
Carnitine metabolism to trimethylamine by an unusual ... · Carnitine metabolism to trimethylamine...
6
Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota Yijun Zhu a,1 , Eleanor Jameson a,1 , Marialuisa Crosatti b,1 , Hendrik Schäfer a , Kumar Rajakumar b , Timothy D. H. Bugg c , and Yin Chen a,2 a School of Life Sciences and c Department of Chemistry, University of Warwick, Coventry CV4 7AL, United Kingdom; and b Department of Infection, Immunity, and Inflammation, University of Leicester, Leicester LE1 9HN, United Kingdom Edited by David W. Russell, University of Texas Southwestern Medical Center, Dallas, TX, and approved January 29, 2014 (received for review September 5, 2013) Dietary intake of L-carnitine can promote cardiovascular diseases in humans through microbial production of trimethylamine (TMA) and its subsequent oxidation to trimethylamine N-oxide by hepatic flavin-containing monooxygenases. Although our microbiota are re- sponsible for TMA formation from carnitine, the underpinning mo- lecular and biochemical mechanisms remain unclear. In this study, using bioinformatics approaches, we first identified a two-component Rieske-type oxygenase/reductase (CntAB) and associated gene cluster proposed to be involved in carnitine metabolism in repre- sentative genomes of the human microbiota. CntA belongs to a group of previously uncharacterized Rieske-type proteins and has an unusual “bridging” glutamate but not the aspartate residue, which is believed to facilitate intersubunit electron transfer be- tween the Rieske center and the catalytic mononuclear iron center. Using Acinetobacter baumannii as the model, we then demon- strate that cntAB is essential in carnitine degradation to TMA. Het- erologous overexpression of cntAB enables Escherichia coli to produce TMA, confirming that these genes are sufficient in TMA formation. Site-directed mutagenesis experiments have confirmed that this unusual “bridging glutamate” residue in CntA is essential in catalysis and neither mutant (E205D, E205A) is able to produce TMA. Taken together, the data in our study reveal the molecular and biochemical mechanisms underpinning carnitine metabolism to TMA in human microbiota and assign the role of this novel group of Rieske-type proteins in microbial carnitine metabolism. methylated amine metabolism | comparative genomics | gut microbiota I t is increasingly clear that the human microbiota plays an es- sential role in our health and disease (1–5). Understanding the metabolic potential of the human microbiota and its interaction with and regulation by the host and the environment holds the key to unravel the dynamic relationship between ourselves and our associated microbes (4–6). Over the last decade, our knowledge of the human microbiota has been significantly improved thanks to technological advances, including high-throughput sequencing, de- velopment of powerful bioinformatics, and the use of germ-free animal models (7–13). We can now not only characterize the tax- onomic composition, species richness and dynamics, but com- bine the genetic potential encoded in human microbiota and establish the core metabolic pathways enabled by direct se- quencing of the human microbiome (14, 15). Advances in our understanding of the microbiome functions, however, do not match the pace of taxonomic characterization of species diversity and dynamics (16–18). Fully resolving the functional capacity encoded in the human microbiome and the dynamic effects on health and disease still remains a great challenge (11, 16–18). Direct sequencing of the human micro- biome generates datasets dominated by genes encoding pathways for central metabolism (e.g., transcriptional and translational machinery, ATP synthesis, and so forth) (14, 15), therefore contributing little to our understanding of the variable functional capacity encoded in human microbiota between individuals (7, 8). Furthermore, a large fraction of the genes encoded in the human microbiome remain to be functionally characterized (14, 15). Assigning functions encoded in the human microbiome using existing databases can be problematic. For example, the Pfam protein database currently contains over 25% of protein families with no assigned functions (release 26.0) (19). Lack of functional characterization of key microbial functions in our microbiota is exemplified by very recent studies on car- diovascular diseases (20–23). These studies have shown that the human microbiota is responsible for the production of trime- thylamine N-oxide (TMAO), which is believed to promote ath- erogenesis through its interaction with macrophages and lipid metabolism (20–23). TMAO is derived from the microbial me- tabolism of dietary quaternary amines [e.g., choline, L-carnitine, glycine betaine (GBT), and phosphatidylcholine] to trimethyl- amine (TMA), which is subsequently oxidized to TMAO by the host hepatic flavin monooxygenases (21, 22). L-Carnitine (hereafter referred to as carnitine, unless otherwise specified) is considered an important nutrient for human health, playing a key role in mitochondrial fatty acid β-oxidation (24, 25). Al- though carnitine can be acquired through endogenous bio- synthesis, our daily demand is largely met through dietary intake from carnitine-containing food (26). It is known that a significant proportion of dietary carnitine can be further metabolized by microbiota before absorption (20, 27). This microbial-mediated metabolic pathway not only diverts carnitine away from the host, causing conditional carnitine deficiency in certain human pop- ulations, but promotes TMAO formation and subsequent in- creased risk of atherosclerosis (20, 21). However, the underlying Significance Metabolism of L-carnitine, a compound abundant in human diet, to trimethylamine by human microbiota has been shown to promote atherosclerosis and subsequent development of heart disease. However, the underpinning molecular and bio- chemical mechanisms remain unknown. In this study, we reveal that a previously unidentified Rieske-type protein is responsible for carnitine transformation to trimethylamine from human microbiota. Knowledge gained in our study provides the op- portunity not only to explore Rieske protein inhibitors in pre- venting trimethylamine formation in animal studies and clinical trials, but also for its use as a functional genetic marker to better understand human microbiota and their dynamics in our health and disease in future epidemiological studies and dietary interventions. Author contributions: Y.C. designed research; Y.Z., E.J., M.C., and Y.C. performed re- search; Y.Z., E.J., M.C., H.S., K.R., T.D.H.B., and Y.C. analyzed data; and Y.Z., E.J., M.C., H.S., K.R., T.D.H.B., and Y.C. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. Freely available online through the PNAS open access option. 1 Y.Z., E.J., and M.C. contributed equally to this work. 2 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1316569111/-/DCSupplemental. 4268–4273 | PNAS | March 18, 2014 | vol. 111 | no. 11 www.pnas.org/cgi/doi/10.1073/pnas.1316569111 Downloaded by guest on January 1, 2020
Transcript of Carnitine metabolism to trimethylamine by an unusual ... · Carnitine metabolism to trimethylamine...

Carnitine metabolism to trimethylamine by an unusualRieske-type oxygenase from human microbiotaYijun Zhua,1, Eleanor Jamesona,1, Marialuisa Crosattib,1, Hendrik Schäfera, Kumar Rajakumarb, Timothy D. H. Buggc,and Yin Chena,2
aSchool of Life Sciences and cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, United Kingdom; and bDepartment of Infection, Immunity,and Inflammation, University of Leicester, Leicester LE1 9HN, United Kingdom
Edited by David W. Russell, University of Texas Southwestern Medical Center, Dallas, TX, and approved January 29, 2014 (received for reviewSeptember 5, 2013)
Dietary intake of L-carnitine can promote cardiovascular diseases inhumans through microbial production of trimethylamine (TMA)and its subsequent oxidation to trimethylamine N-oxide by hepaticflavin-containing monooxygenases. Although our microbiota are re-sponsible for TMA formation from carnitine, the underpinning mo-lecular and biochemical mechanisms remain unclear. In this study,using bioinformatics approaches, we first identified a two-componentRieske-type oxygenase/reductase (CntAB) and associated genecluster proposed to be involved in carnitine metabolism in repre-sentative genomes of the human microbiota. CntA belongs to agroup of previously uncharacterized Rieske-type proteins and hasan unusual “bridging” glutamate but not the aspartate residue,which is believed to facilitate intersubunit electron transfer be-tween the Rieske center and the catalytic mononuclear iron center.Using Acinetobacter baumannii as the model, we then demon-strate that cntAB is essential in carnitine degradation to TMA. Het-erologous overexpression of cntAB enables Escherichia coli toproduce TMA, confirming that these genes are sufficient in TMAformation. Site-directed mutagenesis experiments have confirmedthat this unusual “bridging glutamate” residue in CntA is essentialin catalysis and neither mutant (E205D, E205A) is able to produceTMA. Taken together, the data in our study reveal the molecularand biochemical mechanisms underpinning carnitine metabolismto TMA in human microbiota and assign the role of this novelgroup of Rieske-type proteins in microbial carnitine metabolism.
methylated amine metabolism | comparative genomics | gut microbiota
It is increasingly clear that the human microbiota plays an es-sential role in our health and disease (1–5). Understanding the
metabolic potential of the human microbiota and its interactionwith and regulation by the host and the environment holds thekey to unravel the dynamic relationship between ourselves andour associated microbes (4–6). Over the last decade, our knowledgeof the human microbiota has been significantly improved thanks totechnological advances, including high-throughput sequencing, de-velopment of powerful bioinformatics, and the use of germ-freeanimal models (7–13). We can now not only characterize the tax-onomic composition, species richness and dynamics, but com-bine the genetic potential encoded in human microbiota andestablish the core metabolic pathways enabled by direct se-quencing of the human microbiome (14, 15).Advances in our understanding of the microbiome functions,
however, do not match the pace of taxonomic characterizationof species diversity and dynamics (16–18). Fully resolving thefunctional capacity encoded in the human microbiome and thedynamic effects on health and disease still remains a greatchallenge (11, 16–18). Direct sequencing of the human micro-biome generates datasets dominated by genes encoding pathwaysfor central metabolism (e.g., transcriptional and translationalmachinery, ATP synthesis, and so forth) (14, 15), thereforecontributing little to our understanding of the variable functionalcapacity encoded in human microbiota between individuals (7,8). Furthermore, a large fraction of the genes encoded in thehuman microbiome remain to be functionally characterized
(14, 15). Assigning functions encoded in the human microbiomeusing existing databases can be problematic. For example, thePfam protein database currently contains over 25% of proteinfamilies with no assigned functions (release 26.0) (19).Lack of functional characterization of key microbial functions
in our microbiota is exemplified by very recent studies on car-diovascular diseases (20–23). These studies have shown that thehuman microbiota is responsible for the production of trime-thylamine N-oxide (TMAO), which is believed to promote ath-erogenesis through its interaction with macrophages and lipidmetabolism (20–23). TMAO is derived from the microbial me-tabolism of dietary quaternary amines [e.g., choline, L-carnitine,glycine betaine (GBT), and phosphatidylcholine] to trimethyl-amine (TMA), which is subsequently oxidized to TMAO bythe host hepatic flavin monooxygenases (21, 22). L-Carnitine(hereafter referred to as carnitine, unless otherwise specified)is considered an important nutrient for human health, playinga key role in mitochondrial fatty acid β-oxidation (24, 25). Al-though carnitine can be acquired through endogenous bio-synthesis, our daily demand is largely met through dietary intakefrom carnitine-containing food (26). It is known that a significantproportion of dietary carnitine can be further metabolized bymicrobiota before absorption (20, 27). This microbial-mediatedmetabolic pathway not only diverts carnitine away from the host,causing conditional carnitine deficiency in certain human pop-ulations, but promotes TMAO formation and subsequent in-creased risk of atherosclerosis (20, 21). However, the underlying
Significance
Metabolism of L-carnitine, a compound abundant in humandiet, to trimethylamine by human microbiota has been shownto promote atherosclerosis and subsequent development ofheart disease. However, the underpinning molecular and bio-chemical mechanisms remain unknown. In this study, we revealthat a previously unidentified Rieske-type protein is responsiblefor carnitine transformation to trimethylamine from humanmicrobiota. Knowledge gained in our study provides the op-portunity not only to explore Rieske protein inhibitors in pre-venting trimethylamine formation in animal studies and clinicaltrials, but also for its use as a functional genetic marker tobetter understand human microbiota and their dynamics inour health and disease in future epidemiological studies anddietary interventions.
Author contributions: Y.C. designed research; Y.Z., E.J., M.C., and Y.C. performed re-search; Y.Z., E.J., M.C., H.S., K.R., T.D.H.B., and Y.C. analyzed data; and Y.Z., E.J., M.C.,H.S., K.R., T.D.H.B., and Y.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1Y.Z., E.J., and M.C. contributed equally to this work.2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316569111/-/DCSupplemental.
4268–4273 | PNAS | March 18, 2014 | vol. 111 | no. 11 www.pnas.org/cgi/doi/10.1073/pnas.1316569111
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020
genetic and biochemical mechanisms of carnitine-dependentTMA production in human microbiota have not been uncovered.
In this report, we describe the identification of the genetic andbiochemical mechanisms for TMA production from carnitine in
CntA CntB
Klebsiella pneumoniae subsp. pneumoniae MGH 78578; subsp. pneumoniae HS11286; subsp. rhinoscleromatis ATCC 13384
Klebsiella pneumoniae 342; NTUH-K2044; KCTC 2242
Klebsiella sp. 1_1_55; sp. MS 92-3; sp. 4_1_44FAAAcinetobacter baumannii ATCC 19606; sp. 6013150;
sp. 6013113; sp. 6014059Acinetobacter sp. SH024; sp. RUH2624Escherichia coli SE11; MS 198-1; MS 84-1; MS 115-1;
MS 182-1; MS 146-1; MS 69-1; MS 187-1; MS 21-1; MS 78-1; MS 116-1; MS 175-1; MS 196-1; MS 124-1; MS 119-1; MS 107-1; MS 145-7;
Escherichia sp. 4_1_40BCitrobacter youngae ATCC 29220Citrobacter freundii 4_7_47CFAACitrobacter sp. 30_2Shigella boydii; Shigella dysenteriae; Shigella sonnei; Shigella sp. D9;
Gam
map
rote
obac
teri
a
CaiT
Providencia stuartii ATCC 25827Providencia rettgeri DSM 1131
Achromobacter piechaudii ATCC 43553
Sporosarcina newyorkensis 2681
BetaprobacteriaFirmicutes
Oxygenase (CntA)
Reductase (CntB)
CntA
Group I
Q44256_C
baA
980.1
M64747_X
ylX
Acylcarnitinehydrolase
(CH3)3N
OH
COO-+
(CH3)3N +
Carnitine
Trimethylamine Malic semialdehyde
Malic acid
Acetylcarnitine
Carnitine oxygenase/ reductase, CntAB
Malic semialdehydedehydrogenase
Malate dehydrogenase
To the tricarboxylic acid cycle
Carnitine
Carnitine transporterCaiT
O
OHO
OH
OHO
OHO
HO
H
(CH3)3N
OH
COO-+
O
[O]
A
B
C D
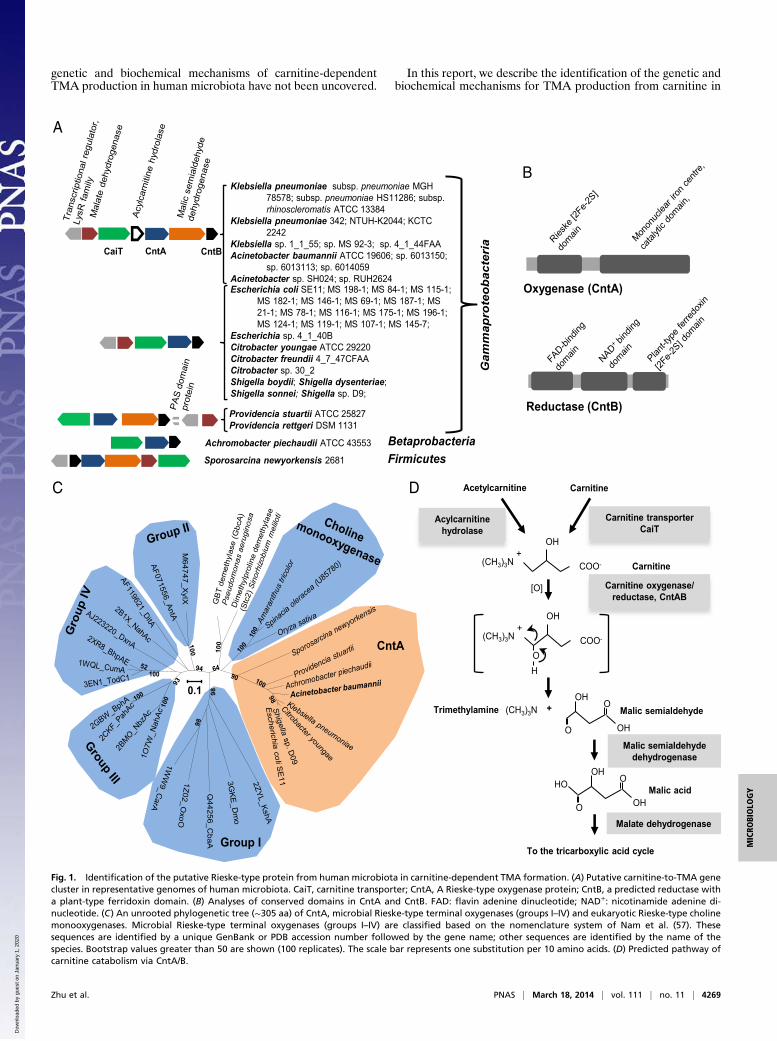
Fig. 1. Identification of the putative Rieske-type protein from human microbiota in carnitine-dependent TMA formation. (A) Putative carnitine-to-TMA genecluster in representative genomes of human microbiota. CaiT, carnitine transporter; CntA, A Rieske-type oxygenase protein; CntB, a predicted reductase witha plant-type ferridoxin domain. (B) Analyses of conserved domains in CntA and CntB. FAD: flavin adenine dinucleotide; NAD+: nicotinamide adenine di-nucleotide. (C) An unrooted phylogenetic tree (∼305 aa) of CntA, microbial Rieske-type terminal oxygenases (groups I–IV) and eukaryotic Rieske-type cholinemonooxygenases. Microbial Rieske-type terminal oxygenases (groups I–IV) are classified based on the nomenclature system of Nam et al. (57). Thesesequences are identified by a unique GenBank or PDB accession number followed by the gene name; other sequences are identified by the name of thespecies. Bootstrap values greater than 50 are shown (100 replicates). The scale bar represents one substitution per 10 amino acids. (D) Predicted pathway ofcarnitine catabolism via CntA/B.
Zhu et al. PNAS | March 18, 2014 | vol. 111 | no. 11 | 4269
MICRO
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020
representative human microbiota (Gammaproteobacteria, Beta-proteobacteria, Firmicutes) through a synthesis of bioinformatics,genetic, and biochemical approaches. These in vivo experimentswith microbial isolates prove valuable for exploring the meta-bolic attributes of a particular group of microbes in carnitinemetabolism guided by bioinformatics (10). This recently identi-fied carnitine-to-TMA enzyme is composed of an oxygenasecomponent (CntA) and a reductase component (CntB). CntAbelongs to a previously uncharacterized group of Rieske-typeproteins, which are best known for ring-hydroxylation of aro-matic hydrocarbons (28). CntA has an unusual alteration of theso-called “bridging” aspartate (substituted by a glutamate resi-due, E205), which plays an essential role in electron transfer incatalysis in the Rieske-type protein family (29–32).
Results and DiscussionDiscovery of a Carnitine Utilization Gene Cluster and Identificationof a Candidate Carnitine Oxygenase (cntAB). We used the HumanMicrobiome Project (HMP) reference genomes (15) for miningfor carnitine-degrading enzymes guided by the following hy-potheses. Microbial conversion of carnitine to TMA has beenstudied in bacteria isolated from humans, Acinetobacter calcoa-ceticus (33) and Serratia marcescens (34). It is known that thecleavage of the carbon (C)–nitrogen (N) bond of carnitine pro-duces TMA and a four-carbon (C4) molecule, which likely entersthe central tricarboxylic acid cycle in the form of malate orsuccinate. We therefore hypothesize that the enzyme responsiblefor splitting the C-N bond is clustered in the genome withenzymes responsible for the synthesis of malate and succinatebecause the C4 unit is further used as a carbon source in thesebacteria (33, 34).We further reasoned that a transporter is needed for microbial
carnitine transport. Two types of bacterial carnitine transportersare known, a BCCT-type permease (35) and an ABC-type activetransporter of the choline/betaine/carnitine family (36). Wetherefore used a BLASTP algorithm to search the HMP refer-ence genomes (754 were available as of February 2013) for genesencoding either CaiT (a BCCT type carnitine-specific antiporter)or CaiX (the carnitine-specific substrate binding protein of theABC transporter cassette). Our BLASTP search data revealedthat many gammaproteobacterial HMP genomes contain CaiTbut not CaiX homologs (Table S1). CaiT is also found in someBetaproteobacteria and Firmicutes.We then inspected the neighborhood of caiT for genes in-
volved in malate or succinate metabolism and this resulted in theidentification of five groups of gene clusters in 39 HMP refer-ence genomes (Fig. 1A), with representative isolates from vari-ous sites of the body, including skin, airway, gastrointestinaltract, and feces (Table S2). We focused our analyses on Acine-tobacter spp. because they are known to degrade carnitine toTMA (33). A close investigation of the caiT neighborhoodrevealed the presence of genes likely to be involved in C4 me-tabolism (Fig. 1A). These include genes coding for a malicsemialdehyde dehydrogenase and a malate dehydrogenase, to-gether channeling the C4 carbon into the central tricarboxylicacid cycle. A conserved lysR-type transcriptional regulator ispresent in these gene clusters. Immediately downstream of thecarnitine transporter caiT is the gene encoding an acylcarnitinehydrolase, which is required for growth and hydrolysis of acyl-carnitine to carnitine (37). The functions of two other genes—which are always present in the caiT gene cluster—are not ex-plicit; we designate these as cntA and cntB, respectively.Bioinformatic analyses predicted that cntB encodes a NAD
(P)-dependent reductase, containing a flavin-binding domain aswell as a plant-type ferredoxin [2Fe-2S] domain (Fig. 1B and Fig.S1). CntA is predicted to be a member of the Rieske-type pro-tein family, which is characterized by two histidine and twocysteine residues coordinating the [2Fe-2S] cluster (28). Se-quence analyses of CntA from HMP bacterial genomes showedconserved Rieske motif and a catalytic mononuclear iron domain(Fig. 1B). To date, most characterized microbial Rieske-type
proteins are involved in the catabolism of ring-structured aromaticcompounds (Table S3) (38). However, recent bioinfomatic andevolutional analyses suggest that Rieske-type proteins are muchmore diverse than previously thought (38–40). Indeed, eukary-otic Rieske-type terminal oxygenases have also been identified[e.g., choline monooxygenase (41, 42)], and recent studies haveconfirmed that Rieske-type proteins can carry out nonring hy-droxylation reactions, such as oxidative demethylation (43) andoxidative carbocyclization (44). Furthermore, many novel Rieske-type proteins have been identified with no assigned function, manyof which originated from newly sequenced microbial genomes(38). Our phylogenetic analyses reveal that CntA forms a distinctgroup in the Rieske-type protein family, which is more closelyrelated to the eukaryotic choline monooxygenases (Fig. 1C). Ourbioinformatic and phylogenetic analyses therefore suggestthat cntAB may encode a novel enzyme that catalyses the initialstep of carnitine degradation to TMA (and a C4 compound),although C-N bond cleavage by Rieske and Rieske-type enzymeshas not previously been reported. The predicted pathway forcarnitine metabolism to TMA is shown in Fig. 1D.
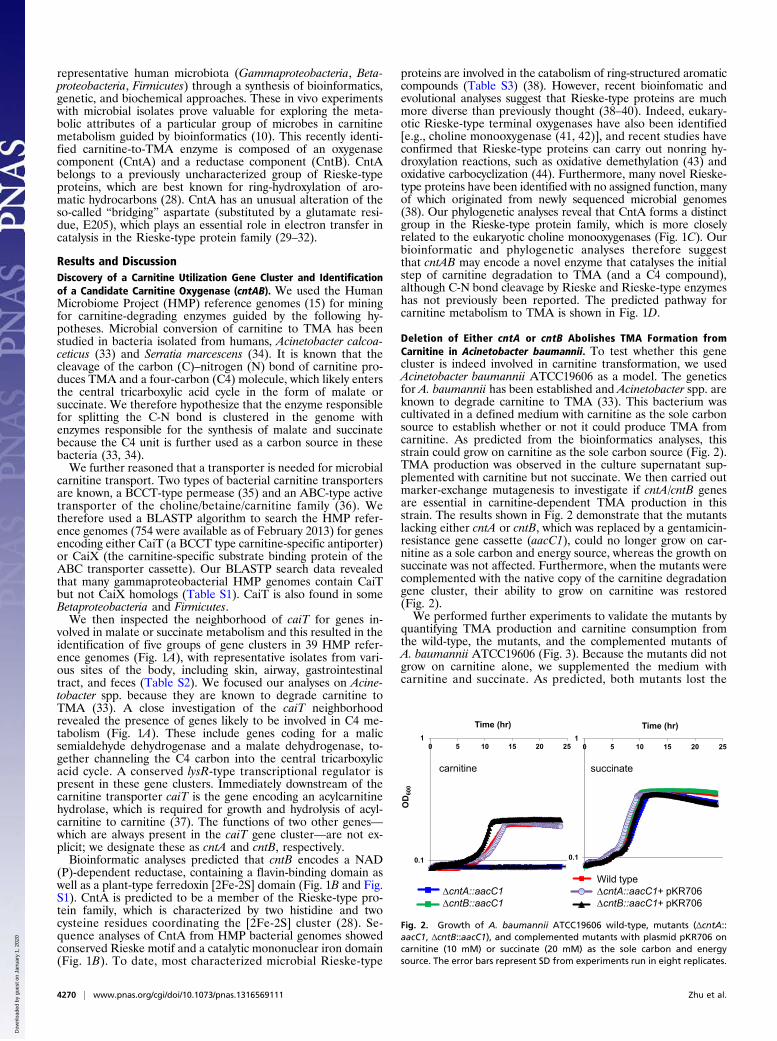
Deletion of Either cntA or cntB Abolishes TMA Formation fromCarnitine in Acinetobacter baumannii. To test whether this genecluster is indeed involved in carnitine transformation, we usedAcinetobacter baumannii ATCC19606 as a model. The geneticsfor A. baumannii has been established and Acinetobacter spp. areknown to degrade carnitine to TMA (33). This bacterium wascultivated in a defined medium with carnitine as the sole carbonsource to establish whether or not it could produce TMA fromcarnitine. As predicted from the bioinformatics analyses, thisstrain could grow on carnitine as the sole carbon source (Fig. 2).TMA production was observed in the culture supernatant sup-plemented with carnitine but not succinate. We then carried outmarker-exchange mutagenesis to investigate if cntA/cntB genesare essential in carnitine-dependent TMA production in thisstrain. The results shown in Fig. 2 demonstrate that the mutantslacking either cntA or cntB, which was replaced by a gentamicin-resistance gene cassette (aacC1), could no longer grow on car-nitine as a sole carbon and energy source, whereas the growth onsuccinate was not affected. Furthermore, when the mutants werecomplemented with the native copy of the carnitine degradationgene cluster, their ability to grow on carnitine was restored(Fig. 2).We performed further experiments to validate the mutants by
quantifying TMA production and carnitine consumption fromthe wild-type, the mutants, and the complemented mutants ofA. baumannii ATCC19606 (Fig. 3). Because the mutants did notgrow on carnitine alone, we supplemented the medium withcarnitine and succinate. As predicted, both mutants lost the
0.1
10 5 10 15 20 25
OD
600
Time (hr) Time (hr)
carnitine succinate
0.1
10 5 10 15 20 25
Wild type∆cntA::aacC1 ∆cntA::aacC1+ pKR706∆cntB::aacC1 ∆cntB::aacC1+ pKR706
Fig. 2. Growth of A. baumannii ATCC19606 wild-type, mutants (ΔcntA::aacC1, ΔcntB::aacC1), and complemented mutants with plasmid pKR706 oncarnitine (10 mM) or succinate (20 mM) as the sole carbon and energysource. The error bars represent SD from experiments run in eight replicates.
4270 | www.pnas.org/cgi/doi/10.1073/pnas.1316569111 Zhu et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020
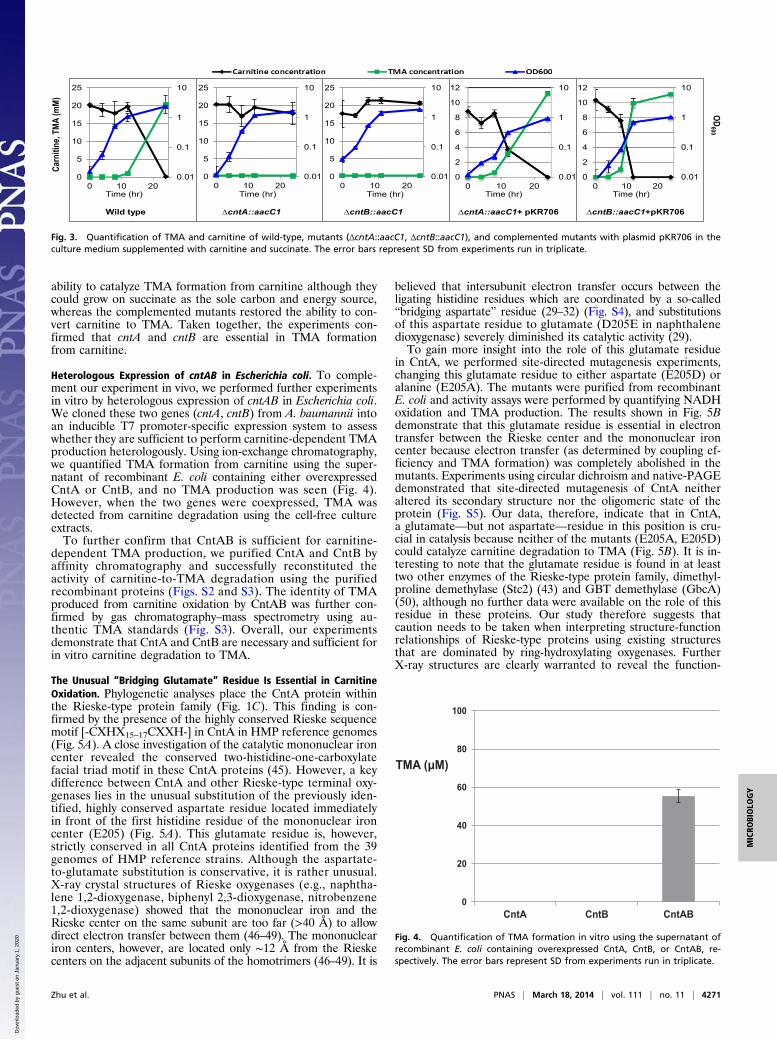
ability to catalyze TMA formation from carnitine although theycould grow on succinate as the sole carbon and energy source,whereas the complemented mutants restored the ability to con-vert carnitine to TMA. Taken together, the experiments con-firmed that cntA and cntB are essential in TMA formationfrom carnitine.
Heterologous Expression of cntAB in Escherichia coli. To comple-ment our experiment in vivo, we performed further experimentsin vitro by heterologous expression of cntAB in Escherichia coli.We cloned these two genes (cntA, cntB) from A. baumannii intoan inducible T7 promoter-specific expression system to assesswhether they are sufficient to perform carnitine-dependent TMAproduction heterologously. Using ion-exchange chromatography,we quantified TMA formation from carnitine using the super-natant of recombinant E. coli containing either overexpressedCntA or CntB, and no TMA production was seen (Fig. 4).However, when the two genes were coexpressed, TMA wasdetected from carnitine degradation using the cell-free cultureextracts.To further confirm that CntAB is sufficient for carnitine-
dependent TMA production, we purified CntA and CntB byaffinity chromatography and successfully reconstituted theactivity of carnitine-to-TMA degradation using the purifiedrecombinant proteins (Figs. S2 and S3). The identity of TMAproduced from carnitine oxidation by CntAB was further con-firmed by gas chromatography–mass spectrometry using au-thentic TMA standards (Fig. S3). Overall, our experimentsdemonstrate that CntA and CntB are necessary and sufficient forin vitro carnitine degradation to TMA.
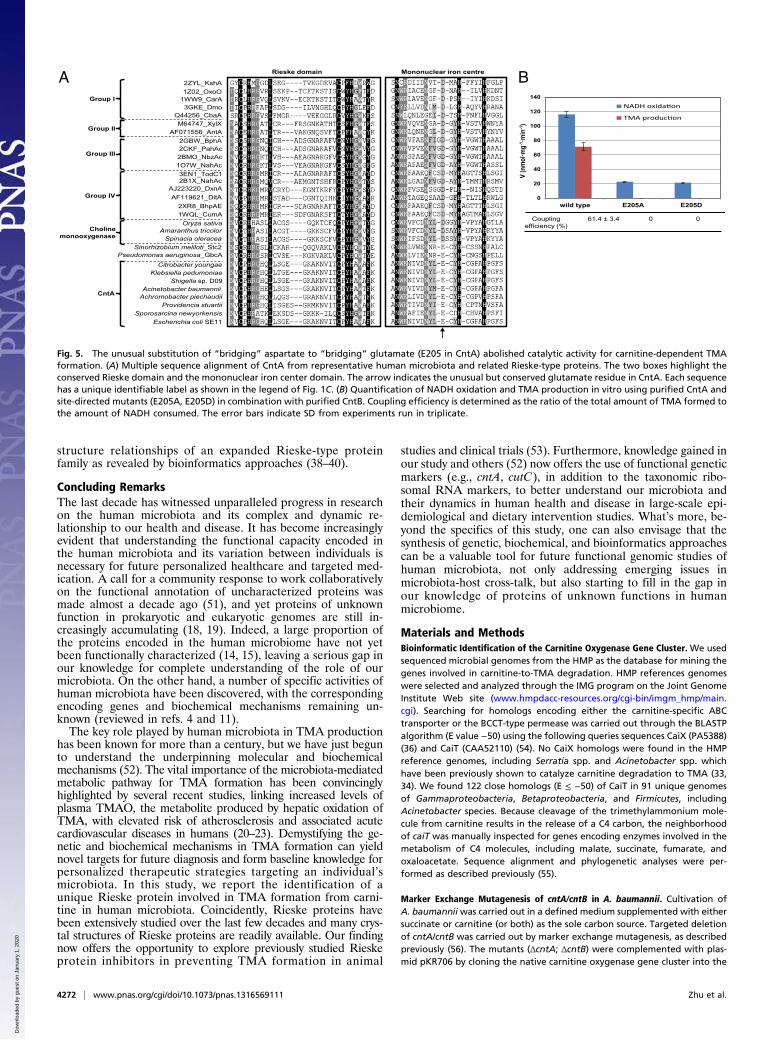
The Unusual “Bridging Glutamate” Residue Is Essential in CarnitineOxidation. Phylogenetic analyses place the CntA protein withinthe Rieske-type protein family (Fig. 1C). This finding is con-firmed by the presence of the highly conserved Rieske sequencemotif [-CXHX15–17CXXH-] in CntA in HMP reference genomes(Fig. 5A). A close investigation of the catalytic mononuclear ironcenter revealed the conserved two-histidine-one-carboxylatefacial triad motif in these CntA proteins (45). However, a keydifference between CntA and other Rieske-type terminal oxy-genases lies in the unusual substitution of the previously iden-tified, highly conserved aspartate residue located immediatelyin front of the first histidine residue of the mononuclear ironcenter (E205) (Fig. 5A). This glutamate residue is, however,strictly conserved in all CntA proteins identified from the 39genomes of HMP reference strains. Although the aspartate-to-glutamate substitution is conservative, it is rather unusual.X-ray crystal structures of Rieske oxygenases (e.g., naphtha-lene 1,2-dioxygenase, biphenyl 2,3-dioxygenase, nitrobenzene1,2-dioxygenase) showed that the mononuclear iron and theRieske center on the same subunit are too far (>40 Å) to allowdirect electron transfer between them (46–49). The mononucleariron centers, however, are located only ∼12 Å from the Rieskecenters on the adjacent subunits of the homotrimers (46–49). It is
believed that intersubunit electron transfer occurs between theligating histidine residues which are coordinated by a so-called“bridging aspartate” residue (29–32) (Fig. S4), and substitutionsof this aspartate residue to glutamate (D205E in naphthalenedioxygenase) severely diminished its catalytic activity (29).To gain more insight into the role of this glutamate residue
in CntA, we performed site-directed mutagenesis experiments,changing this glutamate residue to either aspartate (E205D) oralanine (E205A). The mutants were purified from recombinantE. coli and activity assays were performed by quantifying NADHoxidation and TMA production. The results shown in Fig. 5Bdemonstrate that this glutamate residue is essential in electrontransfer between the Rieske center and the mononuclear ironcenter because electron transfer (as determined by coupling ef-ficiency and TMA formation) was completely abolished in themutants. Experiments using circular dichroism and native-PAGEdemonstrated that site-directed mutagenesis of CntA neitheraltered its secondary structure nor the oligomeric state of theprotein (Fig. S5). Our data, therefore, indicate that in CntA,a glutamate—but not aspartate—residue in this position is cru-cial in catalysis because neither of the mutants (E205A, E205D)could catalyze carnitine degradation to TMA (Fig. 5B). It is in-teresting to note that the glutamate residue is found in at leasttwo other enzymes of the Rieske-type protein family, dimethyl-proline demethylase (Stc2) (43) and GBT demethylase (GbcA)(50), although no further data were available on the role of thisresidue in these proteins. Our study therefore suggests thatcaution needs to be taken when interpreting structure-functionrelationships of Rieske-type proteins using existing structuresthat are dominated by ring-hydroxylating oxygenases. FurtherX-ray structures are clearly warranted to reveal the function-
0.01
0.1
1
10
0
5
10
15
20
25
0 10 20
Wild type ∆cntA::aacC1 ∆cntB::aacC1 ∆cntA::aacC1+ pKR706 ∆cntB::aacC1+pKR706
0.01
0.1
1
10
0
5
10
15
20
25
0 10 200.01
0.1
1
10
0
5
10
15
20
25
0 10 20
Carn
itine
, TM
A (m
M)
OD600
Time (hr) Time (hr) Time (hr)
0.01
0.1
1
10
0
2
4
6
8
10
12
0 10 200.01
0.1
1
10
0
2
4
6
8
10
12
0 10 20Time (hr) Time (hr)
Fig. 3. Quantification of TMA and carnitine of wild-type, mutants (ΔcntA::aacC1, ΔcntB::aacC1), and complemented mutants with plasmid pKR706 in theculture medium supplemented with carnitine and succinate. The error bars represent SD from experiments run in triplicate.
0
20
40
60
80
100
CntA CntB CntAB
TMA (μM)
Fig. 4. Quantification of TMA formation in vitro using the supernatant ofrecombinant E. coli containing overexpressed CntA, CntB, or CntAB, re-spectively. The error bars represent SD from experiments run in triplicate.
Zhu et al. PNAS | March 18, 2014 | vol. 111 | no. 11 | 4271
MICRO
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020
structure relationships of an expanded Rieske-type proteinfamily as revealed by bioinformatics approaches (38–40).
Concluding RemarksThe last decade has witnessed unparalleled progress in researchon the human microbiota and its complex and dynamic re-lationship to our health and disease. It has become increasinglyevident that understanding the functional capacity encoded inthe human microbiota and its variation between individuals isnecessary for future personalized healthcare and targeted med-ication. A call for a community response to work collaborativelyon the functional annotation of uncharacterized proteins wasmade almost a decade ago (51), and yet proteins of unknownfunction in prokaryotic and eukaryotic genomes are still in-creasingly accumulating (18, 19). Indeed, a large proportion ofthe proteins encoded in the human microbiome have not yetbeen functionally characterized (14, 15), leaving a serious gap inour knowledge for complete understanding of the role of ourmicrobiota. On the other hand, a number of specific activities ofhuman microbiota have been discovered, with the correspondingencoding genes and biochemical mechanisms remaining un-known (reviewed in refs. 4 and 11).The key role played by human microbiota in TMA production
has been known for more than a century, but we have just begunto understand the underpinning molecular and biochemicalmechanisms (52). The vital importance of the microbiota-mediatedmetabolic pathway for TMA formation has been convincinglyhighlighted by several recent studies, linking increased levels ofplasma TMAO, the metabolite produced by hepatic oxidation ofTMA, with elevated risk of atherosclerosis and associated acutecardiovascular diseases in humans (20–23). Demystifying the ge-netic and biochemical mechanisms in TMA formation can yieldnovel targets for future diagnosis and form baseline knowledge forpersonalized therapeutic strategies targeting an individual’smicrobiota. In this study, we report the identification of aunique Rieske protein involved in TMA formation from carni-tine in human microbiota. Coincidently, Rieske proteins havebeen extensively studied over the last few decades and many crys-tal structures of Rieske proteins are readily available. Our findingnow offers the opportunity to explore previously studied Rieskeprotein inhibitors in preventing TMA formation in animal
studies and clinical trials (53). Furthermore, knowledge gained inour study and others (52) now offers the use of functional geneticmarkers (e.g., cntA, cutC), in addition to the taxonomic ribo-somal RNA markers, to better understand our microbiota andtheir dynamics in human health and disease in large-scale epi-demiological and dietary intervention studies. What’s more, be-yond the specifics of this study, one can also envisage that thesynthesis of genetic, biochemical, and bioinformatics approachescan be a valuable tool for future functional genomic studies ofhuman microbiota, not only addressing emerging issues inmicrobiota-host cross-talk, but also starting to fill in the gap inour knowledge of proteins of unknown functions in humanmicrobiome.
Materials and MethodsBioinformatic Identification of the Carnitine Oxygenase Gene Cluster. We usedsequenced microbial genomes from the HMP as the database for mining thegenes involved in carnitine-to-TMA degradation. HMP references genomeswere selected and analyzed through the IMG program on the Joint GenomeInstitute Web site (www.hmpdacc-resources.org/cgi-bin/imgm_hmp/main.cgi). Searching for homologs encoding either the carnitine-specific ABCtransporter or the BCCT-type permease was carried out through the BLASTPalgorithm (E value −50) using the following queries sequences CaiX (PA5388)(36) and CaiT (CAA52110) (54). No CaiX homologs were found in the HMPreference genomes, including Serratia spp. and Acinetobacter spp. whichhave been previously shown to catalyze carnitine degradation to TMA (33,34). We found 122 close homologs (E ≤ −50) of CaiT in 91 unique genomesof Gammaproteobacteria, Betaproteobacteria, and Firmicutes, includingAcinetobacter species. Because cleavage of the trimethylammonium mole-cule from carnitine results in the release of a C4 carbon, the neighborhoodof caiT was manually inspected for genes encoding enzymes involved in themetabolism of C4 molecules, including malate, succinate, fumarate, andoxaloacetate. Sequence alignment and phylogenetic analyses were per-formed as described previously (55).
Marker Exchange Mutagenesis of cntA/cntB in A. baumannii. Cultivation ofA. baumannii was carried out in a defined medium supplemented with eithersuccinate or carnitine (or both) as the sole carbon source. Targeted deletionof cntA/cntB was carried out by marker exchange mutagenesis, as describedpreviously (56). The mutants (ΔcntA; ΔcntB) were complemented with plas-mid pKR706 by cloning the native carnitine oxygenase gene cluster into the
2ZYL_KshA1Z02_OxoO
1WW9_CarA3GKE_Dmo
Q44256_CbaA
AF071556_AntAM64747_XylX
1O7W_NahAc2BMO_NbzAc2CKF_PahAc2GBW_BphA
3EN1_TodC1
1WQL_CumA2XR8_BhpAE
2B1X_NahAcAJ223220_DxnAAF119621_DitA
Spinacia oleraceaAmaranthus tricolor
Oryza sativa
Sinorhizobium meliloti_Stc2
Sporosarcina newyorkensisProvidencia stuartii
Achromobacter piechaudiiAcinetobacter baumannii
Klebsiella pedumoniaeCitrobacter youngae
Shigella sp. D09
Escherichia coli SE11
Group I
Group II
Group III
Group IV
Choline monooxygenase
CntA
Rieske domain Mononuclear iron centre
Pseudomonas aeruginosa_GbcA
Coupling efficiency (%)
61.4 ± 3.4 0 0
V (n
mol
·mg- 1
·min
-1)
0
20
40
60
80
100
120
140
wild type E205A E205D
NADH oxida on
TMA produc on
A B
Fig. 5. The unusual substitution of “bridging” aspartate to “bridging” glutamate (E205 in CntA) abolished catalytic activity for carnitine-dependent TMAformation. (A) Multiple sequence alignment of CntA from representative human microbiota and related Rieske-type proteins. The two boxes highlight theconserved Rieske domain and the mononuclear iron center domain. The arrow indicates the unusual but conserved glutamate residue in CntA. Each sequencehas a unique identifiable label as shown in the legend of Fig. 1C. (B) Quantification of NADH oxidation and TMA production in vitro using purified CntA andsite-directed mutants (E205A, E205D) in combination with purified CntB. Coupling efficiency is determined as the ratio of the total amount of TMA formed tothe amount of NADH consumed. The error bars indicate SD from experiments run in triplicate.
4272 | www.pnas.org/cgi/doi/10.1073/pnas.1316569111 Zhu et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020

vector pMQ300 (SI Materials and Methods). Bacterial strains, plasmids andprimers used in this study are listed in Tables S4 and S5.
Heterologous Overexpression of cntA/cntB, Site-Directed Mutagenesis andCharacterization of the CntA Mutants. The cntA gene was amplified from A.baumannii and inserted into the vector pET28a (Novagen). Coexpression ofcntA/cntB was achieved by insertion into pCOLADuet-1 under the BamHI/HindIII and the NdeI/KpnI sites, respectively. The CntA mutants (E205D,E205A) were chemically synthesized by GenScript and inserted into the ex-pression vector pET28a under the NdeI/HindIII sites. The resulting plasmidswere then transformed into the expression host E. coli BLR(DE3) pLysS(Merck Biosciences). Cultivation of recombinant E. coli, protein induction byisopropyl β-D-1-thiogalactopyranoside and further purification using His-tagaffinity chromatography are detailed in SI Materials and Methods. Enzymeassays were performed at room temperature (∼22 °C) by quantifying NADH
oxidation and carnitine-dependent TMA production. A 1-mL enzyme assaymixture contained 10 mM Hepes buffer (pH 7.6), 60 μg purified CntA andCntB, respectively, 0.25 mM carnitine, and 0.25 mM NADH. Coupling effi-ciency was determined as the ratio of the total amount of TMA formed tothe amount of NADH consumed.
Analytical Methods. Carnitine and TMA were quantified by a cation-exchange ion chromatography (Metrohm 881 Compact IC Pro) equippedwith a Metrosep C4/250 mm separation column and a conductivity detector(Metrohm). NADH oxidation was quantified using a Shimadzu UV-VIS 1800spectrophotometer by following decrease of absorbance at 340 nm (e =6.2 mM−1·cm−1).
ACKNOWLEDGMENTS. This work is supported by the Royal Society (RG2011/R1) and partially by the Natural Environment Research Council (NE/I027061/1).
1. Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: Human gut mi-crobes associated with obesity. Nature 444(7122):1022–1023.
2. Turnbaugh PJ, et al. (2006) An obesity-associated gut microbiome with increasedcapacity for energy harvest. Nature 444(7122):1027–1031.
3. Gill SR, et al. (2006) Metagenomic analysis of the human distal gut microbiome. Sci-ence 312(5778):1355–1359.
4. Nicholson JK, et al. (2012) Host-gut microbiota metabolic interactions. Science336(6086):1262–1267.
5. Koren O, et al. (2011) Human oral, gut, and plaque microbiota in patients with ath-erosclerosis. Proc Natl Acad Sci USA 108(Suppl 1):4592–4598.
6. Wu GD, et al. (2011) Linking long-term dietary patterns with gut microbial enter-otypes. Science 334(6052):105–108.
7. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012) Diversity, stabilityand resilience of the human gut microbiota. Nature 489(7415):220–230.
8. Tremaroli V, Bäckhed F (2012) Functional interactions between the gut microbiotaand host metabolism. Nature 489(7415):242–249.
9. Weinstock GM (2012) Genomic approaches to studying the human microbiota. Nature489(7415):250–256.
10. Spor A, Koren O, Ley R (2011) Unravelling the effects of the environment and hostgenotype on the gut microbiome. Nat Rev Microbiol 9(4):279–290.
11. Marchesi JR (2011) Human distal gut microbiome. Environ Microbiol 13(12):3088–3102.12. Arumugam M, et al.; MetaHIT Consortium (2011) Enterotypes of the human gut
microbiome. Nature 473(7346):174–180.13. Yatsunenko T, et al. (2012) Human gut microbiome viewed across age and geogra-
phy. Nature 486(7402):222–227.14. Qin J, et al. (2012) A metagenome-wide association study of gut microbiota in type 2
diabetes. Nature 490(7418):55–60.15. HumanMicrobiome Project Consortium (2012) Structure, function and diversity of the
healthy human microbiome. Nature 486(7402):207–214.16. Proctor LM (2011) The human microbiome project in 2011 and beyond. Cell Host
Microbe 10(4):287–291.17. Morgan XC, Segata N, Huttenhower C (2013) Biodiversity and functional genomics in
the human microbiome. Trends Genet 29(1):51–58.18. Galperin MY, Koonin EV (2010) From complete genome sequence to ‘complete’ un-
derstanding? Trends Biotechnol 28(8):398–406.19. Punta M, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40(Da-
tabase issue):D290–D301.20. Koeth RA, et al. (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in
red meat, promotes atherosclerosis. Nat Med 19(5):576–585.21. Bennett BJ, et al. (2013) Trimethylamine-N-oxide, a metabolite associated with ath-
erosclerosis, exhibits complex genetic and dietary regulation. Cell Metab 17(1):49–60.22. Wang Z, et al. (2011) Gut flora metabolism of phosphatidylcholine promotes car-
diovascular disease. Nature 472(7341):57–63.23. Tang WH, et al. (2013) Intestinal microbial metabolism of phosphatidylcholine and
cardiovascular risk. N Engl J Med 368(17):1575–1584.24. Friedman S, Fraenkel G (1955) Reversible enzymatic acetylation of carnitine. Arch
Biochem Biophys 59(2):491–501.25. Fritz IB, McEwen B (1959) Effects of carnitine on fatty-acid oxidation by muscle. Sci-
ence 129(3345):334–335.26. Rebouche CJ (1992) Carnitine function and requirements during the life cycle. FASEB J
6(15):3379–3386.27. Rebouche CJ, Chenard CA (1991) Metabolic fate of dietary carnitine in human adults:
Identification and quantification of urinary and fecal metabolites. J Nutr 121(4):539–546.28. Ferraro DJ, Gakhar L, Ramaswamy S (2005) Rieske business: Structure-function of
Rieske non-heme oxygenases. Biochem Biophys Res Commun 338(1):175–190.29. Parales RE, Parales JV, Gibson DT (1999) Aspartate 205 in the catalytic domain of
naphthalene dioxygenase is essential for activity. J Bacteriol 181(6):1831–1837.30. Pinto A, Tarasev M, Ballou DP (2006) Substitutions of the “bridging” aspartate 178
result in profound changes in the reactivity of the Rieske center of phthalate diox-ygenase. Biochemistry 45(30):9032–9041.
31. Jiang H, Parales RE, Lynch NA, Gibson DT (1996) Site-directed mutagenesis of con-served amino acids in the alpha subunit of toluene dioxygenase: potential mono-nuclear non-heme iron coordination sites. J Bacteriol 178(11):3133–3139.
32. Beharry ZM, et al. (2003) Histidine ligand protonation and redox potential in therieske dioxygenases: Role of a conserved aspartate in anthranilate 1,2-dioxygenase.Biochemistry 42(46):13625–13636.
33. Seim H, et al. (1982) Splitting of the C-N bond in carnitine by an enzyme (trime-
thylamine forming) from membranes of Acinetobacter calcoaceticus. FEMS Microbiol
Lett 15(3):165–167.34. Unemoto T, Hayashi M, Miyaki K, Hayashi M (1966) Formation of trimethylamine
from DL-carnitine by Serratia marcescens. Biochim Biophys Acta 121(1):220–222.35. Ziegler C, Bremer E, Krämer R (2010) The BCCT family of carriers: From physiology to
crystal structure. Mol Microbiol 78(1):13–34.36. Chen C, Malek AA, Wargo MJ, Hogan DA, Beattie GA (2010) The ATP-binding cassette
transporter Cbc (choline/betaine/carnitine) recruits multiple substrate-binding pro-
teins with strong specificity for distinct quaternary ammonium compounds. Mol Mi-
crobiol 75(1):29–45.37. Meadows JA, Wargo MJ (2013) Characterization of Pseudomonas aeruginosa growth
on O-acylcarnitines and identification of a short-chain acylcarnitine hydrolase. Appl
Environ Microbiol 79(11):3355–3363.38. Capyk JK, Eltis LD (2012) Phylogenetic analysis reveals the surprising diversity of an
oxygenase class. J Biol Inorg Chem 17(3):425–436.39. Schmidt CL, Shaw L (2001) A comprehensive phylogenetic analysis of Rieske and
Rieske-type iron-sulfur proteins. J Bioenerg Biomembr 33(1):9–26.40. Kweon O, et al. (2008) A new classification system for bacterial Rieske non-heme iron
aromatic ring-hydroxylating oxygenases. BMC Biochem 9:11.41. Rathinasabapathi B, et al. (1997) Choline monooxygenase, an unusual iron-sulfur
enzyme catalyzing the first step of glycine betaine synthesis in plants: Prosthetic
group characterization and cDNA cloning. Proc Natl Acad Sci USA 94(7):3454–3458.42. Mitsuya S, et al. (2011) Isolation and characterization of a novel peroxisomal choline
monooxygenase in barley. Planta 234(6):1215–1226.43. Daughtry KD, et al. (2012) Quaternary ammonium oxidative demethylation: X-ray
crystallographic, resonance Raman, and UV-visible spectroscopic analysis of a Rieske-
type demethylase. J Am Chem Soc 134(5):2823–2834.44. Sydor PK, et al. (2011) Regio- and stereodivergent antibiotic oxidative carbocycliza-
tions catalysed by Rieske oxygenase-like enzymes. Nat Chem 3(5):388–392.45. Hegg EL, Que L, Jr. (1997) The 2-His-1-carboxylate facial triad—An emerging struc-
tural motif in mononuclear non-heme iron(II) enzymes. Eur J Biochem 250(3):625–629.46. Karlsson A, et al. (2003) Crystal structure of naphthalene dioxygenase: Side-on
binding of dioxygen to iron. Science 299(5609):1039–1042.47. Ferraro DJ, et al. (2007) Structural investigations of the ferredoxin and terminal
oxygenase components of the biphenyl 2,3-dioxygenase from Sphingobium yanoi-
kuyae B1. BMC Struct Biol 7:10.48. Friemann R, et al. (2005) Structural insight into the dioxygenation of nitroarene com-
pounds: The crystal structure of nitrobenzene dioxygenase. J Mol Biol 348(5):1139–1151.49. Martins BM, Svetlitchnaia T, Dobbek H (2005) 2-Oxoquinoline 8-monooxygenase
oxygenase component: Active site modulation by Rieske-[2Fe-2S] center oxidation/
reduction. Structure 13(5):817–824.50. Wargo MJ, Szwergold BS, Hogan DA (2008) Identification of two gene clusters and
a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine
catabolism. J Bacteriol 190(8):2690–2699.51. Roberts RJ (2004) Identifying protein function—A call for community action. PLoS Biol
2(3):E42.52. Craciun S, Balskus EP (2012) Microbial conversion of choline to trimethylamine re-
quires a glycyl radical enzyme. Proc Natl Acad Sci USA 109(52):21307–21312.53. Link TA, Haase U, Brandt U, von Jagow G (1993) What information do inhibitors
provide about the structure of the hydroquinone oxidation site of ubihydroquinone:
Cytochrome c oxidoreductase? J Bioenerg Biomembr 25(3):221–232.54. Eichler K, Bourgis F, Buchet A, Kleber HP, Mandrand-Berthelot MA (1994) Molecular
characterization of the cai operon necessary for carnitine metabolism in Escherichia
coli. Mol Microbiol 13(5):775–786.55. Chen Y, Patel NA, Crombie A, Scrivens JH, Murrell JC (2011) Bacterial flavin-containing
monooxygenase is trimethylamine monooxygenase. Proc Natl Acad Sci USA 108(43):
17791–17796.56. Kochar M, et al. (2012) Deletion of TnAbaR23 results in both expected and un-
expected antibiogram changes in a multidrug-resistant Acinetobacter baumannii
strain. Antimicrob Agents Chemother 56(4):1845–1853.57. Nam JW, et al. (2001) New classification system for oxygenase components involved in
ring-hydroxylating oxygenations. Biosci Biotechnol Biochem 65(2):254–263.
Zhu et al. PNAS | March 18, 2014 | vol. 111 | no. 11 | 4273
MICRO
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Janu
ary
1, 2
020


















