
Cardiac Troponins in Patients with Suspected or …170456/FULLTEXT01.pdf · Cardiac Troponins in...
74
ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2007 Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 269 Cardiac Troponins in Patients with Suspected or Confirmed Acute Coronary Syndrome New Applications for Biomarkers in Coronary Artery Disease KAI EGGERS ISSN 1651-6206 ISBN 978-91-554-6924-5 urn:nbn:se:uu:diva-7945
-
Upload
duongkhanh -
Category
Documents
-
view
215 -
download
0
Transcript of Cardiac Troponins in Patients with Suspected or …170456/FULLTEXT01.pdf · Cardiac Troponins in...

ACTAUNIVERSITATISUPSALIENSISUPPSALA2007
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 269
Cardiac Troponins in Patients withSuspected or Confirmed AcuteCoronary Syndrome
New Applications for Biomarkers in Coronary ArteryDisease
KAI EGGERS
ISSN 1651-6206ISBN 978-91-554-6924-5urn:nbn:se:uu:diva-7945


LIST OF PAPERS
I Eggers KM, Oldgren J, Nordenskjöld A, Lindahl B. Diagnostic value of serial measurement of cardiac markers in patients with chest pain: Limited value of adding myoglobin to troponin I for exclusion of myocardial infarction. Am Heart J 2004; 148: 574-81.
II Eggers KM, Ellenius J, Dellborg M, Groth T, Oldgren J, Swahn E, Lindahl B.Artificial neural network algorithms for early diagnosis of acute myocardial infarction and prediction of infarct size in chest pain patients.Int J Cardiol 2007; 114: 366-74.
III Eggers KM, Oldgren J, Nordenskjöld A, Lindahl B. Combining different biochemical markers of myocardial ischemia does not improve risk stratification compared to troponin I alone. Coron Artery Dis 2005; 16: 315-19.
IV Eggers KM, Dellborg M, Oldgren J, Swahn E, Venge P, Lindahl B. Early risk prediction in patients with chest pain by biochemical markers including estimates of renal function. Accepted for publication in Int J Cardiol 2007.
V Eggers KM, Lagerqvist B, Venge P, Wallentin L, Lindahl B. Persistent cardiac troponin I elevation in stabilized patients after an episode of acute coronary syndrome predicts adverse long-term outcome. Submitted.
VI Eggers KM, Lagerqvist B, Venge P, Wallentin L, Lindahl B. Pathophysiologic mechanisms and prognostic value of persistent cardiac troponin I elevation in stabilized patients after an episode of acute coronary syndrome. Manuscript.
Reprints were made with permissions from the publishers.


CONTENTS
INTRODUCTION ..........................................................................................9
BACKGROUND ..........................................................................................11The molecular biology of cardiac troponin ..............................................11Analytical issues.......................................................................................13Diagnosis of acute myocardial infarction in patients with chest pain ......14Biochemical markers for risk assessment ................................................17
AIMS ............................................................................................................22
MATERIAL AND METHODS....................................................................23Patients .....................................................................................................23Study protocols.........................................................................................25Biochemical analysis................................................................................27Description of the applied ANN-algorithms ............................................28Other examinations ..................................................................................29Definition of the index diagnosis .............................................................30Follow-up and endpoints ..........................................................................31Statistical analysis ....................................................................................32
RESULTS .....................................................................................................35Paper I ......................................................................................................35Paper II .....................................................................................................38Paper III....................................................................................................40Paper IV ...................................................................................................42Papers V and VI .......................................................................................44
DISCUSSION...............................................................................................50Cardiac troponin I for early assessment of patients with suspectedacute coronary syndrome .........................................................................51
Early diagnosis of acute myocardial infarction (papers I and II).........51Early risk prediction (papers III and IV) .............................................52Early prediction of infarct size (paper II) ............................................53

Cardiac troponin I in patients stabilized after an episode of acute coronary syndrome .........................................................................54
Prevalence of cardiac troponin I elevation (paper V) ..........................54Possible causes of cardiac troponin I elevation (papers V and VI) .....54Prognostic importance of cardiac troponin I elevation (papers V and VI) ................................................................................55
Clinical implications and future directions ..............................................56
CONCLUSIONS ..........................................................................................59
SUMMARY IN SWEDISH (SAMMANFATTNING PÅ SVENSKA).......60
ACKNOWLEDGEMENTS..........................................................................62
REFERENCES .............................................................................................64

ABBREVIATIONS
ACC American College of Cardiology ACS Acute coronary syndrome AMI Acute myocardial infarction ANN Artificial neural network CCU Coronary care unit CK Creatine kinase CRP C-reactive protein cTnI Cardiac troponin I cTnT Cardiac troponin T CV Coefficient of variation ESC European Society of Cardiology FAST Fast Assessment of Thoracic Pain FASTER Fast Assessment of Thoracic Pain by nEuRal networks FRISC FRagmin and Fast Revascularization during InStability in
Coronary artery disease GFR Glomerular filtration rate LV-EF Left ventricular ejection fraction NPV Negative predictive value NT-pro BNP N-terminal pro-brain natriuretic peptide POC Point of care PPV Positive predictive value ROC Receiver operator characteristics TIMI Thrombolysis In Myocardial Infarction WHO World Health Organization

8

9
INTRODUCTION
Ischemic heart disease is a major cause of mortality worldwide. In almost all cases it begins with atherosclerotic plaque development in the coronary vessels and clinically manifests with the effects of limited coronary flow. In case of a sudden plaque rupture, an acute coronary syndrome (ACS) occurs with exposure of highly procoagulant contents of the plaque to circulating platelets and coagulatory factors. This results in the formation of a thrombus at the site of rupture which, when occlusive, causes a transmural myocardial infarction in the territory supplied by the artery accompanied by ST-segment elevation on the ECG. However, in the majority of cases, the thrombus is only partially occlusive. In this situation, distal ischemia or necrosis is caused by microembolization of thrombotic particles and is often restricted to the subendocardial layers of the myocardium.
In case of myocardial necrosis, intracellular macromolecules are released from the cardiomyocytes into the blood stream. Among these, the troponins which form part of the myocardial contractile apparatus are entirely specific for cardiac damage. The troponins are therefore regarded as markers of choice for the diagnosis of acute myocardial infarction (AMI) and prognostic assessment of patients with suspected or ongoing ACS according to guide-lines for the management of non ST-segment ACS published by the Euro-pean Society of Cardiology (ESC) and the American College of Cardiology (ACC) in 2000 1 .
However, during the recent years, the analytical qualities of assays for troponin measurement have been constantly improved. This has enabled the reliable detection of quantitatively small but clinically relevant troponin elevations in patients who otherwise would have been regarded as troponin-negative 2 . In addition, the increased emphasis on rapid troponin testing has a lead to the implementation of patient-near point of care (POC)-systems which, when integrated to computerized systems, could offer early and con-venient information on troponin results together with clinical decision sup-port. Finally, troponin results need to be considered in conjunction to novel markers of cardiovascular risk as multimarker approaches might improve prognostication. Taken together, these issues emphasize the need for (re)-evaluation of the role of troponin measurements using new technologies and in the context of other established and emerging biochemical markers.

10
This is particularly important in the large group of patients presenting with chest pain suggestive of an ACS but also in patients who have been stabi-lized after an episode of ACS. In chest pain patients for example, an acceler-ated troponin sampling protocol using a sensitive assay on a POC instrument might allow the early selection of troponin-positive patients to appropriate levels of health care while in troponin-negative patients found to be at high risk, other tests to establish a definite diagnosis could be initiated early. Tro-ponin-negative patients at low risk also need early identification for rapid transfer to stepdown units or early discharge. In stable patients who have had an ACS, high-sensitive assays could detect ongoing troponin leakage from the injured myocardium. In analogy to the adverse prognosis associated with troponin positivity in other conditions than ACS 3-6 , elevated troponin levels in post-ACS patients might indicate a high risk for cardiovascular events and thus, identify subjects in whom further cautious investigation would be mandatory.

11
BACKGROUND
The molecular biology of cardiac troponin The contractile apparatus of striated muscle consists of myofibrils as its basic building unit. Myofibrils are organized by myosin-based thick fila-ments and thin filaments which consist of two strands of actin. The contrac-tion of the myofibrils occurs by interaction of the thick and thin filaments. This process is regulated by tropomyosin, a protein located on the thin fila-ment which blocks the active sites of actin, thereby preventing myosin from binding to it.
The troponin complexes are bound at regular intervals to the tropomyosin molecule and consist of three subunits: Troponin T, a binding protein, that attaches the troponin complex to tropomyosin, troponin I which binds to actin and modulates the interaction of actin and myosin, and troponin C which binds to calcium, thereby causing a change of the confirmation of tropomyosin which mediates muscle contraction 7 . Troponin T and I have three isoforms each: one is specific to myocardial tissue (cardiac troponin T cTnT and cardiac troponin I cTnI ) whereas the two others are found in
the skeletal muscle1. Apart from the troponin bound to the myofibrils in the cardiomyocytes (‘structural pool’), a small amount of structurally unbound troponin also exists in the cytosol (‘cytosolic pool’; 5% for cTnT, 3.7% for cTnI) 8, 9 . After cardiomyocyte necrosis, the troponins are released into the circulation as free proteins or complexed forms, initially due to leakage from the cytosolic pool after loss of cell membrane integrity and thereafter due to an ongoing degradation of the damaged myofibrils (figure 1). Once released to the blood stream, cTnI and, to a lesser extent, cTnT are suscepti-ble to various biochemical modifications including phosphorylation, oxida-tion and proteolysis.
Even though troponin elevation indicates cardiac damage it does not define its nature. Apart from thrombotic coronary events, troponin elevation may also occur in case of increased myocardial oxygen demand (e.g. tachyar-rhythmia, hypertensive crisis), decreased oxygen supply (e.g. hypotension), increased myocardial wall tension because of volume or pressure overload (e.g. heart failure) or disturbance of cardiomyocyte cell integrity (e.g. sepsis)
1 If not stated otherwise, the term troponin used in this work refers to cardiac troponin.

12
Figure 1. Schematic representation of the structure of the myofibrillar thin filament and the distribution of troponins in the cardiac myocyte.
Reprinted with permission from reference 11.
Table 1. Causes and mechanisms of troponin elevation.
Diagnosis Mechanism
AMI Coronary intervention (PCI, CABG)
Thrombotic/embolic coronary occlusion
Coronary vasospasm Increased sympathetic activity (cocaine, catecholamine storm)
Coronary vasoconstriction
Left ventricular hypertrophy Hypertensive crisis Prolonged tachyarrhythmia Sepsis
Increased oxygen demand
Hypotension Aortic dissection
Decreased oxygen supply
Congestive heart failure Pulmonary embolism Pulmonary hypertension Strenuous exercise
Myocardial strain
Cardiac trauma Direct current cardioversion Infiltrative diseases (amyloidosis, sarcoidosis) Myocarditis SepsisDrugs (chemotherapy, alcohol)
Direct myocardial damage
Adapted from references 10-12.

13
10-12 . An overview of the possible causes for troponins elevation is given in table 1. Troponin elevation has also been reported in patients with renal failure 13 although the causes are not completely understood. Current hy-potheses include clinically silent coronary microinfarctions 14 , myocardial stretch due to volume overload 15 and an association to inflammation and oxidative stress 16, 17 .
Analytical issues Troponin is measured with two-site ‘sandwich’ immunoassays with a cap-ture antibody to bind it and a detection antibody for the determination of the quantity of bound troponin. As the reactivity between the antibodies and the targeted troponin epitopes determines the performance of the immunoassays, antibodies directed against the stable part of the troponin molecule and not affected by modifications, i.e. degradation processes after release to the bloodstream or occurring in vitro, should be preferred [18]. This is particu-larly important for cTnI owing to the multitude of different degradation products detectable after release to the blood.
Several critical issues concerning measurement of troponin deserve com-ment. As any detectable troponin level is indicative of adverse outcome 19 ,assays should provide a high analytical precision at the low end of their range. This will result in the reliable identification of a higher proportion of patients at risk 2 . However, the vast majority of commercially available troponin assays perform with insufficient precision at the low decision levels recommended by current guidelines 20 . Another problem is the presence of heterophilic antibodies which by binding to the antitroponin antibodies may generate a false-positive signal 21 . Even autoantibodies to the central part of the cTnI molecule exist which can cause negative test results for some cTnI immunoassays 22 . A third important issue is the need for standardiza-tion of the different available troponin assays. Due to patent restrictions, there is only one commercially available cTnT assay. For cTnI in contrast, a number of assays exists with different normal ranges, detection limits and medical decision cut-offs owing to the utilization of antitroponin antibodies directed against different cTnI subunit complexes. Results from one cTnI assay can thus, not be extrapolated to another. Therefore, clinicians need to understand which assay is being used in their centre in order to be able to interpret the results correctly.

14
Diagnosis of AMI in patients with chest pain The cardiac troponins are the primary markers of choice for the assessment of patients with suspected ACS 23 . Chest pain suggestive of an ACS is the cause of > 8 million emergency department visits per year in the U.S. 24and of 150 000 annual emergency department visits in Sweden 25 . The complaints of chest discomfort encompasses many varying conditions, rang-ing from insignificant to high-risk, e.g. ACS, pulmonary embolism or aortic dissection. The overall objective of early evaluation of this large and hetero-geneous patient population is to target high-risk patients to appropriate levels and types of health care and to timely start-up treatments aiming at reducing morbidity and mortality. Patients found to be at low risk also need identifica-tion for rapid transfer to non-intensive care units or early discharge.
The electrocardiogram The standard 12-lead ECG is a cornerstone of the initial evaluation of pa-tients with suspected ACS. In case of localized ST-segment elevation, the diagnosis of a transmural AMI is immediately established. However, the diagnostic and prognostic accuracy of the ECG in patients with non ST seg-ment-elevation ACS is limited because of several issues. ECG changes in-dicative of subendocardial ischemia, i.e. ST-segment depression or T-wave inversion may also occur in other conditions. Confounding ECG patterns such as pacing or bundle branch block can obscure ischemic ECG changes. Furthermore, a single ECG is only a momentary sample and therefore unable to mirror dynamic coronary blood flow variations occurring in ACS. Thus, the ECG often is non-diagnostic in unselected chest pain populations 26which is why biochemical markers are needed to establish the diagnosis of AMI in the vast majority of the patients.
Biochemical markers for the diagnosis of AMI
Cardiac troponin I and T cTnI and cTnT were first recognized as markers of myocardial injury in the late 1970s and 1980s 27, 28 . After the implementation of the first-generation cTnT assays, it soon became clear that unstable angina patients with normal CK-MB but elevated troponin levels had an increased cardiac risk 29, 30 . The facts that troponin is cardiospecific and that any detectable troponin level is associated with adverse outcome 19 were anticipated by the ESC/ACC joint committee in 2000, recommending cardiac troponin re-sults with a significant rise and fall in an appropriate clinical context as marker of choice for the diagnosis of AMI. For clinical decision making, a cut-off corresponding to the 99th percentile in healthy reference populations

15
has been proposed 1 and re-emphasized in recently published guidelines from the National Academy of Clinical Biochemistry 31 . However, as most troponin assays do not perform with an acceptable analytic precision at these low concentrations, the use of the lowest concentration measurable with a < 10% coefficient of variation (10% CV) has been suggested as alter-native decision level 20 .
Using conventional cut-offs, troponin levels rise 4-8 hours after an ischemic event, reach a peak after 12-36 hours and remain elevated for 3-10 days. However, with sensitive assays and low detection limits, small amounts of troponin released from the cytosolic pool may be detected ear-lier. Despite some differences in biochemical and analytical characteristics, there is no convincing evidence that one troponin is superior to the other. Assay related issues such as analytical sensitivity and imprecision appear to determine the information obtained from troponin results to a greater extent than the chosen troponin.
Creatine kinase-MB Creatine kinase (CK) is an enzyme present in the cytoplasma of muscle cells, regulating the production of intracellular adenosine triphosphate in order to meet increased energy demands. Measurements of total CK for the detection of AMI was proposed in the early 1960s 32 . In the myocardium, the MB isoenzyme of CK accounts for ~ 20% of the total CK content. With regard to its higher cardiospecificity, CK-MB has been used as diagnostic marker for AMI for many years, often implemented into diagnostic criteria established by the World Health Organization (WHO) including typical symptoms, ischemic ECG changes and elevated biomarkers 33 . In AMI, CK-MB rises after 3-6 hours, peaks after 12-20 hours and returns to normal after 48-72 hours. However, the use of CK-MB for the diagnosis of AMI is confounded by its occurrence in skeletal muscle, resulting in a constant se-rum level which increases in skeletal muscle disorders. Elevation of CK-MB also occurs in renal failure. To overcome these drawbacks, the use of the relative index relying on the percentage of CK-MB with respect to total CK has been proposed. In addition, the implementation of immunoassays allow-ing the determination of CK-MB concentration (CK-MB mass) instead of its catalytic activity has substantially improved the diagnostic utility of CK-MB.
Myoglobin Myoglobin is a small heme binding protein present in both cardiac and skeletal muscle 34 . Due to its small molecular weight and presence in the cytoplasma, it is rapidly released in case of cell membrane disturbance. My-oglobin levels increase within 2 hours after onset of myocardial necrosis, peak in 6-9 hours and return to normal after 24-36 hours. However, myoglo-

16
bin has shortcomings as its sensitivity for AMI is low in patients presenting late while its specificity is insufficient due to elevations occurring in skeletal muscle damage or renal failure. For cardiac low-risk patients with a low incidence of ACS, the frequency of false positive myoglobin results often exceeds that of the true positives, thereby substantially limiting its diagnostic value 35 . Thus, myoglobin has been regarded most useful as a non-specific screening marker in patients presenting very early or for the detection of re-infarction in patients with a recent AMI who still have elevated troponin levels.
Combinations of different biochemical markers Because biochemical cardiac markers have different characteristics and each potentially offers certain advantages, a logical approach would be to use some of them in combination. This has been extensively assessed in a large number of studies 36 . In particular algorithms based on early results for troponin and myoglobin have been recommended for rapid rule-out of AMI protocols 37, 38 .
Point of care testing for cardiac markers Testing of biochemical markers of myocardial damage is usually a time-consuming procedure due to infrequent analyses at the central laboratory and transport-related delays. Even though guidelines have recommended that marker results should be available within 30 minutes 39 , this goal at pre-sent is not met in a large proportion of hospitals 40 . In emergency depart-ments overwhelmed with patients, clinicians therefore might tend to admit chest pain patients who basically are at low risk for an ACS. The increased emphasis on rapid and reliable cardiac marker testing has led to the imple-mentation of POC systems for measurement of troponin. Several studies have demonstrated that, by achieving a shorter turnaround time, POC testing allows faster triaging of patients, thereby improving hospital resource use and providing economic savings 41-43 . It therefore might be suspected that accelerated and serial biochemical marker monitoring using a POC device could furthermore improve clinical decision-making due to an increased speed at which results are available.
Decision tools The early assessment of chest pain patients may be facilitated by the use of advanced statistical techniques for the calculation of the probability of an ACS. Prediction tools such as the Goldman Chest Pain Protocol 44 or the ACI-TIPI (Acute Coronary Ischemia Time-Insensitive Predictive Instru-ment) 45 applying clinical and ECG data have been widely assessed and have demonstrated clinical usefulness. However, a major drawback of these models is that none of them applies biochemical marker results which in part

17
may depend on the unavailability of POC testing systems at the time of model development.
Another option for early assessment of patients with suspected ACS is the use of artificial neural networks (ANN), sophisticated computer algorithms which are able to recognize complex patterns in measured clinical parame-ters (‘input variables’) not apparent to other forms of analyses 46 . ANN have been successfully applied to a broad range of biomedical problems 47and previous studies have demonstrated that ANN approaches can identify AMI accurately 48-52 . However, in most of these studies, a large variety of input variables has been used which is practically demanding in a busy unit and therefore might limit the acceptance of an ANN. Further limitations of previous studies are the restriction to ECG findings on admission 48, 51, 52 and inappropriate standards for the diagnosis of AMI because of the use of WHO criteria 48-51 or historic markers such as CK and lactate dehy-drogenase 49 .
Biochemical markers for risk assessment in patients with suspected or confirmed ACS
The purpose of early evaluation of patients with suspected or confirmed ACS by troponin measurements is not only to detect myocardial necrosis but more importantly, to identify patients who are at high risk for future cardiac events. Of note, prognostication depends on what risk one aims to predict and when. In the emergency department setting, the greatest challenge is to identify lower-risk patients without ACS who can be transferred to non-intensive care units or discharged to their home with confidence, thus avoid-ing the risks and costs of unnecessary procedures and therapies. In patients with confirmed ACS in contrast, the rationale of risk stratification is to iden-tify patients who are most likely to benefit from specific therapeutic inter-ventions aimed at reducing mortality, e.g. medical treatment with glycopro-tein IIb/IIIa inhibitors, low-molecular weight heparin or an early invasive approach. In the late phase after an ACS, prognostic assessment should focus on the detection of left ventricular dysfunction and ventricular arrhythmias, gradually becoming the most important contributors to poor outcome 53 ,and the modification of cardiovascular risk factors in order to halt the pro-gression of atherosclerosis. Taken together, these issues illustrate that model-ing cardiac risk is a complex issue.
The initial risk prediction in patients with ACS has to be based on the lim-ited information which is available at the time of evaluation, often only the patients history, clinical findings and ECG data. Biomarker results improve

18
prognostication and are clinically advantageous as they often are rapidly available at low costs, can be measured serially, thereby demonstrating dy-namic changes in disease state and offer insights in different components of the pathophysiology of ACS when used as a panel. Not surprisingly, scien-tific work has focused much on biochemical markers for the improvement of prognostic strategies applicable in the various phases of an ACS, from its early manifestations to its end-stage. Apart from the troponins, also markers of hemodynamic stress, inflammation and renal dysfunction have gained much attention and will be treated shortly in the following paragraphs.
Markers of myocardial necrosis According to current guidelines, the troponins are regarded as the bio-chemical markers of choice for prognostication in patients with ACS 23 .With increasing troponin levels there is a significant gradient of risk 19 as the amount of troponin leakage correlates with more severe coronary culprit lesions 54, 55 , impaired myocardial tissue perfusion 56 and a larger in-farct size 57-61]. Accordingly, a series of studies has confirmed the power-ful role of troponin testing for the identification of chest pain patients at high risk 29, 62 and patients with non-ST segment elevation ACS who are likely to benefit from intensive therapies 63-65 . Even CK-MB and myoglobin have been suggested as markers of risk 66-68 and some authors have advo-cated their use along with cardiac troponin results for improving early risk stratification 69, 70 . However, as both CK-MB and myoglobin lack cardio-specificity, elevated levels in critically ill patients may reflect other prognos-tically adverse mechanisms than myocardial necrosis, thereby limiting their prognostic value regarding cardiac events.
Up until now, it has not been clear whether troponin testing has a role for prognostication in the stable phase after an ACS. According to the current concept, cardiac troponins should not be detectable in subjects without evi-dence of ongoing coronary ischemia. However, considering the high preva-lence of troponin elevation in patients with other pathologies than ACS 10,11 , it might be suspected that troponins also are detectable with the use of highly sensitive assays in stable post-ACS patients. Thus, troponin testing in these patients might allow the identification of high-risk subjects in whom further investigation would be mandatory.
The natriuretic peptides Brain natriuretic peptide (BNP) and the N-terminal fragment of its pro-hormone, NT-pro BNP, are elevated in congestive heart failure in response to increased myocardial wall tension 71 . Even myocardial hypoxia regard-less of pre-existing left ventricular dysfunction can induce BNP release 72 .

19
The physiologic actions of BNP include natriuresis, vasodilatation and inhi-bition of the renin-angiotensin-aldosterone axis and the sympathetic nervous system 71 . As NT-pro BNP has a greater relative increase above baseline values, it seems to be a better candidate for prognostication than BNP 73 .However, the relation of NT-pro BNP levels to age, gender and kidney func-tion need to be considered 74, 75 and up until now, differences regarding the clinical utility of both markers have not been found 76 .
In case of acute myocardial ischemia, the concentration of BNP rises rap-idly, peaks at 2-4 hours with a second peak occurring after 5 days in case of pronounced left ventricular remodeling 77 . The amount of BNP and NT-pro BNP elevation correlates with the extent of myocardial damage 78 and predicts mortality regardless of troponin levels both in ACS 79-81 and in the stable phase after the index event 82, 83 . Levels of natriuretic peptides carry strong prognostic information even in general populations and lower risk patients with stable angina or chest pain 84-86 . However, at present there are only sparse data demonstrating prognostic benefits provided by an approach guided by natriuretic peptide levels. This may in part explain why measurements of these markers so far have not been widely adopted into clinical routine in ACS.
Inflammatory markers / C-reactive protein The role of inflammation for the pathogenesis of atherosclerosis from le-sion initiation through to progression and thrombotic complications is well established 87 . Inflammatory processes are involved in vascular endothe-lial injury leading to the development of a mature atherosclerotic plaque. Inflammatory cells and mediators participate in processes weakening the fibrous cap that covers the atheroma core of the plaque, thereby making it prone to rupture. As a consequence, inflammatory markers have been exten-sively studied for the prediction of cardiovascular risk. In particular the acute-phase reactant C-reactive protein (CRP) has received much attention. CRP is synthesized in the hepatocytes as an unspecific physiological re-sponse to inflammatory stimuli and under control of circulating interleukin-6. Besides reflecting the degree of inflammation, CRP may also directly promote vascular inflammation by stimulation of tissue factor production in monocytes 88 , expression of cell adhesion molecules 89 and activation of the complement system 90 . CRP has shown strong associations with sub-sequent cardiovascular events in healthy individuals and patients with chronic atherosclerotic disease 91-93 . However, its attributive prognostic value beyond traditional cardiovascular risk factors appears only to be mod-erate 94, 95 .

20
In ACS, a significant upregulation of intramyocardial cytokines including interleukin-6 occurs 96 , resulting in a pronounced CRP response which reaches a peak after 48-54 hours 97 . Numerous studies have shown that in ACS patients, CRP levels independently predict mortality and provide in-cremental prognostic information to troponin results 98-100 . Surprisingly, only few authors have evaluated CRP for risk prediction in chest pain pa-tients with suspected ACS. In these studies, CRP offered only limited prog-nostic value which in part may depend on small sample sizes 101, 102 .Even in the long-term after an ACS, CRP levels are indicative of adverse events 103, 104 , possibly as they reflect a cytokine activation in response to myocardial injury, triggering adverse remodeling processes which pave the way to left ventricular dilatation and heart failure 105 .
Markers of renal failure Chronic kidney disease is an independent risk factor for cardiovascular disease, even at a moderate reduction of the glomerular filtration rate (GFR) 106 . This depends on an increased prevalence of atherosclerotic risk fac-
tors in patients with renal failure and a direct promotion of atherosclerosis via changes in parameters such as lipids 107 , inflammatory and coagula-tory factors 107, 108 and endothelial function 109 . The GFR can be measured by the clearance of exogenous substances such as inulin, iohexol or Cr-EDTA. However, as these methods are time-consuming and costly, surrogate markers for the estimation of GFR are more commonly applied. Among these, creatinine is widely used but has shortcomings as its serum levels vary with age, gender, muscle mass and protein intake 110 . Estima-tion of the GFR can be improved by creatinine-based equations such as the Cockcroft-Gault formula 111 and the Modification of Diet in Renal Dis-ease (MDRD) formula 112 (table 2).
Table 2. Recommended estimations of glomerular filtration.
Name Equation
Cockcroft-Gault formula ( 140-age x weight kg ) / (72 x serum creatinine mg/dl x 0.85 for women)
MDRD formula 186 x serum creatinine mg/dl -1.154 x age-0.203 x 0.742 for women x 1.21 for blacks
Cystatin C, an inhibitor of the elastolytic cysteine proteases cathepsin S and K which are involved in vascular injury 113 , has recently emerged as a new marker of renal function. Cystatin C is produced in all nucleated cells at a constant rate, is freely filtered by the renal glomerulus and metabolized by the proximal tubule without subsequent reabsorption to the bloodflow 114 .Several studies have demonstrated that cystatin C mirrors GFR more ade-

21
quately than the creatinine level and at least as good as the Cockcroft-Gault formula 114-116 . Cystatin C is also a better indicator of cardiovascular risk than the estimated creatinine-clearance 117-119 . This may be related to the fact that cystatin C levels are a more adequate measure of renal function but even its association to vascular inflammation has been suggested by some authors as possible cause for its prognostic superiority 118, 120 .
Multimarker strategies As for diagnostic purposes, an algorithm applying a panel of biochemical markers reflecting different aspects in ACS appears to be a promising option for improving risk stratification. There is strong evidence demonstrating that the natriuretic peptides and CRP work synergistically along with the tropon-ins in patients with chest pain 86 and ACS 79-81, 98, 99 . Multimarker strategies incorporating CK-MB or myoglobin in combination with troponin results in contrast have only been evaluated by few authors 69, 70 . How-ever, at present there is no consensus whether a multimarker approach should be used for prognostication and which biomarker combination should be preferred at the different stages of an ACS.
Biochemical markers for the assessment of infarct size The extent of AMI is a major determinant of prognosis 121, 122 . At pre-sent, contrast-enhanced magnetic resonance imaging is the most accurate method for quantification of infarct size 123 but may not be available at a very early stage, i.e. when infarct-limiting therapy still can be initiated 124 .An alternative approach is to estimate infarct size from peak or cumulative serum concentrations of biochemical markers of myocardial damage 125 .The first markers of interest were total CK and CK-MB which after a series of studies demonstrated good correlation to infarct size and cardiac compli-cations post AMI 121, 126 . However, the usefulness of CK and CK-MB is limited by their lack of cardiospecificity and altered kinetics depending on coronary reperfusion status 127 . Even myoglobin levels may serve as a relative early measure of the extent of AMI 128, 129 but their applicability is restricted due to similar reasons as for CK-MB. cTnI and cTnT concentra-tions after 48-96 hours correlate well with infarct size and left ventricular function 57-61 regardless of reperfusion status 57, 58 and may therefore be more suitable, also given their relative slow increase and long persistence after myocardial damage. However, almost all studies assessing biochemical markers for estimation of infarct size are based on late results, i.e. when the myocardial damage has become irreversible. Accordingly, the value of very early measurements for assessment of infarct size is not known. This draw-back may be overcome by the use of selected statistical methods, e.g. ANN 50 .

22
AIMS
The aim of this work was to evaluate the importance of cTnI testing for assessment of patients with suspected or confirmed ACS in the light of im-proved analytical methods, new technologies for measurement and interpre-tation of cTnI levels and in the context of other novel and established bio-chemical markers.
Specific aims In patients with chest pain suggestive of an ACS,
- to compare the early diagnostic performance of different cTnI thresholds relative to CK-MB, myoglobin and different multimarker strategies when analyzed by early frequent POC measurements (paper I),
- to validate pre-specified ANN-algorithms based on POC cTnI and my-oglobin results for the early diagnosis of AMI and the prediction of ma-jor infarct size (paper II), and
- to assess the prognostic value of an approach combining early POC cTnI results with other markers of myocardial damage (paper III) and markers of left ventricular dysfunction, inflammation or renal function (paper IV).
In patients stabilized after an episode of ACS,
- to evaluate the prevalence of cTnI elevation detectable during 6 months after the index event (papers V and VI), and
- to examine the prognostic importance of persistent cTnI elevation and its relationship to left ventricular function, coronary status, treatment strat-egy during the index hospitalization and dynamic changes of biochemi-cal markers of left ventricular dysfunction and inflammation (papers V and VI).

23
MATERIAL AND METHODS
Patients
Papers I and III In the FAST II (Fast Assessment of Thoracic Pain)-study, 197 patients admitted to the coronary care unit (CCU) at the Uppsala University Hospital between 2000 and 2001 were included. The inclusion criterion was chest pain with a duration 15 minutes within the last 24 hours, warranting the suspicion of an ACS. Pathological ST-segment elevation on the admission 12-lead ECG was the only exclusion criterion. The analysis in paper I is based on all 197 patients whereas for paper III, only the 191 patients with first time admission were considered.
Paper II The FASTER I (Fast Assessment of Thoracic Pain by nEuRal networks)-study was conducted between 2002 and 2003 at three investigational centers in Sweden. The inclusion criterion was admittance to the CCU because of chest pain with a duration 15 minutes within the last 8 hours, suspective of an ACS. The study exclusion criteria were pathological ST-segment eleva-tion on the admission 12-lead ECG or strong suspicion of acute myocarditis.
A total of 380 patients were enrolled in the study. Of these, 16 patients did not fulfill the inclusion criterion or had at least one exclusion criterion. Thirty-nine patients were excluded because of inappropriate biochemical marker sampling. Patients with confounding diseases (previously known renal failure with creatinine > 150 mol/L, severe skeletal muscle trauma within the last 7 days or known AMI within the last 10 days; n=15) were excluded for the aim of the analysis presented in paper II. Thus, a total of 310 patients was assessed.
Paper IV For this analysis, patients from the FAST II- and the FASTER I-studies with symptom onset within the last 8 hours, first time admission and ade-

24
quate blood sampling were pooled to one sample population. The population consisted of 452 patients, 127 from the FAST II-study and 325 from the FASTER I-study.
The allocation of the patients from the FAST II- and FASTER I-studies to the analyses presented in papers I-IV is demonstrated in figure 2.
Figure 2. Patient population in papers I-IV.
Papers V and VI The FRISC II (FRagmin and Fast Revascularization during InStability in Coronary artery disease)-trial was a prospective multicentre study in which totally 3489 patients with non ST-segment elevation ACS were randomized between 1996 and 1998 in a factorial design to an early invasive or a non-invasive strategy and to 3 months treatment with dalteparin or placebo [130]. The inclusion criteria were symptoms of unstable coronary artery disease with objective signs of myocardial ischemia such as electrocardiographic changes (ST-segment depression 0.1 mV or T-wave inversion 0.1 mV) or elevated biochemical markers of myocardial necrosis. Exclusion criteria were increased risk for bleeding, anemia, indication for treatment with thrombolysis in the past 24 hours or percutaneous coronary intervention within the last 6 months. Patients with a history of previous open heart sur-gery, advanced age or poor general health, and those included after comple-tion of recruitment of patients to the invasive vs. non-invasive arm were treated primarily non-invasively and randomized only regarding 3 month treatment with dalteparin or placebo. In the invasive strategy, the aim was to

25
perform coronary angiography and, if appropriate, revascularization within 7 days from admission. Patients randomized to the non-invasive arm under-went coronary angiography only in case of refractory or recurrent angina or if they showed signs of severe ischemia on a pre-discharge exercise test.
The population of the analysis presented in paper V consisted of 1092 pa-tients from the FRISC II-trial who had been enrolled at selected study cen-ters. The analysis in paper VI was based on the 898 patients who had cTnI results available at the three measurement instances assigned in the study protocol (823 patients for analyses regarding the endpoint of AMI; see sec-tion on specific statistics).
For all studies, verbal and written informed consent was obtained from all patients and the study protocols were approved by the local ethics commit-tees.
Study protocols
Papers I and III In the FAST II-study, whole blood samples for analysis of cTnI, CK-MB and myoglobin were obtained at the time of enrolment, thereafter every 30 minutes during the first 2 hours, then at 3, 6, 12 and in the case of AMI at 24 hours (cTnI and CK-MB only). Analyses were performed on local Stratus CS instruments (Dade Behring, Deerfield, IL, USA). Additional samples of all three markers were sent at pre-specified time-points to the central labora-tory for analysis on an AxSYM platform (Abbot Diagnostics, Abbot Park, IL, USA).
Paper II Blood sampling was performed in the FASTER I-study at the time of en-rolment, after 40 minutes, 80 minutes, 2, 3, 6 and 12 hours. cTnI was meas-ured in all samples. Myoglobin was measured in all early samples up until 3 hours. CK-MB was measured at the time of enrolment, after 6 and 12 hours. In case of any cTnI elevation 0.1 g/L within the first 12 hours, a last sample for analysis of cTnI and CK-MB was drawn 24 hours after enrol-ment. Analyses were performed on Stratus CS instruments situated at the CCU of each participating study centre.

26
Paper IV For the pooled population of FAST II- and FASTER I-patients, Stratus CS cTnI results obtained at the time of enrolment, 30/40 min (FAST II-/FASTER I-study, respectively), 90/80 min, 2, 3, 6, 12 and 24 hours were used. NT-pro BNP, high sensitivity CRP and cystatin C were retrospectively analyzed in frozen plasma samples drawn at enrolment. Creatinine was sys-tematically measured in the FASTER I-study at enrolment while for the FAST II-study, results obtained at hospital admission were used (median delay until first blood sample 0.8 hours 25th, 75th percentiles 0.0-1.4 hours ).
Papers V and VI cTnI was measured at 6 (4-7) weeks, 3 and 6 months after randomization in frozen samples of EDTA plasma using the recently refined AccuTnI assay (Beckman Coulter, Fullerton, CA, USA) in all patients without an AMI or a revascularization procedure during the preceeding 14 days. NT-pro BNP and high sensitivity CRP were determined in samples obtained at 6 (4-7) weeks and 6 months. Creatinine was measured at randomization. The sample sizes regarding the different analyses in papers V and VI are given in figure 3.
Figure 3. Sample sizes in papers V and VI.

27
Biochemical analysis The Stratus CS analyzer is a fluorometric enzyme immunoassay analyzer for POC measurement of cTnI, CK-MB and myoglobin using whole blood samples anticoagulated with lithium heparin. The lower detection limit for the cTnI assay is 0.03 g/L, the 99th percentile among healthy individuals is 0.07 g/L and the lowest concentration assuring a 10% CV is 0.1 g/L131. The upper reference level for CK-MB is 3.5 g/L and 98 g/L and 56 g/L for myoglobin in males and females, respectively. The CV intervals for
cTnI were < 5.2% (at 0.45-18.0 g/L) in the FAST II-study and < 4.6% (at 0.39-8.44 g/L) in the FASTER I-study.
The Access AccuTnI assay is a sandwich immunoassay with a 99th percen-tile of 0.04 g/L among healthy subjects regardless of age and 0.021 g/Lamong subjects < 60 years according to previous evaluations 131-133 . The lowest concentration measurable with a 10% CV was 0.06 g/L 131, 133 .However, recent modifications of the assay in terms of a change of the manufacturing process combined with an enhanced signal to dose relation-ship have resulted in more robust results at the low end of its range. Accord-ingly, the validation at the Department of Clinical Chemistry, Uppsala Uni-versity Hospital has revealed 10% and 20% CVs for the modified assay of 0.014 g/L and 0.008 g/L, respectively.
NT-pro BNP was determined in all studies using the Elecsys pro BNP sandwich immunoassay on Elecsys 2010 instruments (Roche Diagnostics, Mannheim, Germany). The analytical range of this assay extends from 5 to 35 000 ng/ml. High-sensitive CRP was measured in all studies using the Immulite CRP assay (Diagnostic Products Corp., Los Angeles, CA, USA). The measurable range for this assay is 0.1 to 500 mg/L. Cystatin C was de-termined in the FAST II- and FASTER I-studies by a latex-enhanced reagent (N Latex Cystatin C, Dade Behring) using a Behring BN ProSpec analyzer (Dade Behring) with a range of detection of 0.20 to 7.33 mg/L. Creatinine was analyzed using the Advia 1650 system (Bayer Diagnostics, Tarrytown, NY, USA) in the FAST II- and FASTER I-studies and on local systems situ-ated at the respective study centres in the FRISC II-trial. Creatinine-clearance was calculated according to the Cockcroft-Gault formula: ( 140-age x weight kg ) / (72 x serum creatinine mg/dl x 0.85 for women) 111 .

28
Description of the applied ANN-algorithms An ANN is a computer network consisting of different processing units (‘nodes’) which simulate neurons. The nodes are hierarchically arranged in different layers and highly interconnected in analogy to synaptic connections (figure 4) 46 . Data are fed into the ANN via the nodes in the input layer, one node for each input variable. After transformation of the data within each node, the resulting value is multiplied with a specific number (‘weight’) which has been determined during the training phase of ANN development. The weighted data are then passed to the next network layer which can be nodes in a hidden layer or the node in the output layer. In each node, the weighted sum of all incoming signals is compared with a pre-determined threshold. If the signal exceeds the threshold, the node is activated. If not, the node remains quite. When finally the output node is activated, it pro-duces a numerical result in the range from 0 to 1, indicating a specific cate-gory within a given classification.
Figure 4. Flow schematic of an artificial neural network.
Reprinted with permission from reference 134.
ANN achieve their performance by modification of the weights assigned to the various connections. This is done during training sessions in which data from a representative population with defined input variables and a known outcome are fed to the network. The ANN begins its first training epoch with a set of arbitrary weights. The output of the network then is compared with the desired output, which is given by the known outcomes. If there is a dis-crepancy between the calculated and actual output, the error is back-propagated through the ANN by adjusting the weights in an iterative manner until the error discrepancy is within an allowed tolerance.

29
The most widely used ANN is the multi-layer perceptron which consists of one input and one output layer linked by a hidden layer of nodes. Another commonly applied ANN is the single-layer perceptron which essentially is a multi-layer perceptron without hidden layer. In the FASTER I-study, the multi-layer perceptron with a hidden layer of two nodes, and the single-layer perceptron were used. Two different sets of input quantities were chosen for both ANN models: normalized serial measurements of cTnI and myoglobin, either alone or in combination with their respective rates of change (Table 3). Normalization was performed by division of the measurement values with their respective median values among healthy individuals according to the manufacturer: 0.02 g/L for cTnI, and 42 g/L and 30 g/L for myoglo-bin in men and women, respectively. The results presented in the analysis are from the ANN-algorithms with the highest predictive values, calculated at the determined decision thresholds.
Table 3. The various ANN-structures and sets of input variables used for the three classification tasks in the FASTER I-study.
Classification task ANN-structure Sets of input variables
Rule-in and rule-out of ‘TnI 0.1 AMI’
SLP cTnI and myoglobin
Rule-in and rule-out of ‘TnI 0.4 AMI’
MLP with 2 hidden units cTnI and myoglobin; cTnI and myoglobin and rates of change
Prediction of ‘major AMI’ SLP cTnI and myoglobin; cTnI and myoglobin and rates of change
SLP: single-layer perceptron. MLP: multi-layer perceptron. ‘TnI 0.1 AMI’: acute myocardial infarction, decision limit cTnI 0.1 g/L. ‘TnI 0.4 AMI’: acute myocardial infarction, deci-sion limit cTnI 0.4 g/L.
Other examinations In the FAST II- and FASTER I-studies, resting 12-lead ECGs were docu-mented on admission, after 12 hours and additionally after 24 hours in those patients who, by the attending physician, were regarded as having an AMI. The ECGs were interpreted by an independent evaluator blinded to outcome. Patients with non-sinus rhythm, evidence of Q-waves, bundle branch block or significant ST-segment changes were regarded as having an abnormal ECG.
Echocardiographies were performed in the FRISC II-study before hospital discharge. The left ventricular ejection fraction (LV-EF) was visually as-sessed and considered depressed when it was < 0.45. The presence and grade

30
of any coronary stenosis and the Thrombolysis In Myocardial Infarction (TIMI) flow grade were assessed in the 453 patients randomized to the inva-sive arm by coronary angiography according to a detailed evaluation form. A
50% diameter obstruction was considered significant.
Definition of the index diagnosis The index events in the FAST II- and FASTER I-studies were classified by independent endpoint evaluators with access to all clinical and laboratory data.
Papers I and III The diagnosis of AMI was considered present in the FAST II-study if one of the following criteria was fulfilled in conjunction with the qualifying chest pain:
- development of a pathological Q-wave (duration > 0.03 seconds and amplitude > 25% of the following R-wave amplitude) in 2 con-tiguous leads in the 12-lead ECG within 24 hours
- cTnI 1.3 g/L within 24 hours (AxSYM; 10% CV level 131 ) at least at one measurement instance without obvious reasons for non-ischemic causes.
In addition, a second classification based on WHO-criteria for the defini-tion of AMI was applied, using elevation of CK-MB > 10 g/L (AxSYM; double upper reference level) within 24 hours as biochemical criterion 33 .
Paper II The criteria for AMI classification in the FASTER I-study were 2 ele-vated cTnI levels within 24 hours from admission together with the qualify-ing chest pain and at least one of the following criteria:
- development of a pathological Q-wave (duration 0.03 s and 0.1 mV in depth) in 2 contiguous leads in the 12-lead ECG within 24 hours
- new ST-segment depression 0.1 mV in 2 contiguous leads- new ST-segment elevation at the J point in 2 contiguous leads with
the cut-off points 0.2 mV in leads V1-V3 and 0.1 mV in all other leads

31
Two different sets of biochemical criteria for the definition of AMI were used in order to validate the ANN-algorithms even when different diagnostic standards were applied. The first set of criteria employed a decision limit of cTnI 0.1 g/L (‘TnI 0.1 AMI’), corresponding to the lowest concentration measurable with a 10% CV 131 . The second set of criteria employed a decision limit of cTnI 0.4 g/L (‘TnI 0.4 AMI’), corresponding to the de-cision limit for AMI recommended in Sweden at the time of investigation.
The study patients with a ‘TnI 0.4 AMI’ were further classified with regard to the extent of infarct size which was prospectively defined using peak CK-MB levels within 24 hours. A major AMI was indicated if peak CK-MB was
35 g/L, i.e. 10 times the upper reference level.
Paper IV For the pooled population assessed in paper IV, FASTER I-criteria includ-ing cTnI 0.1 g/L at 2 measurement instances as biochemical criterion were applied for the definition of AMI. Accordingly, 4 FAST II-patients had their final index diagnosis changed from AMI to unstable angina.
Follow-up and endpoints
Papers I-IV Patients were followed up in the FAST II- and FASTER I-studies by re-search nurses with telephone contacts at 30 days and 6 months (± 2-4 weeks) after discharge. The study endpoints were total and cardiac mortality and AMI. Information regarding death was obtained from the Swedish National Registry on Mortality. Information regarding AMI was obtained from the hospitals diagnosis registries and patient records.
Papers V and VI Patients were followed up in the FRISC II-study by outpatient visits after 6 (4-7) weeks, 3 and 6 months, and by telephone contacts after 12 and 24 months. Thereafter, and up to 5 years after randomization, all information on events was based on National Registries run by the Swedish Health Author-ity. The study endpoints were total mortality and AMI.

32
Statistical analysis For all diagnostic estimates, the sensitivities, specificities, negative predic-tive values (NPV) and positive predictive values (PPV) were calculated. Receiver operator characteristics (ROC)-curve analysis was applied for si-multaneous assessment of sensitivities and specificities and for the calcula-tion of optimal cut-offs. Continuous variables were described as medians, 25th and 75th percentiles. Comparisons of medians were performed using the Mann-Whitney U test or Kruskal-Wallis test, as appropriate. Categoric vari-ables were expressed as frequencies and percentages. Differences between categoric variables were analyzed with the Pearson 2 test. The McNemar test was used for comparison of paired categorical data. To assess the corre-lation between the levels of biochemical markers, the Spearman rank corre-lation coefficients were calculated. Variables independently associated with the biochemical markers and endpoints were identified by multivariate logis-tic regression analysis. Kaplan-Meier plots were used to illustrate the timing of events. In all analyses, a p value < 0.05 was considered significant. All statistics were calculated using the Statistical Package for Social Sciences software program versions 10.0.0, 11.5, 12.0.1 and 14.0 (SPSS Inc., Chi-cago, IL, USA).
Specific statistics
Paper I The sensitivities of the biochemical markers were first compared using ROC-curve analysis at a fixed specificity of 95% and second, as cumulative diagnostic estimates using cTnI 0.07 g/L, 0.1 g/L and 0.4 g/L and the respective upper reference levels of CK-MB and myoglobin as cut-offs.
In a post-hoc analysis, the diagnostic performance of the following multi-marker strategies within 6 hours from admission was studied:
- Rule 1: cTnI 0.1 g/L or cTnI 0.07 g/L + myoglobin 98 g/L (males) / 56 g/L (females),
- Rule 2: cTnI 0.07 g/L or myoglobin 98 g/L (males) / 56 g/L (females).

33
Paper II Pre-defined criteria based on previous experiences 135 were used to vali-date the diagnostic usefullness of the ANN-algorithms for diagnosis of AMI: NPV > 94% and PPV > 78% at 2 hours.
The ANN-algorithm for the prediction of a ‘major AMI’ (CK-MB defined) was evaluated in all patients with an AMI according to the cTnI 0.4 g/Ldecision limit. The information obtained from the ANN-indication of a ‘ma-jor AMI’ at 2 hours was assessed by comparing its sensitivity with a simple ROC-curve derived cTnI cut-off.
Paper III cTnI 0.1 g/L, CK-MB 3.5 g/L and myoglobin 98 g/L (males) / 56 g/L (females) within 6 hours from admission were used for prognostica-tion, both as single markers and incorporated into the following multimarker strategies:
- Rule 1: TnI 0.1 g/L or myoglobin 98 g/L (males) / 56 g/L(females),
- Rule 2: TnI 0.1 g/L or CK-MB 3.5 g/L,- Rule 3: TnI 0.1 g/L or CK-MB 3.5 g/L or myoglobin 98
g/L (males) / 56 g/L (females).
Paper IV Median values and optimal ROC-curve derived cut-offs of the tested bio-chemical markers were used for risk prediction. For cTnI, peak values within the 24 hour sampling schedule were used. Comparisons of the prognostic value of the biomarkers were performed in patients with results for all mark-ers available.
Different marker combinations for prognostication were then defined, combining peak cTnI 0.1 g/L within 2 hours as pre-selected time point and ECG findings (abnormal or normal ECG) with either NT-pro BNP, CRP, cystatin C or creatinine-clearance. The prognostic information pro-vided by these multimarker combinations was evaluated by ROC-curve analysis and multivariate logistic regression analysis.

34
Papers V and VI The prevalence and prognostic importance of cTnI was assessed applying the following cut-offs: cTnI > 0.04 g/L, > 0.02 g/L and > 0.01 g/L which is in the range measurable with a 10-20% CV by the refined version of the AccuTnI assay. The prognostic importance of cTnI elevation relative to the respective cut-offs was evaluated both on each measurement instance and throughout the 6 month sampling period. For the latter purpose, all patients who had cTnI results available at all measurement instances were divided into subgroups according to cTnI levels over time, i.e. negative cTnI at all measurement instances, elevated cTnI at 1-2 instances (‘temporary cTnI elevation’) and elevated cTnI at all instances (‘persistent cTnI elevation’). For prognostication regarding AMI, only samples of patients without an AMI during follow-up until respective measurement instance were used since only the first AMI after blood sampling were counted as endpoint.
Variables independently associated with the prevalence of persistent cTnI elevation were identified by multivariate logistic regression analysis. Differ-ent models were constructed with adjustment for age (10 year increments), gender, diabetes, congestive heart failure, previous AMI and creatinine-clearance on admission (10 ml/min increments) in model 1. A previous AMI was defined as a history of AMI prior to the index event or a cTnT level > 0.035 g/L at randomization (Elecsys 2010; 10% CV level 20 ). Additional adjustment was made for LV-EF < 0.45 during hospitalization and levels of NT-pro BNP and CRP at 6 months (model 2), randomization to the invasive vs. non-invasive arm (model 3) and angiographic findings (model 4).
The prognostic importance of persisting cTnI elevation was evaluated by univariate and multivariate logistic regression analysis applying both unad-justed models and a similar approach as for the analysis regarding the preva-lence of persistent cTnI elevation but with inclusion of persistent cTnI eleva-tion as a co-variate.

35
RESULTS
Paper I
General findings The baseline characteristics and final index diagnoses of the study popula-tions assessed in papers I, II and IV are shown in table 4. The allocation of the patients from the FAST II- and FASTER I-studies to the different analy-ses is demonstrated in figure 2 in the method section of this work.
Table 4. Clinical characteristics and final index diagnoses of the patient populations from the FAST II- and FASTER I-studies.
FAST II n=197
(Paper I)
FASTER I n=310
(Paper II)
pooled population n=452
(Paper IV)
Delay to first blood sample (h) 5.5 (3.4-9.6) 4.7 (3.4-6.1) 4.5 (3.1-5.9)* Age (years) 66 (55-75) 65 (57-76) 65 (56-75) Male 130 (66%) 200 (65%) 298 (66%) Hypertension 91 (46%) 116 (37%) 185 (41%) Diabetes 31 (16%) 51 (17%) 77 (17%) Current smoking 30 (15%) 54 (17%) 80 (18%) Previous AMI 72 (37%) 95 (31%) 154 (34%) Congestive heart failure 35 (20%) 46 (15%) 76 (16%) Previous revascularisation 56 (28%) 88 (28%) 131 (29%)
Final index diagnosis AMI 43 (22%) - - cut-off cTnI 0.1 g/L AMI - 102 (33%) 140 (31%) unstable angina - 62 (20%) 82 (18%)*
cut-off cTnI 0.4 g/L AMI - 73 (24%) - unstable angina - 91 (29%) -
unstable angina 30 (15%) - - other heart disease 43 (22%) 11(4%) 37 (8%)** non-cardiac disease 19 (10%) 14 (4%) 26 (6%)* unspecified chest pain 62 (31%) 121 (39%) 167 (37%) Medians given with 25th, 75th percentiles. Asterisks refer to differences between patient sub-groups from the FAST II- and FASTER I-study: * p < 0.05; ** p < 0.001. AMI: myocardial infarction.
36
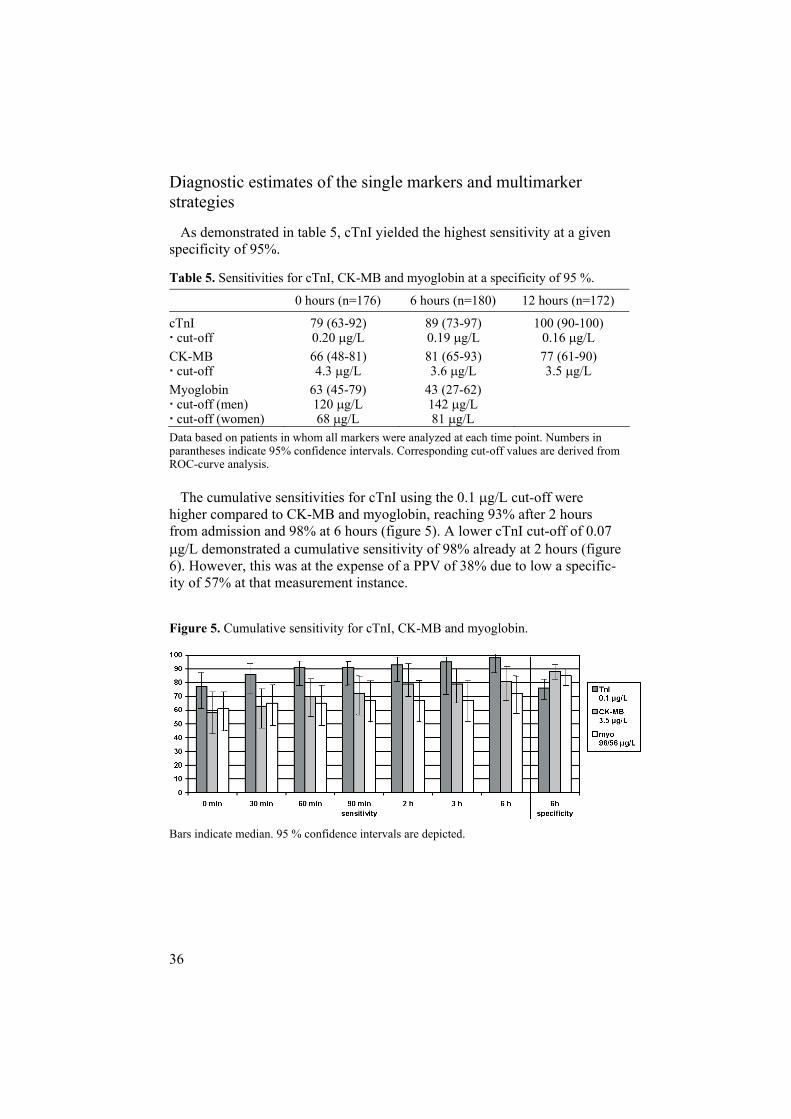
Diagnostic estimates of the single markers and multimarker strategies
As demonstrated in table 5, cTnI yielded the highest sensitivity at a given specificity of 95%.
Table 5. Sensitivities for cTnI, CK-MB and myoglobin at a specificity of 95 %.
0 hours (n=176) 6 hours (n=180) 12 hours (n=172)
cTnI cut-off
79 (63-92) 0.20 g/L
89 (73-97) 0.19 g/L
100 (90-100) 0.16 g/L
CK-MB cut-off
66 (48-81) 4.3 g/L
81 (65-93) 3.6 g/L
77 (61-90) 3.5 g/L
Myoglobin cut-off (men) cut-off (women)
63 (45-79) 120 g/L 68 g/L
43 (27-62) 142 g/L 81 g/L
Data based on patients in whom all markers were analyzed at each time point. Numbers in parantheses indicate 95% confidence intervals. Corresponding cut-off values are derived from ROC-curve analysis.
The cumulative sensitivities for cTnI using the 0.1 g/L cut-off were higher compared to CK-MB and myoglobin, reaching 93% after 2 hours from admission and 98% at 6 hours (figure 5). A lower cTnI cut-off of 0.07
g/L demonstrated a cumulative sensitivity of 98% already at 2 hours (figure 6). However, this was at the expense of a PPV of 38% due to low a specific-ity of 57% at that measurement instance.
Figure 5. Cumulative sensitivity for cTnI, CK-MB and myoglobin.
Bars indicate median. 95 % confidence intervals are depicted.

37
Figure 6. Cumulative sensitivity and specificity of different cut-off levels for cTnI.
Bars indicate median. 95 % confidence intervals are depicted.
Rule 1 and rule 2 demonstrated cumulative sensitivities of 98% at 2 hours which however, were not significantly higher than that of cTnI 0.1 g/L.At the same time point, the cumulative specificity of rule 1 was lower com-pared to cTnI 0.1 g/L (77% vs 81%; n.s.) as was the cumulative specific-ity of rule 2 compared to cTnI 0.1 g/L (50% vs 81%; p <0.001) and com-pared to cTnI 0.07 g/L (50% vs 57%; p=0.002).
Using the alternative CK-MB standard for defining AMI gave increased sensitivities of CK-MB at a given specificity of 95% which exceeded those of cTnI both on admission and at 6 hours. The sensitivities of myoglobin at this level of specificity were in the same range regardless the chosen stan-dard.

38
Paper II
General findings The patient characteristics and the final index diagnoses of the selected patients from the FASTER I-study are given in table 2. Twenty-eight pa-tients had a major AMI as determined by peak CK-MB levels.
Diagnostic indications of AMI by the ANN-algorithms The ANN-algorithms provided high diagnostic estimates after 2 hours of monitoring with sensitivities and specificities in the range of 95-100% and 90-97%, respectively. As demonstrated in table 6, the ANN indications of ‘TnI 0.1 AMI’ and ‘TnI 0.4 AMI’ resulted in highly significant PPV and NPV when tested against the required pre-defined values.
Table 6. Performance measures of the ANN-algorithms for detection and exclusion of ‘TnI 0.1 AMI’ and ‘TnI 0.4 AMI’ after 2 hours of biochemical monitoring.
Estimates p-value
‘TnI 0.1 AMI’ PPV NPV
87% 99%
0.009 0.0001
‘TnI 0.4 AMI’ PPV NPV
90% 99%
0.006 0.0004
PPV: positive predictive value, tested against the required pre-defined value of 78%. NPV: negative predictive value, tested against the required pre-defined value of 94%. ‘TnI 0.1 AMI’: acute myocardial infarction, decision limit cTnI 0.1 g/L. ‘TnI 0.4 AMI’: acute myocardial infarction, decision limit cTnI 0.4 g/L.
The ANN indications produced higher cumulative sensitivities for AMI at 2 hours compared to the respective cTnI based standard (‘TnI 0.1 AMI’: 99% vs 94%; p=0.06; ‘TnI 0.4 AMI’ 96% vs 86%; p=0.02) while their cu-mulative specificities were lower (‘TnI 0.1 AMI’: 93% vs 97%; p=0.008; ‘TnI 0.4 AMI’ 97% vs 99%; p=0.03).

39
Prediction of ‘major infarct’ size by the ANN-algorithm In the 73 patients with an AMI according to the cTnI 0.4 g/L decision limit, a ROC-derived cTnI cut-off of 1.76 g/L corresponded to the ANN-indication of ‘major infarct’ size in terms of identical diagnostic specificities after 2 hours. As demonstrated in figure 7, the ANN-indication allowed the prediction of a ‘major AMI’ with a significantly higher sensitivity at 2 hours compared to cTnI 1.76 g/L (96% vs 68%; p=0.008).
Figure 7. Cumulative sensitivities for the ANN-indication of a ‘major AMI’ com-pared to sensitivities of cTnI 1.76 g/L.
Diagnostic specificities after 2 hours monitoring were 78% for both methods. Bars indicate median. Estimated 95% confidence intervals are depicted. Number of patients in each group is shown in each bar (n=28 ‘major AMI; n=45 non-‘major AMI’).
40
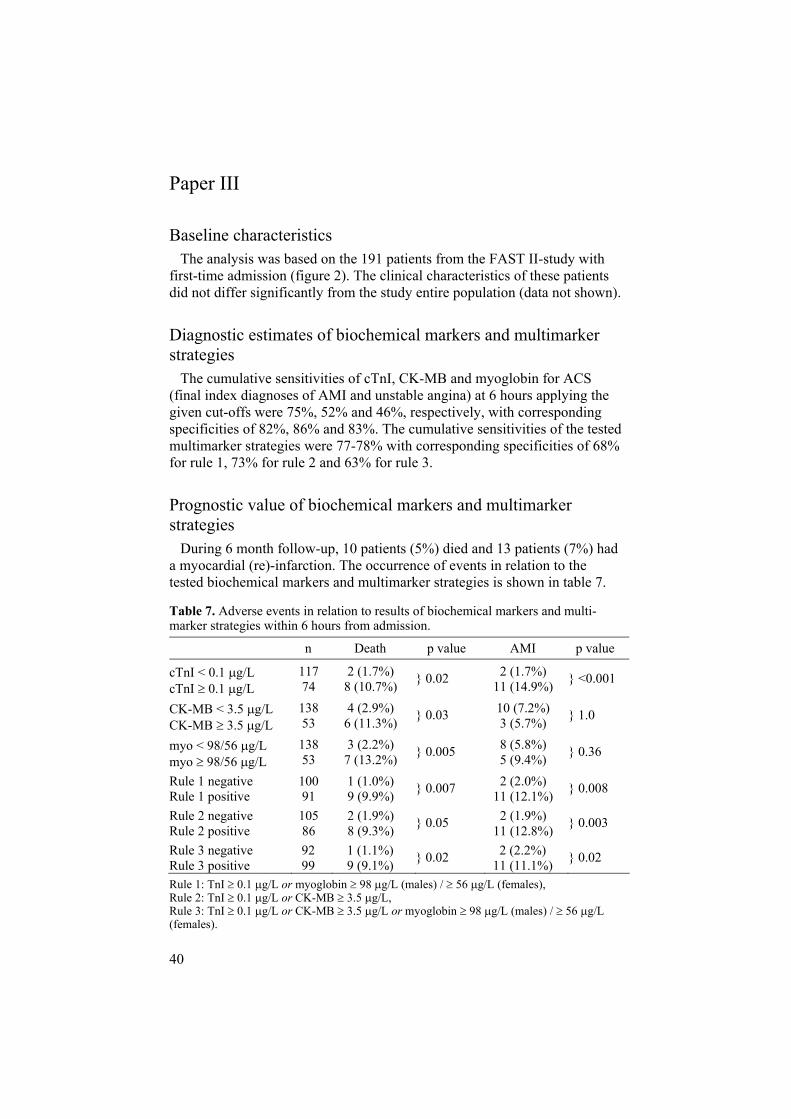
Paper III
Baseline characteristics The analysis was based on the 191 patients from the FAST II-study with first-time admission (figure 2). The clinical characteristics of these patients did not differ significantly from the study entire population (data not shown).
Diagnostic estimates of biochemical markers and multimarker strategies The cumulative sensitivities of cTnI, CK-MB and myoglobin for ACS (final index diagnoses of AMI and unstable angina) at 6 hours applying the given cut-offs were 75%, 52% and 46%, respectively, with corresponding specificities of 82%, 86% and 83%. The cumulative sensitivities of the tested multimarker strategies were 77-78% with corresponding specificities of 68% for rule 1, 73% for rule 2 and 63% for rule 3.
Prognostic value of biochemical markers and multimarker strategies During 6 month follow-up, 10 patients (5%) died and 13 patients (7%) had a myocardial (re)-infarction. The occurrence of events in relation to the tested biochemical markers and multimarker strategies is shown in table 7.
Table 7. Adverse events in relation to results of biochemical markers and multi-marker strategies within 6 hours from admission.
n Death p value AMI p value
cTnI < 0.1 g/L cTnI 0.1 g/L
117 74
2 (1.7%) 8 (10.7%) } 0.02 2 (1.7%)
11 (14.9%) } <0.001
CK-MB < 3.5 g/L CK-MB 3.5 g/L
138 53
4 (2.9%) 6 (11.3%) } 0.03 10 (7.2%)
3 (5.7%) } 1.0
myo < 98/56 g/L myo 98/56 g/L
138 53
3 (2.2%) 7 (13.2%)
} 0.005 8 (5.8%) 5 (9.4%)
} 0.36
Rule 1 negative Rule 1 positive
100 91
1 (1.0%) 9 (9.9%)
} 0.007 2 (2.0%) 11 (12.1%)
} 0.008
Rule 2 negative Rule 2 positive
105 86
2 (1.9%) 8 (9.3%) } 0.05 2 (1.9%)
11 (12.8%) } 0.003
Rule 3 negative Rule 3 positive
92 99
1 (1.1%) 9 (9.1%) } 0.02 2 (2.2%)
11 (11.1%) } 0.02
Rule 1: TnI 0.1 g/L or myoglobin 98 g/L (males) / 56 g/L (females), Rule 2: TnI 0.1 g/L or CK-MB 3.5 g/L, Rule 3: TnI 0.1 g/L or CK-MB 3.5 g/L or myoglobin 98 g/L (males) / 56 g/L(females).

41
cTnI provided the highest prognostic value regarding myocardial (re)-infarction and cardiac mortality (0 vs. 8 patients 10.7%; p<0.001 ). My-oglobin was predictive for the endpoint of total mortality. The overall prog-nostic capacity of the tested multimarker strategies was non-superior to cTnI
0.1 g/L apart from rule 1 considering total mortality.

42
Paper IV
General findings The clinical characteristics of the pooled population from the FAST II- and FASTER I-studies are shown in table 4. The admission ECG was abnormal in 227 patients (50%). During 6 month follow-up, 14 patients (3%) died and 21 patients (5%) suffered a myocardial (re)-infarction.
In total, 173 patients (38%) had a peak cTnI level 0.1 g/L within 24 hours from admission. The median levels for the other tested markers were NT-pro BNP 190 ng/L (59-774 ng/L 25th, 75th percentiles ), CRP 2.1 mg/L (1.0-54.9 mg/L), cystatin C 1.17 mg/L (1.06-1.33 mg/L) and creatinine-clearance 56.7 ml/min (70.9-44.4 ml/min). NT-pro BNP was strongly corre-lated to creatinine-clearance (r=-0.54; p<0.001) and cystatin C (r=0.48; p<0.001) and moderately correlated to peak cTnI within 24 hours (r=0.39; p<0.001). Even creatinine-clearance and cystatin C were strongly correlated (r=-0.55; p<0.001). All other tested biochemical markers were moderately correlated (r=0.10-0.30) apart from CRP in relation to creatinine-clearance (r=-0.08; n.s.).
According to ROC-curve analysis, the optimal cut-off values for the pre-diction of the combined endpoint of death or (re)-infarction were cTnI 0.1
g/L (area under the curve AUC 0.73; 95% confidence interval CI 0.66-0.80), NT-pro BNP 550 ng/L (AUC 0.80; 95% CI 0.73-0.87), CRP 3.7 mg/L (AUC 0.68; 95% CI 0.57-0.78), cystatin C 1.28 mg/L (AUC 0.75; 95% CI 0.66-0.85) and creatinine-clearance 47.5 ml/min (AUC 0.76; 95% CI 0.66-0.86).
Prognostic value of single biochemical markers On univariate analysis, cTnI, NT-pro BNP and cystatin C were highly pre-dictive for both the endpoints of death and myocardial (re)-infarction (table 8). Independent predictors of the combined endpoint of death or myocardial (re)-infarction were peak cTnI 0.1 g/L within 24 hours (Odds ratio OR3.9 95% CI 1.5-10.4 ; p=0.007), cystatin C 1.28 mg/L (OR 5.6 95% CI 1.9-16.3 ; p=0.002) and NT-pro BNP 550 ng/L (OR 2.7 95% CI 1.0-7.3 ;p=0.045).

43
Table 8. Adverse events at 6 months in relation to peak cTnI levels within 24 hours and baseline levels of NT-pro BNP, CRP, cystatin C and creatinine-clearance.
n Death p value AMI p value
Median levels
NT-pro BNP < 190 ng/L NT-pro BNP 190 ng/L
210 205
010 (4.9%) } 0.001 3 (1.4%)
14 (6.8%) } 0.006
CRP < 2.1 mg/L CRP 2.1 mg/L
209 206
2 (1.0%) 8 (3.9%) } 0.06 7 (3.3%)
10 (4.9%) } 0.47
Cystatin C < 1.17 mg/L Cystatin C 1.17 mg/L
201 214
1 (0.5%) 9 (4.2%) } 0.02 3 (1.5%)
14 (6.5%) } 0.01
Creat.-cl. > 56.7 ml/min Creat.-cl. 56.7 ml/min
210 205
010 (4.9%) } 0.001 6 (2.9%)
11 (5.4%) } 0.22
ROC cut-offs
cTnI < 0.1 g/L cTnI 0.1 g/L
268 147
2 (0.7%) 8 (5.4%) } 0.005 4 (1.5%)
13 (8.8%) } 0.001
NT-pro BNP < 550 ng/L NT-pro BNP 550 ng/L
293 122
1 (0.3%) 9 (7.4%) }<0.001 6 (2.0%)
11 (9.0%) } 0.002
CRP < 3.7 mg/L CRP 3.7 mg/L
283 132
4 (1.4%) 6 (4.5%) } 0.08 9 (3.2%)
8 (6.1%) } 0.19
Cystatin C < 1.28 mg/L Cystatin C 1.28 mg/L
286 129
1 (0.3%) 9 (7.0%) }<0.001 4 (1.4%)
13 (10.1%) }<0.001
Creat.-cl. > 47.5 ml/min Creat.-cl. 47.5 ml/min
288 127
2 (0.7%) 8 (6.3%) } 0.002 9 (3.1%)
8 (6.3%) } 0.18
Analysis performed in patients with results available for all biochemical markers (n=415).
Risk stratification by different combinations of prognostic markers On multivariate logistic regression analysis, both cTnI 0.1 g/L within 2 hours (OR 3.6 95% CI 1.5-8.7 ; p=0.005) and an abnormal admission ECG (OR 3.3 95% CI 1.2-9.1 ; p=0.02) independently predicted the combined endpoint. When added as continuous variables to this model, both NT-pro BNP (OR 1.6 95% CI 1.2-2.2 p=0.002) and cystatin C (OR 5.6 95% CI 1.7-18.6 p=0.005) emerged as independent predictors and resulted in an improved prognostication as expressed as increasing areas under the ROC curves from 0.73 to 0.80-0.81 (figure 8).

44
Figure 8. ROC-curves for the prediction of death or myocardial (re)-infarction at 6 months by a combination of cTnI 0.1 g/L within 2 hours, a pathologic ECG and NT-pro BNP or cystatin C.
Analysis performed in patients with results available for all biochemical markers (n=415).
The combination of cTnI 0.1 g/L within 2 hours, an abnormal admis-sion ECG and cystatin C 1.28 mg/L appeared to be clinically most valu-able as it allowed the identification of the highest proportion of patients without adverse events during follow-up (table 9). All non-ACS patients suffering the combined endpoint were identified by this multimarker combi-nation due to at least one abnormal result. The event rates among the strata at same risk defined by the various marker combinations however, differed not significantly.
Table 9. Death or myocardial (re)-infarction at 6 months in relation to cTnI 0.1 g/L within 2 hours, a generally abnormal ECG and a third biochemical marker.
all negative 1 positive 2 positive p-value cTnI 0.1 g/L/ECG 3/161 (1.9%) 7/160 (4.4%) 15/94 (16.0%) <0.001 Median levels + NT-pro BNP 190 ng/L 1/131 (0.8%) 3/87 (3.4%) 21/197 (10.7%) 0.001 + Cystatin C 1.17 mg/L 0/103 (0.0%) 3/123 (2.4%) 22/189 (11.6%) <0.001 ROC-derived cut-offs + NT-pro BNP 550 ng/L 3/155 (1.9%) 2/105 (1.9%) 20/155 (12.9%) <0.001 + Cystatin C 1.28 mg/L 0/140 (0.0%) 4/121 (3.3%) 21/154 (13.6%) <0.001 Analysis performed in patients with results available for all biochemical markers (n=415).

45
Papers V and VI
General findings The demographic data and the baseline characteristics of the study popula-tion assessed in papers V and VI are given in table 10. In total, 453 patients had been randomized to the invasive strategy, 442 to the non-invasive strat-egy and 197 patients had not been randomized regarding the invasive vs. non-invasive strategy. Five-hundred forty-six patients (50%) had been ran-domized to 3 months treatment with dalteparin.
Table 10. Clinical characteristics of the subpopulation from the FRISC II-trial.
total (n=1092)
cTnInegative (n=377)
temporary cTnI
elevation(n=288)
persistent cTnI
elevation(n=233)
p-value*
median age 67.4 (59.3-73.9)
64.6 (56.3-71.2)
68.1 (60.1-73.9)
69.8 (63.2-76.0)
<0.001
Males 780 (71%) 251 (67%) 204 (71%) 180 (77%) 0.01 Hypertension 362 (33%) 107 (28%) 102 (35%) 87 (37%) 0.11 previous AMI 733 (67%) 219 (58%) 208 (72%) 176 (76%) 0.002 Diabetes 146 (13%) 45 (12%) 33 (12%) 30 (13%) 0.64 Smoking 645 (59%) 224 (59%) 174 (60%) 135 (58%) 0.64 previous stroke 54 (5%) 9 (2%) 20 (7%) 13 (6%) 0.47 previous PCI/CABG
116 (11%) 39 (10%) 22 (8%) 30 (13%) 0.13
Treatment at discharge
Aspirin 1051 (96%) 362 (96%) 293 (98%) 217 (93%) 0.02 Betablockers 936 (86%) 328 (87%) 250 (87%) 194 (83%) 0.19 ACE inhibitors 211 (19%) 48 (13%) 54 (19%) 61 (26%) <0.001 Ca antagonist 199 (18%) 63 (17%) 56 (19%) 48 (21%) 0.38 Digitalis 47 (4%) 5 (1%) 12 (4%) 18 (8%) 0.001 Diuretics 213 (20%) 50 (13%) 59 (21%) 54 (23%) 0.02 Nitrates 387 (35%) 117 (31%) 106 (37%) 80 (34%) 0.87 LLD 460 (42%) 176 (47%) 131 (46%) 82 (35%) 0.007
Medians given with 25th, 75th percentiles. Previous AMI defined as a history of myocardial infarction and/or cardiac Troponin T > 0.035 g/L at randomization. LLD: lipid lowering drugs. *P-values refer to comparison between patient cohorts with negative cTnI
0.01 g/L/temporary cTnI elevation > 0.01 g/L and persistent cTnI elevation > 0.01 g/L.
46
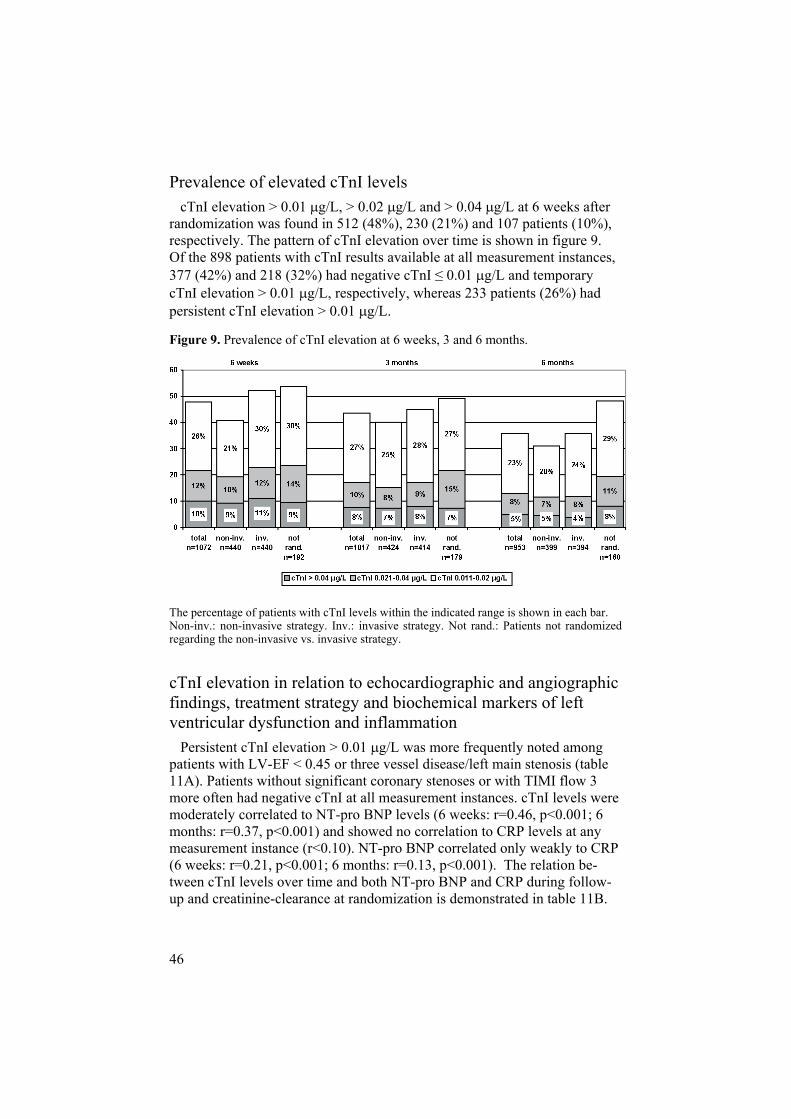
Prevalence of elevated cTnI levels cTnI elevation > 0.01 g/L, > 0.02 g/L and > 0.04 g/L at 6 weeks after randomization was found in 512 (48%), 230 (21%) and 107 patients (10%), respectively. The pattern of cTnI elevation over time is shown in figure 9. Of the 898 patients with cTnI results available at all measurement instances, 377 (42%) and 218 (32%) had negative cTnI 0.01 g/L and temporary cTnI elevation > 0.01 g/L, respectively, whereas 233 patients (26%) had persistent cTnI elevation > 0.01 g/L.
Figure 9. Prevalence of cTnI elevation at 6 weeks, 3 and 6 months.
The percentage of patients with cTnI levels within the indicated range is shown in each bar. Non-inv.: non-invasive strategy. Inv.: invasive strategy. Not rand.: Patients not randomized regarding the non-invasive vs. invasive strategy.
cTnI elevation in relation to echocardiographic and angiographic findings, treatment strategy and biochemical markers of left ventricular dysfunction and inflammation Persistent cTnI elevation > 0.01 g/L was more frequently noted among patients with LV-EF < 0.45 or three vessel disease/left main stenosis (table 11A). Patients without significant coronary stenoses or with TIMI flow 3 more often had negative cTnI at all measurement instances. cTnI levels were moderately correlated to NT-pro BNP levels (6 weeks: r=0.46, p<0.001; 6 months: r=0.37, p<0.001) and showed no correlation to CRP levels at any measurement instance (r<0.10). NT-pro BNP correlated only weakly to CRP (6 weeks: r=0.21, p<0.001; 6 months: r=0.13, p<0.001). The relation be-tween cTnI levels over time and both NT-pro BNP and CRP during follow-up and creatinine-clearance at randomization is demonstrated in table 11B.

47
Table 11. cTnI levels over time in relation to (A) results of echocardiography and coronary angiography and (B) biochemical markers.
(A) n cTnI negative
temporary cTnI
elevation
persistent cTnI
elevation
p-value*
LV-EF < 0.45 110 28 (25.5%) 33 (30.0%) 49 (44.5%) <0.001 3 vd/LM 127 28 (22.1%) 45 (35.4%) 54 (42.5%) <0.001 0 vd 47 28 (59.6%) 15 (31.9%) 4 (8.5%) 0.001 TIMI flow 0-1 113 35 (31.0%) 40 (35.4%) 38 (33.6%) 0.17 TIMI flow 3 211 97 (46.0%) 68 (32.2%) 46 (21.8%) 0.002
(B) n cTnI negative
temporary cTnI
elevation
persistent cTnI
elevation
p-value*
NT-pro BNP (6 w) 888 213 ng/ml 340 ng/ml 703 ng/ml <0.001 NT-pro BNP (6 m) 888 171 ng/ml 238 ng/ml 488 ng/ml <0.001 CRP (6 w) 896 2.36 mg/L 2.57 mg/L 2.63 mg/L 0.10 CRP (6 m) 896 2.01 mg/L 2.45 mg/L 2.40 mg/L 0.19 Creat-Cl. (rand.) 898 66.1 ml/min 62.4 ml/min 61.4 ml/min 0.001 3 vd/LM: Three vessel disease/left main stenosis. 0 vd: no significant coronary stenoses. Creat.-Cl. Creatinine-clearance. *P-values refer to comparison between patient cohorts with negative cTnI
0.01 g/L/temporary cTnI elevation > 0.01 g/L and persistent cTnI elevation > 0.01 g/L.
Patients randomized to the invasive strategy had a higher prevalence of persistent cTnI elevation > 0.01 g/L than patients randomized to the non-invasive strategy (107 patients 28% vs. 72 patients 19% ; p=0.005). Pa-tients in the non-invasive arm, on the other hand, more often had negative cTnI at all measurement instances compared to patients in the invasive arm (180 patients 48% vs. 147 patients 39% ; p=0.01). The prevalence of per-sistent cTnI elevation did not differ between patients randomized to pro-longed treatment with dalteparin or placebo (106 patients 24% vs. 127 patients 28% ; p=0.26).
According to model 3 in the multivariate logistic regression analysis, the only factors independently associated to persistent cTnI elevation were NT-pro BNP at 6 months (OR 2.5 95% CI 2.0-3.1 ; p<0.001), male gender (OR 2.2 95% CI 1.4-3.7 ; p=0.001) and randomization to the invasive strategy (OR 1.8 95% CI 1.2-2.7 ; p=0.004).

48
Prognostic importance of elevated cTnI levels In total, 106 patients (10%) died and 258 patients (24%) suffered an AMI during 5-year follow-up. Among the patient cohort eligible for the evaluation of cTnI results over time, death and AMI occurred in 66 patients (7%) and 110 patients (13%), respectively. The event rates in patients with and with-out elevation of cTnI relative to the tested cut-off values are shown in table 12, demonstrating that the 0.01 g/L cut-off provided the best risk stratifica-tion.
Table 12. Total mortality and myocardial infarction at 5 years in relation to cTnI levels over time.
cTnI negative temporary cTnI elevation
persistent cTnI elevation
p value
> 0.01 g/L Death 17/377 (4.5%) 18/288 (6.3%) 31/233 (13.3%) <0.001 AMI 39/362 (10.8%) 32/259 (12.4%) 39/202 (19.3%) 0.01 > 0.02 g/L Death 38/632 (6.0%) 20/207 (9.7%) 8/59 (13.6%) 0.04 AMI 71/596 (11.9%) 28/174 (16.1%) 11/53 (20.8%) 0.10 > 0.04 g/L Death 56/774 (7.2%) 9/108 (8.3%) 1/16 (6.3%) 0.91 AMI 93/718 (13.0%) 14/91 (15.4%) 3/14 (21.4%) 0.55
The mortality and rate of AMI during follow-up did not differ significantly between patients with negative cTnI 0.01 g/L and temporary cTnI eleva-tion > 0.01 g/L. Compared to these cohorts considered as one group, the unadjusted OR for death among patients with persistent cTnI elevation > 0.01 g/L was 2.8 (95% CI 1.7-4.6; p<0.001) and 1.9 (95% CI 1.2-2.8; p=0.005) for AMI. The occurrence of events is illustrated in figure 10.
Persistent cTnI elevation was independently predictive for mortality even after adjustment according to model 1 (OR 2.1 95% CI 1.3-3.6 ; p=0.005) but lost its independent prognostic value when NT-pro BNP and CRP at 6 months were entered as co-variates (model 2). In model 3, the only inde-pendent predictors for mortality were NT-pro BNP (OR 2.2 95% CI 1.5-3.3 ; p<0.001) and CRP (OR 1.5 95% CI 1.1-2.1 ; p=0.006). Even AMI in that model was only predicted by NT-pro BNP (OR 1.6 95% CI 1.2-2.2 ;p=0.002) and CRP (OR 1.5 95% CI 1.2-2.0 ; p=0.001). The prognostic im-portance of persistent cTnI elevation according to unadjusted multivariate logistic regression analysis in selected subgroups is illustrated in figures 11A and B.

49
Figure 10. Probability of death and myocardial infarction in relation to cTnI levels over time.
cTnI negative: cTnI < 0.01 g/L at all measurement instances. Temporary cTnI elevation: cTnI 0.01 g/L at 1-2 measurement instances. Persistent cTnI elevation: cTnI 0.01 g/L at all measurement instances. P-value regarding mortality = 0.001. P-value regarding AMI = 0.014.

50
Figure 11. Odds ratios for (A) mortality and (B) myocardial infarction associated with persistent cTnI elevation in relation to other risk indicators. (A)
(B)
Creatinine-clearance: median level at randomization applied as cut-off.NT-pro BNP and CRP: median levels at 6 months applied as cut-offs.

51
DISCUSSION
cTnI for early assessment of patients with suspected ACS
Early diagnosis of AMI (papers I and II) With regard to their high cardiospecificity, the troponins are acknowledged as markers of choice for the diagnosis of AMI and for risk stratification in patients with suspected and confirmed ACS 23 . For clinical decision-making, a troponin cut-off corresponding to the 99th percentile among healthy individuals has been proposed or alternatively, the lowest concentra-tion measurable with a < 10% CV 1, 20 . The results from the FAST II-study support this recommendation, firstly as cTnI outperformed CK-MB and myoglobin in terms of early AMI diagnosis, and secondly, as the 10% CV level of the applied assay (Stratus CS; cTnI 0.1 g/L) provided an overall superior diagnostic performance compared to the other tested cTnI cut-offs. Using an accelerated POC testing protocol and a lower cTnI cut-off allowed reliable ruling out of AMI in all but one AMI patients already 2 hours after admission. Importantly, minor cTnI elevation was detectable at least as early as myoglobin, commonly regarded as the earliest marker of cardiac damage. This is probably related to leakage of cTnI from the cytoso-lic pool after disturbance of cardiomyocyte cell membrane integrity due to ischemic injury.
As cardiac markers have different characteristics and each offer certain advantages, a logical approach would be the use of some of them in combi-nation for diagnostic purposes. Recent studies applying traditional WHO criteria for defining AMI have demonstrated favourable results in particular for panels using troponin and myoglobin results 37, 38 . The tested multi-marker strategies in the FAST II-study however, failed to provide diagnostic value beyond that obtained from cTnI 0.1 g/L which depends on the use of a more appropriate standard for defining AMI (ESC/ACC criteria) com-pared to the aforementioned studies.
However, despite the usefulness of troponin for detecting AMI, the attend-ing clinician needs to be aware that small troponin elevation may occur in

52
non-ischemic cardiac pathologies 10, 11 and to a certain extent in general populations 132, 136 . This has become increasingly important with the implementation of lowered cut-offs and newer-generation assays with high sensitivity. Thus, there is a risk for misclassification and unnecessary CCU admission of troponin-positive patients who do not have an ACS. It therefore must be emphasized that for fulfillment of the diagnosis of AMI, troponin levels should demonstrate a significant rise or fall in an appropriate clinical context 23 . However, it may take hours from the index event until troponin levels rise over currently used thresholds. Clinicians therefore, might tend to admit patients instead for waiting until AMI has definitely been ruled in or out by serial marker testing. In order to speed up clinical decision-making, the use of computer-based decision aids able to detect more subtle but rele-vant changes of troponin levels might be a valuable option.
An ANN is a computational technique able to identify complex multidi-mensional relationships in clinical data not apparent to other forms of analy-sis [46]. Applying early serial cTnI and myoglobin results as input variables, the ANN-algorithms in the FASTER I-study were useful for diagnosis of AMI within 2 hours even when different biochemical criteria for defining AMI were used. However, the early diagnostic value of these ANN-algorithms was not consistently better than of a single cTnI cut-off which probably depends on the use of myoglobin results as input variable given the low cardiospecificity of this marker. A different selection of inputs, i.e. cTnI and its rate of change only, might have resulted in higher diagnostic esti-mates relative to a conventional cTnI cut-off.
Early risk prediction (papers III and IV) The purpose of early evaluation of chest pain patients by troponin meas-urements is not only to detect myocardial damage but more importantly, to identify patients who are at high risk for future cardiac events. Previous stud-ies in ACS patients have demonstrated that any detectable troponin elevation is associated with an impaired prognosis 19 . However, in unselected pa-tient populations, slight troponin elevations may occur in other pathologies than AMI 10, 11 or represent analytical noise, given the relative impreci-sion of the most available assays at the low end of their range. Furthermore, negative troponin results do not equate with the absence of an ACS why other risk markers may be considered for improving risk stratification.
According to the results from the FAST II-study, CK-MB, myoglobin and the tested multimarker strategies did not provide superior prognostic value compared to cTnI apart from myoglobin regarding the endpoint of total mor-tality. This is in line with previous studies 29, 30, 137 and depends on the higher sensitivity of cTnI regarding the detection of patients at increased

53
cardiac risk (i.e. patients with AMI and unstable angina) and the lower car-diospecificity of CK-MB and myoglobin. Taken together, these findings thus emphasize that neither CK-MB nor myoglobin should be considered for the prediction of cardiovascular events in patients with suspected ACS when cTnI results are available.
Biochemical markers of hemodynamic stress, inflammation and renal function have gained much attention for risk prediction. The prognostic value of these markers in conjunction to cTnI results has been closer evalu-ated in the pooled chest pain population from the FAST II- and FASTER I-studies. This analysis demonstrates that, apart from cTnI, also NT-pro BNP and cystatin C independently predicted the combined endpoint of total mor-tality or AMI. Creatinine-clearance was moderately predictive on univariate analysis whereas CRP offered only very limited prognostic information which contrasts to its prognostic importance among healthy individuals and ACS patients [81, 91-93, 98, 99]. This finding probably depends in part on population characteristics as a fairly heterogeneous population was evaluated in the present analysis without exclusion of patients with inflammatory con-ditions.
Adding a third marker to serially measured cTnI results and ECG findings, the conventional cornerstones of the initial patient assessment, allowed the identification of a patient cohort at very low risk already 2 hours after admis-sion. However, given the low event rate in the sample population, the results of the analysis do not permit conclusions which marker combination is supe-rior. As kidney function is assessed in most patients admitted because of chest pain, the findings suggest the use of an adequate renal marker, prefera-bly cystatin C 1.28 mg/L, as a conjunct to cTnI and ECG findings. This cut-off roughly corresponds to a GFR of 57 ml/min 138 and is in the same range as that denoting high risk among elderly persons and patients with coronary artery disease 118, 119 . Routine analyses of NT-pro BNP or CRP in this setting in contrast, are not advocated as they do not provide additional prognostic information.
Early prediction of infarct size (paper II) The purpose of very early prognostication in patients with suspected ACS is not only the identification of low-risk subjects but also the identification of patients at high risk for future events. In particular the extent of AMI is a major determinant of prognosis 121, 122 . Biochemical markers of myocar-dial necrosis correlate well with infarct size 125 . Thus, a crude prediction of the impending infarct size based on very early biochemical marker results could facilitate the decision whether to use infarct-limiting therapy, e.g. per-

54
cutaneous coronary intervention or treatment with glycoprotein IIb/IIIa in-hibitors 124 .
In the FASTER I-study, an ANN-algorithm using early cTnI and myoglo-bin results as input variables indicated a ‘major AMI’ considerably earlier than would have been possible by applying conventional cTnI results. This is probably due to the use of myoglobin and its rate of change as inputs, both good measures of infarct size 128, 129 . Thus, while it is likely that my-oglobin was the reason for the non-superiority of the ANN-algorithms for AMI diagnosis relative to single cTnI cut-offs, it improved the performance of the ANN-algorithm for the prediction of ‘major infarct’ size. However, as ANN only have limited ability to explain possible causal relationships, fur-ther investigation in this field is warranted.
cTnI in patients stabilized after an episode of ACS
Prevalence of cTnI elevation (paper V) While the role of cardiac troponin testing for the diagnosis of AMI and risk
prediction in patients with an ongoing ACS is well established 23 , there is no information regarding the prevalence and importance of elevated troponin levels in the stable phase after an episode of ACS. This depends in part on the unavailability of highly sensitive assays up until now. Using the recently refined version of the AccuTnI assay, a high prevalence of elevated cTnI levels over at least 6 months was detected in stable post-ACS patients who participated in the FRISC II-trial. During this period, the proportion of cTnI levels > 0.01 g/L (i.e. the concentration measurable with a 10-20% CV) subsequently decreased from 48% to 36% and 26% of the patients had per-sistent cTnI elevation > 0.01 g/L.
Possible causes of cTnI elevation (papers V and VI) As in previous studies evaluating populations without ACS, elevated cTnI levels in stable post-ACS patients were related to age, male gender, cardio-vascular high-risk features and co-morbidities such as hypertension, de-pressed left ventricular systolic function or impaired renal function [4, 10, 11, 136, 139-141]. In these conditions, detectable troponin levels may be caused by increased demand ischemia or myocardial strain because of vol-ume and pressure overload 4, 10, 11 . Persistent cTnI elevation in stable post-ACS patients was also strongly associated to a previous AMI which together with the higher prevalence of adverse angiographic findings in per-sistently cTnI positive patients identifies a greater atherosclerotic burden as a

55
contributing factor. One could also speculate that the temporary pattern of cTnI elevation in some patients might depend on acute causes, e.g. clinically silent ruptures of inflammated coronary plaques with thrombus formation and downstream embolization. However, cTnI elevation was not more com-mon in patients with anginal complaints and CRP levels, known to mirror vascular inflammation 142 and more severe coronary lesions 143, 144 ,did not correlate with cTnI results. Thus, it seems not likely that cTnI posi-tivity after an ACS is related to continuing coronary plaque instability or vascular inflammation.
Remodeling and apoptosis could be another explanation for elevated tro-ponin levels in the long-term after an ACS. In this condition, leakage of tro-ponin and its degradation fragments to the blood stream is owing to an ongo-ing disintegration of myofibrils 145 . Apoptotic processes persist for months after an AMI 146, 147 . Accordingly, an elevated rate of cTnI cleavage over several weeks post AMI has been demonstrated in animals 148 . The fact that, besides male gender, NT-pro BNP levels at 6 months
were the most important predictor for persistent cTnI elevation supports this hypothesis. Thus, the association between persistent cTnI positivity and in-creased NT-pro BNP levels probably reflects larger previous or index infarc-tions and/or more pronounced remodeling processes after an ACS. Patients with negative cTnI and those with temporary cTnI elevation and LV-EF 0.45 on the other side, had considerably low NT-pro BNP levels at 6 months indicating an index event with minor myocardial damage and/or a greater myocardial recovery.
Prognostic importance of cTnI elevation (papers V and VI) Any detectable cTnI level in stable post-ACS patients was associated with an impaired prognosis which corresponds to previous studies evaluating both patients with and without an ACS 3-6, 19, 132 . Patients with persistent cTnI elevation > 0.01 g/L had a 2.8 fold higher mortality and a nearly dou-bled rate of AMI compared to patients with negative cTnI and temporary cTnI elevations considered as one group. However, on multivariate logistic regression analysis, both NT-pro BNP and CRP levels at 6 months were stronger predictors for adverse outcome. Patients with persistent cTnI eleva-tion and NT-pro BNP above the 6 month median of 232 ng/ml had a consid-erable high event rate which is consistent with the concept that this cohort also had suffered more pronounced myocardial damage. Patients with NT-pro BNP < 232 ng/ml at 6 months in contrast, had a low event rate despite persistent cTnI elevation. Thus, it appears that the processes causing persis-tent cTnI elevation in this subcohort have less severe consequences. CRP levels at 6 months provided incremental prognostic value to cTnI results regardless whether cTnI was persistently elevated or not. Considering the

56
weak correlation between CRP and NT-pro BNP levels, it is likely that the prognostic value of CRP is determined by a prolonged low-grade inflamma-tion of extra-cardiac origin 103, 104 .
Clinical implications and future directions
Early diagnosis of AMI in chest pain patients by measurement of cTnI Following the introduction of the ESC/ACC guidelines for management of non ST-segment elevation ACS 1 , the troponins have become widely an-ticipated as biochemical markers of choice for the detection of myocardial necrosis. The diagnostic superiority of the troponins relative to other markers of myocardial damage has been recently re-emphasized by recommendations from the National Academy of Clinical Biochemistry 31 . The results from the FAST II-study confirm the higher value of cTnI compared to CK-MB and myoglobin in terms of early AMI diagnosis. This is also supported by more recent findings from Kavsak and colleagues 149 . Thus, CK-MB and myoglobin should be regarded as historic markers and only be considered when troponin results are not available.
Serial cTnI results applying an accelerated POC testing protocol allowed the early and reliable assessment of patients admitted because of chest pain. In particular patients without an AMI and those with a low risk for future cardiovascular events could be identified rapidly. Thus, such a protocol could permit targeting of these patients to appropriate levels of health care, e.g. admission to a stepdown unit or evaluation in the outpatient setting. This might improve hospital resource utilization due to a reduction of the length of hospital stay 41-43 . In the remaining patients, further observation in-cluding biochemical marker monitoring in a controlled clinical setting is advocated.
However, using contemporary decision limits on assays with improved sensitivity, small but prognostically relevant troponin elevation can be de-tected in patients with other pathologies than AMI and even in apparently healthy individuals 10, 11, 132, 136 . It is possible that such troponin re-lease results from temporarily increased cardiomyocyte membrane perme-ability which challenges the paradigm that cardiac troponin is undetectable in the blood stream in the absence of ongoing myocardial damage 150 .This implies a considerable risk for misdiagnosis of AMI in patients with non-ischemic chest pain when assessment only is based on troponin results. It is therefore important to emphasize that for fulfilment of the diagnosis of

57
AMI, a significant rise and/or fall of serially measured troponin results needs to be observed in combination with clinical evidence of ongoing myocardial damage (e.g. ECG changes) [23, 31].
The interpretation of troponin results may be facilitated by the implemen-tation of a relative change criterion or the use of more sophisticated statisti-cal techniques, e.g. ANN. The output of an ANN could also be integrated in an informative way in decision support systems or electronic patient records, thereby providing real-time information assisting the clinician in the decision whether and where to admit the patient. The ANN-algorithms in the FASTER I-study performed fairly good but not considerably better than a single cTnI cut-off which may depend on the selected input variables. The clinical value of an optimized ANN-approach for rapid assessment of chest pain patients thus, remains to be validated prospectively.
cTnI for early risk prediction in patients with chest pain The results presented in this work confirm the role of the cardiac troponins as the preferred biochemical markers for prognostication in patients with suspected ACS. Using a multimarker approach based on cTnI results al-lowed the early identification of patients at very low risk. However, with growing insights in the mechanisms of acute coronary ischemia, the investi-gation of new risk markers will probably continue to accelerate in the next years. At present, markers of plaque destabilization (e.g. myeloperoxidase 151 ) or myocardial ischemia (e.g. ischemia-modified albumin 152 ) gain
much attention. Proteomic and genomic approaches are likely to further ex-tend the list of candidate markers. In this context, it is noteworthy that a new risk marker needs to provide incremental value to established prognostic tools in order to be useful for clinical application. The importance of this issue is illustrated by the results from the pooled lower-risk population from the FAST II- and FASTER I-studies. In this analysis, any of the tested mark-ers provided superior prognostic value compared to risk assessment applying a more conventional approach based on early cTnI results, ECG findings and an adequate marker of kidney function such as cystatin C. Thus, more com-plex approaches such as scoring systems 153, 154 or computational meth-ods 155 probably are needed to improve risk prediction.
Very early risk assessment often depends on limited information. There-fore, much could be gained when the size of the impending infarct, known to be a major determinant of poor prognosis 121, 122 , would be predictable at an early stage. Initiation of infarct-limiting therapy would be an attractive option in these patients 124 . The ANN-algorithm evaluated in the FASTER I-study indicated a ‘major AMI’ under development already after 2 hours and considerably earlier than cTnI results. Thus, this ANN-approach appears

58
to be a useful instrument for prospective studies testing the value of treat-ments for limiting the extent of infarct size.
cTnI for risk prediction in the stable phase after an ACS Contrasting to patients with suspected or ongoing ACS, the role of tro-ponin testing in the long-term after an ACS has not been studied so far which in part depends on the unavailability of highly sensitive assays up until now. The results from the FRISC II-study demonstrate for the first time that an unexpected high proportion of stable post-ACS patients has small but persistent cTnI elevation during at least 6 months from the index event. This appears mainly to reflect an ongoing loss of cardiomyocytes due to remodel-ing processes and apoptosis. As in patients with congestive heart failure 141 , persistent cTnI elevation was associated with poor outcome, in par-
ticular when also NT-pro BNP and CRP levels were elevated. These three markers thus, appear to reflect different adverse pathophysiological mecha-nisms why their implementation as a multimarker panel into follow-up rou-tines after an ACS appears to be promising. This approach might allow the identification of patients at high risk in whom further cautious investigation and intensive secondary preventive and cardioprotective therapies are man-datory.

59
CONCLUSIONS
Based on this work, the following conclusions can be made:
1. Regarding patients with suspected ACS:
- cTnI with a cut-off corresponding to the 10% CV level is the most appropriate biochemical marker for diagnostic and prognostic as-sessment.
- an accelerated POC testing protocol applying serially measured cTnI results and a lower cut-off (99th percentile) allows early exclusion of AMI,
- the early diagnosis of AMI might be facilitated by the use of ANN-algorithms based on cTnI and myoglobin results,
- adding a third biomarker to POC cTnI results and ECG findings al-lows the early identification of a patient cohort at very low risk. An adequate measure of renal function such as cystatin C appears to be most suitable as conjunct to cTnI and the ECG for improvement of risk stratification,
- risk assessment may be further improved by the early identification of an impending major non ST-segment elevation infarction with the use of an ANN-algorithm applying results of cTnI and myoglobin.
2. Regarding stabilized patients after an episode of ACS:
- minor but persistent cTnI elevation can frequently be detected dur-ing a period of at least 6 months after an ACS when a highly sensi-tive assay is used,
- persistent cTnI elevation appears mainly to be related to impaired left ventricular function,
- persistent cTnI elevation is associated with a considerable risk for adverse events during 5 year follow-up. Levels of NT-pro BNP and CRP provide incremental prognostic information to cTnI results.

60
SUMMARY IN SWEDISH (SAMMANFATTNING PÅ SVENSKA)
Ett akut koronärt syndrom (ACS) förorsakas ofta av en ruptur av ett arteri-osklerotiskt plack i hjärtats kranskärl med sekundär bildning av tromber. Dessa tromber kan embolisera nedströms och orsaka celldöd i hjärtmuskeln vilket är likvärdigt med hjärtinfarkt. De känsligaste och mest specifika bio-kemiska infarktmarkörerna är troponinerna (kardiellt troponin I cTnI eller troponin T cTnT ) som också har ett högt prediktivt värde för framtida kar-diovaskulära händelser. Följaktligen anses troponinerna vara en hörnsten för både diagnos av hjärtinfarkt och riskbedömning av patienter med ACS enligt aktuella internationella riktlinjer. Analysmetoden för cTnI har på senare år emellertid förfinats och förenklats vilket bland annat har möjliggjort snabba seriella mätningar på patientnära instrument. Det finns således skäl att un-dersöka det diagnostiska och prognostiska värdet av cTnI mätt med förbätt-rad analysteknik hos oselekterade patientgrupper med bröstsmärtor där ett ACS ännu inte har bekräftats. En snabb och tillförlitlig bedömning av dessa patienter med hjälp av cTnI mätningar skulle öppna möjligheten att redan i ett tidigt skede kunna slussa olika patientgrupper vidare till lämpliga vårdni-våer och behandlingar. Frågan är också om andra riskindikatorer som markö-rer för hjärtfunktion, inflammation eller njurfunktion tillför tilläggsinforma-tion till cTnI resultat och på så sätt förbättrar den tidiga riskbedömningen. Slutligen är det oklart om förhöjda cTnI värden även kan påvisas hos patien-ter som har stabiliserats efter en episod med ACS och om eventuella cTnI stegringar hos dessa patienter är förknippade med en försämrad långtidspro-gnos.
Syftet med denna avhandling var således att undersöka betydelsen av cTnI för snabb utredning av oselekterade bröstsmärtepatienter med misstänkt ACS som deltog i FAST II-studien (n=197) och FASTER I-studien (n=380). Dessutom utvärderades förekomsten av cTnI stegring och dess prognostiska betydelse hos stabiliserade post-ACS patienter som hade inkluderats i FRISC II-studien (n=1092).
Resultat från FAST II- och Faster I-studierna visar att cTnI är mycket vär-defull för tidig diagnostik av hjärtinfarkt och riskvärdering av patienter med bröstsmärtor. Snabba seriella mätningar av cTnI på ett patientnära analysin-strument (Stratus CS) tillät redan 2 timmar efter ankomst ett säkert uteslu-

61
tande av hjärtinfarkt och identifiering av lågrisk patienter om cTnI kombine-rades med EKG fynd och serumnivåer av en markör för njurfunktion, före-trädesvis cystatin C. Analys av andra markörer för hjärtmuskelskada (CK-MB eller myoglobin) förbättrade inte diagnostiken och tillförde ingen ytter-ligare prognostisk information vilket också gällde för markörer för hjärtfunk-tion (NT-pro BNP) och inflammation (CRP).
Den tidiga bedömningen av bröstsmärtepatienter med hjälp av snabba seri-ella cTnI mätningar kan möjligtvis förbättras genom användning av utvalda statistiska metoder som t ex artificiella neurala nätverk (ANN). Ett ANN är en avancerad datorbaserad algoritm som har förmågan att bearbeta och tolka markörresultat och på så vis identifiera komplexa samband som inte kan upptäckas med hjälp av konventionella analyser. I FASTER I-studien utvär-derades olika ANN-algoritmer som baserades på cTnI resultat i kombination med myoglobin. Med denna metod kunde en hjärtinfarkt säkert bekräftas eller uteslutas redan 2 timmar efter ankomst. ANN-algoritmernas prestanda var dock inte avsevärt bättre än infarktdiagnostik med enkla mätningar av cTnI. Däremot tillät tolkningen av tidiga resultat av cTnI och myoglobin med hjälp av en annan ANN-algoritm prediktionen av en stor infarkt under utveckling vid 2 timmar från ankomst. En sådan ANN-algoritm förefaller således att vara ett lämpligt verktyg för framtida utvärderingar av infarktbe-gränsande behandlingar.
Resultaten av analyserna från FRISC II-studien visar att persisterande cTnI stegring kunde detekteras med hjälp av en mycket känslig metod (Access AccuTnI) hos 26% av alla post-ACS patienter under en 6-månaders period från indexhändelsen utan att det fanns tecken till fortsatt instabil kranskärl-sjukdom. Förutom manlig kön var persisterande cTnI stegring framför allt relaterad till NT-pro BNP nivåer efter 6 månader vilket indikerar ett sam-band mellan det pågående cTnI utsläppet och ogynnsamma strukturella pro-cesser i hjärtmuskeln efter ACS episoden. Persisterande cTnI stegring var i högsta grad förenad med en ökad dödlighet och förekomst av hjärtinfarkt under en 5-årig uppföljningstid. Serumnivåer av NT-pro BNP och CRP vid 6 månader tillförde betydelsefull tilläggsinformation. Resultat från denna ana-lysen kan således användas som utgångspunkt för framtida studier med syf-tet att identifiera högriskpatienter under uppföljningstiden efter en episod med ACS. Dessa individer kan potentiell vara betjänta av vidare ingående utredning och intensiv förebyggande läkemedelsbehandling.
Sammanfattningsvis bekräftar undersökningarna i denna avhandling att seriella mätningar av cTnI är ett värdefullt verktyg för tidig diagnostisk och prognostisk bedömning av patienter med misstänkt ACS. Därutöver tillför resultaten ny och så här långt unik kunskap beträffande betydelsen av cTnI hos stabiliserade patienter som har haft en episod med ACS.

62
ACKNOWLEDGEMENTS
I wish to express my sincere gratitude to all those who have contributed to the work leading to this thesis. In particular, I would like to thank:
Bertil Lindahl, my supervisor, for years of never ending and never failing enthusiasm, engaged guidance and always constructive criticism. I guess, it is hard to find someone who, even in the darkest nights of Sunnersta, is so extremely helpful and effective,
Stefan James, my co-supervisor, for stimulating co-operation, for introduc-ing me to cardiology celebs and for guidance through the mysterious world of Swedish grammar,
Lars Wallentin, for support and sharing your knowledge in the field of clinical research,
Johan Ellenius, my co-author, for introducing me into the fancy microcos-mos of biostatistical methods,
my co-authors Jonas Oldgren, Anna Nordenskjöld, Torgny Groth, Mikael Dellborg, Ewa Swahn, Per Venge and Bo Lagerqvist, for sharing your exper-tise and critically reviewing the manuscripts, thereby making them better,
my colleagues and members of the research group on ischemic heart dis-ease Bertil Andrén, Erik Björklund, Christina Christersson, Ziad Hijazi, Nina Johnston, Christoph Varenhorst and Gerhard Wikström, for encouragement and fruitful discussions,
Gunnar Frostfeldt, chief of the Department of Cardiology, for giving me the possibility to work with my thesis far away from the hospital and its duties,
Anna Karin Lindow for skilful help with the preparation of the manu-scripts and this thesis,
the staff at the Uppsala Clinical Research Centre, for supporting the con-duction of the studies and assistance with questions regarding statistics and the preparation of the databases,

63
my parents, for having patience with me and not arguing about geographic distances,
Antje, for much more than encouraging me with this thesis.
This work was supported by grants from the Erik, Karin and Gösta Se-lander Foundation and the Swedish Heart and Lung Foundation.

64
REFERENCES
1. Alpert JS, Thygesen A, Antman EM, et al. Myocardial infarction redefined - a consensus document of the joint European Society of Cardiology / American College of Cardiology Committee for the Redefinition of Myocardial Infarc-tion. J Am Coll Cardiol 2000; 36: 959-69.
2. Venge P, Lagerqvist B, Diderholm E, et al. Clinical performance of three car-diac troponin assays in patients with unstable coronary artery disease (a FRISC II substudy). Am J Cardiol 2002; 89: 1035-41.
3. Kontos MC, Shah R, Fritz LM, et al. Implication of different cardiac troponin I levels for clinical outcomes and prognosis of acute chest pain patients. J Am Coll Cardiol 2004; 43: 958-65.
4. Horwich TB, Patel J, MacLellan WR, et al. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and in-creased mortality rates in advanced heart failure. Circulation 2003; 108: 833-38.
5. Waxman DA, Hecht S, Schappert J, et al. A model for troponin I as a quantita-tive predictor of in-hospital mortality. J Am Coll Cardiol 2006; 48:1755-62.
6. Kavsak PA, Newman AM, Lustig V, et al. Long-term health outcomes associ-ated with detectable troponin I concentrations. Clin Chem 2007; 53: 220-27.
7. Collinson PO, Boa FG, Gaze DC. Measurement of cardiac troponins. Ann Clin Biochem 2001; 38: 423-49.
8. Katus HA, Remppis A, Scheffold T, et al. Intracellular compartmentation of cardiac troponin T and its release kinetics in patients with reperfused and non-reperfused myocardial infarction. Am J Cardiol 1991; 67: 1360-67.
9. Adams JE, Schechtman KB, Landt Y, et al. Comparable detection of acute myocardial infarction by creatine kinase MB isoenzyme and cardiac troponin I. Clin Chem 1994; 40: 1291-95.
10. Jeremias A, Gibson M. Narrative review: Alternative causes for elevated car-diac troponin levels when acute coronary syndromes are excluded. Ann Intern Med 2005; 142: 786-91.
11. Korff S, Katus HA, Giannitsis E. Differential diagnosis of elevated troponins. Heart 2006; 92: 987-93.
12. Collinson PO, Stubbs PJ. Are the troponins confusing? Heart 2003; 89: 1285-87.
13. Freda BJ, Tang WH, Van Lente F, et al. Cardiac troponins in renal insuffi-ciency. J Am Coll Cardiol 2002; 40: 2065-71.
14. Ooi DS, Isolato PA, Veinot JP, et al. Correlation of antemortem serum creatine kinase, creatine-kinase MB, troponin I, and troponin T with cardiac pathology. Clin Chem 2000; 46: 338-44.
15. Sharma R, Gaze DC, Pellerin D, et al. Cardiac structural and functional abnor-malities in end stage renal disease patients with elevated cardiac troponin T. Heart 2006; 92: 804-09.

65
16. Lowbeer C, Stenvinkel P, Pecoits-Filho R, et al. Elevated cardiac troponin T in predialysis patients is associated with inflammation and predicts mortality. J In-tern Med 2003; 253: 153-60.
17. Scott B, Deman A, Peeters P, et al. Cardiac troponin T and malondialdehyde modified plasma lipids in hemodialysis patients. Nephrol Dial Transplant 2003; 18: 737-42.
18. Panteghini M, Gerhardt W, Apple FS, et al. Quality specifications for cardiac troponin assays. Clin Chem Lab Med 2001; 39: 174-78.
19. Lindahl B, Venge P, Wallentin L. Relation between troponin T and the risk of subsequent cardiac events in unstable coronary artery disease. Circulation 1996; 93: 1651-57.
20. Apple FS, Wu AH, Jaffe AS. European Society of Cardiology and American College of Cardiology guidelines for redefinition of myocardial infarction: How to use existing assays clinically and for clinical trials. Am Heart J 2002; 144: 981-86.
21. Yeo KT, Storm CA, Li Y, et al. Performance of the enhanced Abbot AxSYM cardiac troponin I reagent in patients with heterophilic antibodies. Clin Chim Acta 2000; 292: 13-23.
22. Eriksson S, Halenius H, Pulkki K, et al. Negative interference in cardiac tro-ponins I immunoassays by circulating troponins autoantibodies. Clin Chem 2005; 51: 839-47.
23. Bertrand ME, Simoons ML, Fox KA, et al. Management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J 2002; 23: 1809-40. Erratum in: Eur Heart J 2003; 24: 1174-75. Eur Heart J 2003; 24: 485.
24. Storrow AB, Gibler WB. Chest pain centers: Diagnosis of acute coronary syn-dromes. Ann Emerg Med 2000; 35: 449-61.
25. Läkemedelsboken 2005/2006. Stockholm: Apoteket AB, 2005. 26. Pope JH, Selker HP. Acute coronary syndromes in the emergency department:
Diagnostic characteristics, tests and challenges. Cardiol Clin 2005; 23: 423-51. 27. Cummins P, McGurk B, Littler WA. Possible diagnostic use of cardiac specific
contractile proteins in assessing cardiac damage. Clin Sci 1979; 56: 30. 28. Katus HA, Remppis A, Looser, et al. Enzyme linked immunoassay of cardiac
troponin T for the detection of acute myocardial infarction in patients. J Mol Cell Cardiol 1989; 21: 1349-53.
29. Hamm CW, Goldmann BU, Heeschen C, et al. Emergency room triage of pa-tients with acute chest pain by means of rapid testing for cardiac troponin I or T. N Engl J Med 1997; 337: 1648-53.
30. Ohman EM, Armstrong PW, Christenson RH, et al. Cardiac troponin T levels for risk stratification in acute myocardial ischemia. N Engl J Med 1996; 335: 1333-41.
31. Morrow DA, Cannon CP, Jesse RL, et al. National Academy of Clinical Bio-chemistry Laboratory Medicine Practice Guidelines: Clinical characteristics and utilization of biochemical markers in acute coronary syndrome. Clin Chem 2007; 53: 552-74.
32. Dreyfus JC, Schapira G, Rasnais J, et al. La creatine-kinase sérique dans le diagnostic de l’infarctus myocardique. Rev Fran Etud Clin Biol 1960; 5: 386- 389.
33. Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology / World Health Or-ganization Task Force on the Standardization of Clinical Nomenclature. Circu-lation 1979; 3: 607-09.

66
34. Vaidya HC, Vaananen K. Myoglobin and carbonic anhydrase III. In: Wu AH (ed.).Cardiac markers. Totowa NJ: Humana Press Inc., 1998: 103-112.
35. Kontos MC, Anderson FP, Schmidt KA, et al. Early diagnosis of acute myo-cardial infarction in patients without ST-segment elevation. Am J Cardiol 1999; 83: 155-58.
36. Balk EM, Ioannidis JP, Salem D, et al. Accuracy of biomarkers to diagnose acute cardiac ischemia in the emergency department: A meta-analysis. Ann Emerg Med 2001; 37: 478-94.
37. McCord J, Nowak RM, Mc Cullough PA, et al. Ninety-minute exclusion of acute myocardial infarction by use of quantitative point-of-care testing of my-oglobin and troponin I. Circulation 2001; 104: 1483-88.
38. Ng SM, Krishnaswamy P, Morissey R, et al. Ninety-minute accelerated critical pathway for chest pain evaluation. Am J Cardiol 2001; 88: 611-17.
39. Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA guidelines for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2000; 36: 970-1062.
40. Novis DA, Jones BA, Dale JC, et al. Biochemical markers of myocardial injury test turnaround time: a College of American Pathologists Q-probe Study. Arch Path Lab Med 2004; 128: 158-64.
41. Roberts RR, Zalenski RJ, Mensah EK, et al. Costs of an emergency depart-ment-based accelerated diagnostic protocol vs hospitalization in patients with chest pain: a randomized controlled trial. JAMA 1997; 278: 1670-76.
42. Collinson PO, John C, Lynch S, et al. A prospective randomized controlled trial of point-of-care testing on the coronary care unit. Ann Clin Biochem 2004; 41: 397-404.
43. Apple FS, Chung AY, Kogut ME, et al. Decreased patient charges following implementation of point-of-care cardiac troponin monitoring in acute coronary syndrome patients in a community hospital cardiology unit. Clin Chim Acta 2006; 370: 191-95.
44. Goldman L, Weinberg M, Weisberg M, et al. A computer-derived protocol to aid in the diagnosis of emergency room patients with acute chest pain. N Engl J Med 1982; 307: 588-96.
45. Selker HP, Griffith JL, D’Agostino RB. A tool for judging coronary care unit admission appropriateness, valid for both real-time and retrospective use. Med Care 1991; 29: 610-27.
46. Cross SS, Harrison RF, Kennedy RL. Introduction to neural networks. Lancet 1995; 346: 1075-79.
47. Lisboa PJ. A review of evidence of health benefit from artificial neural net-works in medical intervention. Neural Netw 2002; 15: 11-39.
48. Baxt WG. Use of an artificial neural network for the diagnosis of myocardial infarction. Ann Intern Med. 1991; 115: 843-48. Erratum in: Ann Intern Med 1992; 116: 94.
49. Furlong JW, Dupuy ME, Heinsimer JA. Neural network analysis of serial car-diac enzyme data. A clinical application of artificial machine intelligence. Am J Clin Pathol 1991; 96: 134-41.
50. Ellenius J, Groth T, Lindahl B, et al. Early assessment of patients with sus-pected acute myocardial infarction by biochemical monitoring and neural net-work analysis. Clin Chem 1997; 43: 1919-25.
51. Kennedy RL, Harrison RF, Burton AM, et al. An artificial neural network sys-tem for diagnosis of acute myocardial infarction (AMI) in the accident and

67
emergency department: evaluation and comparison with serum myoglobin measurements. Comput Methods Programs Biomed. 1997; 52: 93-103.
52. Baxt WG, Shofer FS, Sites FD, et al. A neural computational aid to the diagno-sis of acute myocardial infarction. Ann Emerg Med 2002; 39: 366-73.
53. Westerhout CM, Fu Y, Lauer MS, et al. Short- and long-term risk stratification in acute coronary syndrome. The added value of quantitative ST-segment de-pression and multiple biomarkers. J Am Coll Cardiol 2006; 48: 939-47.
54. Heeschen C, van den Brand MJ, Hamm CW, et al. Angiographic findings in patients with refractory unstable angina according to troponin status. Circula-tion 1999; 104: 1509-14.
55. Lindahl B, Diderholm E, Lagerqvist B, et al. Mechanisms behind the prognos-tic value of troponin T in unstable coronary artery disease: a FRISC II substudy. J Am Coll Cardiol 2001; 38: 979-86.
56. Wong GC, Morrow DA, Murphy S, et al. Elevations in troponin T and I are associated with abnormal tissue level perfusion: a TACTICS-TIMI 18 substudy. Treat Angina with Aggrastat and Determine Cost of Therapy with an Invasive or Conservative Strategy-Thrombolysis in Myocardial Infarction. Cir-culation 2002; 106: 202-07.
57. Licka M, Zimmermann R, Zehelein J, et al. Troponin T concentrations 72 hours after myocardial infarction as a serological estimate of infarct size. Heart 2002; 87: 520-24.
58. Panteghini M, Cuccia C, Bonetti G, et al. Single-point cardiac troponin T at coronary care unit discharge after myocardial infarction correlates with infarct size and ejection fraction. Clin Chem 2002; 48: 1432-36.
59. Rao AC, Collinson PO, Rose AJ, et al. Prospective evaluation of the role of routine cardiac troponin T measurement to identify left ventricular ejection fraction < 40% after first myocardial infarction. Heart 2003; 89: 559-60.
60. Panteghini M, Bonetti G, Pagani F, et al. Measurement of troponin I 48 h after admission as a tool to rule out impaired left ventricular function in patients with a first myocardial infarction. Clin Chem Lab Med 2005; 43: 848-54.
61. Steen H, Giannitsis E, Futterer S, et al. Cardiac troponin T at 96 hours after acute myocardial infarction correlates with infarct size and cardiac function. J Am Coll Cardiol 2006; 48: 192-94.
62. Heeschen C, Goldmann BU, Langenbrink L, et al. Evaluation of a rapid whole blood ELISA for quantification of troponin I in patients with acute chest pain. Clin Chem 1999; 45: 1789-96.
63. Cannon CP, Weintraub WS, Demopoulos LE, et al. Comparison of early inva-sive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344: 1879-87.
64. Hamm CW, Heeschen C, Goldmann B, et al. Benefit of abciximab in patients with refractory unstable angina in relation to serum troponin T levels. N Engl J Med 1999; 340: 1623-29.
65. Morrow DA, Cannon CP, Rifai N, et al. Ability of minor elevations of troponin I and T to predict benefit from an early invasive strategy in patients with unsta-ble angina and non-ST elevation myocardial infarction: results from a random-ized trial. JAMA 2001; 286: 2405-12.
66. Hoekstra JW, Hedges JR, Gibler WB, et al. Emergency department CK-MB: a predictor of ischemic complications. National cooperative CK-MB project group. Acad Emerg Med 1994; 1: 17-27.

68
67. Störk TV, Wu AH, Müller-Bardorff M, et al. Diagnostic and prognostic role of myoglobin in patients with suspected acute coronary syndrome. Am J Cardiol 2000; 86: 1371-74.
68. De Lemos JA, Morrow DA, Gibson CM, et al. The prognostic value of serum myoglobin in patients with non-ST-segment elevation acute coronary syn-dromes. Results from the TIMI 11B and TACTICS-TIMI 18 studies. J Am Coll Cardiol 2002; 40: 238-44.
69. Newby LK, Storrow AB, Gibler WB, et al. Bedside multimarker testing for risk stratification in chest pain units: The chest pain evaluation by creatine kinase-MB, myoglobin, and troponin I (CHECKMATE) study. Circulation 2001; 103: 1832-37.
70. McCord J, Nowak RM, Hudson MP, et al. The prognostic significance of serial myoglobin, troponin I, and creatine-kinase-MB measurements in patients evaluated in the emergency department for acute coronary syndrome. Ann Emerg Med 2003; 42: 343-50.
71. De Lemos JA, McGuire DK, Drazner MH. B-type natriuretic peptide in cardio-vascular disease. Lancet 2003; 362: 316-22.
72. Tateishi J, Masutani M, Ohyanagi M, et al. Transient increase in plasma brain (B-type) natriuretic peptide after percutaneous transluminal coronary angio-plasty. Clin Cardiol 2000; 23: 776-80.
73. Hunt PJ, Richards AM, Nicholls MG, et al. Immunoreactive amino-terminal pro-brain natriuretic peptide (NT-PROBNP): a new marker of cardiac impair-ment. Clin Endocrinol (Oxf) 1997; 47: 287-96.
74. Johnston N, Jernberg T, Lindahl B, et al. Biochemical indicators of cardiac and renal function in a healthy elderly population. Clin Biochem 2004; 37: 210-16.
75. Luchner A, Hengstenberg C, Löwel H, et al. Effect of compensated renal fail-ure on approved heart failure markers. Direct comparison of Brain natriuretic peptide (BNP) and N-terminal pro-BNP. Hypertension 2005; 46: 118-23.
76. Richards M, Nicholls MG, Espiner EA, et al. Comparison of B-type natriuretic peptides for assessment of cardiac function and prognosis in stable ischemic heart disease. J Am Coll Cardiol 2006; 47: 52-60.
77. Talwar S, Squire IB, Downie PF, et al. Profile of plasma N-terminal proBNP following acute myocardial infarction; correlation with left ventricular systolic function. Eur Heart J 2000; 21: 1514-21.
78. Richards AM, Nicholls MG, Yandle TG, et al. Plasma N-terminal pro-brain natriuretic peptide and adrenomedullin: new neurohormonal predictors of left-ventricular function and prognosis after myocardial infarction. Circulation 1998; 97: 1921-29.
79. De Lemos JA, Morrow DA, Bentley JH, et al. The prognostic value of B-type natriuretic peptide in patients with acute coronary syndromes. N Engl J Med 2001; 345: 1014-21.
80. Omland T, Persson A, Ng L, et al. N-terminal pro-B-type natriuretic peptide and long-term mortality in acute coronary syndromes. Circulation 2002; 106: 2913-18.
81. James SK, Lindahl B, Siegbahn A, et al. N-terminal pro-brain natriuretic pep-tide and other risk markers for the separate prediction of mortality and subse-quent myocardial infarction in patients with unstable coronary artery disease: a Global Utilization of Strategies To Open occluded arteries (GUSTO)-IV substudy. Circulation 2003; 108: 275-81.
82. Lindahl B, Lindbäck J, Jernberg T, et al. Serial analyses of N-terminal pro-type natriuretic peptide in patients with non-ST-segment elevation acute coronary

69
syndromes: a Fragmin and fast Revascularisation during In Stability in Coro-nary artery disease (FRISC)-II substudy. J Am Coll Cardiol 2005; 45: 533-41.
83. Morrow DA, de Lemos JA, Blazing MA, et al. Prognostic value of serial B-type natriuretic peptide testing during follow-up of patients with unstable coro-nary artery disease. JAMA 2005; 294: 2866-71.
84. Kistorp C, Raymond I, Pedersen F, et al. N-terminal pro-brain natriuretic pep-tide, C-rective protein, and urinary albumin levels as predictors of mortality and cardiovascular events in older adults. JAMA 2005; 293: 1609-16.
85. Kragelund C, Grønning B, Køber L, et al. N-terminal pro-B-type natriuretic peptide and long-term mortality in stable coronary artery disease. N Engl J Med 2005; 352: 666-75.
86. Jernberg T, Stridsberg M, Venge P, et al. N-terminal pro brain natriuretic pep-tide on admission for early risk stratification of patients with chest pain and no ST-segment elevation. J Am Coll Cardiol 2002; 40: 437-45.
87. Ross R. Atherosclerosis - an inflammatory disease. N Engl J Med 1999; 340: 115-26.
88. Cermak J, Key NS, Bach RR, et al. C-reactive protein induces human periph-eral blood monocytes to synthesize tissue factor. Blood 1993; 82: 513-20.
89. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation 2000; 102: 2165-68.
90. Bhakdi S, Torzewski M, Klouche M, et al. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activa-tion. Arterioscler Thromb Vasc Biol 1999; 19: 2348-54.
91. Ridker PM, Cushman M, Stampfer MJ, et al. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336: 973-79.
92. Ridker PM, Hennekens CH, Buring JE, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342: 836-42.
93. Zebrack JS, Muhlestein JB, Horne BD, et al. C-reactive protein and an-giographic coronary disease: independent and additive predictors of risk in sub-jects with angina. J Am Coll Cardiol 2002; 39: 632-37.
94. Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350: 1387-97.
95. Wilson PW, Nam BH, Pencina M, et al. C-reactive protein and risk of cardio-vascular disease in men and women from the Framingham Heart Study. Arch Intern Med 2005; 165: 2473-78.
96. Deten A, Volz HC, Briest W, et al. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Car-diovasc Res 2002; 55: 329-40.
97. James SK, Oldgren J, Lindbäck J, et al. An acute inflammatory reaction in-duced by myocardial damage is superimposed on a chronic inflammation in un-stable coronary artery disease. Am Heart J 2005; 149: 619-26.
98. Lindahl B, Toss H, Siegbahn A, et al. Markers of myocardial damage and in-flammation in relation to long-term mortality in unstable coronary artery dis-ease. N Engl J Med 2000; 343: 1139-47.
99. Heeschen C, Hamm CW, Bruemmer J, et al. Predictive value of C-reactive protein and troponin T in patients with unstable angina: a comparative analysis. J Am Coll Cardiol 2000; 35: 1535-42.

70
100. James SK, Armstrong P, Barnathan E, et al. Troponin and C-reactive protein have different relations to subsequent mortality and myocardial infarction after acute coronary syndrome. J Am Coll Cardiol 2003; 41: 916-24.
101. Hillis GS, Terregino C, Taggart P, et al. Soluble intercellular adhesion mole-cule-1 as a predictor of early adverse events in patients with chest pain com-patible with myocardial ischemia. Ann Emerg Med 2001; 38: 223-28.
102. Swinburn JM, Stubbs P, Soman P, et al. Rapid assessment of patients with non-ST-segment elevation acute chest pain: Troponins, inflammatory markers, or perfusion imaging? J Nucl Cardiol 2002; 9: 491-99.
103. Ridker PM, Rifai N, Pfeffer MA, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average choles-terol levels. Circulation 1998; 98: 839-44.
104. Harb TS, Zareba W, Moss AJ, et al. Association of C-reactive protein and se-rum amyloid A with recurrent coronary events in stable patients after healing of acute myocardial infarction. Am J Cardiol 2002; 89: 216-21.
105. Ono K, Matsumori A, Shioi T, et al. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Cir-culation 1998; 98: 149-56.
106. Best PJ, Reddan DN, Berger PB, et al. Cardiovascular disease and chronic kidney disease: Insights and an update. Am Heart J 2004; 148: 230-42.
107. Muntner P, Hamm LL, Kusek JW, et al. The prevalence of non-traditional risk factors for coronary heart disease in patients with chronic kidney disease. Ann Intern Med 2004; 140: 9-17.
108. Shlipak MG, Fried LF, Crump C, et al. Elevations of inflammatory and proco-agulant biomarkers in elderly persons with renal insufficiency. Circulation 2003; 107: 87-92.
109. Blacher J, Safar ME, Guerin AP, et al. Aortic pulse wave velocity index and mortality in end-stage renal disease. Kidney Int 2003; 63: 1852-60.
110. Spencer K. Analytical reviews in clinical biochemistry: the estimation of creatinine. Ann Clin Biochem 1986; 23: 1-25.
111. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31-41.
112. Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461-70.
113. Liu J, Sukhova GK, Sun JS, et al. Lysosomal cysteine proteases in atheroscle-rosis. Arterioscler Thromb Vasc Biol 2004; 24: 1359-66.
114. Newman DJ. Cystatin C. Ann Clin Biochem 2002; 39: 89-104. 115. Laterza OF, Price CP, Scott MG. Cystatin C: an improved estimator of glome-
rular filtration rate? Clin Chem 2002; 48: 699-707. 116. Hoek FJ, Kemperman FA, Krediet RT. A comparison between cystatin C,
plasma creatinine and the Cockcroft and Gault formula for the estimation of glomerular filtration rate. Nephrol Dial Transplant 2003; 18: 2024-31.
117. Jernberg T, Lindahl B, James S, et al. Cystatin C. A novel predictor of outcome in suspected or confirmed non-ST-elevation acute coronary syndrome. Circula-tion 2004; 110: 2342-48.
118. König W, Twardella D, Brenner H, et al. Plasma concentrations of cystatin C in patients with coronary heart disease and risk for secondary cardiovascular events: more than simply a marker of glomerular filtration rate. Clin Chem 2005; 51: 321-27.

71
119. Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352: 2049-60.
120. Luc G, Bard JM, Lesueur C, et al. Plasma cystatin-C and development of coro-nary heart disease: The PRIME Study. Atherosclerosis 2006; 185: 375-80.
121. Grande P, Kiilerich S. Relationship between serum CK-MB-estimated acute myocardial infarct size and clinical complications. Acta Med Scand 1984; 215: 355-62.
122. Miller TD, Christian TF, Hopfenspirger MR, et al. Infarct size after acute myo-cardial infarction measured by quantitative tomographic 99mTc sestamibi imgaing predicts subsequent mortality. Circulation 1995; 92: 334-41.
123. Wagner A, Mahrholdt H, Holly TA, et al. Contrast-enhanced MRI and routine single photon emission computed tomography (SPECT) perfusion imaging for detection of subendocardial myocardial infarcts: an imaging study. Lancet 2003; 361: 374-79.
124. Kastrati A, Mehilli J, Dirschinger J, et al. Myocardial salvage after coronary stenting plus abciximab versus fibrinolysis plus abciximab in patients with acute myocardial infarction: a randomised trial. Stent versus Trombolysis for Occluded Coronary Arteries in Patients with Acute Myocardial Infarction (STOPAMI-2) Study. Lancet 2002; 359: 920-25.
125. Gibbons RJ, Valeti US, Araoz PA, et al. The quantification of infarct size. J Am Coll Cardiol 2004; 44: 1533-42.
126. Hackel DB, Reimer KA, Ideker RE, et al. Comparison of enzymatic and anat-omic estimates of myocardial infarct size in man. Circulation 1984; 70: 824-35.
127. Vatner SF, Baig H, Manders WT, et al. The effects of coronary artery reperfu-sion on myocardial infarct size calculated from creatine kinase. J Clin Invest 1978; 61: 1048-56.
128. Groth T, Hakman M, Sylven C. Prediction of infarct size from early serum myoglobin observations. Scand J Clin Lab Invest 1987; 47: 599-603.
129. Yamashita T, Abe S, Arima S, et al. Myocardial infarct size can be estimated from serial plasma myoglobin measurements within 4 hours of reperfusion. Circulation 1993; 87: 1840-49.
130. The Fragmin and Fast Revascularisation during InStability in Coronary artery disease Investigators. Invasive compared with non-invasive treatment in unsta-ble coronary-artery disease: FRISC II prospective randomised multicentre study. Lancet 1999; 354: 708-15.
131. Panteghini M, Pagani F, Yeo KT, et al. Evaluation of imprecision for cardiac troponin assays at low-range concentrations. Clin Chem 2004; 50: 327-32.
132. Zethelius B, Johnston N, Venge P. Troponin I as a predictor of coronary heart disease and mortality in 70-year old men. Circulation 2006; 113: 1071-78.
133. Uettwiller-Geiger D, Wu AH, Apple FS, et al. Multicenter evaluation of an automated assay for troponin I. Clin Chem 2002; 48: 869-76.
134. Freeman RV, Eagle KA, Bates ER, et al. Comparison of artificial neural net-works with logistic regression in prediction of in-hospital death after percuta-neous transluminal coronary angioplasty. Am Heart J 2000; 140: 511-20.
135. Ellenius J, Groth T. Methods for selection of adequate neural network struc-tures with application to early assessment of chest-pain patients by biochemical monitoring. Int J Med Inf 2000; 57: 181-202.
136. Wallace TW, Abdullah SM, Drazner MH, et al. Prevalence and determinants of troponin T elevation in the general population. Circulation 2006; 113: 1958-65.

72
137. Rao SV, Ohman EM, Granger CB, et al. Prognostic value of isolated troponin elevation across the spectrum of chest pain syndromes. Am J Cardiol 2003; 91: 936-40.
138. Larsson A, Malm J, Grubb A, et al. Calculation of glomerular filtration rate expressed in mL/min from plasma cystatin C values in mg/L. Scand J Clin Lab Invest 2004; 64: 25-30.
139. Hamwi SM, Sharma AK, Weissman NJ, et al. Troponin-I elevation in patients with increased left ventricular mass. Am J Cardiol 2003; 92: 88-90.
140. Chapelle JP, Dubois B, Bovy C, et al. Comparison of plasma cardiac troponins T and I in chronically hemodialyzed patients in relation to cardiac status and age. Clin Chem Lab Med 2002; 40: 240-45.
141. Perna ER, Macin SM, Cimbaro Canella JP, et al. Ongoing myocardial injury in stable severe heart failure. Value of cardiac troponin T monitoring for high-risk patient identification. Circulation 2004; 110: 2376-82.
142. Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest 2003; 111: 1805-12. Erratum in: J Clin Invest 2003; 112: 299.
143. Avanzas P, Arroyo-Espliguero R, Cosin-Sales J, et al. Markers of inflammation and multiple complex stenoses (pancoronary plaque vulnerability) in patients with non-ST segment elevation acute coronary syndrome. Heart 2004; 90: 847-52.
144. Zairis MN, Papadaki OA, Manousakis SJ, et al. C-reactive protein and multiple complex coronary artery plaques in patients with primary unstable angina. Atherosclerosis 2002; 164: 355-59.
145. Labugger R, Organ L, Collier C, et al. Extensive troponin I and T modification detected in serum from patients with acute myocardial infarction. Circulation 2000; 102: 1221-26.
146. Sam F, Sawyer DB, Chang DL et al. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Circ Physiol 2000; 279: H422-8.
147. Palojoki E, Saraste A, Eriksson A, et al. Cardiomyocyte apoptosis and ventricu-lar remodeling after myocardial infarction in rats. Am J Physiol Circ Physiol 2001; 280: H2726-31.
148. Chandrashekhar Y, Sen S, Anway R, et al. Long-term caspase inhibition ame-liorates apoptosis, reduces myocardial troponin I cleavage, protects left ven-tricular function, and attenuates remodeling in rats with myocardial infarction. J Am Coll Cardiol 2004; 43: 295-301.
149. Kavsak PA, Mac Rae AR, Newman AM, et al. Effects of contemporary tro-ponin assay sensitivity on the utility of the early markers myoglobin and CKMB isoforms in evaluating patients with possible acute myocardial infarc-tion. Clin Chim Acta 2007; 380: 213-16.
150. Wu AH, Fukushima N, Puskas R, et al. Development and preliminary clinical validation of a high sensitivity assay for cardiac troponin using a capillary flow (single molecule) fluorescence detector. Clin Chem 2006; 52: 2157-59.
151. Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxi-dase in patients with chest pain. N Engl J Med 2003; 349: 1595-1604.
152. Collinson PO, Gaze DC, Bainbridge K, et al. Utility of admission cardiac tro-ponin and ”Ischemia modified albumin” measurements for rapid evaluation and rule out of suspected acute myocardial infarction in the emergency department. Emeg Med J 2006; 23: 256-61.
153. Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic de-cision making. JAMA 2000; 284: 835-42.

73
154. Eagle KA, Lim MJ, Dabbous OH, et al. A validated prediction model for all forms of acute coronary syndrome: estimating the risk of 6-month postdis-charge death in an international registry. JAMA 2004; 291: 2727-33.
155. Voss R, Cullen P, Schulte H, et al. Prediction of risk of coronary events in middle-aged men in the Prospective Cardiovascular Münster Study (PROCAM) using neural networks. Int J Epidemiol 2002; 31: 1253-62.

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 269
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-7945
ACTAUNIVERSITATISUPSALIENSISUPPSALA2007


















