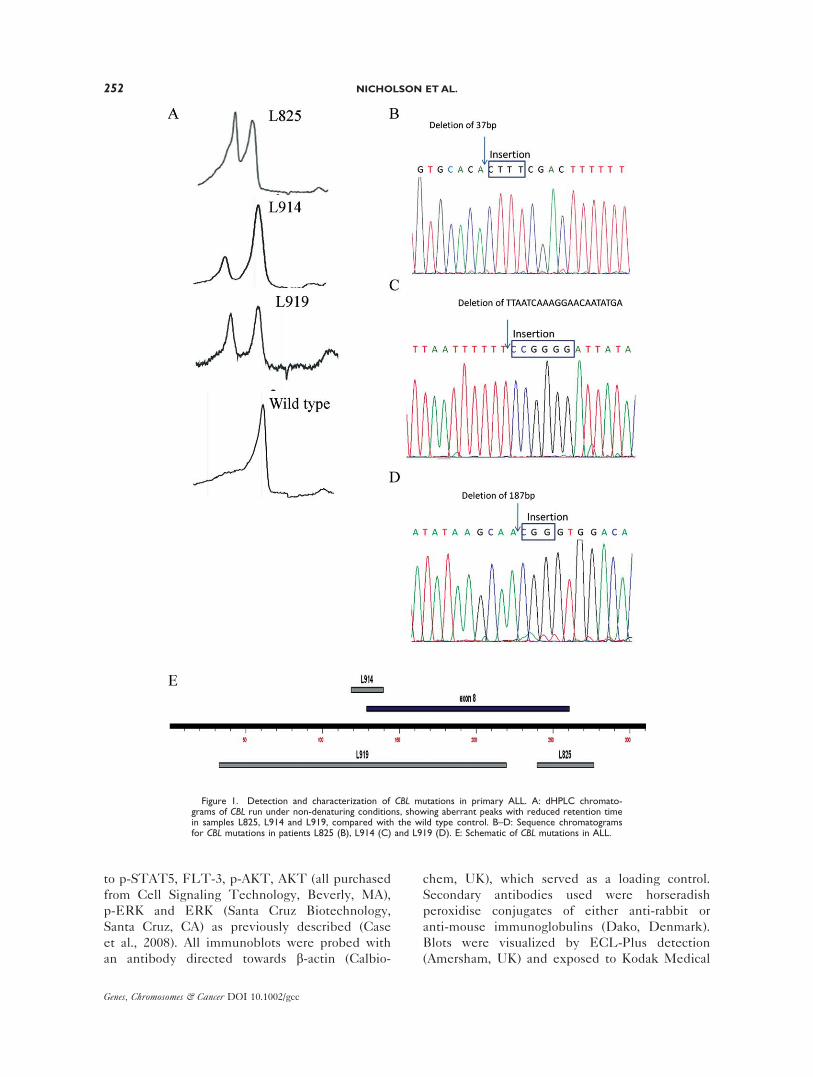
RT-PCR of CBL with primers positioned in
exons 5 and 10 showed a wild type product of
752 bp in addition to a smaller 620 bp product in
bone marrow cells from patient L825 and L914
which was absent in a bone marrow aspirates
taken in clinical remission and also absent in the
PreB 697 cell line, which served as a wild type
control (Fig. 2A). Sequencing of the excised
smaller band in both patients identified CBLsequence, minus exon 8, which results in an in-
frame deletion (D8), disrupting the linker-RING
domain that is responsible for E3 ligase activity
(Fig. 2B). There was insufficient material to
Figure 2. Expression of mutant CBL in primary ALL. A: Agarosegel electrophoresis of CBL RT-PCR products generated from patientL825 and L914 diagnostic and remission samples, with the PreB 697cell line serving as a wild type control. B: Sequence chromatogramsof the mutant RT-PCR product of L825 showing splicing of CBL exon7 to exon 9.
CBL MUTATIONS AND CHILDHOOD ALL 253
Genes, Chromosomes & Cancer DOI 10.1002/gcc
perform this analysis for L919 but given the posi-
tion of the genetic lesion it is likely that this
patient too has exon 8 skipping. The CBL D8mutation has been previously reported in AML
and in a BAF3 cell line model it induced growth
factor-independence when cotransfected with
FLT3, causing autophosphorylation of FLT3 and
constitutive activation of the downstream targets
STAT5 and AKT (Caligiuri et al., 2007; Reindl
et al., 2009). FLT3 ligand-dependent hyperprolif-
eration of CBL mutant cells could be abrogated
by treatment with the FLT3 PTK inhibitor
PKC412, suggesting that CBL D8 AML may be
responsive to this class of targeted agent (Reindl
et al., 2009).
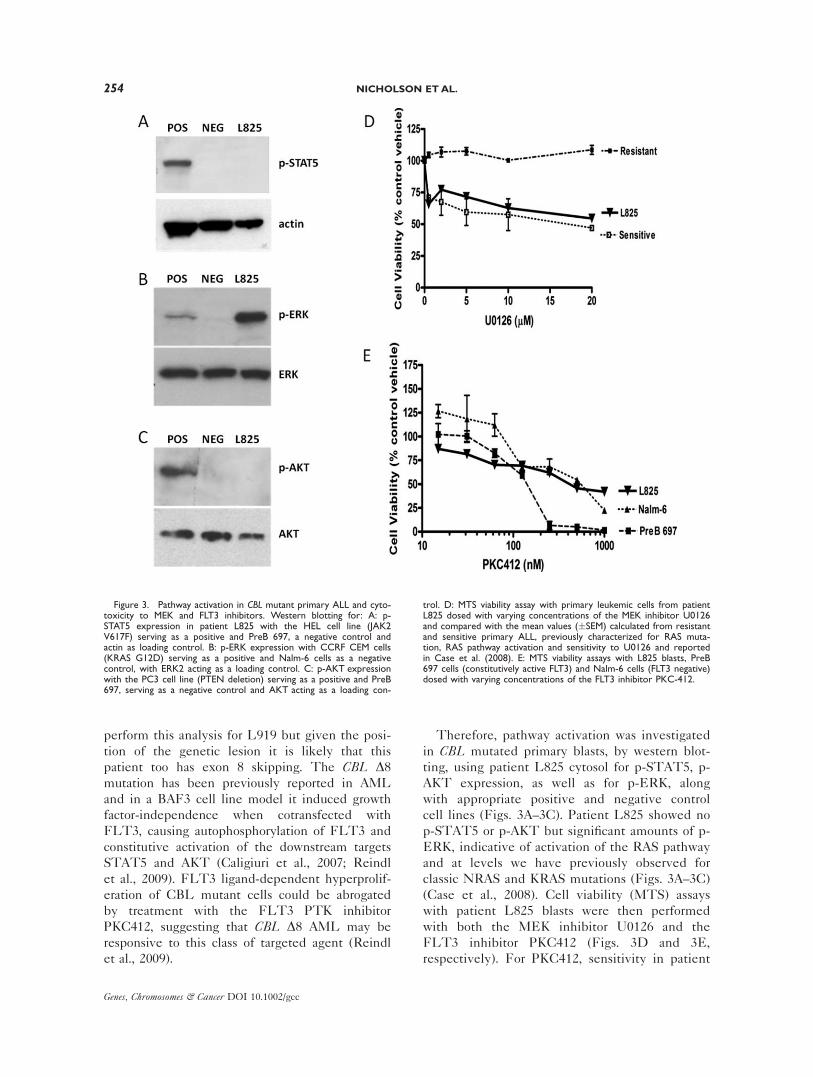
Therefore, pathway activation was investigated
in CBL mutated primary blasts, by western blot-
ting, using patient L825 cytosol for p-STAT5, p-
AKT expression, as well as for p-ERK, along
with appropriate positive and negative control
cell lines (Figs. 3A–3C). Patient L825 showed no
p-STAT5 or p-AKT but significant amounts of p-
ERK, indicative of activation of the RAS pathway
and at levels we have previously observed for
classic NRAS and KRAS mutations (Figs. 3A–3C)
(Case et al., 2008). Cell viability (MTS) assays
with patient L825 blasts were then performed
with both the MEK inhibitor U0126 and the
FLT3 inhibitor PKC412 (Figs. 3D and 3E,
respectively). For PKC412, sensitivity in patient
Figure 3. Pathway activation in CBL mutant primary ALL and cyto-toxicity to MEK and FLT3 inhibitors. Western blotting for: A: p-STAT5 expression in patient L825 with the HEL cell line (JAK2V617F) serving as a positive and PreB 697, a negative control andactin as loading control. B: p-ERK expression with CCRF CEM cells(KRAS G12D) serving as a positive and Nalm-6 cells as a negativecontrol, with ERK2 acting as a loading control. C: p-AKT expressionwith the PC3 cell line (PTEN deletion) serving as a positive and PreB697, serving as a negative control and AKT acting as a loading con-
trol. D: MTS viability assay with primary leukemic cells from patientL825 dosed with varying concentrations of the MEK inhibitor U0126and compared with the mean values (�SEM) calculated from resistantand sensitive primary ALL, previously characterized for RAS muta-tion, RAS pathway activation and sensitivity to U0126 and reportedin Case et al. (2008). E: MTS viability assays with L825 blasts, PreB697 cells (constitutively active FLT3) and Nalm-6 cells (FLT3 negative)dosed with varying concentrations of the FLT3 inhibitor PKC-412.
254 NICHOLSON ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

L825 primary blasts was compared with cell line
controls that have been previously characterized
for FLT3 receptor activation and responsiveness
to FLT3 inhibition, i.e., PreB 697 serving as a
positive control (constitutively active FLT3; re-
sponsive) and Nalm-6 serving as a negative con-
trol (FLT3 receptor negative; nonresponsive)
(Brown et al., 2005). Patient L825 showed little
sensitivity to the drug, particularly when com-
pared with a cohort of MLL rearranged, FLT3
high, ALL primary samples reported by Stam
et al. (2005). However, L825 cells when dosed
with U0126 showed a significant cytotoxic
response and when compared with our published
primary ALL cohort characterized for RAS muta-
tion status, pathway activation and U0126 sensi-
tivity, were as sensitive as those with classic RAS
mutations and the highest levels of p-ERK
expression.
In terms of clinical details, CBL mutant posi-
tive patients were all B cell precursor ALL but
heterogeneous in terms of age, presenting white
cell count, CD10 expression, cytogenetics, and
clinical response (Table 1). For example, patient
L825, a 14-year-old girl, presented with a high
peripheral white cell count, apparently normal
cytogenetics and had a poor response to therapy
as gauged by an exceptionally high end of induc-
tion minimal residual disease (MRD) level of
1.3% and thus is at very high risk of relapse (Cou-
stan-Smith et al., 2000). The other two patients
were younger, presented with a low white cell
count, one pseudodiploid, one high hyperdiploid,
with the latter responding well to induction ther-
apy, with an end of induction MRD level of
<0.01%. In a large study of myeloid neoplasms,
the presence of CBL family mutations was associ-
ated with a poorer prognosis, particularly for indi-
viduals with homozygous mutations (Makishima
et al., 2009); however, other studies have found no
such relationship (Sanada et al., 2009).
In summary, this is the first report of somatic
mutation of CBL in ALL with a frequency of
1.7%, a similar incidence to AML. While uncom-
mon, it emphasizes the importance of RAS path-
way activation in the molecular pathology of
ALL and may identify children who, along with
other RAS pathway mutant positive cases, are
candidates for MEK inhibitor clinical trials.
REFERENCES
Bandi SR, Brandts C, Rensinghoff M, Grundler R, TickenbrockL, Kohler G, Duyster J, Berdel WE, Muller-Tidow C, Serve H,
Sargin B. 2009. E3 ligase-defective Cbl mutants lead to a gen-eralized mastocytosis and myeloproliferative disease. Blood114:4197–4208.
Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, AdenisA, Escudero P, Kim TY, Pover GM, Morris CD, DouillardJY. 2011. A Phase II, open-label, randomised study toassess the efficacy and safety of the MEK1/2 inhibitorAZD6244 (ARRY-142886) versus capecitabine monotherapyin patients with colorectal cancer who have failed one ortwo prior chemotherapeutic regimens. Invest New Drugs29:1021–1028.
Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D.2005. FLT3 inhibition selectively kills childhood acute lympho-blastic leukemia cells with high levels of FLT3 expression.Blood 105:812–820.
Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ,Marburger TB, Wen J, Perrotti D, Bloomfield CD, WhitmanSP. 2007. Novel c-CBL and CBL-b ubiquitin ligase mutationsin human acute myeloid leukemia. Blood 110:1022–1024.
Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N,Bailey S, Vormoor J, Hall AG, Irving JA. 2008. Mutation ofgenes affecting the RAS pathway is common in childhood acutelymphoblastic leukemia. Cancer Res 68:6803–6809.
Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG,Raimondi SC, Sandlund JT, Rivera GK, Rubnitz JE, RibeiroRC, Pui CH, Campana D. 2000. Clinical importance of minimalresidual disease in childhood acute lymphoblastic leukemia.Blood 96:2691–2696.
Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, RataulMS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA,Maciejewski JP. 2008. 250K single nucleotide polymorphismarray karyotyping identifies acquired uniparental disomy andhomozygous mutations, including novel missense substitutionsof c-Cbl, in myeloid malignancies. Cancer Res 68:10349–10357.
Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuireC, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A,Brandts C, Serve H, Reiter A, Chase AJ, Cross NC. 2009.Frequent CBL mutations associated with 11q acquired unipar-ental disomy in myeloproliferative neoplasms. Blood 113:6182–6192.
Irving J, Jesson J, Virgo P, Case M, Minto L, Eyre L, Noel N,Johansson U, Macey M, Knotts L, Helliwell M, Davies P,Whitby L, Barnett D, Hancock J, Goulden N, Lawson S. 2009.Establishment and validation of a standard protocol for thedetection of minimal residual disease in B lineage childhoodacute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica 94:870–874.
Kales SC, Ryan PE, Nau MM, Lipkowitz S. 2010. Cbl and humanmyeloid neoplasms: The Cbl oncogene comes of age. CancerRes 70:4789–4794.
Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archam-beault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, Emanuel PD, Hasle H, Issa JP, van den Heuvel-Eibrink MM, Locatelli F, Stary J, Trebo M, Wlodarski M,Zecca M, Shannon KM, Niemeyer CM. 2009. Mutations inCBL occur frequently in juvenile myelomonocytic leukemia.Blood 114:1859–1863.
Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J,Muramatsu H, O’Keefe C, Hsi E, Paquette RL, Kojima S, ListAF, Sekeres MA, McDevitt MA, Maciejewski JP. 2009. Muta-tions of e3 ubiquitin ligase cbl family members constitute anovel common pathogenic lesion in myeloid malignancies. JClin Oncol 27:6109–6116.
Muramatsu H, Makishima H, Jankowska AM, Cazzolli H,O’Keefe C, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H,Takahashi Y, Kato K, Manabe A, Kojima S, Maciejewski JP.2010. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2mutations are pathogenic in juvenile myelomonocytic leukemia.Blood 115:1969–1975.
Nicholson L, Hall AG, Redfern CP, Irving J. 2010. NFkappaBmodulators in a model of glucocorticoid resistant, childhoodacute lymphoblastic leukemia. Leuk Res 34:1366–1373.
Ogawa S, Shih LY, Suzuki T, Otsu M, Nakauchi H, KoefflerHP, Sanada M. 2010. Deregulated intracellular signaling bymutated c-CBL in myeloid neoplasms. Clin Cancer Res16:3825–3831.
Rathinam C, Thien CB, Flavell RA, Langdon WY. 2010. Myeloidleukemia development in c-Cbl RING finger mutant mice isdependent on FLT3 signaling. Cancer Cell 18:341–352.
CBL MUTATIONS AND CHILDHOOD ALL 255
Genes, Chromosomes & Cancer DOI 10.1002/gcc

Reindl C, Quentmeier H, Petropoulos K, Greif PA, Benthaus T,Argiropoulos B, Mellert G, Vempati S, Duyster J, Buske C,Bohlander SK, Humphries KR, Hiddemann W, Spiekermann K.2009. CBL exon 8/9 mutants activate the FLT3 pathway andcluster in core binding factor/11q deletion acute myeloid leuke-mia/myelodysplastic syndrome subtypes. Clin Cancer Res15:2238–2247.
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S,Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, OdaH, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N,Onodera M, Nobuyoshi M, Hayashi Y, Harada H, Kurokawa M,Chiba S, Mori H, Ozawa K, Omine M, Hirai H, Nakauchi H,Koeffler HP, Ogawa S. 2009. Gain-of-function of mutated
C-CBL tumour suppressor in myeloid neoplasms. Nature460:904–908.
Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R,Rensinghoff M, Thiessen C, Tickenbrock L, Schwable J,Brandts C, August B, Koschmieder S, Bandi SR, Duyster J, Ber-del WE, Muller-Tidow C, Dikic I, Serve H. 2007. Flt3-depend-ent transformation by inactivating c-Cbl mutations in AML.Blood 110:1004–1012.
Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M,Beverloo HB, van der Voort E, Valsecchi MG, de Lorenzo P,Sallan SE, Armstrong SA, Pieters R. 2005. Targeting FLT3 inprimary MLL-gene-rearranged infant acute lymphoblastic leu-kemia. Blood 106:2484–2490.
Genes, Chromosomes & Cancer DOI 10.1002/gcc
256 NICHOLSON ETAL.













![𝛾𝛿T-Cell Acute Lymphoblastic Leukemia/Lymphoma: Discussion of … · 2019. 7. 30. · [7]E.D.Merrill,R.Agbay,R.N.Mirandaetal.,“Primarycutaneous T-celllymphomasshowinggamma-delta(𝛾𝛿)phenotypeand](https://static.fdocuments.net/doc/165x107/60aa64bf856b015a425f2e48/t-cell-acute-lymphoblastic-leukemialymphoma-discussion-of-2019-7-30.jpg)




