
Caracterização de um fragmento recombinante scFab anti Z...
126
Universidade de Brasília Faculdade de Medicina Programa de Patologia Molecular C C a a r r a a c c t t e e r r i i z z a a ç ç ã ã o o d d e e u u m m f f r r a a g g m m e e n n t t o o r r e e c c o o m m b b i i n n a a n n t t e e s s c c F F a a b b a a n n t t i i Z Z - - D D N N A A p p r r o o d d u u z z i i d d o o e e m m P P i i c c h h i i a a p p a a s s t t o o r r i i s s . . Rafael Trindade Burtet Orientador: Prof. Dr. Marcelo de Macedo Brígido Co-orientador: Prof. Dr. Marcos Antônio Santos Silva Brasília - DF Abril de 2006
Transcript of Caracterização de um fragmento recombinante scFab anti Z...

Universidade de Brasília
Faculdade de Medicina
Programa de Patologia Molecular
CCaarraacctteerriizzaaççããoo ddee uumm ffrraaggmmeennttoo rreeccoommbbiinnaannttee ssccFFaabb aannttii ZZ--DDNNAA
pprroodduuzziiddoo eemm PPiicchhiiaa ppaassttoorriiss..
Rafael Trindade Burtet
Orientador: Prof. Dr. Marcelo de Macedo Brígido Co-orientador: Prof. Dr. Marcos Antônio Santos Silva
Brasília - DF Abril de 2006

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

CCaarraacctteerriizzaaççããoo ddee uumm ffrraaggmmeennttoo rreeccoommbbiinnaannttee ssccFFaabb aannttii ZZ--DDNNAA pprroodduuzziiddoo
eemm PPiicchhiiaa ppaassttoorriiss..
Rafael Trindade Burtet Orientador: Prof. Dr. Marcelo de Macedo Brígido Co-orientador: Prof. Dr. Marcos Antônio Santos Silva
Universidade de Brasília
Faculdade de Medicina
Programa de Patologia Molecular
Dissertação apresentada ao
Programa de pós-graduação em
Patologia Molecular da Faculdade de
Medicina da Universidade de Brasília
como requisito parcial à obtenção do
grau de mestre em Patologia Molecular.
Brasília - DF Abril de 2006

ii
Banca examinadora
Dr. Elíbio Leopoldo Rech – EMBRAPA/CENARGEN
Dr. Fernado Araripe Gonçalves Torres – UnB
Dra. Ildinete Silva-Pereira – UnB
Dr. Marcelo de Macedo Brígido – UnB
Dr. Marcos Antônio Santos Silva – UnB
Trabalho desenvolvido no
Laboratório de Biologia Molecular
da Universidade de Brasília, sob a
orientação da Prof. Dr. Marcelo de
Macedo Brígido.

iii
Este trabalho é dedicado a Maria Carlinda
Trindade (in memmorian) avó para sempre
querida e amada e Gustavo Trindade (in
memmorian) saudoso primo, precocemente
levado do nosso convívio.

iv
AGRADECIMENTOS
Primeiramente gostaria de agradecer ao meu orientador, Profº. Marcelo pela
oportunidade de poder trabalhar no laboratório de Biologia Molecular, e principalmente pelo
voto de confiança, por acreditar em meu potencial quando cheguei em Brasília, apenas com o
diploma de licenciado na mão. Sinceramente Muito obrigado! Espero não ter te decepcionado.
A Profª. Andréa pela amizade e por ter sempre um conselho certo na hora certa, flexível
quando podia e firme quando devia. Sem falar que às vezes fica difícil entender o Marcelo
sem as tuas traduções. Ao Professor Marcos, meu co-orientador, por pacientemente me
agüentar na bancada no início do projeto. Esse trio realmente merece um agradecimento
muito especial por estar sempre disposto a me auxiliar no que fosse possível.
A Profª. Ildinete pela simpatia sempre presente e a Profª. Lídia pelas infindáveis dicas
sobre a levedura Pichia pastoris e o processo fermentativo.
Aos demais professores do Laboratório de Biologia Molecular, Élida, Márcio, Sueli e
Fernando sempre prontos para qualquer questionamento, por contribuírem para meu
aprendizado e pelo apoio.
Aos queridos colegas do Laboratório de Imunologia Molecular. Aqueles que me
ajudaram no início da minha formação como pesquisador, por agüentarem minhas bagunças,
minhas dúvidas, muitas vezes primárias. Pat Lu a nossa querida e amada Bruxa do Norte,
Hernandez essa figuraça que demorou mas voltou pro lab., Chrisinha com certeza a pessoa
mais serena que já conheci, Diorge (Didi) pelas conversas memoráveis sobre a Biologia
Molecular, Gabi por me ajudar a entender melhor essa tal de maxiprep.
Aos colegas sempre presentes, Flavinha (Glamurosa, a Rainha do Funk) substituta da
Pat Lu em ter que agüentar minha bagunça na bancada ao lado. A dupla dinâmica Bárbara e
Mariany, o lab. não seria tão divertido sem vocês. A Cecília, pelas caronas intermináveis
quando eu morava na asa sul, e por ser esse amor de pessoa.
Ao grande veterinário Pedro e Mauro (Joselito), meus grandes amigos e companheiros
de cachaça, pra esquecer ou comemorar, sempre prontos pra marcar um tempinho no “Sun
Set”.
Aos demais colegas de Lab, 01 Gina, Luane, Izabel, Carol, Henrique, Liziani
(arenguera), Lelê (xuxinha), Mariana, Victor e Paulo sempre agüentando ou acompanhando
minhas doideras, reclamações e momentos de inspiração e descontração.

v
Túlio pelas dicas, antes e depois do seu tour pela Europa. A Alessandra Dantas que
todos adoram perturbar, eu principalmente. Ao sempre prestativo Davi, o portador do canguru
vermelho, o almoxarifado nunca mais será o mesmo sem você, e ao seu fiel escudeiro Tiago.
Vivis, a dentista mais bióloga que conheci, a sua calma é diretamente proporcional a sua
prontidão em ajudar um amigo. Ao grande amigo Saulo, essa criatura estranha que é tão
pirado quanto eu. Ao grande amigo Alex, o cearense mais dengoso do mundo, sempre pronto
pra qualquer explicação à cerca da biologia molecular ou qualquer outro assunto, utilizando
seu vocabulário altamente rebuscado. A Nádia, se agressão também é uma forma de carinho,
então a gente se gosta muito, vê se volta logo, estamos com saudades. Ao grande filósofo
Hugo. Sérgio (“seu puliça”) e Taisa um casal super gente boa. Tiago e Tati vocês dois juntos
quebram um alambique. Guilherme, obrigado pela força com o fermentador, não teria domado
esse aparelhinho sem a tua ajuda. Marciano, meu conterrâneo, esse bagual que veio direto de
Caxias pra tocar o terror no DF. A propósito, conversa com o Batmam! Luciano, esse
Cuiabano tranqüilo que perdeu um pouco o eixo na época da defesa. Para de escrever e vamos
tomar umas bereja Bacural! Ao Junior Popó que só não é mais Goiano por falta de espaço.
Aos demais colegas do laboratório Rose, Maria José, Bruno B., Janice, Fabrício,
Rafael Ajuz, Cristiano, Vanessa, Natália, Basti (Made in Suécia), Plínio, Lorena, Vera,
Larissa, Alice, Marcus, André Nicola, Pat Girl, Pat Vet, Eduardo, Sócrates, Leandro, Luciana
e Tatiana. Todos vocês, que me ajudaram e apoiaram, com sua amizade e companheirismo.
Aos funcionáios, Celso, Fátima e Ivanildes, pelas longas conversas na sala de lavagem
enquanto lavava as ponteiras.
Aos queridos amigos da enzimologia Gil MSU (Mestre Supremo do Universo, ou o
que quer que isso signifique) e Daniel Paiva, companheiros de RU, um mais maluco que o
outro.
Aos amigos da Colina, Wady, Lucas (BH), Javier (made in Colombia) e Michiel
Huiskens (made in Holanda). Aos amigos do superpopuloso 203, Keninho pelas séries filmes
e etc. Baiano, George, Luquinhas (onipresente na Colina), Paulo e Hugo. E aos colegas de ap.
Frederico, Arquimedes e Rosevel.
As queridas secretarias do Programa em Patologia Molecular Carol e Érica, sempre,
extremamente simpáticas e prestativas.
A você Camilinha, pelo seu companheirismo e compreensão. Pela sua paciência, pois
mais do que ninguém, você teve que agüentar meu mau humor, minhas explosões e minhas
frustrações. Foste meu ombro quando precisei chorar e a companhia constante nas horas de
alegria. Obrigado por estar ao meu lado sempre quando precisei, na ajuda para organizar não

vi
só os tópicos e as referências, mas alguns até experimentos. Se hoje tenho sucesso devo muito
a você. Amo você gatinha.
Aos grandes amigos Artur, Bianca, Luciano e Geraldo, figuras incríveis que conheci
por intermédio da Camila.
Mãe, você foi meu porto seguro, só pude alçar vôo, só pude ir tão longe porque sabia
que se caísse o teu abraço me seguraria. Apesar da distância moras no meu coração.
Pai, pode ser difícil pra ti compreenderes todas as coisas que fiz até agora, mas saiba
que fui atrás do meu sonho e espero te deixar orgulhoso de minhas conquistas. Você é meu
herói, meu exemplo e tenho muitas saudades.
Mano, meu querido padrasto, você foi a imagem em que me espelhei, um exemplo de
integridade, obrigado por ter sido criado como um filho teu.
Aos meus irmãos, Juca, Thais e Rafaela, estou muito orgulhoso de vocês, estão
começando uma fase difícil, muito trabalho e muito estudo. Pena que não estou por perto pra
poder acompanhá-los e ajudá-los. Aos pequenos Fernanda, Luiza, Gabriel e Mariana, vocês
tem um longo caminho pela frente, saibam que estarei sempre disponível quando precisarem
desse irmão mais velho, mesmo a distancia.
Tia Magda pelos maravilhosos ensinamentos que levarei pro resto da vida.
Tia Marisa e Tereza, meus exemplos de vida dedicada a cultura e a educação, Valeu
pelo apoio.
Tia Malba e Tio Luciano, não tenho palavras para agradecer a força que vocês me
deram em Brasília, por terem me recebido de braços abertos com tanto carinho.
Tia Teca, Tio Paulo e seus “bebês”, Giovani e Rodrigo, obrifgado pelo carinho e
apoio.
Agradeço a CAPES e ao CNPq por ter me patrocinado nessa jornada.
Aos criadores do Google e do pubmed.com não sei o que seria de mim sem essas
ferramentas.
Ao índio tupi-guarani que inventou o chimarrão, porque senão fosse a erva-mate me
acompanhar durante as madrugadas, não terminaria a dissertação a tempo.
A todos aqueles que de certa forma contribuíram com esse trabalho.
E ao meu querido e bom Deus que colocou todas essas pessoas maravilhosas em meu
caminho. Por me dar força, fé e esperança.
Muito Obrigado a Todos!

vii
ÍNDICE
ÍNDICE DE TABELAS xi
ÍNDICE DE FIGURAS xii
LISTA DE TERMOS E ABREVIATURAS xvi
RESUMO xix
ABSTRACT xx
1 - INTRODUÇÃO 1
1.1 - PROPRIEDADES GERAIS DO SISTEMA IMUNE 1
1.2 - ANTICORPOS 2
1.2.1 - Estrutura dos Anticorpos 4
1.2.1.1 - Fragmentação Enzimática dos Anticorpos 5
1.2.1.2 - Obtenção e Aplicações Biotecnológicas de Anticorpos
Recombinantes 7
1.3 - EXPRESSÃO HETERÓLOGA DE ANTICORPOS 9
1.3.1 - Sistema de expressão em Pichia pastoris 9
1.4 - FERMENTAÇÃO 15
1.4.1 - O processo de cultivo 16
1.4.2 - Controle dos parâmetros do processo fermentativo 17
1.4.3 - Sistemas de transferência de Oxigênio 17
1.4.4 - Concentração celular 19
1.4.5 - Temperatura e pH 19
1.5 - ANTICORPOS E Z-DNA 20
1.6 - O VETOR DE EXPRESSÃO DE scFab 21
2 - JUSTIFICATIVA 23
3 - OBJETIVOS 24
4 - MATERIAIS E MÉTODOS 25
4.1 - CONSIDERAÇÕES INICIAIS 25

viii
4.2 - MATERIAIS 27
4.2.1 - Células 27
4.2.1.1 - Linhagem Bacteriana 27
4.2.1.2 - Linhagem de Pichia pastoris 27
4.2.2 - Plasmídio Utilizado 27
4.2.3 - Meios de Cultura para bactérias 27
4.2.4 - Meios de cultura e Soluções estoque para leveduras 28
4.2.5 - Soluções estoques de Inibidores de Proteases 31
4.2.6 - Soluções e tampões de uso geral 31
4.2.7 - Concentradores Amicon® 32
4.2.8 - Soluções para extração de DNA plasmidial 32
4.2.9 - Enzimas e Tampões 33
4.2.10 - Soluções para Eletroforese em Gel de Agarose 33
4.2.11 - Soluções para Eletroforese em Gel de Poliacrilamida 34
4.2.12 - Soluções para coloração com Coomassie Brillant Blue (G-250) 35
4.2.13 - Soluções para coloração com Prata 35
4.2.14 - Soluções para os Ensaios Imunológicos (Imunoprecipitação,
ELISA, Western, Colony e Dot blotting) 36
4.2.15 - Inibidores utilizados no ELISA 37
4.2.16 - Marcador de massa molecular (MM) para proteínas 38
4.2.17 - Anticorpos 38
4.2.18 - Soluções para coluna de Proteína A-Sepharose™ 39
4.2.19 - Soluções para coluna de Gel Filtração 39
4.2.20 - Soluções para coluna de Proteína L 39
4.2.21 - Soluções para coluna de Troca Iônica 40
4.2.22 - Membrana de Nitrocelulose 40
4.3 - MÉTODOS 41
4.3.1 - Preparação de DNA Plasmidial através do método de lise alcalina
em larga escala 42
4.3.2 - Digestão do DNA plasmidial com Endonucleases de Restrição 42
4.3.3 - Análise de DNA em Gel de Agarose 42
4.3.4 - Eluição de fragmentos de DNA 42
4.3.5 - Preparação de DNA para transformação de Pichia pastoris 42

ix
4.3.6 - Transformação da levedura P. pastoris por eletroporação 43
4.3.7 - Análise dos transformantes de P. pastoris por Colony blotting ou
Western de colônia 43
4.3.8 - Expressão da Proteína Recombinante em P. Pastoris 44
4.3.8.1 -Fermentação em frascos do tipo Erlenmeyer 44
4.3.8.2 - Produção em larga escala do scFab recombinante em frascos do
tipo Erlenmeyer 45
4.3.8.3 -Sistema de Fermentação em Bioreator 45
4.3.9 - Processamento do Sobrenadante de Cultura em Colunas Amicon 47
4.3.10 - Análise de Proteínas em Gel de SDS-PAGE 48
4.3.11 - Análise de Proteínas por Western Blotting 48
4.3.12 - Análise de Proteínas por Dot Blotting 49
4.3.13 - Purificação do Corante Coomassie Brillant Blue G-250 49
4.3.14 - Coloração com Coomassie Briliant Blue G-250 49
4.3.15 - Coloração com Prata 50
4.3.16 - ELISA (Enzyme-linked immunosorbent assay) 50
4.3.16.1 - ELISA - Ensaio de ligação direta 50
4.3.16.2 - ELISA - Ensaio de Inibição 51
4.3.17 - Purificação dos scFabs recombinantes por cromatografia de
afinidade 52
4.3.17.1 - Proteína A-Sepharose™ 52
4.3.17.2 - Proteína L agarose 52
4.3.18 - Purificação dos scFabs recombinantes por Cromatografia de
Gel Filfração 53
4.3.19 - Quantificação de Proteínas Utilizando o Kit BCA 54
4.3.20 - Imunoprecipitação 54
4.3.21 - Coluna de Troca Iônica MonoQ 55
4.3.22 - Deglicosilação 55
5 - RESULTADOS e DISCUSSÃO 56
5.1 – PREPARAÇÃO DO VETOR pPIg Fab 56
5.2 – TRANSFORMAÇÃO DA LEVEDURA e SELEÇÃO DE CLONES
RECOMBINANTES 56

x
5.3 - PRODUÇÃO DO FRAGMENTO RECOMBINANTE e CINÉTICA
DE INDUÇÃO 58
5.3.1 - Quantificação do sobrenadante – BCA 59
5.4 - CARACTERIZAÇÃO DO scFab RECOMBINANTE 61
5.4.1 - Análise do sobrenadante de cultura por Western Blotting 61
5.4.2 – Análise da Atividade Ligante do Fragmento scFab 63
5.4.2.1 – Ensaio de Ligação Direta 63
5.4.2.2 - Ensaio de inibição 65
5.5 – FERMENTAÇÃO EM BIOREATOR 67
5.6 – PURIFICAÇÃO 73
5.6.1 – Cromatografia de Gel Filtração 73
5.6.2 - Imunoafinidade Proteína A 79
5.6.3 - Imunoafinidade Proteína L 81
5.6.4 – Imunoprecipitação 83
5.6.5 – Cromatografia de Troca Iônica 86
5.7 – DEGLICOSILAÇÃO 90
6 – CONCLUSÃO e PERSPECTIVAS 94
7 - REFERÊNCIAS BIBLIOGRÁFICAS 95
8 - ANEXO I 101

xi
ÍNDICE DE TABELAS
Tabela 1. Lista parcial de proteínas heterólogas que foram expressas em
P. pastoris com sucesso. 10
Tabela 2. Inibidores utilizados nos ensaios de inibição. 38
Tabela 3. Anticorpos utilizados. 38
Tabela 4. Marcadores Moleculares Padrão usados na Gel Filtração. 39
Tabela 5. Proteínas possíveis no sistema pPIg Fab. 62
Tabela 6. Coletas dos tempos na fase de indução por metanol. 69
Tabela 7. Concentração das amostras coletadas nas Cromatografias de Gel
Filtração e aplicadas no gel SDS-PAGE e Western Blotting. 78
Tabela 8. Amostras utilizadas para análise da Imunoprecipitação aplicadas
no gel SDS-PAGE e Western Blotting. 85

xii
ÍNDICE DE FIGURAS
Figura 1. Estrutura de uma molécula de anticorpo. 3
Figura 2. Representação esquemática da molécula de anticorpo e seus
principais fragmentos. 5
Figura 3. Fragmentação enzimática da molécula de anticorpo. 6
Figura 4. Via metabólica da oxidação do metanol em formaldeído na
levedura P. pastoris. 12
Figura 5. Substituição gênica por duplo crossover no locus AOX1 em
P. pastoris. 14
Figura 6. Inserção gênica por simples crossover no locus his4 em
P. pastoris. 14
Figura 7. Imagens geradas por computador das três estruturas conhecidas
do DNA. 21
Figura 8. Representação esquemática do vetor utilizado para a expressão
dos scFab em P. pastoris. 25
Figura 9. Representação esquemática da construção do cassete de expressão
do pPIg Fab. 26
Figura 10. Foto do sistema de fermentação em biorreator utilizado. 46
Figura 11. Seleção de clones de P. pastoris recombinantes produtores de scFab
recombinante por colony blotting em membrana de nitrocelulose. 57

xiii
Figura 12. Análise da produção de proteínas recombinantes em células de
P. pastoris em diversos tempos de indução. 59
Figura 13. Linha de tendência da quantificação por BCA das proteínas totais
do sobrenadante de cultura da levedura P. pastoris em frasco 60
Figura 14. Análise da expressão de proteínas recombinantes em
P. pastoris. 62
Figura 15. Imunoensaio enzimático (ELISA) de ligação direta ao Z-DNA 64
comparando a ligação do fragmento recombinante scFab ao mAbZ22. 64
Figura 16. Imunoensaio enzimático (ELISA) de ligação direta ao Z-DNA
comparando a ligação do fragmento recombinante scFab ao mAb Z22 e FvFc. 64
Figura 17. Imunoensaio enzimático (ELISA) de inibição do fragmento
recombinante scFab. 66
Figura 18. Imunoensaio enzimático (ELISA) de inibição do fragmento FvFc. 66
Figura 19. Análise por SDS-PAGE da produção do fragmento scFab
recombinante durante a fase de indução na fermentação em bioreator. 69
Figura 20. Análise por Western Blotting da produção do fragmento scFab
recombinante por fermentação em bioreator. 70
Figura 21. Linha de tendência da quantificação por BCA das proteínas totais
do sobrenadante de cultura da levedura P. pastoris em bioreator 71

xiv
Figura 22. Quantificação estimada de scFab, no sobrenadante da cultura de
P. pastoris em bioreator, em relação às diluições de BSA. 71
Figura 23. Gráfico da análise por Gel filtração da calibração da coluna
com os padrões controle de massa molecular conhecida. 74
Figura 24. Gráfico da análise por Gel filtração do fragmento scFab
recombinante produzido pela levedura P. pastoris. 75
Figura 25. Análise por Dot Blotting da Gel Filtração 1 do fragmento
scFab recombinante produzido pela levedura P. pastoris. 75
Figura 26. Gráfico da análise por Gel filtração do fragmento scFab
recombinante produzido pela levedura P. pastoris. 76
Figura 27. Análise por Dot Blotting das frações resultantes da Gel
Filtração 3 do fragmento scFab recombinante produzido pela
levedura P. pastoris. 76
Figura 28. Análise por SDS-PAGE da purificação do fragmento
scFab recombinante por Cromatografia de Gel Filtração, coloração com
Coomassie Brillant Blue G-250. 78
Figura 29. Análise por Western Blotting da purificação do fragmento
scFab recombinante por Cromatografia de Gel Filtração. 79
Figura 30. Análise por Dot Blotting das amostras recolhidas durante
a purificação do fragmento scFab recombinante produzido em P. pastoris
por Imunoafinidade a Proteína A. 80

xv
Figura 31. Análise por Dot Blotting das amostras recolhidas durante
a purificação do fragmento scFab recombinante produzido em P. pastoris
por Imunoafinidade a Proteína L. 82
Figura 32. Análise por Dot Blotting das amostras recolhidas durante
a purificação do anticorpo mAbZ22 por Imunoafinidade a Proteína L. 83
Figura 33. Análise por SDS-PAGE e Western Blotting da purificação
do fragmento scFab recombinante por Imunoprecipitação. 85
Figura 34. Análise por Dot Blotting da purificação do fragmento scFab
recombinante por Cromatografia de Troca Iônica. 87
Figura 35. Análise por SDS-PAGE da purificação do fragmento scFab
recombinante por cromatografia de Troca Iônica, coloração com prata. 88
Figura 36. Análise por Western Blotting da purificação do fragmento
scFab recombinante por cromatografia de Troca Iônica. 89
Figura 37. Análise por Western Blotting da deglicosilação do fragmento
scFab recombinante. 91
Figura 38. Análise por SDS-PAGE da comparação da deglicosilação
do fragmento scFab recombinante e IgG humana, coloração com prata. 92
Figura 39. Análise da seqüência do scFab pelo programa NetNGlyc
para a averiguação de possíveis sítios de N-Glicosilação. 93
Figura 40. Análise da seqüência do scFab pelo programa NetOGlyc
para a averiguação de possíveis sítios de O-glicosilação. 93

xvi
LISTA DE TERMOS e ABREVIATURAS
ADCC Citotoxicidade celular mediada por anticorpos.
AmpR Gene de resistência à ampicilina (β-lactamase).
APS Persulfato de Amônia.
AOX1, 2 Genes da álcool oxidase 1 e 2.
Asn Asparagina.
Asp Ácido aspático.
BCIP 5-Bromo-4-Cloro-indolil fosfato.
BrEt Brometo de Etídeo.
BSA Albumina bovina sérica.
ºC Graus Celsius.
CD Marcador de superfície de célula (Cluster of diferentiation).
cDNA Ácido desoxirribonucléico complementar.
CDR Região determinante de complementaridade.
cm Centímetros.
ColE1 Origem de replicação de E. coli.
C-terminal Extremidade Carboxi-terminal.
Da Dalton.
DNA Ácido Desoxirribonucléico.
dsDNA DNA fita dupla.
EDTA Ácido Etilenodiaminotetracético.
Fab Fragmento de anticorpo de ligação ao antígeno.
Fc Fragmento cristalizável de anticorpo (porção constante).
FcR-II Receptor de Fc tipo II.
FL Fluorescência.
FR Arcabouço (Framework).
Fv Fragmento variável do anticorpo.
FvFc Fragmento variável fusionado ao Fc
g Grama.
GalNac N-acetilgalactosamina.
GlcNAc N-acetilglicosamina.

xvii
h Hora.
HIS 4 Gene histidinol desidrogenase.
Ig Imunoglobulina.
IL Interleucina.
KCl Cloreto de potásio.
kb Kilobase.
kDa Kilodalton.
L Litro.
M Molar.
µg Micrograma.
µl Microlitro.
μm Micrômetro.
μM Micromolar.
mA Miliamper.
mAb Anticorpo monoclonal.
mg Miligrama.
min Minutos.
mL Mililitro.
mM Milimolar.
MgCl2 Cloreto de Magnésio.
MgSO4 Sulfato de Magnésio.
mL Mililitro.
mM Milimolar.
MM Massa molecular.
mRNA Ácido Ribonucléico mensageiro.
NaCl Cloreto de Sódio.
NaOH Hidróxido de Sódio.
NBT Nitro Blue Tetrazole.
ng Nanograma.
OD600 Densidade ótica a 600nm.
OKT3 Anticorpo monoclonal anti-CD3.
Ori Origem de replicação.
Pb Pares de base.

xviii
PBS Tampão Fosfato - Salina.
PCR Reação em cadeia da polimerase.
pep 4 Linhagem protease menos.
pH Potencial hidrogeniônico.
PMSF Fluoreto de fenilmetilsulfonato.
PpA Proteína A
PpL Proteína L
Pro Prolina.
p/v Peso/Volume.
q.s.p. Quantidade suficiente para.
RNA Ácido Ribonucléico.
RNAse Ribonuclease.
rpm Rotações por minuto.
scFv Fragmento variável de anticorpo cadeia única (Single chain Fragment
Variable).
SDS Sódio Dodecil Sulfato.
SDS-PAGE Gel de poliacrilamida desnaturante (com SDS).
Ser Serina.
ssDNA Fita simples de DNA.
TEMED N,N,N’,N’- tetrametil etilenodimetilamina.
Thr Treonina.
Tris-Base Tris(hidroximetil)aminometano.
U Unidade enzimática.
UV Ultravioleta.
v Volume.
VH Domínio variável da cadeia pesada de um anticorpo.
VL Domínio variável da cadeia leve de um anticorpo.
v/v Volume/Volume.
YNB Yeast Nitrogen Base.

xix
RESUMO
A produção de anticorpos e seus fragmentos para o uso na medicina é um recurso cada
vez mais visado por profissionais dessa área. Porém, alguns fatores limitam sua utilização e,
principalmente, a produção dos mesmos. Com intuito de contribuir para o avanço na
expressão heteróloga desses fragmentos, propomos o uso de um vetor monocistrônico para a
expressão de anticorpos recombinantes. Com esse objetivo, foi feita a expressão e
caracterização do scFab recombinante, derivado do anticorpo anti Z-DNA mAbZ22, na
levedura metilotrófica Pichia pastoris. Verificou-se que é possível a expressão do Fab em
gene monocistrônico pois a molécula mostrou-se ativa e funcional, além de apresentar uma
provável dimerização mostrando montagem e processamento corretos. Estabeleceram-se as
condições de fermentação em bioreator para produção em larga escala e de possíveis métodos
de purificação da proteína. Foram produzidas uma quantidade de proteína total de 435 mg/L,
no sistema de fermentação em frasco, e 8,4 g/L, em biorreator. Estimou-se que, no biorreator,
aproximadamente 0,25 g/L do total de proteínas do sobrenadante correspondem ao fragmento
scFab recombinante. A purificação do scFab foi tentada por várias metodologias, sendo que a
coluna de troca iônica mostrou-se a mais eficiente. Sendo a purificação bem estabelecida e o
processamento correto inteiramente confirmado, o cassete de expressão desse vetor pode ser
substituído para a produção de fragmentos de anticorpos de interesse clínico.

xx
ABSTRACT
The production of antibodies and its fragments for medical applications it’s a goal
more and more aimed at for health agents. Nevertheless, many factors limit their use and,
especially, their production. In order to contribute to this quest, we propose the use of a
monocistronic vector for the expression of recombinant antibodies fragments. With this
purpose, we performed the expression and characterization of a recombinant scFab, derived
from the anti Z-DNA mAbZ22 antibody, by the methylotrophic yeast Pichia pastoris. We
found that the expression of the Fab monocistronic gene was perfectly achieved. The product
was functional and active, probably correctly processed and folded as a dimer. We established
parameters for high-yield expression in bioreactor fermentation and purification methods. A
total protein amount of 435 mg/L was produced in shake flask cultures, and 8,4 g/L in
bioreactor. In the bioreactor we estimated that around 0,25 g/L corresponds to recombinant
scFab. Purification of the protein was tried in several ways, and the most efficient process was
found to be the Ion Exchange Chromatography. When the purification and correct folding is
done, replacement of this expression cassette of the vector to produce antibodies of clinical
interest can be done.

1
1 – INTRODUÇÃO
1.1 – PROPRIEDADES GERAIS DO SISTEMA IMUNE
Historicamente, o termo imunidade é derivado do latim immunitas, significa isenção
ou dispensa, referia-se à desobrigação de vários deveres cívicos e processos legais oferecida
aos senadores romanos durante seus mandatos. Biologicamente, imunidade constitui a
proteção contra determinado tipo de enfermidade, seu estudo surgiu pela observação de certas
pessoas, que após se recuperarem de certas infecções, ficavam posteriormente “imunes” a
doença, ou seja, não a desenvolviam novamente. O sistema imune é constituído pelas células
e moléculas responsáveis por essa imunidade, e a resposta coordenada e coletiva à introdução
de substancias estranhas no organismo, recebe o nome de resposta imune (Abbas et al, 2003).
A função fisiológica do sistema imune é a resistência natural ou adquirida que o
organismo apresenta a sua invasão por agentes estranhos, sejam microorganismos infecciosos,
parasitas ou simplesmente substâncias como venenos e toxinas. Todavia mesmo substâncias
não infecciosas podem estimular respostas imunes. Além disso, em alguns casos, os próprios
mecanismos que normalmente protegem contra a infecção e eliminam as substâncias
estranhas são capazes e causar lesão tecidual e doença. Portanto, uma definição mais inclusiva
da imunidade é a de uma reação a invasores estranhos, incluindo microorganismos bem como
macromoléculas, como proteínas e polissacarídeos independente das conseqüências
fisiológicas ou patológicas dessa reação. Imunologia é o estudo da imunidade num sentido
mais amplo incluindo eventos celulares e moleculares que ocorrem após o organismo
encontrar substâncias estranhas (Abbas et al, 2003).
A defesa contra os microorganismos e macromoléculas é mediada por reações iniciais
que constituem a imunidade inata e por respostas mais tardias, a imunidade adquirida. A
imunidade inata (também chamada natural ou nativa) consiste de mecanismos que existem
antes da infecção, capazes de respostas rápidas a microorganismos e que reagem
essencialmente do mesmo modo às infecções repetidas. Já a imunidade adquirida desenvolve-
se como uma resposta à infecção e se adapta a ela, suas principais características são a grande
especificidade para as distintas macromoléculas e a capacidade de responder mais
vigorosamente a repetidas exposições ao mesmo antígeno, por isso é também designada
imunidade específica.

2
As respostas imunes inata e adquirida podem ser classificadas ainda como celular ou
humoral, dependendo dos diferentes componentes do sistema imune que nelas atuam,
funcionando para eliminar diferentes tipos de antígenos. A resposta imune celular inata é
mediada pelas células fagocitárias em geral, já a imunidade celular adquirida é executada
basicamente pelos linfócitos T.
A imunidade humoral é mediada, essencialmente por todos os componentes não
celulares do sistema imune, como os membros do sistema complemento e outros mediadores
de inflamação, por exemplo, as chamadas citocinas, que regulam e coordenam muitas
atividades celulares. A imunidade humoral adquirida é basicamente mediada por proteínas
plasmáticas do grupo das globulinas, chamadas anticorpos (Ac), que são produzidas pelos
linfócitos B. Os anticorpos reconhecem os antígenos especificamente, neutralizam sua
infectividade marcando-os para posterior eliminação pelos vários mecanismos efetores. Esta é
a principal defesa contra microorganismos extracelulares e suas toxinas. (Abbas et al, 2003).
1.2 – ANTICORPOS
Os anticorpos ou imunoglobulinas (Ig) são glicoproteínas de elevada massa molecular
capazes de se ligar especificamente em um determinado antígeno e ativar os sistemas efetores
celulares, desencadeando uma resposta imune. São produzidos pelos linfócitos B ativados
(plasmócitos) e encontram-se em abundância no soro dos vertebrados, podendo estar solúveis
no plasma ou ancorados na superfície dos linfócitos B como receptores de membrana.
A molécula de anticorpo é de natureza tetramérica podendo ser separada em duas
cadeias pesadas (H) idênticas, cada uma com aproximadamente 55 kD, e duas cadeias leves
(L) idênticas, cada uma com 25 kD, unidas por pontes dissulfeto e por ligações não covalentes
(Kuby, 2002) (figura 1A). Tanto as cadeias leves quanto as cadeias pesadas contém uma série
de unidades homólogas repetidas, cada uma com cerca de 110 resíduos de aminoácidos que se
enovelam independentemente em um motivo globular classificado como Domínio Imune
(figura 1B). Cada domínio consiste de um arranjo estável de fitas β antiparalelas ligadas por
pontes de hidrogênio, as quais formam uma estrutura em bicamada, estabilizada por uma
ponte dissulfeto (Padlan, 1994).
Cada cadeia leve e pesada de uma imunoglobulina apresenta regiões variáveis (V)
aminoterminais (N-terminal) e regiões constantes (C) carbóxi-terminais. As regiões
constantes das cadeias leves (CL - Constant Light) podem apresentar dois tipos de domínio
segundo suas seqüências de aminoácidos. São designadas capa (κ) ou lâmbda (λ), e

3
determinam o subtipo da cadeia leve. Já as regiões constantes das cadeias pesadas (CH –
Constant Heavy) são constituídas de três ou quatro domínios (CH1, CH2, CH3 e CH4)
agrupando-se em cinco padrões diferentes de seqüências de aminoácidos designadas pelas
letras do alfabeto grego α, δ, ε, γ e μ. Esse último é o critério determinante da classe ou
isotipo ao qual o anticorpo pertence, podendo este ser IgA, IgD, IgE, IgG ou IgM,
respectivamente correspondendo aos cinco tipos de região constante. Estes cinco isotipos
apresentam capacidade de multimerização distinta e funções efetoras próprias (Paul, 2003).
Figura 1. Estrutura de uma molécula de anticorpo. A - Representação esquemática de uma molécula de IgG. Neste desenho os sítios de ligação do antígeno (antigen binding) são formados pela justaposição de domínios variáveis de cadeias leves (VL) e domínios variáveis de cadeias pesadas (VH); B - Organização espacial dos domínios de uma IgG revelada pela cristalografia de raio X. A estrutura secundária predominante nos anticorpos é a folha β antiparalela (β-Sheet) com pequenas α-hélices em algumas alças (Little et al, 2000; Padlan, 1994). Fc, fragmento cristalizável; Fab, fragmento de ligação ao antígeno; Fv, fragmento variável.
A região variável é denominada VL (variable light) nas cadeias leves e VH (variable
heavy) nas cadeias pesadas. A superfície de ligação ao antígeno é formada pela justaposição
dos domínios VH e VL, que juntos formam o fragmento variável (Fv). Essa superfície é
estruturalmente complementar a uma região de contato no antígeno, o epítopo. Por sua
complementaridade ao epitopo, essa superfície de contato é também conhecida como
paratopo.
A maioria das diferenças de seqüência, nas regiões variáveis, entre os distintos
anticorpos é confinada a três curtas extensões nas regiões V das cadeias leves e pesadas, já

4
que os domínios VH e VL não são uniformemente variáveis em toda sua extensão. Essas
regiões são chamadas hipervariáveis ou Regiões Determinantes de Complementaridade (CDR
– Complementarity-Determining Region) em uma alusão a estrutura tridimensional
complementar à estrutura do epítopo (Abbas, 2003).
As CDR são fundamentais para a definição de especificidade dos anticorpos. Elas são
denominadas de acordo com a sua localização a partir do amino terminal: CDR-L1; CDR-L2;
CDR-L3; CDR-H1; CDR-H2; e CDR-H3, para as cadeias VL e VH, respectivamente. As
regiões não hipervariáveis que intercalam as CDRs são chamadas de arcabouço ou framework
(FR) (Wu e Kabat, 1970).
1.2.1 – Estrutura dos Anticorpos
Devido à estrutura em domínios da molécula de anticorpo (figura 1B), é possível a sua
divisão em subunidades funcionais. Estas podem ser utilizadas separadamente, ou combinadas
com as de outros anticorpos para formar novas moléculas com propriedades específicas
(Haydem et al., 1997).
Estruturalmente, podemos dividir a molécula de anticorpo em três principais
fragmentos, dois Fabs (fração que se liga ao antígeno), que são idênticos entre si, contendo as
cadeias leves e dois domínios da cadeia pesada (CH1 e VH), e um Fc (fração cristalizável),
contendo os demais domínios constantes das cadeias pesadas, CH2 e CH3, e, dependendo do
isotipo do anticorpo, também CH4. Os Fabs estão ligados ao Fc por uma região em forma de
dobradiça (hinge), que varia em extensão e flexibilidade nas diferentes classes de anticorpos
(figura 2). Os fragmentos Fab carregam a atividade de ligação ao antígeno, enquanto o
fragmento Fc contribui para as funções efetoras, como a ligação ao receptor Fc, a meia vida
no soro e a fixação do complemento. Desta forma, o recrutamento do sistema imune é
mediado pelas regiões constantes, porém essas funções são desencadeadas pela ligação do
antígeno ao sítio de combinação, espacialmente distante, na região variável. A região
dobradiça contém duas ou três pontes disulfeto, esse peptídeo de ligação é altamente
susceptível a proteólise, o que permite o desmembramento da molécula em suas partes
funcionais Fab e Fc (Plünckthun e Skerra, 1989).
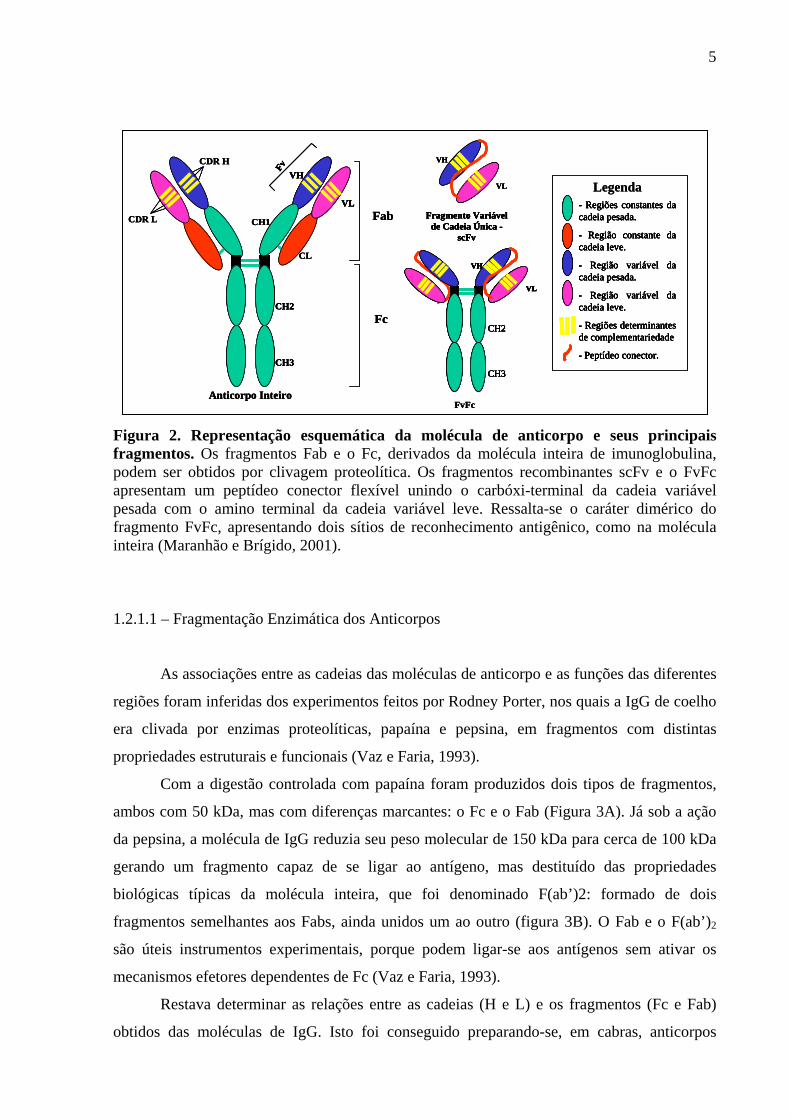
5
Fab
CH3
CH2
CH1
CL
CDR H
CDR L
Anticorpo Inteiro
Fc
VH
VL
Fv
Fragmento Variável de Cadeia Única -
scFv
VH
VL
VH
VL
CH3
CH2
FvFc
Fab
CH3
CH2
CH1
CL
CDR H
CDR L
Anticorpo Inteiro
Fc
VH
VL
Fv
Fab
CH3
CH2
CH3
CH2
CH1
CL
CDR H
CDR L
Anticorpo Inteiro
Fc
VH
VL
Fv
VH
VL
Fv
VH
VL
Fv
Fragmento Variável de Cadeia Única -
scFv
VH
VL
Fragmento Variável de Cadeia Única -
scFv
VH
VL
Fragmento Variável de Cadeia Única -
scFv
Fragmento Variável de Cadeia Única -
scFv
VH
VL
VH
VL
CH3
CH2
FvFc
VH
VL
CH3
CH2
VH
VL
VH
VL
VH
VL
CH3
CH2
CH3
CH2
CH3
CH2
FvFc
Legenda- Regiões constantes da cadeia pesada.
- Região constante da cadeia leve.
- Região variável da cadeia pesada.
- Região variável da cadeia leve.
- Regiões determinantes de complementariedade
- Peptídeo conector.
Legenda- Regiões constantes da cadeia pesada.
- Região constante da cadeia leve.
- Região variável da cadeia pesada.
- Região variável da cadeia leve.
- Regiões determinantes de complementariedade
- Peptídeo conector.
- Regiões constantes da cadeia pesada.
- Região constante da cadeia leve.
- Região variável da cadeia pesada.
- Região variável da cadeia leve.
- Regiões determinantes de complementariedade
- Peptídeo conector.
- Regiões constantes da cadeia pesada.
- Região constante da cadeia leve.
- Região variável da cadeia pesada.
- Região variável da cadeia leve.
- Regiões determinantes de complementariedade
- Peptídeo conector.
- Regiões constantes da cadeia pesada.
- Região constante da cadeia leve.
- Região variável da cadeia pesada.
- Região variável da cadeia leve.
- Regiões determinantes de complementariedade
- Peptídeo conector.
Figura 2. Representação esquemática da molécula de anticorpo e seus principais fragmentos. Os fragmentos Fab e o Fc, derivados da molécula inteira de imunoglobulina, podem ser obtidos por clivagem proteolítica. Os fragmentos recombinantes scFv e o FvFc apresentam um peptídeo conector flexível unindo o carbóxi-terminal da cadeia variável pesada com o amino terminal da cadeia variável leve. Ressalta-se o caráter dimérico do fragmento FvFc, apresentando dois sítios de reconhecimento antigênico, como na molécula inteira (Maranhão e Brígido, 2001).
1.2.1.1 – Fragmentação Enzimática dos Anticorpos
As associações entre as cadeias das moléculas de anticorpo e as funções das diferentes
regiões foram inferidas dos experimentos feitos por Rodney Porter, nos quais a IgG de coelho
era clivada por enzimas proteolíticas, papaína e pepsina, em fragmentos com distintas
propriedades estruturais e funcionais (Vaz e Faria, 1993).
Com a digestão controlada com papaína foram produzidos dois tipos de fragmentos,
ambos com 50 kDa, mas com diferenças marcantes: o Fc e o Fab (Figura 3A). Já sob a ação
da pepsina, a molécula de IgG reduzia seu peso molecular de 150 kDa para cerca de 100 kDa
gerando um fragmento capaz de se ligar ao antígeno, mas destituído das propriedades
biológicas típicas da molécula inteira, que foi denominado F(ab’)2: formado de dois
fragmentos semelhantes aos Fabs, ainda unidos um ao outro (figura 3B). O Fab e o F(ab’)2
são úteis instrumentos experimentais, porque podem ligar-se aos antígenos sem ativar os
mecanismos efetores dependentes de Fc (Vaz e Faria, 1993).
Restava determinar as relações entre as cadeias (H e L) e os fragmentos (Fc e Fab)
obtidos das moléculas de IgG. Isto foi conseguido preparando-se, em cabras, anticorpos

6
contra s cadeias pesadas (anti-H) e contra as cadeias leves (anti-L) e testando-os contra os
fragmentos Fc e Fab. O soro de cabra anti-cadeia pesada (anti-H) reagia tanto com o Fab
como com o Fc, enquanto que o soro anti-cadeia leve (anti-L) não reagia com o Fc. Com base
nessas informações, Porter propôs um modelo para a estrutura da IgG que, em linhas gerais, é
o modelo aceito até hoje, e graças a este modelo da estrutura química dos anticorpos o Dr.
Porter recebeu junto com o pesquisador estadunidense Gerald M. Edelman o prêmio Nobel
em 1972 (Vaz e Faria, 1993).
Os resultados da proteólise limitada por papaína ou por pepsina sobre outros isotipos,
ou das IgGs de outras espécies, nem sempre reproduzem os resultados obtidos originalmente.
Entretanto, a organização básica da molécula da IgG que Porter deduziu de seus experimentos
é comum a todas as espécies de Ig de todos os isotipos e de todas as espécies. De fato, esses
experimentos de proteólise proporcionaram a primeira evidência de que as funções de
reconhecimento do antígeno e as funções efetoras das moléculas de Ig estavam espacialmente
segregadas (Abbas et al., 2003).
Figura 3. Fragmentação enzimática da molécula de anticorpo. Representação esquemática dos fragmentos obtidos por meio da digestão proteolítica controlada com as enzimas (A) papaína e (B) pepsina. A cadeia Leve (L) está representada em cinza e cadeia pesada (H) em preto. (adaptado de Kuby, 2002).

7
1.2.1.2 – Obtenção e Aplicações Biotecnológicas de Anticorpos Recombinantes
A utilização de fragmentos de anticorpos no tratamento de enfermidades é uma
perspectiva atrativa para a medicina. Para tanto se faz necessária sua produção e purificação
em larga escala. A digestão proteolítica e subseqüente purificação dos fragmentos consistem
em procedimentos complexos e inviáveis para a produção em larga escala, onde a eficiência
da digestão e processo de redobramento da molécula in vivo declina com a quantidade de
domínios de imunoglobulina e pontes dissulfeto internas. Os Fvs dissociam a VH e VL em
condições fisiológicas, e por isso não são úteis em terapias. Porém, com o surgimento da
tecnologia do DNA recombinante, assim como o desenvolvimento de técnicas baseadas na
reação polimerásica em cadeia (PCR) para a amplificação e clonagem de genes de
imunoglobulinas (Orlandi et al., 1989), a engenharia de anticorpos evoluiu rapidamente.
Um dos maiores avanços na tecnologia de engenharia de anticorpos foi a construção
de moléculas de fragmentos de anticorpos de cadeia única (scFv - single-chain Fv) (Bird et
al, 1988). Os genes que codificam para os domínios VL e VH são clonados e fusionados por
um peptídeo flexível, normalmente (Gly4Ser)3, a fim de estabilizar a interação entre os
mesmos, criando um único gene que codifica toda molécula (figura 2). Os fragmentos scFv
formam derivados funcionais de anticorpos que conservam a especificidade monovalente de
ligação do anticorpo intacto original (Bird e Walker, 1991).
Outra técnica muito importante é a utilização de hibridomas para produção de
anticorpos com as características desejadas. Os hibridomas são células obtidas por meio da
fusão de uma célula tumoral com linfócitos maduros (plasmócitos), oriundos de um animal
imunizado (Maranhão e Brígido, 2001). Após essa fusão, os hibridomas gerados passarão a
produzir anticorpos monoclonais, e serão a base para a engenharia de anticorpos, objetivando
o melhoramento estrutural ou ainda a obtenção de seqüências para a produção de outros
fragmentos de anticorpos com variadas finalidades.
Por meio da seleção do isotipo da cadeia pesada do anticorpo selecionado para
utilização é possível o direcionamento de uma resposta imune efetora específica, gerada pela
porção Fc. Outra perspectiva é a manipulação da imunoglobulina para a utilização de porções
menores da molécula, normalmente Fab ou scFv. Essa característica é visada quando se
pretende que a molécula tenha alta penetrabilidade, no entanto, esse tipo de mini-molécula
apresenta uma meia vida reduzida no soro do paciente. Quando se busca um maior tempo de
circulação na corrente sangüínea, a opção mais frequente é a clonagem da molécula inteira e
sua expressão em sistemas eucarióticos, para obtenção da montagem correta da estrutura

8
tetramérica (com duas cadeias leves e duas cadeias pesadas) no retículo endoplasmático das
células utilizadas. Formas intermediárias entre o fragmento variável de cadeia única e o
anticorpo inteiro, como o FvFc (Figura 2), também têm sido propostas recentemente
(Andrade et. al., 2000; De Lorenzo et al., 2005).
Uma importante aplicação de anticorpos recombinantes é na localização de tecidos
tumorais, incluindo a utilização de fragmentos de anticorpos marcados com radioisótopos. Por
isso, a obtenção de anticorpos monoclonais ou fragmentos de origem humana é de interesse
na clínica médica. Os avanços na engenharia de anticorpos permitiram o transplante de CDRs
entre domínios variáveis de diferentes anticorpos com finalidades diversas, como a
humanização de anticorpos. Neste caso, CDRs humanas são introduzidas em anticorpos
monoclonais de origem murina, disponibilizando seu uso sem a reação de sensibilidade, à
porção murina, induzida no paciente.
Além das modificações estruturais, os fragmentos de anticorpo passaram a ser
produzidos em diversos sistemas de expressão (fermentação bacteriana, células de mamíferos,
animais e plantas geneticamente modificados, sistema de expressão em P. pastoris) que
abriram novas perspectivos para o desenvolvimento, inovação terapêutica e produção de
agentes diagnósticos (Mountain e Adair, 1992 e Kipriyanov e Little, 1999).
A maleabilidade da manipulação de sistemas de expressão para anticorpos
recombinantes permite que aplicações biotecnológicas sejam direcionadas para utilização
desses produtos em diversos processos tecnológicos e na terapêutica. Os fragmentos
recombinantes podem ser expressos sozinhos ou fusionados a toxinas, enzimas, receptores de
adesão, citocinas, gerando proteínas bioespecíficas.
Anticorpos podem ser desenvolvidos para se tornarem catalisadores de determinada
reação química. Este é o caso dos anticorpos catalíticos que são normalmente produzidos para
um análogo estável de um intermediário em uma reação química. A afinidade pelo análogo e
pelo intermediário permite a este reagente funcionar como um catalisador, diminuindo a
energia de ativação da reação através da estabilização energética do intermediário (Zhu et al.,
2006; Kato, et al., 2005; Uda e Hifumi, 2004).

9
1.3 – EXPRESSÃO HETERÓLOGA DE ANTICORPOS
A escolha de um determinado sistema de expressão depende da aplicação e da
complexidade dos fragmentos de anticorpo recombinantes (Hayden et al, 1997). Uma série de
sistemas de expressão têm sido relatados (Fromer & Ninnemann, 1995), para em E. coli,
células de insetos, plantas, leveduras e células de mamíferos.
Para que o anticorpo heterólogo possa ser aplicado em pacientes, ele deve ser
produzido preferencialmente em células animais, para melhor processamento, visando,
sobretudo uma glicosilação mais adequada ao organismo humano, uma vez que carboidratos
são muito imunogênicos. Porém, para testes iniciais com o antígeno, o anticorpo humanizado
pode ser produzido de modo mais simples, em organismos que já tenham sua maquinaria
gênica bem conhecida, além de um acúmulo de dados práticos de manipulação. Neste caso, o
organismo mais indicado seria a bactéria E. coli, um dos organismos mais utilizados e
conhecidos na engenharia genética e de proteínas. Uma outra vantagem deste organismo é sua
alta taxa de crescimento, o que otimiza sua utilização. Entretanto, o sistema de expressão que
tem se mostrado mais eficiente para a produção de fragmentos de anticorpos funcionais é o da
levedura metilotrófica Pichia pastoris (Cregg et al, 1985). Sua utilização para produção de
biofármacos cresce com a expectativa de desenvolvimento de linhagens modificadas para
reproduzir padrões de glicosilação humanos (Li et al., 2006).
1.3.1 – Sistema de expressão em Pichia pastoris
O uso da levedura P. pastoris tem sido uma alternativa eficaz na produção de
anticorpos e de várias outras proteínas heterólogas. As vantagens de sua utilização vão desde
a simplicidade das técnicas necessárias para a manipulação genética até a habilidade desta
levedura para produção de altos níveis de proteínas heterólogas (Cereghino e Cregg, 2000;
Daly e Hearn, 2005). Várias proteínas já foram expressas com sucesso em P. pastoris, com
nível de expressão satisfatório, como podemos observar na tabela 1.
O sistema de expressão em levedura tem uma série de vantagens frente ao sistema
bacteriano como a capacidade de fazer muitas modificações pós-traducionais tipicamente
associadas com eucariotos superiores, incluindo o processamento de peptídeo sinal, a
formação de pontes dissulfeto, o dobramento correto da proteína recombinante, a adição de
lipídeos e uma glicosilação (O- e/ou N- ligadas) mais próxima, mesmo que não exatamente
igual, a humana (Cereghino e Cregg, 2000; Daly e Hearn, 2005).

10
Tabela 1. Lista parcial de proteínas heterólogas que foram expressas em P. pastoris com sucesso (adaptado de Cereghino e Cregg, 2000; Eldin et al., 1997; Emberson et al., 2005; Hu et al., 2005; Lange et al., 2001; Manual do Kit de Expressão em Pichia, 2002; Marty et al., 2001; Ning et al., 2003; Ning et al., 2005)
Proteína Expressão (g/L)
Forma de expressão Fenótipo
Enzimas Invertase 2.3 Secretada Mut+ Lisozima Bovina C2 0.55 Secretada Mut+ Streptoquinase (ativa) 0.08 Intracelular * Alfa amilase 2.5 Secretada MutS Pectato Liase 0.004 Secretada MutS Fosfo-ribuloquinase de Espinafre 0.1 Intracelular MutS
Antígenos Antígeno de superfície de Hepatite B 0.4 Intracelular MutS Antígeno P69 Perforina 3.0 Intracelular MutS Fragmento C da Toxina Tetânica 12.0 Intracelular Mut+/MutSgp120 de HIV-1 1.25 Intracelular Mut+ Proteína anticoagulante de Carrapato 1.7 Secretada MutS Glicoproteína Bm86 de intestino de Carrapato 1.5 Secretada *
Proteínas Regulatórias Fator de necrose tumoral (TNF) 10.0 Intracelular MutS Fator de crescimento epidermal de Camundongo (EGF) 0.45 Secretada MutS Interferon Humano (IFN) α2b 0.4 Intracelular MutS
Proteínas de Membrana CD38 humano (parte solúvel) 0.05 Secretada MutS Receptor de serotonina de Camundongo 0.001 Secretada Mut+
Proteases e Inibidores de Proteases Carboxipeptidase B 0.8 Secretada Mut+/MutSEnteroquinase 0.021 Secretada Mut+ Inibidor de Proteinase 6 Humano 0.05 Intracelular Mut+
Fragmentos de Anticorpos Diabody bivalente anti-antígeno carcino-embriônico (CEA), co-receptor de célula T CD2 >0.1 Secretada * Fab anti-HBsAg 0,420 Secretada * Fab atrazine específico (K411B) 0,040 Secretada Mut+ scFv (anti carcinoma ovariano) 0,05 Secretada * scFv anti domínio ED-B da B-fibronectina (B-Fn) 0,020 Secretada * scFv anti-CD33 0,048 Secretada Mut+ scFv anti-Fator de inibição recombinante da Leucemia Humana, feito em Coelho 0.1 Secretada * scFv de coelho >0.1 Secretada MutS scFv anti-serpina 0,025 Secretada MutS scFv anti-ErbB2 0,015 Secretada Mut+ * Não informado

11
Além disso, a viabilidade do sistema de expressão como “Kit” comercialmente
disponível torna a P. pastoris o agente ideal para a produção de anticorpos em laboratório
como uma alternativa às células de mamífero. Apesar de ser a tecnologia de expressão
protéica mais utilizada para a produção comercial de anticorpos terapêuticos (Verna et al.,
1998), a manutenção de cultura de células de mamífero é um processo laborioso, caro e de
capacidade limitada. Estima-se que, em 2010, a demanda do mercado de anticorpos
monoclonais seja pelo menos cinco vezes superior a capacidade máxima de produção destes
fármacos neste tipo de cultura (Gomord et al., 2004).
Outra vantagem do sistema de expressão em P. pastoris é a presença da protease Kex2
que é utilizada para o processamento do peptídeo sinal e, no caso deste trabalho, também da
proteína heteróloga expressa. O peptídio sinal do fator alfa, presente no vetor utilizado,
consiste em uma seqüência de 19 aminoácidos (pré-peptídio) seguidos de uma seqüência de
60 aminoácidos hidrofóbicos (pró-peptídio). Este sinal é responsável pelo transporte da pró-
proteína (sem o pré-peptídio) pelo RE e sua subseqüente clivagem. A pró-proteína é
transportada pelo Golgi onde o pró-sinal é clivado pela Kex2 liberando a proteína madura.
(Daly e Hearn, 2005)
O processamento do peptídeo sinal envolve três passos. Primeiro, a remoção do pré-
sinal pela peptidase no RE. Segundo, a endopeptidase Kex2 reconhece o duplo par de
resíduos Lys- Arg no pró-sinal. Este processo é rapidamente seguido pela clivagem da
repetição Glu-Ala por Ste13. A eficiência desse processo pode ser afetada pela seqüência de
aminoácidos presentes em torno do sitio de clivagem. Por exemplo, é sabido que a eficiência
de clivagem, tanto de Kex2 quanto de Stel3, pode ser influenciada pela proximidade de
resíduos de prolina. Além do mais, a estrutura terciária formada pela proteína exógena pode
proteger os sitios de clivagem de suas respectivas proteases (Cereghino e Cregg, 2000).
No presente trabalho foi utilizado um sítio diferenciado para o processamento da
proteína recombinante. Trabalhos anteriores mostraram a eficiência de processamento de uma
seqüência kex2-like, ou seja um sítio de clivagem similar ao da endopeptidase kex2
(Contreras et al., 1991). O sítio kex2-like consiste na seqüência Arg-Met-Asp-Lys-Arg-Ala-
Pro, onde a clivagem ocorre logo após o par de resíduos Lys-Arg seguido do processamento
da seqüência Ala-Pro. Espera-se que esse processamento ocorra junto com a remoção do pré-
peptídeo sinal, no retículo endoplasmático da levedura, onde o ambiente redutor é propício
para a formação de pontes dissulfeto para a montagem correta das cadeias leve e pesada do
fragmento Fab.

12
A P. pastoris é uma levedura metilotrófica, ou seja, é capaz de utilizar metanol como
única fonte de carbono. Essa característica é um dos fatores responsáveis por sua capacidade
de produção em larga escala de proteínas heterólogas. A primeira enzima da via de utilização
de metanol, álcool oxidase (AOX), é codificada por dois genes: AOX1 e AOX2 (Macauley-
Patrick et al, 2005; Daly e Hearn, 2005). Embora as proteínas Aox1 e Aox2 possuam 97% de
identidade de seqüência e atividade enzimática equivalente, mais de 95% da atividade de
álcool oxidase é atribuída à AOX1. Isso ocorre, pois o promotor AOX1 é mais forte. Após a
adição de metanol, transcritos do gene AOX1 são rapidamente induzidos a altos níveis,
perfazendo 5% do RNA poliA+ total (Cereghino e Cregg, 2000: Daly e Hearn, 2005). A
proteína Aox1 é superexpressa e chega a 30% das proteínas totais intracelulares. Dessa forma,
genes heterólogos são então clonados sob o controle do promotor AOX1 para permitir uma
indução rápida, forte e controlada pelo metanol.
Figura 4. Via metabólica da oxidação do metanol em formaldeído na levedura P. pastoris. A enzima Álcool Oxidase (1) catalisa a oxidação de metanol em formaldeído, sendo este produto utilizado para geração de energia e biomassa. 2, Catalase; 3, Formaldeído deidrogenase; 4, Formato deidrogenase, 5, Diidroxiacetona sintase; 6, Diidroxiacetona quinase; 7, Frutose 1,6-Bifosfato aldolase; 8, Frutose 1,6-bisfosfatase. (adaptado de Cereghino e Cregg, 2000).
O metanol é oxidado para a formação de formaldeído pela a enzima álcool oxidase
(AOX) no peroxissomo (figura 4). Essa reação requer O2 e forma H2O2 e formaldeído. Essa
enzima não é especifica para metanol e pode oxidar outros álcoois primários, contudo quanto
mais maior o número de carbonos no álcool mais decresce a atividade da enzima. O H2O2
produzido pela reação da AOX é convertido em água pela peroxissomo catalase. A conversão
do metanol em formaldeído é o passo limitante dessa via, sendo regulado pelo maior acúmulo

13
da enzima AOX na célula. Isso é observado, por exemplo, quando o rápido aumento da
concentração de metanol no meio causa uma inibição do crescimento celular. O formaldeído
produzido nesta reação é mais oxidado tanto pela via dissimulatória citosólica, o que aumenta
a energia, quanto pela via assimilatória para assimilação de biomassa. (Daly e Hearn, 2005)
Os vetores de expressão em P. pastoris são geralmente do tipo integrativo, inserindo
os genes heterólogos no genoma da levedura para maximizar a estabilidade do transgene. São
bifuncionais, desenvolvidos tanto para a transformação em E. coli quanto P. pastoris
(Cereghino e Cregg, 2000). Para a manutenção em E. coli, os plasmídios contêm uma origem
de replicação bacteriana e o gene de resistência a ampicilina. Possuem um cassete de
expressão formado pelo promotor e pela região terminadora de transcrição do gene AOX1,
além de uma marca de seleção, sendo a mais utilizada a de seleção auxotrófica pelo gene
histidinol deidrogenase (HIS4), usada em combinação com linhagens his4-. Um dos vetores
mais utilizados é o plasmídio pPIC9 (Invitrogen), que possui como sinal de secreção o
peptídeo sinal do fator α de S. cerevisiae (Torres e Moraes, 2000 e Daly e Hearn, 2005).
A linhagem SMD1168 possui uma mutação no gene HIS4 tornando a levedura
deficiente na síntese de histidina. Quando transformadas com plasmídio de expressão que
carregam o gene HIS4 para complementar o gene his4 da hospedeira, os transformantes
podem ser facilmente selecionados pela habilidade de crescer em meio sem histidina.
A recombinação homóloga para a integração dos plasmídios pode ocorrer no locus
HIS4 ou no locus AOX1. Quando a recombinação ocorre no locus AOX1 por meio de um
duplo crossover entre regiões do vetor e do genoma, a região codificadora AOX1 é
completamente removida (figura 5). O fenótipo resultante MutS (Methanol utilization slow) é
causado pela perda de atividade de álcool oxidase codificada pelo gene AOX1, restando
apenas o AOX2. Isto resulta num fenótipo de crescimento lento em meio com metanol.
Quando a recombinação homóloga ocorre através de um crossover simples, levando à
inserção gênica no locus HIS4 ou no locus AOX1, o fenótipo resultante é Mut+ (Methanol
utilization plus). Este fenótipo se refere à habilidade de metabolizar metanol como única fonte
de carbono comparável às linhagens selvagens (figura 6). Algumas proteínas são melhor
expressas em um tipo de fenótipo do que em outro (MutS ou Mut+).

14
Figura 5. Substituição Gênica por duplo crossover no locus AOX1 em P. pastoris. A figura acima mostra a substituição gênica no locus AOX1. Numa cepa his4- como a SMD1168, a substituição gênica (inserção ômega) é originada por um duplo crossover, do promotor AOX1 e da região 3’AOX1, entre o vetor e o genoma. Isto resulta numa remoção completa da região codificadora do AOX1 genômico. O fenótipo resultante é His+ MutS. Os transformantes His+ podem ser facilmente localizados pelo seu fenótipo e o MutS serve como indicador da integração via substituição do locus AOX1. O resultado deste tipo de substituição gênica é a perda do locus AOX1 (MutS) e o ganho de um cassete de expressão contendo PAOX1, o gene de interesse e HIS4. (adaptado do Manual do Kit de expressão em Pichia, Invitrogen, 2002)
Figura 6. Inserção Gênica por simples crossover no locus his4 em P. pastoris. A figura acima mostra o resultado da inserção de um plasmídio, com uma cópia HIS4, no locus do gene his4, provocando a ruptura do mesmo e a reversão do fenótipo por complementação gênica. Tanto na cepa SMD1168 (Mut+) quanto KM71 (MutS), os eventos de inserção gênica no locus his4 resultam do simples crossover entre esse locus e do gene HIS4 do vetor, culminando na inserção de uma ou mais cópias do vetor. Desde que o AOX1 genômico ou os loci aox::ARG4 não estejam envolvidos neste evento de recombinação, o fenótipo de cada transformante His+ será o mesmo fenótipo Mut da cepa original. Com a linearização do vetor recombinante em sítios de restrição enzimática localizados nos genes HIS4, os fenótipos Mut+
ou MutS podem ser convenientemente gerados dependendo da cepa hospedeira utilizada (adaptado do Manual do Kit de expressão em Pichia, Invitrogen, 2002).

15
Em relação a outras leveduras utilizadas para a expressão de genes, como
Saccharomyces cerevisiae, P. pastoris tem a vantagem de não realizar a hiperglicosilação da
proteína secretada, como acontece na primeira, fato que poderia tornar a proteína mais
imunogênica em humanos e/ou interferir no enovelamento da mesma, tornando-a
biologicamente inapta (Cregg, 1999). Tanto S. cerevisiae quanto P. pastoris apresentam
padrões de glicosilação do tipo manose N-ligada. Porém, em S. cerevisiae, as proteínas
costumam apresentar mais de 50 resíduos de manose, caracterizando assim a condição de
hiperglicosilação. Em P. pastoris, a N-glicosilação se inicia no retículo endoplasmático (ER)
com a transferência da unidade de oligossacarídeo lipídio ligado, Glc3ManGlc (Glc, glicose;
GlcNAc, N-acetilglicosamina) para asparagina na seqüência de reconhecimento Asn-X-
Ser/Thr. Além disso, esta levedura não é capaz de realizar glicosilações terminais do tipo α1,3
(Cereghino e Cregg, 2000). Há evidências que este tipo de ligação seja responsável pela
hiper-imunogenicidade apresentada por algumas proteínas expressas em S. cerevisiae (Cregg,
1993).
A expressão heteróloga em P. pastoris pode ser feita de forma intracelular ou via
secreção. A vantagem da segunda é que P. pastoris secreta poucas proteínas nativas,
facilitando, portanto a posterior purificação (Daly e Hearn, 2005).
1.4 – FERMENTAÇÃO
A fermentação é uma transformação bioquímica provocada num substrato por
fermento vivo ou por um principio extraído deste fermento. A importância deste processo esta
diretamente relacionada com diversos setores da agroindústria, com destaque para a
alcooleira.
A fermentação é conhecida pela humanidade desde a época pré-histórica, mesmo que
ainda não entendida, sendo originalmente utilizada para preparação e conservação de
alimentos e bebidas. Entre os primeiros alimentos que surgiram da utilização desse processo
se encontram os pães, queijos e algumas bebidas fermentadas das quais descendem a cerveja e
o vinho, por exemplo (Amorin e Leão, 2005).
O nome “fermento” surgiu da observação do processo, que libera uma espécie de
fumaça (CO2) e aquece o material que esta sofrendo a transformação. Assim sendo, os antigos
pensavam que o composto estava “fervendo” surgindo daí a denominação.
A fabricação de pães é uma técnica milenar, sendo sabidamente conhecida por povos
da Mesopotâmia e Egito há pelo menos 6.000 anos. Ninguém sabe precisar ao certo a data da

16
invenção do queijo. A teoria mais conhecida é que por volta de 10.000 a.C. os pastores
aprenderam a separar a coalha do leite fermentado, surgindo daí os primeiros processos de
preparação desse alimento. Na opinião de muitos, a cerveja e o vinho não foram as primeiras
bebidas obtidas por meio da fermentação, e sim o hidromel. Esta bebida era obtida pelo
abandono de soluções aquosas de mel silvestre. A cerveja, por sua vez, teria sido uma das
bebidas mais consumidas nos tempos primitivos, sendo encontrados resíduos e registro em
tábuas que datam de 8.000 a.C. (Amorin e Leão, 2005).
O vinho, é conhecido pela humanidade há mais de 8.000 anos e deve ter surgido pela
fermentação espontânea da uva na região do Cáucaso. Muitos autores consideram que se
extraiu o álcool, pela primeira vez, por meio do vinho. Outros acreditam que ele foi obtido de
outras bebidas fermentadas, como a cerveja. O fato é que as bebidas fermentadas foram muito
consumidas até se perceber que era possível extrair delas a parte da qual lhes provinha o
“espírito” por meio da destilação (do latim dis, que significa separar, e sitllo, derramar gota a
gota) (Amorin e Leão, 2005).
O processo fermentativo só foi entendido após a descoberta do mundo microscópico.
Foi Lavoisier, em seu Traité Élémentaire de Chimie (1789), quem realizou uma análise
sistemática sobre as substâncias envolvidas na fermentação, analisando as relações ponderais
que ligam uma substância fermentescível - o açúcar - aos produtos da fermentação: o álcool e
o dióxido de carbono. Também foi Lavoisier quem estabeleceu o balanço da reação,
mostrando que o açúcar era desdobrado em álcool e anidro carbônico e, ainda, que era
possível recombiná-los em açúcar (revisado por Amorin e Leão, 2005).
Anos depois, com o melhoramento sensível das técnicas de investigação científica, foi
possível a observação de fermentados e, por volta de 1880, a fermentação alcoólica como
sinal de atividade fisiológica de leveduras já era aceita. A equação global da fermentação
alcoólica foi enunciada em 1908 por Harden e Young, sendo ela:
C6H12O6 2CH3CH2OH +CO2
Dessa forma foi possível caracterizar as fermentações como transformações químicas
cujos agentes são os microorganismos, sendo estabelecidas suas reações (revisado por Amorin
e Leão, 2005).
1.4.1 - O processo de cultivo
Há vários tipos de biorreatores para o cultivo microbiano ou de células animais. Os
fermentadores podem ser classificados pelo fato das células estarem suspensas ou

17
imobilizadas. Algumas células, principalmente as animais, podem ser dependentes de
ancoramento, necessitando assim de um suporte físico para crescerem, como por exemplo,
microcarregadores ou garrafas “Roller”. Outra forma de se manter as células imobilizadas é
pelo uso de membranas fibrosas ocas (hollow fiber) (Tonso, 2000). Já o cultivo de células
microbianas, é usualmente feito com as mesmas suspensas, utilizando-se principalmente
biorreatores tipo tanque agitado, com ou sem algum sistema de separação de células, e em
vários casos, reatores do tipo “air-lift”, com a agitação feita pela própria aeração (Tonso,
2000).
1.4.2 - Controle dos parâmetros do processo fermentativo
São muitas as variáveis que afetam o crescimento e a produção em processos
fermentativos em biorreatores. Por meio do desenvolvimento de diferentes técnicas, a
capacidade de se observar um processo em biorreator tem evoluído muito nos últimos anos.
Quando possível, os parâmetros como o pH, a temperatura e o oxigênio dissolvido
(DO) devem ser acompanhados simultaneamente, isto é, no próprio biorreator, sem a
necessidade de amostragem (observação do cultivo "on-line", durante a linha de produção),
pois traz uma série de vantagens para a manutenção do processo. Porém, a simples ação da
retirada de amostras muitas vezes prejudica o processo, por exemplo, devido à diminuição do
volume de reação no caso de pequenos reatores ou a possibilidade de contaminação. Além
disso, a utilização dos resultados de uma análise para fins de controle depende da sua
disponibilidade em tempo real. Desta forma, o desenvolvimento de novas ferramentas de
análise "on-line”, como determinação da concentração de substrato e de produtos inibitórios,
permitem o melhor controle do processo, aumentando a produtividade (Thiry e Cingolani,
2002; Tonso, 2000). Idealmente, se deseja também o monitoramento "on-line" da
concentração de produto, objeto final do processo fermentativo.
1.4.3 - Sistemas de transferência de Oxigênio
Para a manutenção do crescimento celular de cultivos aeróbicos microbianos, a
concentração de oxigênio dissolvido é fundamental. Sendo assim, o oxigênio pode ser
considerado como um “nutriente” do meio de cultura. Portanto, uma variável a ser
acompanhada e, se possível, controlada numa faixa adequada, pois tanto valores em excesso
como em falta acarretam na perda de produtividade (Macauley-Patrick et al, 2005).

18
Mas, o oxigênio tem a solubilidade muito baixa quando comparado com a fonte de
carbono, sendo ainda mais reduzida em meios de cultura. Devido a essa baixa solubilidade,
não pode ser fornecido como os demais nutrientes, junto ao meio em quantidade suficiente
para todo o cultivo. Contudo, é um componente que deve estar em solução para ser utilizado
pelo microorganismo e a cultura deve ser suplementada com uma taxa suficiente de Oxigênio
durante o crescimento, para suprir a demanda (Zhang, 2005).
As formas de transferência de oxigênio ao meio não diferem muito entre os cultivos
microbianos, sendo a demanda do processo fermentativo, normalmente, suprida pela aeração
e agitação do meio de cultura. Porém, devido à velocidade de crescimento e a grande
dependência de oxigênio da levedura metilotrófica P. pastoris, em cultivos aerados por
aspersão, a oxigenação do meio costuma ser limitante. Valores típicos da velocidade
específica de respiração situam-se entre 1 e 24 µg de oxigênio por 106 células por horas
(revisado por Tonso, 2000). Considerando a dificuldade de medida destes valores, uma vez
que são muito pequenos, a estimativa da velocidade de consumo de oxigênio é baseado na
análise do sinal da sonda de oxigênio dissolvido em resposta a um pulso de ar ou oxigênio
injetado no reator.
Uma outra forma de se obter uma medida da concentração de oxigênio dissolvido é
através da Taxa de Transferência de Oxigênio (OTR - oxygen mass transfer rate), sendo a
medida da mudança de concentração de oxigênio dissolvido em um período de tempo. A OTR
no fermentador depende de vários fatores como a geometria do biorreator (tipo de biorreator,
distribuidor e desing do agitador), propriedades líquidas da solução (viscosidade, tensão
superficial) e a dispersão de energia no fluido que depende do fluxo de ar e velocidade de
agitação. Desta forma, a composição e propriedades do meio mudam com o tempo e têm um
importante efeito na taxa de transferência (Zhang, 2005).
Diversos trabalhos foram publicados estudando os efeitos da concentração de oxigênio
dissolvido em diferentes sistemas de cultivo (Lee et al., 2003; Damasceno et al., 2004; Zhang,
2005). De modo geral, o que se observa é que em concentrações extremas, próximas de 0% e
100%, o crescimento e produção são prejudicados. Porém, existe uma ampla faixa de
concentrações nas quais os parâmetros do cultivo são pouco influenciados pela concentração
de oxigênio, o que facilita o processo como um todo.

19
1.4.4 - Concentração celular
Certamente, a concentração celular é uma das variáveis mais importantes em todo
processo fermentativo. Dela depende a estimativa das velocidades específicas de crescimento,
do consumo de substrato e da produção. Existem várias técnicas para o acompanhamento da
concentração celular em um cultivo com células em suspensão (Konstantinov, 1996), sendo
classificadas em:
a) Medidas diretas da concentração: densitometria de ressonância acústica,
espectroscopia magnética nuclear em tempo real, condutividade, capacitância, fluorescência,
medida da luz absorvida ou espalhada e análise de imagens.
b) Medidas indiretas: estimativas baseadas na velocidade de respiração, de produção
de gás carbônico e de produção de ATP.
A técnica mais empregada é o uso de um espectrofotômetro. Ele fornece a densidade
ótica (DO) como base para estimativa da concentração celular, aproveitando-se do fato que os
meios de cultura são geralmente límpidos e as células crescem geralmente isoladas uma das
outras. Previamente, ou ainda durante o processo, deve-se proceder a calibração, que permita
transformar o valor de DO em concentração celular. Tal calibração pode ser desde um simples
fator DO/concentração celular, como uma equação de reta (com dois parâmetros, portanto) ou
polinomial.
1.4.5 - Temperatura e pH
Em qualquer processo fermentativo existe uma faixa de temperatura e pH ideais para o
organismo utilizando. Essa faixa é definida de acordo com as necessidades fisiológicas do
organismo e com a solubilidade do oxigênio no meio, uma vez que a temperatura influencia
esse parâmetro, já que o O2 é mais solúvel em baixas temperaturas. O ideal para a produção
em biorreator é a utilização das temperaturas mais baixas dentro da faixa ideal do
microorganismo em questão.
Outra questão a ser considerada com relação à temperatura é sua importância na
solubilidade e no melhoramento da expressão da proteína. Por exemplo, diminuir a
temperatura de 30ºC para 25ºC durante a fase de indução com metanol resultou em um
aumento do rendimento da produção de quatro vezes mais de galactose oxidase clonada em P.
pastoris. (revisado por Thiry e Cingolani, 2002). Outro fator a ser considerado é uma menor

20
evaporação do metanol em temperaturas mais baixas, melhorando a produção já que a
levedura aproveita melhor metanol do meio.
A otimização do pH do meio de cultura é crucial para a secreção de proteínas
produzidas pela levedura, uma vez que as mesmas têm a capacidade de crescer em um grande
espectro de pHs (3,0-7,0) (Macauley-Patrick et al., 2005), sendo assim, a escolha do pH
depende da estabilidade da proteína recombinante expressa. O cultivo em meios com pH
relativamente baixo e metanol, torna improvável que haja contaminação com a maioria dos
outros microorganismos (Cereghino e Cregg 2000). Além disso, o uso de um baixo pH pode-
se evitar a degradação do produto de fermentação por proteases. (Thiry e Cingolani, 2002).
1.5 – ANTICORPOS e Z-DNA
A molécula de DNA fita dupla pode apresentar três possíveis conformações, sendo
elas A-DNA e B-DNA (forma mais comum), ambas com a hélice voltada para direita, e Z-
DNA, com a hélice voltada para esquerda (Figura 7). A conformação alternativa do DNA
voltada para a esquerda aparece em condições de alternância de purinas/pirimidinas, como na
seqüência (dG-dC)n. Essa conformação é facilmente obtida in vitro, enquanto que in vivo ela
depende de uma tensão torcional negativa elevada.
Em condições experimentais a conformação Z-DNA está bem definida, bem como sua
obtenção por meio de manipulação química. Entretanto, a correlação do Z-DNA com os
processos biológicos do ácido nucléico, ao longo do ciclo celular, ainda permanecem como
objeto de especulação. O mais provável é que esteja relacionado com a recombinação e a
regulação da expressão gênica, pelo hiper-enovelamento da fita de DNA na heterocromatina
(Oh et al., 2002; Majewski e Ott, 2000). Modelo esse que vem sendo re-valorizado com
recentes descobertas do efeito do Z-DNA sobre a cromatina (Liu et al., 2006).
O anticorpo monoclonal Z22, modelo do estudo realizado, foi obtido inicialmente por
Möller e colaboradores em 1982 (revisado por Andrade, 2000) a partir de hibridomas
construídos com células de camundongos imunizados com poli (dG-dC) brominado (Z-DNA).
Posteriormente, fragmentos de anticorpos Z22 na forma de FvFc e scFv (moléculas
quiméricas de IgG1 Z22) foram utilizadas por integrantes do grupo de Imunologia Molecular
do Laboratório de Biologia Molecular da Universidade de Brasília para caracterização dos
mecanismos de reconhecimento ao Z-DNA (Andrade et al. 2000; Ruggiero, 2002; Andrade et
al. 2005). Com descrições moleculares, estruturais e termodinâmicas, o Z-22 apresenta- se

21
hoje como um excelente repórter em experimentos de expressão heteróloga pela facilidade de
identificação e caracterização.
Figura 7. Imagens geradas por computador das três estruturas conhecidas do DNA. À esquerda, B-DNA; ao centro, A-DNA; e à direita, Z-DNA (Adaptado de Brown, 2002).
1.6 – O VETOR DE EXPRESSÃO DE scFab
Para a expressão de anticorpos heterólogos deve-se considerar cuidadosamente a
escolha do vetor utilizado, uma vez que disso dependem vários fatores que poderão interferir
na obtenção desses produtos. Além da preocupação com a manutenção da afinidade original e
com a eliminação da imunogenicidade da molécula em si, outro aspecto relevante é a
otimização da produção do anticorpo recombinante. Os vetores utilizados para tal propósito
servem para dirigir a síntese da cadeia protéica do anticorpo recombinante em um sistema
heterólogo (bactérias, leveduras ou mesmo células de mamíferos).

22
Esses vetores são normalmente plasmídios, contendo um cassete de expressão,
compatíveis com o tipo celular utilizado como hospedeiro. Muitas vezes são montados
contendo os genes que codificam as cadeias constantes leve e pesada, flanqueadas por sítios
de endonucleases de restrição que facilitam a introdução dos domínios variáveis
recombinantes. Possibilitam também a manipulação da atividade efetora por meio da
clonagem de cadeias constantes de isotipos específicos. A escolha do isotipo, bem como
manipulações pontuais para aumentar a meia vida desses produtos na circulação sangüínea
são exemplos de modificações trabalhadas diretamente no vetor (Maranhão e Brígido, 2001).
O vetor utilizado neste trabalho foi inicialmente construído para posterior produção de
anticorpos humanizados de interesse clínico em substituição a anticorpos monoclonais de
camundongo, seguindo o contexto da participação do grupo no Instituto do Milênio/MCT de
Ivestigação Imunológica.
Esse vetor foi construído na forma monocistrônica para evitar um possível problema
de dosagem que ocorre na expressão de fragmentos de imunoglobulinas. Quando as cadeias
são produzidas separadamente pode haver um desbalanço da estequiometria da montagem
resultando na formação de homodímeros ou estruturas não funcionais. Com a utilização de
um único promotor (AOX1) para o controle da expressão gênica de ambas as cadeias leve e
pesada, obtém-se uma molécula de Fab de cadeia única (scFab) com um conteúdo equimolar
das cadeias. A molécula de scFab deve ser clivada separando as cadeias leve e pesada. Isso é
conseguido com a utilização de um peptídeo conector contendo o sítio de processamento da
protease, de retículo endoplasmático, Kex2. Isso facilitará o processamento e montagem da
molécula de Fab para sua posterior secreção.
A escolha da molécula do Fab foi devido ao fato dela possuir uma alta penetrabilidade
nos tecidos, e um melhor desempenho farmacodinâmico, pois não possui o fragmento Fc e,
portanto não induzirá uma resposta efetora, podendo ser utilizada no mapeamento de tumores,
neutralização de toxinas ou para a desintoxicação de fármacos.
Neste projeto propomos um novo vetor para a expressão de anticorpos recombinantes,
com a expressão e caracterização do scFab recombinante produzido pelo vetor pPIg Fab na
levedura metilotrófica P. pastoris. O scFab possui os domínios variáveis, pesado e leve,
derivados do mAbZ22.

23
2 – JUSTIFICATIVA
A produção de anticorpos e/ou seus fragmentos para o uso na medicina é um recurso
cada vez mais visado por profissionais dessa área. Porém, vários fatores limitam sua
utilização e, principalmente, a produção dos mesmos.
Com intuito de contribuir para o avanço das pesquisas neste ramo, propomos o uso de
um vetor monocistrônico para a expressão do fragmento recombinante correspondente ao Fab
de um anticorpo. Com a produção, em uma única molécula, do fragmento como um todo, e
seu posterior processamento, esperamos obter uma solução simples e viável para este
problema que ocorre na expressão de fragmentos de imunoglobulinas.
O vetor utilizado neste trabalho foi construído visando sua posterior utilização na
produção de anticorpos humanizados de interesse clínico em substituição a anticorpos
monoclonais de camundongo, indo ao encontro dos objetivos gerais do grupo de Imunológica
Molecular da UnB na sua participação no Instituto de Investigação em Imunológica.
Com esse objetivos, foi feita a expressão e caracterização do scFab recombinante
produzido pelo vetor pPIg Fab na levedura metilotrófica Pichia pastoris. Além disso,
procuramos estabelecer condições de fermentação em bioreator, para produção em larga
escala, e de possíveis métodos de purificação da proteína.

24
3 – OBJETIVOS
3.1 – OBJETIVO GERAL
Expressão e caracterização de um fragmento de Fab recombinante de cadeia única
(scFab) derivado do anticorpo anti Z-DNA mAb Z22 produzido na levedura metilotrófica
Pichia pastoris.
3.2 – OBJETIVOS ESPECÍFICOS
- Transformar a levedura P. pastoris com o vetor de expressão pPIg Fab;
- Expressar o scFab recombinante e determinar o melhor tempo de produção;
- Purificar a proteína recombinante por meio de colunas cromatográficas;
- Analisar a atividade ligante do fragmento scFab por meio de ensaios imunoenzi-
máticos (ELISA) de ligação direta e inibição;
- Produzir o scFab em larga escala por fermentação em Biorreator de Tanque agitado.
- Analisar a presença de glicosilação, e processamento correto da molécula de Fab

25
4 - MATERIAIS E MÉTODOS
4.1 – CONSIDERAÇÕES INICIAIS
No presente trabalho, os scFabs recombinantes foram expressos em células de Pichia
pastoris linhagem SMD1168. O vetor de expressão pPIg Fab utilizado foi cedido pelo Prof.
Dr. Marcelo de Macedo Brígido do Laboratório de Biologia Molecular da Universidade de
Brasília.
Dentre os elementos constituintes mais importantes deste plasmídeo destacam-se: a
origem de replicação de E. coli, o gene da β-lactamase (que confere resistência à ampicilina),
o promotor AOX1 que dirige a transcrição do gene de interesse, o sinal de secreção do fator α
que dirige a secreção da proteína expressa, o fragmento 3’AOX1 de terminação da transcrição
e a fase de leitura aberta do gene HIS4.
O pPIg Fab foi construído de forma a originar o cassete de expressão VH-CH1-KEX-
VL-Cκ. O esquema geral do vetor utilizado é mostrado na figura 8.
Figura 8. Representação esquemática do vetor utilizado para a expressão dos scFab em Pichia pastoris. CH1, domínio constante pesado; Cκ, domínio constante leve; KEX, peptídeo de ligação com o sito de reconhecimento para a protease Kex; VH, domínio variável pesado; VL, domínio variável leve; His4, fase aberta de leitura do gene HIS4; 5’ AOX1, promotor AOX1; 3’ AOX1, fragmento 3’ AOX1; 3’ AOX1 (TT), Fragmento 3`AOX1 de terminação da transcrição; BLA, gene da β-lactamase que confere resistência à ampicilina; S, sinal de secreção do fator α; ORI, origem de replicação de E. coli. O esquema também mostra os sítios de clivagem de endonucleases de restrição.

26
O fragmento CH1 foi isolado de um banco de cDNA de baço humano com primers
específicos. Estes continham a seqüência de um peptídeo de ligação com o sítio de
reconhecimento para a protease Kex2, presente no retículo endoplasmático da levedura
(figura 9A). A construção foi isolada e clonada no vetor pIg16 Ck que continha o fragmento
scFv do anticorpo anti Z-DNA Z22 ligado ao domínio Cκ humano, originando o vetor pIg16
Fab (figura 9B). O cassete de expressão completo foi então tranferido para o vetor de
expressão em P. pastoris pPIg 22 (figura 9C) originando o vetor pPIg Fab utilizado.
Figura 9. Representação esquemática da construção do cassete de expressão do pPIg Fab. (A) A partir da biblioteca de cDNA de baço humano o fragmento CH1 foi clonado no vetor de bactéria pIg16 Cκ juntamente com o sítio de ligação Kex2 (B). O cassete de expressão foi tranferido para o vetor de expressão em levedura pPIg22 (C) gerando o vetor pPIg Fab utilizado no presente trabalho. CH1, domínio constante pesado; Cκ, domínio constante leve; KEX, peptídeo de ligação com o sito de reconhecimento para a protease Kex; VH, domínio variável pesado; VL, domínio variável leve.

27
4.2 - MATERIAIS
4.2.1 – Células
4.2.1.1 - Linhagem Bacteriana
XL1-Blue (Stratagene®) → recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac
[F´ proAB lacIqZ M15Tn10 (Tetr)] (Sambrook et al., 1989).
A linhagem de Escherichia coli foi utilizada basicamente para a amplificação do vetor
de expressão.
4.2.1.2 - Linhagem de Pichia pastoris
SMD 1168 → Δpep4::URA3 Δkex1: :SUC2 his4 ura3 (Cereghino e Cregg, 2000). Fenótipo: His-, Mut+
Esta linhagem de P. pastoris foi utilizada para a integração, no lócus HIS4 genômico,
do plasmídeo com o gene de interesse, para a expressão do scFab anti-Z-DNA recombinante
sob controle do promotor AOX1.
4.2.2 - Plasmídeo Utilizado
pPIg Fab – 9.3kB, VH1-CH1-KEX-VL-Ck (Fab), fragmentos 5´e 3´do promotor
AOX1, sinal de secreção do fator α, múltiplos sítios de clonagem, fragmento 3´AOX1 TT,
HIS4 ORF, ori ColE1, AmpR, derivado do pIg16 (ver tópico 4.1)
4.2.3 - Meios de Cultura para bactérias
a. Meio LB (Luria-Bertani)
Peptona de caseína 1,0% (p/v)
Extrato de levedura 0,5% (p/v)
NaCl 1,0% (p/v)
Ajustar pH para 7,0.

28
b. Meio LB ágar
Meio LB adicionado de ágar bacteriológico a 1,4% (p/v).
c. Meio SOB
Bacto-triptona 2,0% (p/v)
Extrato de levedura 0,5% (p/v)
NaCl 0,06% (p/v)
KCl 0,02% (p/v)
Ajustar pH para 7,0 .
d. Meio SOC
Solução estoque de Mg2+ 2M 1mL
Slução estoque de glicose 2M 1mL
Meio SOB 98mL
Após dissolver os reagentes em água, todos os meios de cultura foram autoclavados a
120°C por 20 minutos. A seguir adicionava-se, quando necessário, o agente antimicrobiano
apropriado: ampicilina, na concentração final de 100 µg/mL.
4.2.4 - Meios de cultura e Soluções estoque para leveduras
a. Meio YPD (Yeast Extract Peptone Dextrose Medium)
Extrato de levedura 1% (p/v)
Peptona 2% (p/v)
Glicose 2% (p/v)
As soluções contendo glicose ou extrato de levedura e peptona foram autoclavada a
120°C por 20 minutos. No momento da utilização eram misturados para as concentrações
acima relacionadas.
b. Meio MD (Minimal Dextrose Medium)
YNB 1,34% (p/v)
Biotina 4x10-5 % (p/v)
Glicose 2% (p/v)
Ágar bacteriológico 1,4% (p/v)

29
Autoclave as soluções e o ágar por 20 minutos. Adicione as soluções estoque ao ágar
(concentração final de 1,4%) e distribua em placas. Para preparar MDH adicione histidina 1X.
c. Meio MM (Minimal Methanol Medium)
YNB 1,34% (p/v)
Biotina 4x10-5 % (p/v)
Metanol 0,5% (v/v)
Ágar bacteriológico 1,4% (p/v)
Autoclave as soluções e o ágar por 20 minutos. Adicione as soluções estoque ao ágar
(concentração final de 1,4%) e distribua em placas. Para preparar MMH adicione histidina 1X.
d. Meio de Crescimento – BMGY (Buffered Glycerol Complex Medium)
2YP 50%
Tampão fosfato, pH 6,0 100 mM
YNB 1,34% (p/v)
Biotina 4x10-5 % (p/v)
Glicerol 1% (v/v)
e. Meio de Indução – BMMY (Buffered Methanol Complex Medium)
2YP 50%
Tampão fosfato, pH 6,0 100 mM
YNB 1,34% (p/v)
Biotina 4x10-5 % (p/v)
L-Arginina 200mM
Casaminoácidos 2%
EDTA 5mM
Metanol 1% (v/v)
Para preparo dos meios BMGY e BMMY, uma solução de extrato de levedura e
peptona de caseína (2YP) autoclavade separadamente por 20 minutos e depois adicione os
demais reagentes previamente esterilizados por filtração (YNB, Biotina, EDTA) ou por
autoclavagem (L-arginina, casaminoácidos, glicerol e Tampão Fosfato de Potássio),
adicionando água estéril p/a completar o volume.

30
f. YNB (Yeast Nitrogen Base) - Solução estoque de 10X
YNB* 3,4% (p/v)
Sulfato de amônio 10% (p/v)
Esterilizada por filtração. Estocar a 4 °C.
*(s/ aminoácidos e s/ sulfato de amônio).
g. Tampão Fosfato de Potássio 1M, pH6,0
K2HPO4 132mM
KH2PO4 686mM
Esterilizada em autoclave por 20 minutos e estocada a temperatura ambiente.
h. 2YP - Solução estoque 2X de Extrato de Levedura e Peptona
Extrato de levedura 2% (p/v)
Peptona de caseína 4% (p/v)
Esterilizada em autoclave por 20 minutos e estocada a temperatura ambiente.
i. PMT4 - Solução de elementos traços
CuSO4 · 5H2O 2,0 g
Iodeto de Sódio (NaI) 0,08 g
MnSO4 · H2O 3,0 g
Molibidato de Sódio (Na2MoO4 · H2O) 0,2 g
Ácido Bórico (H3BO3) 0,02 g
CaSO4 · 2H2O 0,5 g
Cloreto de cobalto (CoCl) 0,5 g
Cloreto de Zinco (ZnCl2) 7,0 g
Sulfato ferroso (FeSO4 · 7H2O) 22 g
Biotina 0,2 g
Ácido Sulfúrico (H2SO4) 1 mL
Os elementos traços foram preparados para 1 Litro de volume final (solução estoque) e
filtrados. Foi adiconado 1mL/L para o meio de cultura utilizado, BMGY (crescimento),
1mL/L a solução de Glicerol 50%. Além de 4mL/L ao suprimento de metanol, quando estas
soluções foram utilizadas no biorreator.

31
j. Antiespumante
Antifoam A Emulsion. Sigma® Número de catálogo A5738
k. Biotina - Solução estoque 500X
Biotina 0,02% (p/v)
Esta solução filtrar ção em membrana microbiológica com poros de 0,22 µm e
estocada a 4°C.
4.2.5 - Soluções estoques de Inibidores de Proteases
a. PMSF (Phenilmethylsulfonyl Fluoride) 0,2 M
Dissolvido em isopropanol e estocado a temperatura ambiente por até 1 ano. É um
inibidor de serino e tiol proteases como por exemplo, quimiotripsina, trombina, papaína etc.
Concentração de uso 100µg/mL.
b. EDTA (Ácido Tetracético Etilenodiamina) 0,5M
Dissolvido em água em pH 8-9 estocada a 4°C por até 6 meses. É um inibidor de
metaloproteases. Concentração de uso 5mM.
4.2.6 - Soluções e tampões de uso geral
a. Azida Sódica – Solução estoque 100X
Azida sódica 5% (p/v)
Esta solução era utilizada na conservação dos tampões PBS e PBST e nas soluções
estoque dos anticorpos em concentração final de 0,05%.
b. Tampão TE
Tris-HCl pH 8,0 10mM
EDTA pH 8,0 1mM
c. Glicerol – Solução estoque
Glicerol 50% (v/v)

32
d. Tampão PBS (Phosphate-Buffered Saline) 10X, pH 7,4
NaCl 1,37M
KCl 27mM
Na2HPO4 100mM
KH2PO4 20mM
e. Tampão PBST 1X, pH 7,4
NaCl 0,137M
KCl 0,0027M
Na2HPO4 0,01M
KH2PO4 0,002mM
Tween 20 0,5% (v/v)
f. Resinas Cromatograficas
Ion Exchange, Econo-Pac High Q Cartridges Bio Rad® (nº. cat. 4.871.463)
Proteína A-Sepharose™ Amersham Biosciences® (nº. cat. 17-0780-01)
Proteína L - Immunopure® Immobilized on Agarose Pierce® (nº. cat. 20510)
Sephacryl ™ S-200 High Resolution, Sigma® (nº. cat. S-200-HR)
g. Kit BCA – Ácido Bicincrônico (Pierce®) Nº. Catálogo 23225
4.2.7 - Concentradores Amicon® Bioseparations
Centricon YM-10 (nº. cat. 4206) e YM-100 (nº. cat. 4212)
Centriprep YM-10 (nº. cat. 4305) e YM-30 (nº. cat. 4307)
Concentrador: Stirred Ultrafiltration Cell Millipore, Modelo 8400 (nº. cat. 5124)
Membrana: Ultrafiltration Membrane. NMWL: 10.000 (nº. cat. 13642)
4.2.8 - Soluções para extração de DNA plasmidial
a. Solução I
Tris-HCl pH 8,0 25mM
EDTA pH 8,0 10mM
Glicose 50mM

33
b. Solução II
NaOH 0,2N
SDS 1,0% (p/v)
c. Solução III
Acetato de potássio 3M
Ácido Acético 2M
pH ajustado para 4,8 - 5,0
d. RNAse A
RNAse A 10mg/mL
Acetato de sódio pH 4,8 50mM
Fervida em banho-maria por 20 minutos.
4.2.9 – Enzimas e Tampões
a. Endonuclease de restrição SalI
SalI Promega® 10U/µL
Buffer D pH 7,9 Promega®
b. Enzima de Deglicosilação PNGase
PNGase F, 15,000 unidades 500,000 U/mL Biolabs®
10X Glicoprotein Denaturing Buffer Biolabs®
10X G7 Reaction Buffer Biolabs®
10% NP-40 Biolabs®
4.2.10 - Soluções para Eletroforese em Gel de Agarose
a. Tampão de Corrida para Gel de Agarose (TEB) 10X
Trizma base 0,89M
Ácido bórico 0,89M
EDTA 0,02M
Ajustar pH para 8,0

34
b. Tampão de Amostra para Gel de Agarose 10X
Tampão de corrida TEB 20X 50% (v/v)
Glicerol 50% (v/v)
Azul de bromofenol 0,1% (p/v)
Xileno cianol 0,1% (p/v)
c. Brometo de etídio – Solução estoque 20.000X
Brometo de etídio 10mg/mL
4.2.11 - Soluções para Eletroforese em Gel de Poliacrilamida
a. Tampão de Corrida para SDS-PAGE
Trizma base 125mM
Glicina 960mM
SDS 0,5% (p/v)
Ajustar pH para 8,3
b. Tampão de Amostra 2X para SDS-PAGE
Tris-HCl pH 6,8 62,50mM
SDS 5% (p/v)
Glicerol 25% (v/v)
ß-mercaptoetanol 5% (v/v)
Azul de bromofenol 0,01% (p/v)
c. Gel Concentrador SDS-PAGE
Solução Acrilamida/Bis-acrilamida (29:1) 4% (p/v)
Tris-HCl pH 6,8 125mM
SDS 0,1% (p/v)
d. Gel Separador SDS-PAGE
Solução Acrilamida/Bis-acrilamida (29:1) 12% (p/v)
Tris-HCl pH 8,8 400mM
SDS 0,1% (p/v)

35
4.2.12 - Soluções para coloração com Coomassie Brillant Blue (G-250)
a. Soluções Estoque para a Coloração
Ácido acético 7,5% (v/v)
Ácido tricloro acético (TCA) 12,5% (v/v)
Sulfato de amônia 20% (p/v)
b. Solução Corante Coomassie Brillant Blue G-250 (500mL)
Sulfato de amônia 30g
Ácido fosfórico 2% (v/v)
Comassie Brilliant Blue (G-250)* 0.5g
*O Comassie Brilliant Blue (G-250) deve ser previamente purificado, conforme
Harlow e Lane (1988), e só adicionado na solução quando o sulfato de amônia estiver
completamente dissolvido no ácido fosfórico.
c. Solução Descorante para Gel SDS-PAGE
Metanol 40% (v/v)
Ácido Acético 10% (v/v)
4.2.13 - Soluções para coloração com Prata
a. Fixação e Parada/Preservação
Metanol 40% (v/v)
Ácido Acético 10% (v/v)
b. Lavagem
Etanol 50% (v/v)
c. Sensibilização (50mL)
Tiosulfato de sódio 10mg
Solução fotosensível e deve ser usada em, no máximo, 15 dias.

36
d. Coloração (50mL)
Nitrato de prata 100mg
Formaldeído 37µL
e. Revelação (50mL)
Tiosulfato de sódio – 0,2mg/mL (p/v) 1mL
Carbonato de sódio 2g
Formaldeído 25µL
Deve ser preparada na hora e usada gelada, para estabilizar a reação.
f. Solução de Secagem (50mL)
Metanol 50% (v/v)
4.2.14 - Soluções para os Ensaios Imunológicos
(Imunoprecipitação, ELISA, Western, Colony e Dot blotting)
a. Tampão de Fosfatase Alcalina (APB)
Tris-HCl pH 9,5 100mM
NaCl 100mM
MgCl2 50mM
b. Tampão para Transferência Semi-Seca de Proteínas, pH 8.3
Trizma-base 48mM
Glicina 39mM
SDS 0,037% (p/v)
Metanol 20% (v/v)
c. Bloqueio - Western, Colony e Dot blotting
Leite em pó desnatado Molico 5% (p/v)
Dissolvido em PBST 1X
d. Bloqueio - ELISA
BSA 3% (p/v)
Dissolvido em PBS 1x

37
e. Sensiblilização - ELISA – Solução estoque 5.000X
Avidina 5µg/mL
Dissolvido em PBS 1x
f. Z-DNA biotinilado – Solução estoque 425X
Z-DNA biotinilado 0.85µg/µL
Dissolvido em PBS 1x
g. Estreptoavidina Imobilizada
Estreptoavidina imobilizada em agarose 4
(Sigma®
, Número de catálogo: S 1638)
h. Solução Reveladora para ELISA
pNPP (para-nitro-fenil-fosfato) 1mg/µL
Dissolvido em APB
i. Solução Reveladora para Western, Colony e Dot blotting
O NBT (Nitro Blue Tetrazole) e o BCIP (5-Bromo-4-Cloro-indolil fosfato) foram
preparados numa solução estoque de 50mg/mL, dissolvidos em N,N-dimetil formamida. Para
preparar 5 mL da solução reveladora adicionavam-se 33µL do estoque de NBT e 66µL do
estoque de BCIP em 5 mL de APB, nesta ordem para se evitar a precipitação dos reagentes.
4.2.15 - Inibidores utilizados no ELISA
O Z-DNA foi preparado por halogenação do polinucleotídeo poli(dG-dC)
(Pharmacia®) por Andrade et al. (2005). Nesta halogenação o átomo N8 da guanina é
modificado pela adição de um átomo de bromo, impedindo estericamente a formação do B-
DNA e conseqüentemente favorecendo a formação de Z-DNA.
Para a preparação de B-DNA, DNA de esperma de Arenque (Sigma®) foi dissolvido
em tampão TE em uma concentração 10 mg/mL para solução estoque. Logo em seguida, o
tubo contendo o DNA era agitado vigorosamente em agitador tipo vortex por
aproximadamente 10 min. Após esse processo o ssDNA foi preparado por desnaturação do B-
DNA por fervura em banho maria por 10 minutos a 100ºC.

38
O oligonucleotídeo de seqüência poli(dG-dC) é comercialmente disponível pela
Pharmacia®, nº de catálogo 27-790. Número de pares de bases é aproximadamente 750. Todos
os estoques eram mantidos a -20ºC.
Tabela 2. Inibidores utilizados nos ensaios de inibição.
Inibidores Seqüência Coinicia nucleotídica ncentração
l nos ensaiosZ-DNA poli Br (dG-dC) • poliBr(dG.dC) 1µg/mL B-DN DNA 10µgssDN DNA turado 10µgOligonucleotídeo poliGC poli( 0µg
A de esperma de Arenque /mL A de esperma de Arenque desna /mL
dG-dCG) 1 /mL
.2.16 - Marcador de massa molecular (MM) para proteínas
Bench Marker™ Pre-Stained Protein Ladder (Invitrogen Life Technologies)
4.2.1
icorpos utilizados Titulação
4
7 - Anticorpos
Tabela 3. Ant
Nome em: WesteProduzido
rn Blotting
Dot ELISA Origem
Número do
catálogo Blotting
Anti-IgG Coelho A 3687 (AP)* Cabra NA NA 1:5.000 Sigma®
Anti-γ Cabra NA NA 1:5.000 Pierce® 31160
Anti-IgG de cabra Coelho 1:5.0 1:5.0
A 1 1
IgG na Policlonal 1:10.000 1:10.000 1:10.000 Sigma K 9001
1 1 Kirkegaard & 2
C M l.,
P. pastoris 1:205
Camundongo
(AP)* nti-Cκ Humano
00 00 1:5.000 Pierce® 31300
Cabra :5.000 :5.000 1:10.000 Pierce® 31129
Anti-Fc humano Huma
Coelho NA NA 1:10.000 Pierce® 31142 ®
Anti-IgG humana (H+L) (AP)* Cabra :5.000 :5.000 NA Perry 15-1006
mAbZ22** amundongo NA NA 1:364 öler et a1982
Andrade et
NA
FvFc *** NA NA 3 al., 2000 NA
N plica* A ado co e alc ** , anti mon nal d igem a e para NA.
do segundo Möler et al. (1982) (revisado por Andrade, 2005). scFv do mAbZ22 fusionado ao fragmento Fc de
A: Não se a P, conjug
mAbZ22m Fosfatas
corpo Z22 alinaoclo e or murin specifico Z-D
Preparado e purifica*** FvFc, proteína recombinante dimérica,uma IgG1 humana expresso na forma solúvel na levedura P. pastoris (Andrade et al., 2000).

39
4.2
Ácido acético pH 3,5 0,5 M
b. Tampão de Neutralização
Tris-HCl pH 11,0 1,5 M
4.2.19 - Soluç
NaOH 0,2M
50mM
100mM
Ajustar
padrões
Tabela 4. Marcadores Moleculares Padrão usados na Gel Filtração*
Reagentes Produto Peso
molecular ado
.18 - Soluções para coluna de Proteína A-Sepharose™
a. Tampão de Equilíbrio e Eluição
ões para coluna de Gel Filtração
a. Tampão de Limpeza
b. Tampão de Equilibrio
Tris-HCl
KCl
pH para 7,5.
c. Marcadores e
Número aproxim(kDa)
Blue Dextran D 4772 2.000 β-Amilase de Batata doce Álcool desidrogenase
oro bovino ócito
A8781 A 8656
200 150
Albumina de s A 8531 66 Anidrase carbônica de eritrbovino
A 7025 29
* Si 0)
.2.20 a L
Na2HPO4 0,1 M
NaCl 0,15 M
gma (nº. cat. MW-GF-20®
4 - Soluções para coluna de Proteín
a. Tampão de Ligação

40
Aju
b. T
0,1 M
Ajustar pH para 2,0
c. T
2,0 M
Ajustar pH para 11,0
4.2.21 - Soluç
a. Tampão de Baixo Sal
20,0 mM
Ajustar pH para 8,5. Após o preparo o tampão foi deaerado.
b. T
Feito em Tris HCl 20 mM pH 8,5 deaerado.
c. D
mM):
25-50-100-150-200-250-300-350-400-450-500
4.2.22 – Mem
t. RPN303E)
star pH para 7,5
ampão de Eluição
Glicina
ampão de Neutralização
Tris Base
ões para coluna de Troca Iônica
Tris HCl
ampão de Alto Sal
NaCl 500 mM
iluições do tampão de Eluição
Concentrações crescentes de NaCl (em
brana de Nitrocelulose
Hybond-C Extra Amersham Bioscience® (nº. ca

41
4.3 - MÉTODOS
Os clones de Escherichia coli transformados com o plasmídio de interesse, doados
de Macedo Brígido, foram recuperados em Meio LB para a posterior
xtração do DNA plasmidial (item 4.3.1) e transformação da levedura (item 4.3.6).
eio LB/Amp (100 µg/mL) por 16 horas a 37°C. A
paração das células foi feita por meio de centrifugação a 4.000 rpm por 15 mine descarte
do sob
0,5v de acetato de amônio 7,5M e 2v de etanol 100% (v/v). As amostras
foram
pelo Dr. Marcelo
e
4.3.1 - Preparação de DNA Plasmidial através do método de lise alcalina em larga escala
(adaptado de Sambrook et al., 1989)
Foram coletados 200mL de cultura de células de E. coli, transformadas com o
plasmídeo de interesse, crescidas em m
se
renadante. Ressuspendieu-se o sedimento em 5 mL de Solução I. Foram adicionados
10mL de Solução II e homogeneização das amostras, invertendo-se gentilmente o tubo várias
vezes. Em seguida procedeu-se a uma incubação em banho de água-gelo por 10 min.
Adicionou-se 7,5 mL de Solução III, repetindo-se o mesmo procedimento de homogeneização
e incubando-se novamente no gelo por 20 min. Centrifugou-se a 10.000 rpm por 40 min a
4°C, o sobrenadante foi então transferido para outro recipiente e adicionado 0,6v de
isopropanol e após uma incubação de 5 min, à temperatura ambiente, centrifugou-se a 12.000
rpm por 20 min. Procedeu-se à secagem do sedimento deixando-se o tubo exposto ao ar. O
sedimento foi então resuspendido em 0,5mL de TE e transferidos para um microtubo, no qual
foi adicionado RNAse A a uma concentração final de 2µg/mL. As amostras foram incubadas
a 37°C por 15 min.
Procedeu-se então a separação das proteínas, para uma melhor purificação do DNA
plasmidial, utilizando-se duas vezes 1v de clorofane e uma vez 1v de clorofil. À última fase
aquosa adicionou-se
incubadas a -20°C por pelo menos uma hora. Em seguida procedeu-se a centrifugaçaõ
por 45 min a 12.000 rpm a 4°C. O sedimento foi lavado com 0,5v de etanol 80% (v/v) e em
seguida seco por meio de exposição ao ar. O sedimento final foi ressuspendido em 100µL de
TE. As amostras foram então conservadas a 4ºC.

42
4.3.2 - Digestão do DNA plasmidial com Endonucleases de Restrição
As digestões dos plasmídeos com enzimas de restrição foram realizadas conforme
struções dos fabricantes. O tempo de incubação, as concentrações de DNA plasmidial e de
mesmas. Em geral,
ram utilizadas 5 a 10 unidades de enzima para cada 1 µg de DNA, sendo incubados por 2 a
4 horas
agarose foi preparada na concentração de 0,8% a 1% em tampão TEB 1X e
ontiendo 0,5 µg/mL de Brometo de Etídeo. As amostras de DNA com tampão de amostra
descrito por
ambrook et al. (1989). Para a visualização do DNA utilizou-se a incidência de luz ultra-
violeta
s de DNA digeridos foram separados por eletroforese em gel de agarose
a fração correspondente ao fragmento desejado foi cortada do gel e transferida para
egidas as instruções do fabricante, Wizard® SV Gel
nd PCR Clean-Up System (Promega® - nº. cat. A9281).
zado por digestão com a enzima de
strição SalI. Esta enzima cliva o plasmídeo no gene HIS4, permitindo a integração do
nte transformação.
A linearização dos plasmídeos foi confirmada por eletroforese em gel de agarose. Ao
sistema acrescentou-se água até o volume 200 μL e adicionado de acetato de sódio 3M pH 4,8
in
enzima variam de acordo com o material a ser digerido e a eficácia das
fo
.
4.3.3 - Análise de DNA em Gel de Agarose (segundo Sambrook et al., 1989)
A
c
para gel de agarose eram aplicadas no gel e submetidas à eletroforese, como
S
no mesmo por um transluminador (Pharmacia-LKB®) e/ou sistema de
fotodocumentação.
4.3.4 - Eluição de fragmentos de DNA
Os fragmento
e
microtubos. Nos passos seguintes foram s
a
4.3.5 - Preparação de DNA para transformação de P. pastoris
Aproximadamente 10 μg do plasmídeo foi lineari
re
plasmídeo no locus do gene his4 defectivo da levedura, após eficie

43
na con
70% e
novam
8 em 5 mL de meio YPD em um
rlenmayer de 125 mL que foi então incubada sob agitação a 30ºC durante a noite. Inoculou-
.
nas mesmas condições durante a noite ou até que a cultura atingisse uma
ensidade óptica à 600nm de 1,3 a 1,5. As células foram então centrifugadas a 1500 rpm por
10 min
guintes condições (eletroporador da BioRad): 1500 V; 400 Ω
e 25 µ
centração final de 10%, 2 volumes de etanol 100% e 1 μL de glicogênio 10 mg/mL
(como carreador). Este sistema foi incubado no freezer -20°C por 1 hora e subseqüentemente
centrifugado a 12.000 rpm por 30 minutos. O sedimento foi lavado com etanol
ente centrifugado a 12.000 rpm por 10 minutos. O sobrenadante foi descartado e o
sedimento ressuspendido em 10 μL de água miliQ estéril.
4.3.6 - Transformação da levedura P. pastoris por eletroporação (adaptado de Cregg et
al., 1985)
Inoculou-se uma colônia de P. pastoris SMD116
e
se de 0,1 a 0,5 mL da cultura acima em 500 mL de meio YPD em um frasco de 2 litros
Incubou-se
d
utos a 4ºC, sendo o sobrenadante descartado. O sedimento foi ressuspendido em 200
mL de água gelada estéril. Repetiu-se a centrifugação, duas vezes, ressuspendo-se o
sedimento em 100 mL de água gelada estéril e em 20 mL de sorbitol 1M gelado estéril,
respectivamente. Centrifugou-se as células novamente, e ressuspendeu-se o sedimento em 200
- 400 μL de sorbitol 1M gelado.
Misturou-se 80 µL de células competentes com 5 a 10 µg de DNA linearizado
(ressuspendidos em 5 a 10 µL de água), e adicionaram-se 320 µL de sorbitol 1M.
Transferiram-se então as células para uma cubeta de eletroporação de 0,2 cm gelada. Incubou-
se a cubeta com as células e o DNA plasmidial linearizado por 5 minutos no gelo. As mesmas
foram então eletroporadas nas se
F. O tempo de eletroporação foi de aproximadamente 10 ms com um campo de 7500
V/cm. Imediatamente após o choque, adicionou-se 1 mL de sorbitol 1M gelado na cubeta.
Transferiu-se o conteúdo para um microtubo estéril. Semeou-se de 200 a 600 µL de células
em placas de meio MD ágar. As placas foram mantidas em uma estufa a 30°C até o
aparecimento das colônias.
4.3.7 – Análise dos transformantes de P. pastoris por Colony blotting ou Western de
colônia

44
Uma pequena porção de massa celular de alguns dos clones obtidos nas placas de
transformação foi repicada serialmente, com o auxílio de palitos estéreis, em placas de meio
ínimo contendo metanol (MM) e glicose (MD). As placas foram incubadas a 30°C por 48
com
.
Transcorrido o tempo de indução, montava-se um “sanduíche” sobre as placas MM
conten
células removidas com os dedos.
jugado a fosfatase alcalina, anti-IgG de cabra
feito em
4.3.8 -
As colônias que se mostraram positivas no colony blotting foram inoculadas em 25
nóculo a 28-30ºC sob agitação
tensa (250-300 rpm) até que a cultura atingisse uma densidade óptica à 600nm (OD600) de 2
por centrifugação a 1500-3000 xg por 5
m
horas. E a cada 24 horas foi adicionado 500 µL de metanol 100% na tampa das placas
meio MM
do: membrana de nitrocelulose diretamente em contato com as colônias, 3 folhas de
papel de filtro Watmann® 3 mm sobrepostas por várias camadas de papel toalha. Colocou-se
um peso sobre o “sanduíche” que foi incubado a 30°C por 1 hora. Passado o tempo de
transferência, desmontou-se o sanduíche e a membrana foi bem lavada com PBST 1X, sendo
o excesso de
A membrana foi incubada com a solução de bloqueio por, no mínimo, 1 hora, sob
agitação e a 4ºC. A solução de bloqueio foi então removida e a membrana lavada 3X com
PBST 1X a temperatura ambiente. A membrana foi incubada com o anticorpo primário, anti-
Cκ Humano feito em cabra, diluído em PBS por 1 a 2 horas sob agitação a 4ºC. O anticorpo
primário foi removido e a membrana novamente lavada 3X com PBST 1X. A membrana foi
então incubada com o anticorpo secundário con
coelho, diluído em PBS 1X por mais 1 a 2 horas sob agitação a 4ºC. O anticorpo
conjugado era removido e a membrana lavada 3X com PBST 1X.
Posteriormente lavou-se a membrana com 10mL de APB (tampão da fosfatase
alcalina) e adicionando-se a solução reveladora (NBT/BCIP). O aparecimento das bandas
coloridas foi controlado visualmente. Após reação, lavou-se a membrana com água destilada
até retirar o excesso da solução reveladora e interromper a reação da enzima. Preservou-se a
membrana seca, sobre papel filtro.
Expressão da Proteína Recombinante em P. pastoris
4.3.8.1 –Fermentação em frascos do tipo Erlenmeyer
(adaptado do Manual do Kit de Expressão em P. pastoris)
mL de meio BMGY em um frasco de 250 mL. Incubou-se o i
in
a 6. Coletou-se as células em tubos Falcon de 50 mL

45
minuto uma OD600 de 1,0 em 100
L de meio BMMY para indução da expressão. A cultura foi então transferida para um frasco
de 1 l
cos do tipo Erlenmeyer
s sob as mesmas condições, mas utilizando 5 frascos
erlenm ers contendo 200 mL de cultura em cada. Os sobrenadantes foram separados dos
os
brenadantes adicionou-se os seguintes inibidores de protease: PMSF - Fenilmetilsulfonil
fluorid
culada em 100 mL de
eio BMGY em um frasco do tipo Erlenmeyer de 1L para o crescimento do pré-inóculo.
O biorreator (figura 10) foi esterilizado juntamente com 900 mL do meio BMGY a
120ºC
controladores. Foram utilizados para o controle de pH, hidróxido de amônia 30% (NH4OH) e
s, à temperatura ambiente. Ressuspendeu-se as células para
m
itro e incubavada a 28-30ºC sob agitação intensa. Adicionou-se metanol para uma
concentração final de 1% a cada 24 horas para manter a indução.
Para o experimento de cinética de indução, a cada tempo (0, 24, 48, 72 e 96 horas),
transferiu-se 5mL da cultura para um microtubo e centrifugou-se na velocidade máxima da
microcentrífuga por 2 a 3 minutos. Após essa metodologia as amostras foram processadas
seguindo o tópico 4.3.9 de métodos.
4.3.8.2 – Produção em larga escala do scFab recombinante em fras
Para a produção em larga escala (1 L) realizada em frascos do tipo Erlenmeyer, as
culturas foram crescidas de acordo com o melhor tempo determinado pela cinética de
indução. As mesmas foram mantida
ey
sedimentos por centrifugação e a seguir filtrados em membrana com poro de 0,45 μm. A
so
o (100μg/mL) e EDTA - Ácido Tetracético Etilenodiamina (0,6mg/mL).
O sobrenadante foi concentrado utilizando o sistema Amicon Stirred Ultrafiltration
Cell Millipore com uma membrana de ultrafiltração (NMWL: 10.000).
4.3.8.3 –Sistema de Fermentação em Bioreator (adaptado do Manual do Kit de Expressão em
Pichia pastoris – Invitrogem e Stratton e Chiruvolu, 1998)
Uma colônia crescida em placa com meio MD, cuja correspondente da placa com
meio MM foi selecionada como positiva pelo colony blotting foi ino
m
Incubou-se a 28-30ºC sob agitação intensa (250-300 rpm) até que a cultura atingisse uma
densidade óptica à 600nm (OD600) de 2 a 5.
por 25 min na autoclave. O pH e o oxigênio dissolvido (DO) foram calibrados antes da
esterilização. Após a esterilização, o meio foi resfriado a 30ºC e ambos, DO e pH, foram
recalibrados. O pH e a temperatura foram monitorados e controlados automaticamente por

46
ácido fosfórico 30% (H3PO4). O oxigênio dissolvido foi monitorado somente através da
leitura fornecida pela sonda de O2, sendo o controle da DO realizado manualmente.
stema de fermentação as e acessórios para cont
O m mentação
450rpm
ma a
com os níveis de alimentação. A vazão de ar foi fixada em 1,2 L/min. Também foram
adicionados 0,7 mL de antiespumante.
Figura 10. Foto do sistema de fermentação em biorreator utilizado. O si completo era composto do tanque de crescimento e demais bomb
role e monitoração do processo como um todo.
eio no biorreator foi inoculado com 100 mL do pré-inóculo. A fer
ocorreu a 30ºC, pH 5,4 e pressão atmosférica. Inicialmente a agitação foi ajustada em
e foi alterada até 1050rpm conforme necessário – a agitação foi configurada de for
manter a DO em aproximadamente 30%. Sendo assim, os níveis de DO foram controlados
manualmente pela agitação, acompanhados até que fosse estabelecido um equilíbrio de acordo
Foi iniciada a batelada alimentada, ou seja, alimentação com 15 mL/h de glicerol 50%
acrescido de 1 mL/L da solução de elementos traços (PMT4) até estabilização da DO em
aproximadamente 30%. A alimentação de glicerol foi interrompida por 1 hora para que as
células esgotassem essa fonte de carbono do meio. O esgotamento do glicerol foi indicado por
um aumento da DO do meio até próximo a 100%.

47
Após o período de “inanição” foi iniciada a indução. Assim, a cultura foi alimentada a
principio com 1,8 mL/h de metanol 100% acrescido de 4 mL/L da solução de elementos
traços,
m coletados a cada 12 horas durante o
process
4.3.9 –
eram retidas e as menores, que atravessavam a
membrana, eram coletadas. Nesta etapa as amostras foram centrifugadas a 5.000 xg durante 1
o-se o Centricon
M-10, que retirava as proteínas menores que 10 kDa e ao mesmo tempo concentrava a
mostra
rana, eram
coletadas. Somente foram concentradas as amostras provenientes da fermentação em frasco,
pelas primeiras 4 horas. Após esse período a adição de metanol 100% foi alterada para
2,5 mL/h e foi mantida entre esses valores, de acordo com a leitura do oxigênio dissolvido no
meio. O tempo total de fermentação foi aproximadamente 73 horas sendo que o período de
indução com metanol durou cerca de 40 horas.
Aproximadamente 5 mL de amostra fora
o fermentativo. A concentração celular foi medida no espectrofotômetro a 600nm. As
amostras foram então centrifugadas a 20.000 xg por 10 min e o sobrenadante coletado e
estocado a 4ºC para posterior análise. Aos sobrenadantes adicionaram-se os seguintes
inibidores de protease: PMSF – Fenilmetilsulfonil fluorido (100 μg/mL) e EDTA - Ácido
Tetracético Etilenodiamina (0,6 mg/mL).
Processamento do Sobrenadante de Cultura em Colunas Amicon
O sobrenadante de cultura foi dializado, pré-purificado e concentrado, de acordo com
o tipo de amostra. Para isso utilizaram-se dois tipos de colunas Amicon. Se a amostra
provinha da fermentação em frasco, era primeiramente filtrada no Centricon YM-100, onde as
proteínas que tinham mais que 100 kDa
hora. A amostra que atravessava a membrana era então concentrada utilizand
Y
. A centrifugação era na mesma condição anterior. A coleta do material retido foi
procedida invertendo-se a coluna e centrifugando-a a 1.000 xg durante 5 minutos para ser
recolhida em um microtubo do tipo Eppendorf, onde o volume do material restante era
completado para 500 µL com PBS 1X, sendo que seu voluma inicial foi de 4 mL.
No caso das amostras provenientes da fermentação em bioreator o processamento foi
diferente. Primeiramente o sobrenadante foi processado na coluna Centriprep YM-30 com
capacidade para 15 mL, onde atravessavam a membrana somente proteínas menores que 30
kDa. As amostras foram centrifugadas a 3.000 xg durante 30 minutos a 4ºC. As amostras
eram então dialisadas em tampão PBS 1X. Após esta etapa 4 mL das amostras que ficavam
retidas na membrana foram filtradas na coluna Centricon YM-100, onde as proteínas que
tinham mais que 100 kDa eram retidas e as menores, que atravessavam a memb

48
no caso
lisada
por 0,12% (p/v) de Persulfato de Amônio e 0,2% (v/v) e 4 µL de TEMED. Uma vez vertido o
ços. Após a
limerização, o gel foi acoplado no aparato de eletroforese.
ok et al., 1989)
dois eletrodos de grafite e submetido a
ma corrente elétrica de 0,8 mA/cm2 de membrana por 1h 30 min.
s foi embebida
m solução de bloqueio e incubada por 1 h à temperatura ambiente ou durante a noite a 4°C
sob ag
das amostras provenientes do bioreator seu volume inicial foi mantido. Nas amostras
retiradas do bioreator o processamento foi apenas a dialise e pré-purificação das amostras
utilizando os centricon e centriprep, com escessão do tempo final (T), restante do meio de
cultura do fermentador que era totalmente concentrado após o termino da fermentação.
4.3.10 - Análise de Proteínas em Gel de SDS-PAGE (Sambrook et al., 1989)
As amostras de scFab processadas foram analisadas em gel desnaturante de
poliacrilamida. Inicialmente preparou-se o gel separador em concentração de 12% (p/v),
sendo a polimerização catalisada pela adição de 0,045% (p/v) de Persulfato de Amônio e
0,2% (v/v) e 6µL de TEMED. Após a polimerização do gel separador, verteu-se o gel
concentrador preparado em concentração de 4% (p/v), tendo a sua polimerização cata
gel concentrador, introduziu-se o pente para permitir a formação dos po
po
As amostras foram ressuspendidas em tampão de amostra 1X para SDS-PAGE.
Imediatamente antes da aplicação das mesmas, procedeu-se à fervura em banho-maria a
100°C por 10 minutos. Após a corrida do gel, de 50 a 80mA, o mesmo foi imerso em solução
fixadora e incubado durante a noite sob agitação suave. Em seguida procedeu-se com os
procedimentos de coloração com Comassie Briliant Blue (G-250) ou prata, especificados
respectivamente nos itens 4.3.14 e 4.3.15 de métodos.
4.3.11 - Análise de Proteínas por Western Blotting (Sambro
Após a corrida, o gel de poliacrilamida foi transferido para a membrana de
nitrocelulose utilizando-se o sistema de transferência semi-seca com eletrodos de grafite
(Pharmacia-LKB®
). Conforme instruções do fabricante, fez-se um "sanduíche" de papéis de
filtro, previamente embebidos em tampão de transferência e contendo internamente a
membrana e o gel. O "sanduíche" foi colocado entre os
u
Após este procedimento, a membrana contendo as proteínas transferida
e
itação branda. A membrana saturada foi lavada 3 vezes com PBST 1X antes de ser

49
incubada, por 1 a 2 h, com o anticorpo primário a 4ºC. Após a incubação a membrana foi
lavada 3 vezes com PBST. Procedeu-se então a incubação com anticorpo secundário,
conjugado com fosfatase alcalina em um título de 1/5000 por 1 a 2 h a 4ºC. Ao término da
incubação o anticorpo conjugado foi removido e a membrana lavada 3X com PBST 1X.
étodos).
e em 250 mL de ácido acético 7,5%. A solução
i aquecida a 70ºC. Foram adicionadas 44 g de sulfato de amônia e a mistura foi resfriada
o por filtração e coletado
pós sua secagem.
lue G-250. A solução foi incubada durante a noite sob uma leve agitação em
mperatura ambiente. A solução corante pode ser recuperada para posterior utilização.
Posteriormente lavou-se a membrana com 10 mL de APB (tampão da fosfatase
alcalina) adicionando-se a solução reveladora (NBT/BCIP). Controlou-se, visualmente, o
aparecimento das bandas coloridas. Após a reação, lavou-se a membrana com água destilada
até retirar o excesso da solução reveladora e interromper a reação da enzima. Preservou-se a
membrana seca, sobre papel filtro.
4.3.12 – Análise de Proteínas por Dot Blotting (Sambrook et al., 1989)
Adicionou-se de 5 a 10 µL dos extratos proteicos obtidos diretamente a uma
membrana de nitrocelulose. Com a membrana seca, contendo as proteínas ligadas, foi
realizado o mesmo procedimento de bloqueio e revelação descrito para o experimento de
Western Blotting (tópico 4.3.12 de m
4.3.13 – Purificação do Corante Coomassie Brillant Blue G-250
Para o uso do Coomassie Brillant Blue G-250 (derivado da dimetilação do Coomassie
Brillant Blue R-250) deve-se, primeiramente purificá-lo, já que as fontes comerciais são
cotaminadas com componentes que interferem na resolução da técnica.
Foram dissolvidas 4 gramas do corant
fo
naturalmente até a temperatura ambiente. O corante foi recuperad
a
4.3.14 – Coloração com Coomassie Briliant Blue G-250
Após a eletroforese o gel de poliacrilamida foi transferido para um recipiente limpo de
plástico ou vidro contendo 5 volumes de TCA 12,5% para fixação das proteínas. Após uma hora de
incubação em temperatura ambiente foram adicinados aproximadamente 10v da solução corante de
Coomassie Briliant B
te

50
Revelou-se o gel com a solução reveladora controlando visualmente o surgimento das
andas. Para fixar a coloração no gel foi utilizado 5v de Sulfato de Amônia 20% incubando
durante
gel foi lavado com etanol 50 % por 1 hora com
trocas a cada 20 minutos. Após as lavagens o gel foi sensibilizado por um minuto sob
da após esse período.
Em seguida o gel foi incubado na solução de nitrato de prata por 20 minutos.
Imedia
solução de metanol 50%, durante a
noite, s
ssuem
alguma
foram invertidas sobre uma pilha de papel toalha e batidas
igorosamente contra a mesma a fim de se eliminar todos os resquícios da solução presentes.
ra evitar a evaporação das soluções, e
b agitação. Os anticorpos utilizados estão detalhados na Tabela 2 de Matérias, item 4.2.17.
b
a noite em temperatura ambiente. Após fixar o gel sua resolução pode ser melhorada
repetindo-se o procedimento de coloração.
4.3.15 – Coloração com Prata
Após a corrida o gel de poliacrilamida foi fixado com a mesma solução descorante da
coloração com Coomassie Briliant Blue sob leve agitação. Após duas horas de fixação a
solução descorante foi retirada e guardada para posterior utilização na etapa de parada e
preservação neste mesmo procedimento. O
agitação, sendo a soluça descarta
tamente após, a solução de coloração foi desprezada e o gel lavado rapidamente três
vezes com água destilada. Foram adicionados 50 mL de solução de revelação, acrescida de 25
µL de Formaldeído e de 1mL de Tiossulfato 0,2 mg/mL. Quando a intensidade das bandas
chegou a um ponto satisfatório a solução foi substituída pela solução de parada e preservação.
O gel foi incubado nesta solução à temperatura ambiente, sob agitação, durante a noite.
Após este procedimento o gel foi colocado em
endo posteriormente secado.
4.3.16 – ELISA (Enzyme-linked immunosorbent assay)
Foram realizados ensaios do tipo ELISA de ligação direta e inibição, utilizando como
antígeno o Z-DNA biotinilado e sensibilizando a placa com avidina. Os protocolos po
s parcularidades e estão descritos separadamente.
Após cada lavagem, as placas
v
Durante as incubações as placas permaneceram fechadas, pa
so
4.3.16.1 – ELISA – Ensaio de ligação direta

51
Uma placa de microtitulação (Falcon) foi inicialmente sensibilizada por uma hora a
temperatura ambiente ou uma noite a 4ºC, com 150 µL de tampão PBS contendo 5 µg/mL de
avidina. Após a sensibilização a placa foi bloqueada com 180 µL de solução de bloqueio
(PBST + BSA 3%) por 1 a 2 horas em temperatura ambiente ou durante a noite a 4ºC. Após
ês lavagens com PBST, adicionou-se 150 µL de Z-DNA biotinilado 2 µg/mL incubando a
nte. Três lavagens com PBST foram realizadas,
ndo logo após as mesmas adicionados 150 µL de tampão PBS contendo 0,1 µg/mL de
extrato
tivas diluições de 1:10.000 e 1:5.000 em PBS 1X.
Incubo
minutos.
tr
placa por 1 a 2 horas em temperatura ambie
se
de sobrenadante pré-purificado (scFab), nas linhas iniciais, de onde se fazia diluições
seriais na razão de 1:3 nas demais linhas.
Como controles positivos foram utilizados 150 µL de PBS com 0,3 µL/mL do
fragmento de anticorpo FvFc e/ou mAbZ22 seguindo o mesmo padrão de diluição serial feito
para as amostras. A placa foi então incubada por 1 a 2 horas em temperatura ambiente. Os
poços foram novamente lavados três vezes com PBST e adicionou-se 100 µL do anticorpo
primário anti-Cκ Humano na diluição de 1:10.000 em PBS 1X. Nos poços com os controles
foram adicionados como anticorpos primários o anti-Fc humano para o FvFc e anti-γ
Camundongo para o mAbZ22, nas respec
u-se a placa de 1 a 2 horas em temperatura ambiente ou durante a noite a 4ºC. Os poços
foram então lavados com PBST e logo em seguida, adicionaram-se, nos poços da amostra
scFab e do controle mAbZ22, 100 µL do anticorpo secundário anti-IgG de cabra conjugado à
fosfatase alcalina na diluição de 1:5.000. Já nos poços que continham o controle com o FvFc
foram adicionados 100 µL de Anti-IgG Coelho conjugado com fosfatase alcalina na diluição
de 1:5.000. A incubação com os anticorpos secundários procedeu-se por uma hora a
temperatura ambiente.
Finalmente, os poços foram lavados três vezes com PBST e uma vez com 100 µL de
APB. Adicionou-se 100 µL de solução reveladora (pNPP) e após cerca de 30 minutos de
reação a placa era analisada no leitor de ELISA “Microplate Reader BioRad®” modelo 450 a
um comprimento de onda de 405nm, considerando-se sempre um branco por placa.
4.3.16.2 – ELISA – Ensaio de Inibição
Utilizou-se placas de microtitulação (Falcon) previamente sensibilizadas e bloqueadas
como especificado no tópico ELISA – Ensaio de ligação direta (item 4.3.16.1). Para efetuar a
inibição, 80 µL dos inibidores foram adicionados na linha inicial, de onde foram feitas
diluições seriais crescentes na razão de 1:3, sendo a placa incubadoa durante 10

52
Após esse período as amostras contendo 0,03 µg/mL de extrato de sobrenadante pré-
4.3.9 de métodos, foram adicionadas e incubadas
urante 2 horas em temperatura ambiente juntamente com os inibidores. Em seguida
proced
m seguida lavada com 5 volumes de PBS para a retirada do
tanol (no qual a resina vem estocada). A mesma foi então equilibrada com 3 volumes de
de PBST
X e posteriormente com 3 volumes de PBS 1X.
L de sobrenadante previamente processado (como no
pico 4.3.9 de métodos). A coluna foi em seguida lavada com 20 mL de PBS 1X. Para a
eluição
namento.
4.3.17.
regulado em 2 mL/mim. A coluna foi lavada com 12 mL de tampão de Eluição, para sua
purificado (scFab), seguindindo o item
d
eu-se o ensaio de ligação de acordo com a metodologia do tópico ELISA – Ensaio de
ligação direta (item 4.3.16.1).
4.3.17 - Purificação dos scFabs recombinantes por cromatografia de afinidade
4.3.17.1 – Proteína A - Sepharose™
A coluna cromatográfica foi montada com cerca de 5 mL de resina Proteína A-
Sepharose™ (Amersham®) e e
e
ácido acético 0,5M pH 3,5. Após esse processo a coluna foi lavada com 3 volumes
1
Aplicou-se à coluna 2 a 3 m
tó
, adicionou-se 6 mL de ácido acético 0,5M pH 3,5 e foram coletadas amostras em
microtubos do tipo Eppendorf de 1 em 1 mL. As amostras foram imediatamente neutralizadas
com 100 µL Tris-HCl 1,5M pH 11,0, previamente colocados em cada tubo de coleta, para
neutralização do pH. O volume da coluna foi completado com PBS 1X por três vezes para
trocar o tampão de eluição pelo tampão de armaze
Imediatamente após o fim da coleta, 10 µL de cada amostra foram aplicados em uma
membrana de nitrocelulose para análise por Dot Blotting, seguindo o protocolo do item 4.3.13
de métodos. As amostras onde se detectaram proteínas foram passadas na coluna Centricon
YM-10 (Amicon®), com membrana de 10 kDa, seguindo o item 4.3.9. Desta maneira o
material foi dializado e concentrado para cerca de 500 μL. Após esse processo as amostras
foram congeladas.
2 – Proteína L - Agarose
Aproximadamente 2 a 3 mL da resina de Proteína L (Pierce®) foi montada na coluna.
Logo após a montagem, a coluna foi conectada a uma bomba peristáltica e o fluxo foi

53
ativação. Após esse processo a coluna foi lavada com 3 volumes do tampão de ligação, para
neutralização do pH da coluna. Logo após esse procedimento foram aplicados 4 mL de
sobrenadante previamente processado (como no tópico 4.3.9 de métodos) juntamente com
o. Aplicou-se então mais 12 mL de Tampão de Ligação para
lavagem da coluna sendo as amostras coletadas, em microtubos do tipo Eppendorf, de 1 em
1mL.
s. As amostras onde se detectavam proteínas eram passadas na coluna Centricon YM-
10 (Am
4.3.18
volumes de Tris-HCl 50mM pH 7,5, sendo em seguida lavada
om 3 volumes de PBS 1X .
oi
plicado um mix com 200 µL dos padrões restantes (β-Amilase, Álcool deidrogenase,
Album
mais 4 mL de Tampão de Ligaçã
a
Para a eluição, adicionou-se 6 mL do respectivo tampão, sendo as amostras coletadas,
novamente, em microtubos do tipo Eppendorf, de 1 em 1mL. As amostras foram
imediatamente neutralizadas com 10 µL do tampão de neutralização previamente colocados
em cada tubo de coleta. O volume da coluna foi completado com PBS por três vezes para
trocar o tampão de eluição pelo tampão de armazenamento (PBS 1X).
Imediatamente após a coleta, 10 µL das amostras foram aplicadas em uma membrana
de nitrocelulose para análise por Dot Blotting, seguindo o protocolo do item 4.3.13 de
método
icon®), com membrana de 10 kDa, e o volume da coluna era completado com PBS por
três vezes para trocar o tampão de eluição pelo tampão de armazenamento (PBS 1X). Desta
maneira o material era dializado e concentrado para cerca de 500 μL. Após esse processo as
amostras eram congeladas.
- Purificação dos scFabs recombinantes por Cromatografia de Filfração em Gel
Aproximadamente 300 mL da resina Sephacryl ™ S-200 High Resolution (Sigma®)
foram montados em uma coluna cromatográfica de 90 x 1,6cm. Logo após a montagem, a
coluna foi conectada a uma bomba peristáltica e o fluxo foi regulado para 0,3 mL/mim. A
coluna foi lavada com 3 volumes de NaOH 0,2M e posteriormente 3 volumes de PBS 1X. A
mesma foi equilibrada com 3
c
Aplicou-se à coluna 200 µL do marcador Blue Dextran e, logo após usa entrada, f
a
ina e Anidrase carbônica). Após a corrida com os marcadores aplicou-se novamente na
coluna 200 µL do marcador Blue Dextran e 1 mL de sobrenadante previamente processado
(como no tópico 4.3.9 de métodos). A coluna foi constantemente lavada com de PBS 1X
enquanto as amostras foram coletadas, em microtubos do tipo Eppendorf, com

54
aproximadamente 2,3 mL por microtudo durante a noite pelo coletor automático. O volume da
coluna foi completado com PBS por três vezes para trocar o tampão de eluição pelo tampão
de arm
s, montou-se uma curva padrão
uotando-se a densidade óptica a 562nm dos padrões versus sua respectiva concentração em
a concentração de proteína na
mostra utilizando-se a equação de reta resultante da curva-padrão.
obilizada em agarose. O
icrotubo foi novamente incubado durante 2 horas no agitador do tipo orbital a temperatura
pós esse peridodo de incubação o microtubo foi centrifugado
or cinco minutos na velocidade de 13.000 rpm. Descartou-se o sobrenadante e repetiu-se a
centrifu
item 4.3.10 de métodos.
azenamento (PBS 1X).
A densidade óptica de cada amostra eluída e coletada foi medida na faixa de 230-320
nm para análise da presença de proteínas. Aquelas em que foram detectadas a presença de
proteínas, por Dot Blotting, foram passadas na coluna Centricon YM-10 (Amicon®), com
membrana de 10kDa. Desta maneira o material foi dializado e concentrado para cerca de
500 μL. Após esse processo as amostras foram congeladas.
4.3.19 – Quantificação de Proteínas Utilizando o Kit BCA
Adicionaram-se 100 μL de cada padrão diluído (25, 125, 250, 500, 750, 1000, 1500,
2000 μg/mL) e da amostra a ser quantificada em microtubos ao qual se juntou 2 mL de
reagente WR em cada. Incubaram-se todos os tubos por 30 minutos a 60°C. Os mesmos
foram resfriados à temperatura ambiente e determinou-se as densidades ópticas a 562 nm
utilizando-se PBS 1X como branco. A partir desses dado
q
μg/mL, conforme orientado pelo fabricante. Determinou-se
a
4.3.20 – Imunoprecipitação
Em um microtubo do tipo eppendorf foram adicionados 20 µL das amostras de scFab
previamente processadas (como no item 4.3.9 de métodos) e 10 µg de Z-DNA biotinilado.
Completou-se com PBS 1X para o volume final de 100 µL. O microtubo foi incubado durante
2 horas em um agitador do tipo orbital a temperatura ambiente sob agitação leve.
Em seguida foram adicionados 20 µL de Estreptoavidina im
m
ambiente sob agitação leve. A
p
gação após ressuspensão do pellet em 10 µL de PBS 1X. A amostra foi ressuspendida
em tampão de amostra 2X para SDS-PAGE. Procedeu a analise no Gel SDS-PAGE como no

55
4.3.21 – Coluna de Troca Iônica MonoQ
coluna foi equilibrada com tampão de baixo sal por 10 mim em um
uxo de 6 mL/mim. Após esse procedimento a coluna foi invertida novamente para a posição
/mim para a aplicação da amostra. Após esse
rocedimento a coluna foi lavada com 10 mL com tampão de baixo sal. O volume da coluna
era com
análise no Gel SDS-PAGE como no
pico 4.3.1 de métodos.
A coluna de troca iônica (Econo-Pac High Capacity Íon Exchange Cartridges Mono Q
Cartridge, Bio Rad®, nº. cat. 732-0028) foi montada e acoplada a bomba peristáltica com
fluxo de 2 mL/mim. A coluna foi lavada no tampão de baixo sal por 2 minutos neste fluxo. A
ativação foi feita com o tampão de alto sal por 10 minutos em um fluxo de 6 mL/mim. Para a
retirada de bolhas que possivelmente poderiam estar no topo da coluna, a mesma foi invertida
no decorrer da ativação. A
fl
inicial. O fluxo foi reduzido para 0,5 mL
p
pletado com PBS por três vezes para trocar o tampão de eluição pelo tampão de
armazenamento (PBS 1X).
A eluição foi feita com concentrações crescentes de NaCl (25 a 500mM) enquanto
coletavam-se as amostras de 1 em 1mL, em microtubos do tipo eppendorf. Imediatamente
após a coleta, 10 µL das amostras eram aplicadas em uma membrana de nitrocelulose para
análise por Dot Blotting, seguindo o protocolo do item 4.3.13 de métodos. As amostras onde
se detectavam proteínas foram passadas na coluna Centricon YM-10 (Amicon®), com
membrana de 10kDa. Desta maneira o material era dializado e concentrado para cerca de 500
μL. Após esse processo as amostras foram congeladas.
4.3.22 – Deglicosilação
Foram desnaturadas 20 µg das amostras de scFab previamente processadas (como no
tópico 4.3.9) com 1X do tampão de desnaturação (Glicoprotein Denaturing Buffer) a 100ºC
por 10 mim. Foram adicionados uma proporção de 1/10 de cada tampão G7 Reaction e
NP-40. Após esse procedimento foram adicionados 5 µL da enzima PNGase F e esse sistema
foi incubando a 37ºC durante a noite. Após esse período a amostra foi ressuspendida em
tampão de amostra 2X para SDS-PAGE e procedeu-se a
tó

56
5 – RESULTADOS e DISCUSSÃO
5.1 – PREPARAÇÃO DO VETOR pPIg Fab
O Vetor de expressão pPIg Fab foi construído em trabalhos anteriores visando à
produção de proteínas recombinantes em quantidade suficiente e um dobramento
tridimensional correto. O vetor foi construído de forma a originar o cassete de expressão VH-
CH1-Kex-VL-Cκ de forma monocistrônica, utilizando o promotor AOX1 que dirige a
transcrição do gene de interesse. Também possui o sinal de secreção do fator α que dirige a
secreção da proteína expressa, o fragmento 3`AOX1 de terminação da transcrição e a fase de
leitura aberta do gene HIS4 para a sua integração deste no genoma da levedura Pichia
pastoris. Além disso contém a origem de replicação de E. coli e o gene da β-lactamase que
confere resistência à ampicilina (maiores detalhes na seção 4.1 de Materiais e Métodos ).
Optou-se pelo sistema de expressão dos fragmentos de anticorpos em células de P.
pastoris, uma vez que essa levedura é bastante conhecida e possui varias vantagens que
motivam sua utilização.
O DNA plasmidial que continha o vetor de expressão foi obtido a partir do estoque
congelado a -80C dos clones de E. coli. Após a recuperação das bactérias e verificação das
clonagens (presença e orientação correta do inserto) por perfil de restrição, com as
endonucleases EcoRI, SmaI e BglII, um clone foi selecionado e utilizado para preparação de
DNA plasmidial em larga escala, conforme o tópico 4.3.1 de Métodos, para transformação da
levedura. Foram obtidos de 30 a 40 µg de DNA, e destes foram utilizados 10 µg para a
transformação
5.2 – TRANSFORMAÇÃO DA LEVEDURA E SELEÇÃO DE CLONES RECOMBINANTES
O vetor obtido foi então utilizado para transformação da levedura metilotrófica
P. pastoris, linhagem SMD1168 conforme descrito nos tópicos 4.3.5 e 4.3.6 em Materiais e
Métodos. Esta linhagem foi utilizada por ser defectiva para os genes PEP4 (pep4-) e HIS4
(his4-), não sendo capaz de produzir, respectivamente, as proteases dependentes da ativação
pela proteinase A e a enzima histidinol desidrogenase, sendo dependente da adição de
histidina para cultivo.

57
Para a transformação os plasmídios obtidos foram linearizados com a enzima de
restrição SalI. O sítio de restrição desta enzima localiza-se dentro do gene HIS4,
possibilitando a integração do vetor no locus his4 por meio de recombinação homóloga com
restaurando o fenótipo His+. Este evento faz com que os clones transformantes expressem a
enzima histidinol desidrogenase (gene exógeno), o que restaura a capacidade destas leveduras
de crescerem em meio deficiente em histidina (marca auxotrófica) facilitando a seleção.
Após a eletroporação os transformantes foram semeados em YPD (meio completo
com glicose) para recuperação e rápido crescimento. Após esse procedimento alguns clones
foram novamente semeados, agora em meio MD (meio mínimo com glicose) sem histidina,
para seleção dos clones transformantes pelo fenótipo His+ e replicadas serialmente em placas
MM (meio mínimo com metanol) para posterior indução com metanol da expressão da
proteína recombinante. A partir dessa indução foi feita a seleção das colônias positivas (que
expressaram scFab) por meio de Colony Blotting, conforme o item 4.3.7 de Materiais e
Métodos , como mostrado na Figura 11.
Figura 11. Seleção de clones de P. pastoris recombinantes produtores de scFab recombinante por Colony Blotting em Membrana de nitrocelulose. Colony blotting das colônias de Pichia pastoris (SMD1168 Invitrogen) selecionadas após transformação com o plasmídio pPIg22 Fab, mostrando as colônias que expressam a molécula recombinante scFab. A expressão foi induzida com metanol. Foram utilizados como anticorpo primário o anti- Cκ humano e secundário o anti-IgG de cabra conjugado à fosfatase alcalina.

58
5.3 - PRODUÇÃO DO FRAGMENTO RECOMBINANTE e CINÉTICA DE INDUÇÃO
Foi escolhido um clone dentre as colônias positivas selecionadas acima que foi
utilizado para o experimento de cinética de indução da proteína recombinante, para
determinação do melhor tempo de coleta do sobrenadante da cultura de P. pastoris.
A colônia escolhida foi inoculada e sua indução foi feita conforme o item 4.3.8 de
Materiais e Métodos . Cerca de 5 mL do sobrenadante da cultura foi coletado em cada ponto,
sendo as amostras utilizadas correspondentes a: 0, 24, 48, 72, 96 horas. Foi feita, também, a
concentração do meio que restou após a retirada das amostras. O sobrenadante foi
concentrado e dialisado, segundo a seção 4.3.9 do tópico de Materiais e Métodos , sendo
adicionados inibidores de protease.
Para identificar o melhor ponto da cinética de indução para a coleta dos sobrenadantes,
isto é, o tempo com maior expressão do scFab, as amostras coletadas foram submetidas à
eletroforese em gel desnaturante de poliacrilamida (SDS-PAGE) e analisadas por Western
blotting. Para a detecção do scFab recombinantes utilizou-se anti-Cκ humano como anticorpo
primário, anti-IgG de cabra conjugado à fosfatase alcalina como secundário e a revelação foi
feita com NBT/BCIP, conforme descrito na seção 4.3.11 do tópico de Métodos.
Observou-se uma banda de aproximadamente 25 kDa, que corresponde ao tamanho
esperado para a cadeia leve do scFab (Figura 12), nos tempos de indução. Observou-se uma
maior quantidade de proteína recombinante no tempo de 24 horas de indução, desta maneira,
ele foi escolhido para a produção em larga escala dos scFab recombinantes.
Não foi observada nenhuma atividade proteolítica sobre os scFab nesta etapa,
indicando que a associação dos inibidores de proteases utilizados, aliados ao uso da linhagem
SMD1168 (pep4), resultou na minimização da degradação e conseqüente aumento do
rendimento obtido.

59
Figura 12. Análise da produção de proteínas recombinantes em células de P. pastoris em diversos tempos de indução. O sobrenadante de cultura foi coletado em diversos tempos de indução com metanol, concentrados no centriprep YM-10 e submetidos à eletroforese em gel SDS-PAGE 12% sendo este transferido para uma membrana de nitrocelulose. Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário. T0, T1, T2, T3, T4 e TT: Respectivamente, amostras nos tempos de indução 0, 24, 48, 72, 96 horas e 96 horas concentrado. ( - ): Controle negativo (BSA 2 µg/mL); ( + ) - Controle positivo IgG Humana Policlonal. M: Marcador de massa molecular Bench Marker™ (Invitrogen®)
5.3.1 - Quantificação do sobrenadante - BCA
Diversos fatores influenciam drasticamente na expressão de proteínas heterólogas em
P. pastoris. Entre eles temos: o número de cópias do cassete de expressão integradas no
genoma; o local da integração do cassete no cromossomo; regiões 5´- e 3´- não traduzidas
(UTRs) do mRNA; contexto do códon de iniciação da transcrição (AUG); composição A + T
do cDNA; natureza do sinal de secreção; atividade proteolítica endógena; condições de
crescimento e meio de cultura; etc. Além de características da própria proteína recombinante,
como solubilidade, tamanho, presença de modificações pós-traducionais, complexidade
estrutural, entre outras (Cereghino e Cregg, 2000; Daly e Hearn, 2005; Macauley-Patrick et
al, 2005).
Para estimar a concentração de scFab produzido foi feita a quantificação das proteínas
totais no sobrenadante por BCA (de acordo com o tópico e 4.3.19 de Métodos ). Foi obtido o

60
resultado de 435 μg/mL de proteínas totais presentes a partir da equação da reta apresentada
no gráfico da Figura 13.
x=(y+0,0469)/0,0011R2 = 0,9916
-0,4-0,2
00,20,40,60,8
11,21,41,61,8
22,22,4
0 500 1000 1500 2000 2500
Concentração (ug/mL)
Abs
545
nm
Figura 13. Linha de tendência da quantificação por BCA das proteínas totais do sobrenadante de cultura da levedura P. pastoris em frasco. A partir desse gráfico foi retirada a equação da reta, que foi usada, juntamente com o valor da absorbância do sobrenadante, para estimar a concentração de proteínas totais.
Apesar de se encontrar relatos na literatura de extremos de expressão de proteínas
secretadas, como no caso do fragmento C da toxina do tétano, que apresentou um rendimento
de 12g/L (Clare et al., 1991), temos em contraste a Interleucina-17 humana, para a qual
obteve-se apenas 0,35 mg/L (Murphy et al., 1998). De maneira geral, os níveis de expressão
observados variam entre 10 e 500 mg/L de cultura quando se utiliza fermentação em
bioreatores. As expressões realizadas sob essas condições apresentam tipicamente níveis
muito mais altos de expressão que aqueles alcançados frascos de cultura, devido à otimização
das condições de temperatura, pH, fonte de carbono, aeração entre outros (Cereghino et al.,
2002). Já os níveis máximos relatados para expressão em frascos de cultura são da ordem de
g/L. Como podemos observar nos estudos realizados com aqualisina I de Thermus aquaticus
(1 g/L), tioredoxina de Aliciclobacillus acidocaldarius (0,9 g/L) e somatotrofina de porco (0,9
g/L) (Oledzka et al., 2003, Digilio et al., 2003, Ouyang et al., 2003) além daqueles constando
na Tabela 1 da introdução.
Diversos fragmentos de anticorpos recombinantes já foram produzidos em P. pastoris,
os níveis de produção de scFvs, por exemplo, variam de cerca de 10 a 250 mg/L (Hu et al.,
2005; Emberson et al., 2005; Marty et al., 2001; Eldin et al., 1997) em frasco ou em
biorreatores.

61
Os níveis de produção, especificamente de Fab, em frasco variam de cerca de 5 a 80
mg/L. Enquanto que as fermentações realizadas em biorreatores apresentam melhores níveis
de expressão,de 412 a 420 mg/L. Dentre os Fabs que apresentaram melhores níveis de
expressão destacam-se: anti-HBs – 50 mg/L (Ning et al., 2003) e o fragmento Fab atrazine
específico (K411B) – 40 mg/L (Lange et al., 2001), produzidos em frascos, e o nível máximo
obtido em bioreator foi de 420 mg/L de anti-HBsAg Fab (Ning et al., 2005).
O nível de produção obtido foi considerado bom, tendo-se em vista que esta produção
foi feita em frascos de cultura e não em fermentador. Além disso, não foi realizado nenhum
procedimento de otimização do processo de produção e purificação da proteína recombinante,
já que se objetivava apenas obter quantidades de proteínas suficientes para realizar os
experimentos de ELISA e Western Blotting.
5.4 - CARACTERIZAÇÃO DO scFab RECOMBINANTE
5.4.1 - Análise do sobrenadante de cultura por Western Blotting
A partir da seqüência de aminoácidos do scFab (Anexo 1), foi utilizado o programa
para análises computacionais de seqüência ProtParamTool, localizado no endereço eletrônico
http://www.expasy.org/tools/, para verificar a massa molecular esperada das cadeias leve e
pesada. Foram preditas separadamente em, respectivamente, 23 e 24 kDa (Tabela 5). Com
objetivo de analisar a separação das cadeias, leve da pesada, do scFab recombinante pela ação
da protease Kex2, foi feito um gel no sistema de SDS-PAGE a 15% (Figura 14B) onde a
amostra previamente processada foi aplicada.
Após eletroforese as amostras foram submetidas a Western blotting (Figura 14A)
utilizando anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase
alcalina como secundário.
O Western demonstrou que a banda visualizada na altura aproximada de 25 kDa é a
cadeia leve. As cadeias pesada e leve, provavelmente, estão migrando juntas nesta banda
evidenciada pelo Western Blotting. Já que, segundo as análises computacionais, ambas tem
massas moleculares muito próximas.
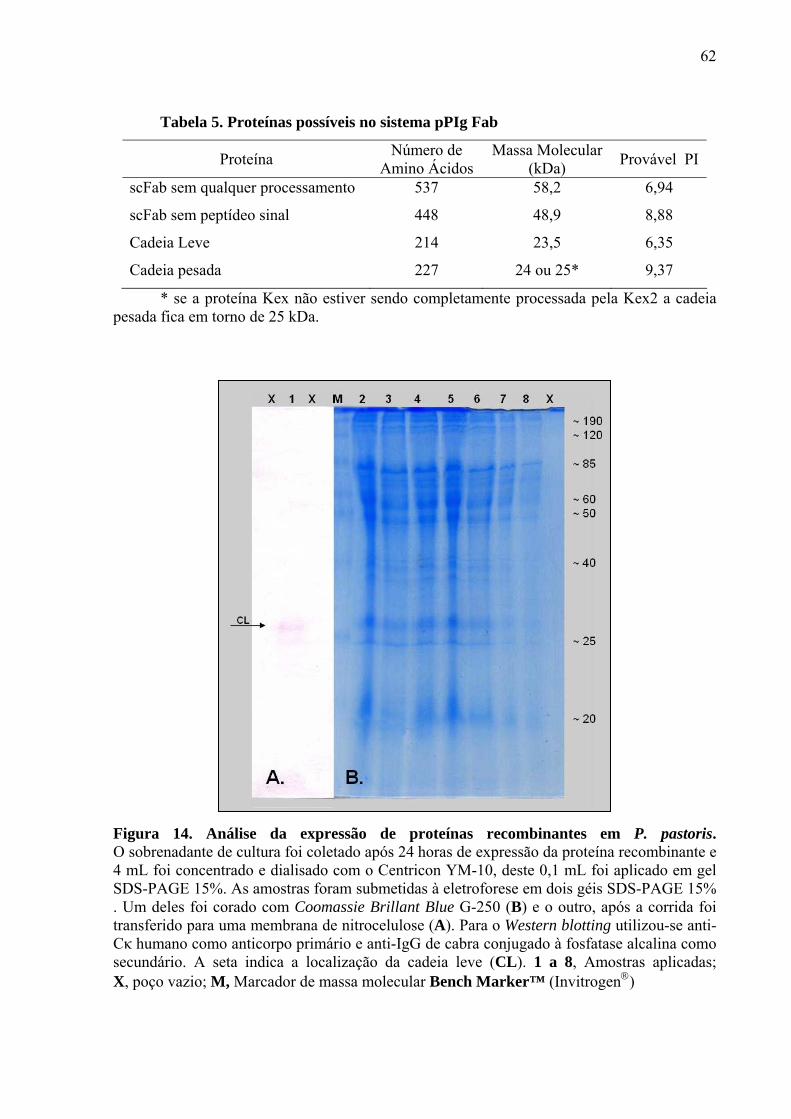
62
Tabela 5. Proteínas possíveis no sistema pPIg Fab
Proteína Número de Amino Ácidos
Massa Molecular (kDa) Provável PI
scFab sem qualquer processamento 537 58,2 6,94
scFab sem peptídeo sinal 448 48,9 8,88
Cadeia Leve 214 23,5 6,35
Cadeia pesada 227 24 ou 25* 9,37
* se a proteína Kex não estiver sendo completamente processada pela Kex2 a cadeia pesada fica em torno de 25 kDa.
Figura 14. Análise da expressão de proteínas recombinantes em P. pastoris. O sobrenadante de cultura foi coletado após 24 horas de expressão da proteína recombinante e 4 mL foi concentrado e dialisado com o Centricon YM-10, deste 0,1 mL foi aplicado em gel SDS-PAGE 15%. As amostras foram submetidas à eletroforese em dois géis SDS-PAGE 15% . Um deles foi corado com Coomassie Brillant Blue G-250 (B) e o outro, após a corrida foi transferido para uma membrana de nitrocelulose (A). Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário. A seta indica a localização da cadeia leve (CL). 1 a 8, Amostras aplicadas; X, poço vazio; M, Marcador de massa molecular Bench Marker™ (Invitrogen®)

63
5.4.2 – Análise da Atividade Ligante do Fragmento scFab
Após a confirmação da massa molecular aparente o sobrenadante de cultura pré-
purificado e dialisado de acordo com o tópico 4.3.9 de Métodos foi analisado quanto a
atividade ligante e seletividade do anticorpo heterólogo produzido pelo antígeno Z-DNA. Para
tanto foram feitos imunoensaio enzimáticos – ELISA, de ligação direta (Figuras 15 e 16) e de
inibição (Figuras 17 e 18), conforme descrito em Métodos nos tópicos 4.3.16.1 e 4.3.16.2
respectivamente.
A ligação ao antígeno pelo fragmento scFab recombinante foi comparada com dois
controles: um fragmento recombinante produzido em P. pastoris (FvFc) e com o mAb Z22,
conhecidamente ligantes de Z-DNA (Andrade, 2000; Andrade, 2005).
5.4.2.1 – Ensaio de Ligação Direta
O ensaio de ligação direta foi realizado com o antígeno (Z-DNA biotinilado)
imobilizado na placa de microtitulação seguindo os procedimentos descritos em Métodos .
Para a revelação foram utilizados como anticorpo primário o anti-Cκ Humano feito em cabra
e como secundário o anti-IgG de cabra feito em coelho conjugado a fosfatase alcalina. Para os
controles da revelação utilizou-se anticorpos diferentes de acordo com o anticorpo sendo os
mesmos especificado em Materiais e Métodos
Na Figura 15 pode-se observar que o anticorpo recombinante scFab apresenta
atividade ligante ao antígeno comparável ao mAbZ22. Entretanto, esperava-se que a ligação
do mAbZ22 fosse superior já que ele, sendo um anticorpo intacto, possui dois sítios de
interação enquanto que o scFab é monovalente, possuindo apenas um sítio de interação com o
antígeno (Figura 2, introdução). Isso pos em dúvida a validade deste controle levando a
repetição do ensaio com um novo controle, o FvFc.
Pode-se observar na Figura 16 que a forma FvFc apresentou uma atividade de ligação
superior, tanto ao scFab quanto ao mAbZ22. Esta atividade afinidade de ligação maior já era
esperada pois, assim como o mAbZ22, o FvFc também possui dois sítios de interação ao
antígeno. Já a baixa atividade do mAbZ22 pode ser devida ao método de detecção utilizado
ou a própria integridade do controle utilizado. Considerando-se esses resultados, optou-se em
realizar os ensaios de inibição usando somente o fragmento FvFc como controle.

64
Ligação Direta scFab
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
10,330,110,0370,0120,0040,00130,0004
Concentração (µg/µL)
Abs
orbâ
ncia
Fab
mAbFab s/ ZDNA
Fab s/ 1ºmAb s/ ZDNA
mAb s/ 1º
Figura 15. Imunoensaio enzimático (ELISA) de ligação direta ao Z-DNA comparando a ligação do fragmento recombinante scFab ao mAbZ22. A placa de microtitulação foi sensibilizada com avidina (5 mg/ml), bloqueada com 3% BSA e logo após foi incubado com o Z-DNA biotinilado (1 µg/ml). O sobrenadante de cultura foi adicionado em diferentes concentrações. A proteína de interesse foi detectada com o anticorpo de anti-Cκ humano feito em cabra, seguido de anti-IgG de cabra conjugado a fosfatase alcalina feito em coelho. No controle mAbZ22 o primário era o anti a cadeia γ de camundongo feito em cabra e o secundário o mesmo usado nas amostras o anti-IgG de cabra conjugado a fosfatase alcalina feito em coelho. A reação colorimétrica foi visualizada adicionando-se 1 mg/ml de pNPP.
Ligação Direta - FvFc / scFab / mAbZ22
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
0,30,10,030,010,0040,0010,00040
Concentração (µg/µL)
Abs
orbâ
ncia
Fab
mAb Z22
FvFc
Fab s/ Ligante
Fab s/ 1º
mAb s/ Ligante
mAb s/ 1º
FvFc s/ Ligante
FvFc s/ 1º
Figura 16. Imunoensaio enzimático (ELISA) de ligação direta ao Z-DNA comparando a ligação do fragmento recombinante scFab ao mAb Z22 e FvFc. A placa de microtitulação foi sensibilizada com avidina (5 mg/ml), bloqueada com 3% BSA e logo após foi incubado com o Z-DNA biotinilado imobilizado (1 mg/ml). O sobrenadante de cultura foi adicionado em diferentes diluições. A proteína de interesse foi detectada com o anticorpo anti-Cκ humano feito em cabra, seguido de de anti-IgG de cabra conjugado a fosfatase alcalina feito em coelho. No controle FvFc o anticorpo primário foi o anti-Fc humano feito em coelho e o secundário o anti-IgG Coelho conjugado a fosfatase alcalina feito em cabra. Já no mAbZ22 o primário foi o anti-γ Camundongo feito em cabra e o secundário o mesmo usado nas amostras, o anti-IgG de cabra conjugado a fosfatase alcalina feito em coelho. A reação colorimétrica foi visualizada adicionando 1 mg/ml de pNPP.

65
5.4.2.2 - Ensaio de inibição
A partir das análises da afinidade relativa da amostra pelo antígeno discutidas acima,
determinou-se a concentração da mesma para a realização dos ensaios de ligação na presença
de competidores relacionados. Os competidores utilizados estão na Tabela 1 em Materiais,
sendo seqüências oligonucleotídicas e polinucleotídicas. Os competidores foram preparados
segundo o tópico 4.2.15 de Materiais.
O ensaio de inibição procedeu-se da mesma forma que o de ligação direta, com a
diferença que os anticorpos recombinantes, tanto o scFab e o FvFc, estavam em concentrações
constantes, 0,03 µg/mL e 0,1 µg/mL, respectivamente. As variáveis nesse ensaios foram as
diluições dos inibidores, feitas de forma serial e crescente na razão de 1:3 conforme o tópico
tópico 4.2.15 de Materiais. Este ensaio visa comparar o nível da capacidade ligante dos
anticorpos, do scFab em relação ao o controle, na presença de um competidor.
Tanto o scFab quanto o FvFc tiveram sua ligação ao antígeno inibida somente pelo
próprio Z-DNA solúvel (Figuras 17 e 18), corroborando com os dados anteriores da literatura
(Andrade et al., 2005).
O fato dos outros antígenos não interferirem na interação do scFab e FvFc com o Z-
DNA, indica seletividade destes anticorpos pelo Z-DNA nas condições analisadas.
Com os resultados obtidos pelos testes imunoenzimático (Figuras 15, 16, 17 e 18)
concluiu-se que o scFab recombinante apresenta capacidade ligante ao antígeno e perfil de
seletividade comparável ao FvFc. Sugere-se então que a conformação estrutural do anticorpo
foi, provavelmente, corretamente montada.

66
Inibição scFab
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
103,331,110,370,120,040Concentração (µg/µL)
Abs
orbâ
ncia Z-DNA
ssDNAB-DNAPoli CGFab
Figura 17. Imunoensaio enzimático (ELISA) de inibição do fragmento recombinante scFab. A placa de microtitulação foi sensibilizada com avidina (5 mg/ml), bloqueada com BSA 3% sendo logo em seguida incubado com o Z-DNA biotinilado imobilizado (1 mg/ml). Foram adicionados 80 µL dos inibidores (Tabela 1 de Materiais) na linha inicial de onde se faziam diluições seriais crescentes na razão de 1:3 e incubados durante 10 minutos. O scFab foi adicionado (0,03 µg/mL) e incubado durante duas horas a temperatura ambiente junto com os inibidores. A detecção foi feita com o anticorpo de anti-Cκ humano feito em cabra, seguido de anti-IgG de cabra conjugado a fosfatase alcalina feito em coelho. A reação colorimétrica foi feita adicionando 1 mg/ml de pNPP. A concentração inicial dos competidores foi de 10 µg/mL, exceto o Z-DNA que foi de 1,11 µg/mL.
Inibição FvFc
0
0,2
0,4
0,6
0,8
1
1,2
1,4
3,31,10,30,120,040Concentração (µg/µL)
Abs
orbâ
ncia Z-DNA
B-DNAssDNApoli CG
Figura 18. Imunoensaio enzimático (ELISA) de inibição do fragmento FvFc. A placa de microtitulação foi sensibilizada com avidina (5 mg/ml), bloqueada com BSA 3% e logo após foi incubada com o Z-DNA biotinilado imobilizado (1 mg/ml). Foram adicionados 80 µL dos inibidores (Tabela 1 de Materiais) na linha inicial de onde se faziam diluições seriais crescentes na razão de 1:3 e incubados durante 10 minutos. O FvFc foi adicionado (0,1µg/mL) e incubado durante 2 horas a temperatura ambiente junto com os inibidores. A detecção foi feita com anti-Fc humano feito em coelho, seguido de anti-IgG de coelho feito em cabra conjugado a fosfatase alcalina. A reação colorimétrica foi feita adicionando 1 mg/ml de pNPP. A concentração inicial dos competidores foi de 10 µg/mL, exceto o Z-DNA que foi de 1,11 µg/mL.

67
5.5 – FERMENTAÇÃO EM BIOREATOR
Para a expressão do scFab recombinante foi utilizado um bioreator de bancada do tipo
tanque agitado, seguindo a metodologia especificada em 4.3.8.3. Durante o processo
fermentativo, várias das condições foram reajustadas (pH, DO, vazão de ar, agitação,
alimentação de glicerol e metanol), principalmente durante a fase de crescimento da levedura
metilotrófica P. pastoris. O pH, a temperatura, a vazão de ar, a DO (oxigênio dissolvido) e
agitação foram constantemente monitorados.
O pré-inóculo foi crescido por 24 horas e observou-se que a OD600 encontrava-se
entre 8 e 9. Como a OD deve estar entre 2 e 5 para a inoculação no fermentador, o mesmo foi
diluído 1:2, com o próprio meio de crescimento (BMGY). Foi feita uma nova leitura de OD
sendo obtido o valor de 4. Para ensaios futuros, o tempo de crescimento do pré-inóculo deverá
ser inferior a 24 horas de forma que não seja necessária a diluição do material.
Durante toda a etapa do processo fermentativo a densidade ótica foi monitorada para
que se mantivesse em 30-35%. Entretanto, esta leitura oscilou consideravelmente ao longo da
fermentação, provavelmente devido à instabilidade da sonda ou mesmo do mecanismo de
difusão do O2 (deficiência de aeração ou variação do tamanho das bolhas). Isso acarretou em
algumas dificuldades nesta etapa e nas posteriores, uma vez que a concentração de oxigênio
dissolvido variou muito, não sendo mantida próxima do valor recomendado (30-35%). A
agitação do meio teve que ser aumentada manualmente de forma progressiva, de acordo com
as observações feitas pelo monitoramento. Durante os períodos em que o bioreator não pode
ser mantido sob observação, o mesmo ficou sob uma condição constante. Dessa forma não foi
possível manter a condição ideal em tempo integral.
Na fase da batelada alimentada foram realizadas análises para verificar se o acumulo
de glicerol no meio estava ou não ocorrendo. O teste, chamado de “spike”, consistia em
desligar a alimentação até que a DO atingisse um valor 10% maior, em relação ao que se
encontrava. O tempo dessa mudança era medido e servia como indicativo se a condição do
glicerol como fator limitante do processo estava ou não sendo mantida. Quanto menor o
tempo mais próximo da saturação (em relação a esse composto) se encontra o sistema, sendo
o tempo ideal de 2 minutos.
É importante observar que este teste (spike) não corresponde integralmente, no
biorreator usado, à condição real. Isso pode ser exemplificado pela condição de DO em que se
encontrava o sistema durante a fase de inanição, que deveria ser entre 100 e 120%, mas ficou
entre 70 e 85%. Isso acontece, provavelmente, porque estes parâmetros foram estabelecidos

68
para biorreatores automatizados profissionais, em que a aeração é feita por O2 puro, sendo sua
difusão no meio a mais alta possível. Tal condição de aeração não corresponde à realidade do
fermentador usado, por questões relacionadas ao design do mesmo.
Durante a fase de crescimento, foi observado excesso de glicerol no meio, nesse dado
momento o spike registrou um tempo de 45 minutos. Neste momento foi executada a parada
do fluxo de alimentação até a estabilização da DO em 30-40%, sendo então reiniciada. Outro
ajuste foi o aumento da agitação, para aproximadamente 1.000 rpm, sendo esta mantida
próxima a esse valor até o fim do processo, para evitar um novo excesso de alimentação.
Quando a condição de glicerol como limitante do crescimento da cultura foi atingida, a
alimentação foi suspensa durante 30 minutos. A fase de inanição, como é chamada, visa obter
uma situação de escassez de fonte de carbono a fim de provocar a “avidez” de consumo do
metanol na fase de indução. Outro aspecto importante dessa fase é a completa eliminação do
glicerol do meio, uma vez que o mesmo inibe a ativação da expressão pelo promotor AOX1,
presente no vetor.
Após o período de inanição, foi iniciada a fase de indução da expressão da proteína
heteróloga. Nesta etapa foi adicionado metanol ao meio de acordo com o descrito em
Métodos. No início dessa fase, foram observadas várias alterações no oxigênio dissolvido,
acreditamos que isso correspondeu a uma fase de adaptação.
Mais uma vez, foram usados os spikes para regular a alimentação com metanol de
forma a mantê-lo como fator limitante da cultura. A medida da DO se manteve, de forma
geral, abaixo dos níveis das fases anteriores.
Foi feito um total de 8 coletas de amostras na fase de indução: 0, A, B, C, D, E, F e G,
respectivamente os tempos de 0, 9, 12, 19, 31, 35, 39 e 41 horas (Tabela 6). Após cada coleta
foi realizada uma leitura da OD600 no espectrofotômetro para o acompanhamento do
crescimento da cultura. A mesma foi então processada (como descrito no tópico 4.3.9 de
métodos) e o sobrenadante foi estocado, após adição de inibidores de protease.
Uma vez terminado todo procedimento foram realizadas análises por eletroforese em
gel de poliacrilamida desnaturante (SDS-PAGE), sendo um dos géis usado para o
procedimento de Western Blotting a fim de confirmar a presença do scFab (figura 20A) e os
demais corados com prata ou Coomassie Blue (Figuras 19 e 20B, respectivamente). De posse
desses resultados verificamos que os melhores tempos de produção de scFab foram 31, 35 e
39 horas, correspondentes às coletas D, E e F respectivamente. Estes resultados indicam que,
neste período a cultura se encontrava nas melhores condições (aeração, alimentação com
metanol e DO) para expressão da proteína heteróloga.

69
Tabela 6. Coletas dos tempos na fase de indução por metanol
Amostra Tempo (horas)
O2 OD600 Metanol (mL/h)
Agitação (rpm)
0 0 115% 70 0,8 1061 A 9 75-85% 85 2,08 960 B 12 45-55% 118 0,8 900 C 19 35-45% 108 0,8 900 D 31 25-35% 105 0,8 900 E 35 30-40% 102 0,8 900 F 39 35-45% 100 0,8 900 G 41 35-45% 98 0,8 900
Figura 19. Análise por SDS-PAGE da produção do fragmento scFab recombinante durante a fase de indução na fermentação em biorreator. O sobrenadante de cultura foi coletado em diversos tempos de indução com metanol. As amostras foram submetidas à eletroforese em gel SDS-PAGE 12% e este foi corado com prata. 0, 9, 12, 19, 31, 35, 39, 41 e T: Amostras nos respectivos tempos de indução 0, 9, 12, 19, 31, 35, 39 e 41 horas e 41 horas concentrado. M: Marcador de massa molecular Bench Marker™ (Invitrogen®).

70
Figura 20. Análise por Western Blotting da produção do fragmento scFab recombinante por fermentação em bioreator. As amostras foram submetidos à eletroforese em dois géis SDS-PAGE 12% . Um deles foi corado com Coomassie Brillant Blue G-250 (B) e o outro, após a corrida foi transferido para uma membrana de nitrocelulose (A). Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário.0, A, C, E e G, Respectivamente, amostras nos tempos de indução 0, 9, 19, 35 e 41 horas; X, poço vazio; M, Marcador de massa molecular Bench Marker™ (Invitrogen®).
Para estimar a concentração de scFab produzido foi feita a quantificação por BCA das
proteínas totais no sobrenadante processado (de acordo com os tópicos 4.3.9 e 4.3.19 de
materiais e métodos, respectivamente). Foi obtido o resultado de 8,4 g/L de proteínas totais
presentes a partir da equação da reta apresentada na Figura 21. Para avaliação de quanto dessa
concentração correspondia à proteína heteróloga expressa foi feito um gel de SDS-PAGE,
com a amostra em várias diluições, e BSA em varias concentrações. Comparando-se a banda
correspondente ao scFab com os padrões de BSA (Figura 22) foi possível estimar que
aproximadamente 250 mg/L do total de proteínas do sobrenadante correspondem ao anticorpo
produzido.

71
y = 0,0004x + 0,084R2 = 0,9992
0,0000,1000,200
0,3000,4000,5000,6000,700
0,8000,9001,000
0,00 500,00 1000,00 1500,00 2000,00 2500,00
Concentração
Abs
Figura 21. Linha de tendência da quantificação por BCA das proteínas totais do sobrenadante de cultura da levedura P. pastoris em bioreator. A partir desse gráfico foi retirada a equação da reta, que foi usada, juntamente com o valor da absorbância do sobrenadante, para estimar a concentração de proteínas totais.
Figura 22. Quantificação estimada de scFab, no sobrenadante da cultura de P. pastoris em bioreator, em relação às diluições de BSA. A cadeia leve do scFab corresponde à banda com tamanho aparente de 28 kDa constando nas colunas 1, 2, 3, e 4. respectivamente 1, 2, 5 e 8 µL de sobrenadante de cultura; (M) marcador de massa molecular Bench Marker™ (Invitrogen®); (5) BSA, 2 µg; (6) BSA, 0,2 µg. Verificamos que a concentração de cadeia leve de scFab no poço 4 tem aproximadamente a mesma concentração de BSA (aproximadamente 70 kDa) no poço 5. Desta forma estima-se que o scFab esteja em uma concentração final de 0,25 g/L.

72
A partir dos dados relatados acima concluímos que a fermentação em bioreator
(8,4 g/L) produziu aproximadamente 19 vezes mais proteínas totais que a fermentação em
frasco (435 μg/mL). Sendo que a fermentação em bioreator gerou aproximadamente 0,25 g/L
de scFab recombinante.
Apesar dos bons resultados obtidos, ainda há a possibilidade de aumentar a quantidade
de proteína recombinante expressa. Uma possibilidade é a otimização do meio de cultura para
o bioreator utilizado (Chena et al., 2004; Boettner et al., 2002), já que o usado foi adaptado a
partir de dois protocolos diferentes. O crescimento foi realizado seguindo as instruções do
Manual do Kit de Expressão em Pichia pastoris (Invitrogem) enquanto que as fases de
batelada e indução seguiram Stratton et al. (1998). Além do mais, existem trabalhos
utilizando uréia como fonte de carbono que resultaram em bons resultados de expressão (324
mg/L) (Kuwae et al., 2005).
Outra maneira de melhorar a expressão seria, na construção do vetor, utilizar o códon
preferencial (códon usage) do microorganismo em questão (Thiry, 2002).
Mesmo os parâmetros obtidos sendo coerentes com Stratton et al. (1998) eles ainda
podem ser aperfeiçoados. Se o biorreator puder ser adaptado para utilização de O2 puro talvez
a densidade ótica de células aumente consideravelmente durante o processo (350-500) (Lee, et
al., 2003; Damasceno et al., 2004)
Outras modificações podem ser feitas no intuito de melhorar o funcionamento do
fermentador como, por exemplo, a automatização do controle de certos parâmetros, como um
sensor de metanol (Hellwig et al., 1999; Cunha et al., 2004) e glicerol (HPLC). A leitura da
OD pode ser obtida on-line com um espectrofotômetro acoplado ao sistema (Tonso, 2000). A
própria contagem de células pode ser determinada através da análise de várias imagens
obtidas em um microscópio. Programas de computador estão cada vez mais adaptados para o
processamento de imagens, permitindo que, após a aplicação de uma seqüência de funções
matemáticas, se determine parâmetros como distribuição de tamanho, grau de agregação, etc.
(Tonso, 2000). A limitação desse procedimento é a necessidade de se preparar várias lâminas
com amostras de cultivo, o que além de tedioso, causa variabilidade de operador para
operador.

73
5.6 - PURIFICAÇÃO
5.6.1 – Cromatografia de Gel Filtração
Na cromatografia de gel filtração (também chamada de cromatografia de exclusão
molecular, permeação em gel ou peneira molecular), as moléculas são separadas de acordo
com seu tamanho e forma. A resina, consiste de esferas de gel contendo poros cujos tamanhos
são relativamente estreitos. O tamanho dos poros é determinado pela quantidade de ligações
cruzadas entre os polímeros do material da resina. Se uma solução aquosa de moléculas de
vários tamanhos for passada através da coluna, as moléculas que forem muito grandes para
entrar nos poros serão excluídas do volume de solvente no interior das esferas, permanecendo
na solução que as contorna. Portanto, as moléculas maiores atravessam a coluna mais
rapidamente que as menores, que percorrem um “caminho” maior pois passam através dos
poros da resina. Pelo fato de os poros das esferas possuírem um certo intervalo de tamanhos, a
filtração em gel pode ser usada para separar uma ampla variedade de moléculas (Voet et al.,
2000).
Dentro do intervalo de tamanhos separados por uma matriz de tamanho médio, existe
uma relação linear entre o volume de eluição relativo de uma substância e o logaritmo de sua
massa molecular (considerando-se que as moléculas tenham formas parecidas). Se uma certa
coluna de filtração em gel for calibrada com várias proteínas de massa molecular conhecida,
então o peso molecular de uma proteína desconhecida poderá ser estimado pela sua posição
de eluição (Voet et al., 2000).
Na tentativa de purificar o scFab presente no sobrenadante de cultura, utilizou-se a
cromatografia por exclusão molecular. Como primeiro procedimento foi feita a corrida dos
padrões, especificados na Tabela 4 em Materiais, a fim de possibilitar a estimativa do
tamanho da molécula de scFab a ser purificada. O resultado dessa corrida está apresentado na
Figura 23. Com base nesses dados estimou-se que a proteína recombinante deve ser recolhida
em torno do microtubo 50, isso porque sua massa molecular estimada é de 50 kDa e a
proteína controle albumina bovina (60 kDa) foi detectada nessa região (Figura 23).
Foram feitas três cromatografias. Na primeira purificação, além do pico produzido
pelo marcador (Blue Dextran) obteve-se um pico entre as frações 45 a 60 (Figura 24). Sendo
confirmada a presença do scFab, por Dot Blotting, entre as frações 45 a 52 (Figura 25).

74
Já a segunda purificação não foi bem sucedida, pois o único pico observado foi o do
Blue Dextran, apesar de que nas frações 52, 53, 54 e 55 foi confirmada a presença da cadeia
leve do scFab por Dot Blotting (dados não mostrados).
Na terceira cromatografia realizada, além do marcador, ocorreu um pico entre as
frações 50 a 70 (Figura 26), sendo o scFab confirmado entre as frações 50 a 57 (Figura 27).
A presença de picos baixos e longos pode indicar que não houve um bom
empacotamento da coluna, mas isso também pode ser devido à grande quantidade de
proteínas produzidas pela levedura P. pastoris, que podem apresentar um tamanho próximo
do scFab montado.
Considerando a estimativa de que o scFab dimerizado e sem o peptídio sinal possui
um tamanho de aproximadamente 50 kDa e que o mesmo foi detectado na região logo antes
do pico da proteína controle Albumina Bovina (60 kDa), inferimos que a montagem correta
do dímero scFab está sendo feita pela levedura P. pastoris.
Cromatografia de Gel Filtração - Controles
-0,100
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 120 125 130 135 140 145 150 155
Amostras coletadas
Abs
Blue Dextran
β-Amilase
Anidrase Carbônica
Albumina Bovina
Álcool deidrogenase
Figura 23. Gráfico da análise por Gel filtração da calibração da coluna com os padrões controle de massa molecular conhecida. As amostras foram aplicadas na coluna, previamente equilibrada, com tampão Tris-HCl 50mM pH 7,5. Em seguida a coluna foi lavada com 3 volumes de PBS 1X a um fluxo de 0,3 mL/mim. Após o procedimento de gel filtração foi medida a densidade ótica na faixa de 230-320 nm de cada uma das frações de coleta.

75
Cromatografia de Gel Filtração 01
-0,100
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100 105
Amostras coletadas
Abs
Blue Dextran
Dot Blotting
scFab
Figura 24. Gráfico da análise por Gel filtração do fragmento scFab recombinante produzido pela levedura P. pastoris. As amostras foram aplicadas na coluna, previamente equilibrada, com tampão Tris-HCl 50 mM pH 7,5. Em seguida a coluna foi eluida com PBS 1X a um fluxo de 0,3 mL/mim. Após o procedimento de gel filtração foi medida a densidade ótica na faixa de 230-320 nm de cada uma das frações de coleta. O quadrado vermelho representa o ensaio de Dot Blotting para a detecção imunoenzimática da proteína recombinante enquanto que o círculo vermelho representa as frações onde sua presença foi confirmada.
45 46 48 50 51
52 54 55 56 58
60 IgG PBS BSA scFab
45 46 48 50 51
52 54 55 56 58
60 IgG PBS BSA scFab
Figura 25. Análise por Dot Blotting da Gel Filtração 1 do fragmento scFab recombinante produzido pela levedura P. pastoris. Logo após o procedimento de gel filtração 5 µL de cada fração foi aplicada em uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação onde utilizou-se anti-Cκ Humano feito em cabra como anticorpo primário e anti-IgG de cabra feito em coelho conjugado à Fosfatase Alcalina como secundário. Cada um dos círculos numerados representa uma fração coletada. O scFab foi detectado entre as frações 45 e 52. Como controles positivos para a revelação foram utilizados: 3 µL de IgG Humana na diluição de 1:10 e 5 µL do próprio scFab. Nos controles negativos utilizou-se 5 µL de BSA (2 µg/mL) e 5 µL de PBST como branco.

76
Cromatografia de Gel Filtração 3
0,000
0,200
0,400
0,600
0,800
1,000
30 35 40 45 50 55 60 65 70 75 80 85 90 95
Amostras coletadas
Abs
Blue Dextran
Dot Blotting
scFab
Cromatografia de Gel Filtração 3
0,000
0,200
0,400
0,600
0,800
1,000
30 35 40 45 50 55 60 65 70 75 80 85 90 95
Amostras coletadas
Abs
Blue Dextran
Dot Blotting
scFab
Figura 26. Gráfico da análise por Gel filtração do fragmento scFab recombinante produzido pela levedura P. pastoris. As amostras foram aplicadas na coluna, previamente equilibrada, com tampão Tris-HCl 50 mM pH 7,5. Em seguida a coluna foi lavada com 3 volumes de PBS 1X a um fluxo de 0,3 mL/mim. Após o procedimento de gel filtração foi medida a densidade ótica na faixa de 230-320 nm de cada uma das frações de coleta. O quadrado vermelho representa o ensaio de Dot Blotting para a detecção imunoenzimática da proteína recombinante enquanto que o círculo vermelho representa onde sua presença foi revelada.
45 46 47 48 49 50 51 52
53 54 55 56 57 58 59 60 61
62 63 64 65 66 67 68 69 70
71 72 73 74 75 BSA IgG PBS
Figura 27. Análise por Dot Blotting das frações resultantes da Gel Filtração 3 do fragmento scFab recombinante produzido pela levedura P. pastoris. Logo após o procedimento de gel filtração 5 µL de cada fração foi aplicada em uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação onde utilizou-se anti-Cκ humano feito em cabra como anticorpo primário e anti-IgG de cabra feito em coelho conjugado à fosfatase alcalina como secundário. Cada um dos círculos numerados representa uma fração coletada. O scFab foi detectado entre as frações 50 e 57. Como controle positivo para a revelação foi utilizado IgG Humana (3 µL) na diluição de 1:100. Para controles negativos utilizou-se 5 µL de PBST, como branco, e 5 µL de BSA (2 µg/mL).

77
Para verificar a presença de outras proteínas em cada purificação, foram feitos dois
géis SDS-PAGE, onde aplicaram-se somente amostras das frações onde foi detectada a
presença da cadeia leve do scFab por Dot Blotting. Sendo que as amostras foram concentradas
de duas em duas, ou seja nos poços A e B estão concentrados as frações de coleta 46, 47, 48 e
49, respectivamente, da Cromatografia 1, sucessivamente, assim como explica a Tabela 7.
No gel SDS-PAGE corado com Coomassie Brillant Blue G-250 (como descrito no
tópico 4.3.14 de Métodos) foi observada a presença de uma banda com massa molecular
aparente de aproximadamente 25 kDa, que corresponde à cadeia leve do scFab, apenas nas
amostras reduzidas E e F, correspondente a Cromatografia 1. As amostras reduzidas C e D
apresentaram uma banda de massa molecular aparente de aproximadamente 30 kDa o que
provavelmente corresponde a alguma outra proteína expressa pela levedura já que, a
purificação da Cromatografia 2 não conseguiu separar eficientemente as moléculas do
sobrenadante.
Nas amostras reduzidas A e B não foi possível detectar a presença de nenhuma
proteína visível neste gel. O que não aconteceu na amostra A não-reduzida onde pode-se
observar uma fraca banda de massa molecular aparente de aproximadamente 50 kDa o que
corresponderia ao scFab dimérico e nativo. Essa banda também foi verificada nas amostras
não-reduzidas C e E.
Podem-se também observar algumas bandas com massa molecular aparente de
aproximadamente 70 kDa nas amostras reduzidas, provavelmente correspondendo a outras
proteínas presentes no sobrenadante de cultura.
Para confirmar a presença do scFab no gel SDS-PAGE foi feita uma análise por
Western Blotting (como descrito no tópico 4.3.11 de Métodos). A cadeia leve do scFab foi
detectada em todas as amostras tanto reduzidas quanto não-reduzidas (Figura 29),
demonstrando a presença do scFab. No entanto, essas bandas não são observadas em todas as
amostras mostradas no gel corado com Coomassie Brillant Blue G-250 (Figura 28), talvez
devido uma baixa resolução alcançada na revelação do mesmo.
Devido a todos esses fatores podemos inferir que a Cromatografia de Gel Filtração não
apresentou o grau de purificação desejado. Por isso optou-se por buscar outros Métodos de
purificação para o scFab recombinante.

78
Tabela 7. Concentração das amostras coletadas nas Cromatografias de Gel Filtração 1, 2 e 3, aplicadas no gel SDS-PAGE e Western Blotting (Figuras 21 e 22 respectivamente)
Amostras Cromatografia Fração
A 1 46-47 B 1 48-49 C 2 52-53 D 2 54-55 E 3 53-54 F 3 55-56
Figura 28. Análise por SDS-PAGE da purificação do fragmento scFab recombinante por Cromatografia de Gel Filtração, coloração com Coomassie Brillant Blue G-250. Logo após o procedimento com a coluna as amostras foram submetidas à eletroforese em gel SDS-PAGE 12% sendo este corado com Coomassie Brillant Blue G-250. Podemos observar a presença de uma banda com massa molecular aparente de aproximadamente 25 kDa nas amostras reduzidas E e F que correspondem à cadeia leve do scFab. E as bandas com massa molecular aparente de aproximadamente 50 kDa nas amostras C e E não-reduzidas que correspondem ao scFab intacto. A e B, Fração 46, 47, 48 e 49 da Cromatografia 1; C e D, Fração 52, 53, 54 e 55 da Cromatografia-2; E e F, Fração 53, 54, 55e 56 da Cromatografia-3; ( - ), Controle negativo (Tampão de Eluição); ( + ), Controle positivo IgG Humana Policlonal; M, Marcador de massa molecular Bench Marker™ (Invitrogen®)

79
Figura 29. Análise por Western Blotting da purificação do fragmento scFab recombinante por Cromatografia de Gel Filtração. Logo após o procedimento com a coluna as amostras foram submetidas à eletroforese em gel SDS-PAGE 12% sendo este transferido para uma membrana de nitrocelulose. Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário. Podemos observar a presença de uma banda com massa molecular aparente de aproximadamente 25 kDa nas amostras reduzidas A, B, C, D, E e F que correspondem à cadeia leve do scFab. E as bandas com massa molecular aparente de aproximadamente 50 kDa nas amostras A, C e E não-reduzidas que correspondem ao scFab intacto. A e B, Fração 46, 47, 48 e 49 da Cromatografia-1; C e D, Fração 52, 53, 54 e 55 da Cromatografia-2; E e F, Fração 53, 54, 55e 56 da Cromatografia-3; ( - ), Controle negativo (BSA 0,02µg); ( + ), Controle positivo IgG Humana Policlonal; M, Marcador de massa molecular Bench Marker™ (Invitrogen®)
5.6.2 - Imunoafinidade Proteína A
A proteína A é um polipeptídeo de 42 kDa que compõe a parede celular da bactéria
Staphylococcus aureus. Contém vários domínios de ligação a IgG que continuam livres
mesmo quando a proteína é imobilizada em uma resina (normalmente agarose). Assim sendo,
uma molécula imobilizada de proteína A pode se ligar à pelo menos 2 moléculas de IgG.
A proteína A tem a capacidade de se ligar a um grande número de moléculas de
superfície celular, além das imunoglobulinas, tais como receptores de Fc do tipo II (FcR-II),
moléculas de adesão, receptor do fator de crescimento do endotélio vascular, receptor de
célula T (TCR), antígenos HLA-AB entre outros. Todas estas moléculas pertencem à
superfamília gênica das Imunoglobulinas e por isso possuem domínios “Ig-like” (regiões

80
homólogas aos domínios constantes ou variáveis das imunoglobulinas), os quais tem
afinidade pela proteína A (Vanamala et al., 2003). Essa proteína liga-se a diferentes partes de
regiões conservadas do arcabouço de imunoglobulinas, na porção Fc das IgGs e em alguns
casos também ao VH do fragmento Fab (Inganas, 1981). Conseqüentemente, pode ser
utilizada na purificação de fragmentos de anticorpos FvFc e Fab.
O procedimento da purificação foi realizado em duplicata de acordo com o protocolo
do fornecedor da resina descrito no tópico 4.3.17.1 de Métodos. Na primeira purificação
(Figura 30A) as amostras foram coletadas somente após o inicio da eluição (tubo 1). Mas nem
mesmo durante a eluição (tubos 1 a 9 – Figura 30A) ou durante a lavagem (tubos 10 a 17 –
Figura 30A) foi detectado o scFab. O mesmo ocorreu na segunda purificação (Figura 30B).
Curiosamente, o scFab não foi detectado nem mesmo durante a sua aplicação (tubos 1 a 11 –
Figura 30B), ou seja, provavelmente deve ter se ligado irreversivelmente à resina, por isso não
foi eluido.
Figura 30. Análise por Dot Blotting das amostras recolhidas durante a purificação do fragmento scFab recombinante produzido em P. pastoris por Imunoafinidade a Proteína A. Logo após o procedimento de imunoafinidade, 5 µL de cada tubo de coleta foi aplicado em uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação onde utilizou-se anti-Cκ humano feito em cabra como anticorpo primário e anti-IgG de cabra feito em coelho conjugado à fosfatase alcalina como secundário. Cada um dos círculos numerados representa um dos microtubos coletados. Não foi detectado o scFab em nenhuma das duas purificações (A e B). A eluição (quadrado) foi feita do tubo 1 ao 9, na primeira purificação (A ) e do 12 ao 20 na segunda (B). Como controles positivos para a revelação foram utilizados: 3 µL de IgG Humana, nas diluições de 1:100, e 5 µL do próprio scFab (1A, 1B, 2A e 2B respectivamente) não passado pela coluna. Como controle negativo (-) utilizou-se 5 µL de BSA (2 µg/mL).

81
5.6.3 - Imunoafinidade Proteína L
A Proteina L (PpL) é uma proteína ligante a imunoglobulinas encontrada na superfície
de Peptostreptococcus magnus e, juntamente com a proteína A (PpA) de Staphylococcus
aureus e a proteína G (PpG) do grupo C e G de Streptococci, é classificada como um
superantígeno para células B. Estes três superantígenos ligam-se a diferentes partes de regiões
conservadas do arcabouço de imunoglobulinas sendo que esta ligação não interfere com a
interação antígeno-anticorpo (Kastern et al., 1992). Conseqüentemente, estes superantígenos
tem sido amplamente usados na purificação, imobilização e detecção de imunoglobulinas.
Particularmente, PpA e PpL têm se demonstrado de uso proveitoso na engenharia de
anticorpos devido à localização de seus sítios de ligação nos domínios das cadeias variáveis
pesada (VH) e leve (VL), respectivamente. Isto permite seu uso na detecção e purificação de
fragmentos de anticorpos como o scFv e o Fab (Enever et al., 2005). No trabalho de Enever
et al. foram descritos os isolamentos de anticorpos ligantes a superantígenos da PpL,
melhorando a afinidade e alterando a especificidade, usando a técnica de phage display.
Demonstraram sua efetividade como agente de captura para os anticorpos e re-direcionamento
das funções efetoras imunes.
Utilizando o BLASTP foi feito um alinhamento da seqüência do VL do scFab,
recombinante expresso em P. pastoris nesse trabalho, com a seqüência do domínio Vκ do
fragmento Fab utilizado no trabalho citado acima (HuVκI). Na comparação das seqüências
houve 76% de identidade, onde 19 dos 21 dos aminoácidos do HuVκI foram positivos (90%),
o que nos levou a concluir que, provavelmente, o scFab possui o domínio de ligação a PpL
possibilitando sua purificação utilizando esta proteína (dados não mostrados).
O procedimento da purificação foi realizado de acordo com o protocolo do fornecedor
da resina descrito no tópico 4.3.17.2 de Métodos. Entretanto, como mostra a Figura 31, a
amostra aplicada na coluna não ficou retida na resina, saindo imediatamente após a sua
aplicação (frações 1 a 12). O mesmo aconteceu com o anticorpo mAbZ22 que foi utilizado
como controle para esse método de purificação (Figura 32). Possivelmente, a conformação
estrutural do anticorpo pode ter se dado de tal forma que o domínio de ligação a Proteína L
ficou enterrado na molécula, impedindo estericamente sua ligação.

82
Figura 31. Análise por Dot Blotting das amostras recolhidas durante a purificação do fragmento scFab recombinante produzido em P. pastoris por Imunoafinidade a Proteína L. Logo após o procedimento de imunoafinidade, 5 µL de cada tubo de coleta foi aplicado em uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação onde utilizou-se anti-Cκ humano feito em cabra como anticorpo primário e anti-IgG de cabra feito em coelho conjugado à fosfatase alcalina como secundário. Cada um dos círculos numerados representa uma das frações coletadas. Foi detectado o scFab nos 12 primeiros. A eluição foi feita do tubo 33 ao 38, onde o mesmo não foi detectado (quadrado). Como controles positivos para a revelação foram utilizados: 3 µL de IgG Humana nas diluições de 1:100 e 1:10 e 5 µL do próprio scFab (+) não passado pelo coluna. Nos controles negativos utilizou-se 5 µL de BSA (2 µg/mL) e 5 µL de PBST como branco.

83
Figura 32. Análise por Dot Blotting das amostras recolhidas durante a purificação do anticorpo mAbZ22 por Imunoafinidade a Proteína L. Logo após o procedimento de imunoafinidade, 5 µL de cada fração de coleta foi aplicado em uma membrana de nitrocelulose, sendo que cada círculo numerado representa uma fração. Procederam-se o bloqueio e a revelação onde utilizou-se anti-γ Camundongo feito em cabra como anticorpo primário e anti-IgG de cabra feito em coelho conjugado à fosfatase alcalina como secundário. Foi detectado o mAbZ22 fortemente nos 3 primeiros tubos. A eluição foi feita do tubo 14 ao 18 onde nada foi detectado (quadrado azul). Como controles positivos para a revelação foram utilizados: 3 µL de IgG Humana nas diluições de 1:100 e 1:10 e 5 µL do próprio mAbZ22 (Z22). No controle negativo foi utilizado 5 µL PBST como branco.
5.6.4 - Imunoprecipitação
A imunoprecipitação é uma técnica muito especifica para a separação analítica de
alvos antigênicos. Pode ser utilizada até mesmo para detecção de proteínas em um estrato
bruto de células lisadas. Quando combinada com outras técnicas, como SDS-PAGE e Western
Blotting, pode ser usada para detectar e quantificar antígenos, determinar a massa molecular
relativa, monitorar o “turnover” de proteínas e modificações pós-trascricionais além de
checar atividade enzimática (Harlow e Lane, 1998).
Anticorpos podem ser usados para purificar proteínas provenientes das soluções assim
como para identificar e caracterizar as mesmas. Os dois Métodos mais comumente usados
para purificar proteínas são a imunoprecipitação e a cromatografia de afinidade.

84
Na imunoprecipitação um anticorpo específico para um antígeno protéico é usado para
isolar este em uma mistura de proteínas. Na maioria dos procedimentos modernos, o anticorpo
é ligado a uma fase sólida (p. ex., uma pérola de agarose), por conjugação química direta ou
indiretamente (Abbas et al., 2003).
No caso da purificação do scFab, o próprio anticorpo é a proteína de interesse, ou seja,
o que seria o “antígeno” no caso das imunoprecipitações usuais. As pérolas de agarose
estavam imobilizando Estreptoavidina, conseqüentemente, o antígeno específico do anticorpo
expresso, o Z-DNA, que estava quimicamente ligado à Biotina.
Após o procedimento da Imunoprecipitação, foram feitos dois géis SDS-PAGE. Um
dos géis foi corado com Coomassie Brillant Blue G-250 (como descrito no tópico 4.3.14 de
Métodos) enquanto que o outro foi transferido e procedeu-se a análise por Western Blotting
como no tópico 4.3.11 de Métodos.
Como controle positivo da imunoprecipitação utilizou-se 1 µg de FvFc que foi
incubado juntamente a 10 µg de Z-DNA biotinilado. O controle negativo foi feito com 20 µL
de extrato de sobrenadante concentrado de scFab sem Z-DNA biotinilado. A amostra a ser
imunoprecipitada consistia de 20 µL de extrato de sobrenadante concentrado de scFab que foi
incubada com 10 µg de Z-DNA biotinilado (Tabela 8).
Todas as amostras foram incubadas com 10 µL de Estreptoavidina Imobilizada em
agarose (Sigma®). As incubações transcorreram como descrito no tópico 4.3.20 de Métodos.
Na análise feita por Western Blotting (Figura 33B) foram observadas a presença de
uma banda,, com massa molecular aparente de aproximadamente 25 kDa, nas amostras 2 e 3.
Esta banda corresponde à cadeia leve do scFab sendo as colunas 2 e 3 correspondentes as
amostras sem e com o Z-DNA biotinilado, respectivamente. Na amostra 1 (FvFc) não foi
detectada nenhuma banda aparente (Figura 33B).
No gel SDS-PAGE corado com Coomassie Brillant Blue G-250 (Figura 33A) em
todas as amostras foram observadas a presença de uma banda, com massa molecular aparente
de aproximadamente 12 kDa, provavelmente correspondendo a estreptoavidina imobilizada
em agarose. Não foi possível verificar a presença de bandas correspondentes a massa esperada
do scFab, isso pode ser devido ao baixo grau de resolução dessa coloração.
Não foi observada nenhuma diferença no padrão de migração no gel, corado com
Coomassie Brillant Blue G-250, não apresentou resolução suficiente para confirmarmos uma
imunoprecipitação eficiente. Assim sendo, optou-se por buscar outros Métodos de purificação
para o scFab recombinante.

85
Tabela 8. Amostras utilizadas para análise da Imunoprecipitação aplicadas no gel SDS-PAGE e Western Blotting (Figura 19A e 19B respectivamente)
Amostras 1 2 3
scFab - + + FvFc + - - Z-DNA Biotinilado + - + Estreptoavidina imobilizada em Agarose + + +
Figura 33. Análise por SDS-PAGE e Western Blotting da purificação do fragmento scFab recombinante por Imunoprecipitação. Logo após o procedimento de Imunoprecipitação as amostras foram submetidas à eletroforese em dois géis SDS-PAGE 12% . Um deles foi corado com Coomassie Brillant Blue G-250 (A) e o outro, após a corrida foi transferido para uma membrana de nitrocelulose (B). Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário. 1, Controle positivo (FvFc); 2, Controle negativo (scFab sem Z-DNA Biotinilado); 3, Amostra de scFab com Z-DNA biotinilado M, Marcador de massa molecular Bench Marker™ (Invitrogen®)

86
5.6.5 – Cromatografia de Troca Iônica
Na cromatografia de troca iônica as moléculas carregadas ligam-se aos grupos de
carga de sinal contrário imobilizados na matriz. Os ânions ligam-se aos grupos catiônicos dos
trocadores de ânions, e os cátions ligam-se aos grupos aniônicos dos trocadores de cátions.
Resinas à base de celulose e agarose são as matrizes mais freqüentemente usadas nesse tipo de
cromatografia para proteínas. A afinidade de ligação de uma proteína depende da presença de
outros íons, que competem com ela pela ligação ao trocador de íons, e do pH da solução, que
influencia na carga líquida da proteína.(Voet et al., 2000).
As proteínas a serem separadas são diluídas em solução-tampão, muitas vezes a
própria solução de lavagem da coluna, que contém o pH e a concentração de sais apropriados.
São em seguida aplicadas na coluna contendo o trocador iônico. A mesma é então lavada com
a solução-tampão. As proteínas com pouca afinidade pelo trocador de íons passam mais
rapidamente do que as proteínas com elevada afinidade.
Esse tipo de coluna tem sido usada para o isolamento de Fabs recombinantes (Prongay
et al., 1990; Tormo et al., 1990; Gololobov et al., 1995). Devido à ausência da porção Fc,
para uma possível ligação a Proteína A-Sefarose (como descrito anteriormente), a troca iônica
torna-se uma excelente opção devido a sua rapidez e facilidade na purificação, já que a
eluição do Fab se dá em frações de baixas concentrações de sal (em torno de 25 a 100 mM).
Após a coleta das frações, 5 µL de cada fração de coleta foi aplicado em uma
membrana de nitrocelulose para que se procede-se à análise por Dot Blotting para verificar a
presença do scFab recombinante.
Durante a lavagem, nas 11 primeiras frações, foi detectada a presença do scFab entre
os tubos 3 e 8 (Figura 34). Isso, provavelmente ocorreu devido um excesso de material
aplicado na coluna, pois o scFab foi novamente detectado entre os tubos 14 e 18 durante, a
eluição com as frações crescentes de NaCl (Figura 34).
Para a confirmação da presença do scFab recombinante, 15 µL das frações 14, 15 e 16
foram aplicados em cada um de dois géis SDS-PAGE, sendo um usado para a coloração com
prata e outra para revelação por Western Blotting.(Figuras 35 e 36, respectivamente)
Nota-se que a melhor fração está na fração 15, eluída com aproximadamente 50 mM
de NaCl, corroborando com os dados da literatura (Figuras 35 e 36).
Este foi o método mais eficiente de purificação, uma vez que a coloração com prata,
que é bastante sensível, não mostrou a presença de outras proteínas na amostra do tubo 15.

87
1 2 3 4 5 6 7 8
9 10 11 12 13 14 15 16
17 18 19 20 21 22 23 24
25 26 27 28 29 30
IgG IgG1:100 1:10 + + + BSA 3% Meio
1 2 3 4 5 6 7 8
9 10 11 12 13 14 15 16
17 18 19 20 21 22 23 24
25 26 27 28 29 30
IgG IgG1:100 1:10 + + + BSA 3% Meio
Figura 34. Análise por Dot Blotting da purificação do fragmento scFab recombinante por Cromatografia de Troca Iônica. Logo após o procedimento com a coluna de troca iônica 5µL de cada tubo de coleta foi aplicado em uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação, sendo que utilizou-se o anti- Cκ humano feito em cabra como anticorpo primário e o anti-IgG de cabra feito em coelho conjugado à fosfatase alcalina como secundário. Cada um dos círculos numerados representa uma das frações coletadas. Foi detectado o scFab entre os tubos 3 e 8 durante a aplicação e lavagem (quadrado). A eluição foi feita com o gradiente de NaCl (25-500 mM) do tubo 14 ao 24, onde foi detectado o scFab do tubo 14 ao 16. Como controles positivos para a revelação foram utilizados: 3 µL de IgG Humana nas diluições de 1:100 e 1:10 e 5 µL do próprio scFab dialisado (+), e não dialisado (+ +). Nos controles negativos utilizaram-se 5 µL de BSA 3% e 5 µL do Meio de Cultura como branco.

88
Figura 35. Análise por SDS-PAGE da purificação do fragmento scFab recombinante por cromatografia de Troca Iônica, coloração com prata. Logo após o procedimento com a coluna de troca iônica 15 µL de amostra foram submetidos à eletroforese em gel SDS-PAGE 12% sendo este corado com prata. Podemos observar a presença de uma banda com massa molecular aparente de aproximadamente 30 kDa que corresponde à cadeia leve do scFab nas colunas 14 e 15. 14, 15 e 16, Frações 14, 15 e 16, eluídas com 25, 50 e 100 mM de NaCl, respectivamente; BSA, 0,02 µg, controle para a revelação; M - Marcador de massa molecular Bench Marker™ (Invitrogen®).

89
Figura 36. Análise por Western Blotting da purificação do fragmento scFab recombinante por cromatografia de Troca Iônica. Logo após o procedimento com a coluna de troca iônica 15 µL de amostra foram submetidos à eletroforese em gel SDS-PAGE 12% sendo este transferido para uma membrana de nitrocelulose. Para o Western blotting utilizou-se anti-Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário. Podemos observar nas colunas 14, 15 e 16 uma banda de massa molecular aparente de 30 kDa correspondente à cadeia leve do scFab. Observamos ainda que esta banda é mais evidente na coluna 15. 14, 15 e 16, Frações 14, 15 e 16, eluídas com 25, 50 e 100 mM de NaCl, respectivamente; BSA, 0,02µg, controle negativo; M - Marcador de massa molecular Bench Marker™ (Invitrogen®)

90
5.7 – DEGLICOSILAÇÃO
A glicosilação é a mais comum modificação pós-traducional que antecede a secreção
protéica. Aproximadamente 0,5–1,0% das proteínas transcritas em genomas eucariotos são
glicoproteínas. Ela ocorre no lúmem do retículo endoplasmático, após a tradução, e os
oligossacarídeos são covalentemente acoplados às proteínas por ligações N-glicosídicas ou O-
glicosídicas (Daly e Hearn, 2005).
Nos oligossacarídeos N-ligados, a N-acetilglicosamina (GlcNAc) é invariavelmente β-
ligada ao nitrogênio amida de um resíduo Asn na seqüência Asn-X-Ser ou Asn-X-Thr, em que
X é qualquer aminoácido, exceto Pro e Asp. A N-Glicosilação ocorre co-traducionalmente, ou
seja, enquanto a proteína está sendo sintetizada. As proteínas que contém oligossacarídeos N-
ligados são normalmente glicosiladas e, em seguida, processadas (Voet et al., 2000).
O acoplamento O-glicosídico mais comum envolve o dissacarídeo central β-galactosil-(1-
3)-α-N-acetilgalactosamina ligado ao grupo OH de Ser ou Thr. A presença de galactose, de
manose e de xilose, formando O-glicosídeos com Ser ou Thr, é menos comum. Os
oligossacarídeos O-ligados variam em tamanho desde um único resíduo de galactose no colágeno
a cadeias de até 1.000 unidades de dissacarídeo nas proteoglicanas (Daly e Hearn, 2005).
Os oligossacarídeos O-ligados são sintetizados no aparelho de Golgi pela adição
seriada de unidades de monossacarídeos a uma cadeia polipeptídica completa. A síntese é
iniciada com a transferência de N-acetilgalactosamina (GalNac) para um resíduo de Ser ou
Thr no polipeptídeo. Os oligossacarídeos N-ligados são transferidos para um Asn em uma
seqüência especifica de aminoácidos, mas os resíduos Ser ou Thr O-glicosilados não são
membros de nenhuma seqüência comum. Em vez disso, a localização dos sítios de
glicosilação são especificados apenas pela estrutura secundaria ou terciária do polipeptídeo. A
O-glicosilação continua com adição passo a passo de açúcares pelas glicosil-transferases
correspondentes (Macauley-Patrick et al., 2005).
Segundo relatos da literatura a P. pastoris possui um sistema de glicosilação muito
parecido com o de células de mamífero, podendo adicionar oligossacarídeos N- ou O-ligados
(Daly e Hearn, 2005). Decidimos testar esse padrão de N-glicosilação da levedura, já que esta
é a que comumente ocorre em anticorpos, mesmo ela não possuindo o sítio padrão de
glicosilação, o amino ácido Asn do fragmento Fc (Li et al., 2006).
Uma vez que foi observada uma diferença entre o peso molecular do scFab, predito pelo
programa ProtParamTool (Anexo 1) e o visualizado nos géis SDS-PAGE, optou-se por uma
análise por deglicosilação. Para verificar o nível de glicosilação da proteína recombinante scFab

91
o sobrenadante de cultura produzido na levedura metilotrófica P. pastoris foi submetido a um
deglicosilação pela enzima PNGase (Biolabs®) que retira as ramificações N-ligadas da proteína.
Aproximadamente 20 µg de amostra previamente processada (como no tópico 4.3.9) foram
utilizados. Após a incubação necessária a amostra foi ressuspendida em tampão de amostra 2X
e procedeu-se à análise no Gel SDS-PAGE como nos tópicos 4.3.11 e 4.3.15 de métodos
(figuras 37 e 38, respectivamente). No Western Blotting além do anticorpo anti-Cκ humano
(figura 37A) utilizou-se também anti-human F(ab’)2 (H+L) feito em cabra conjugado a fosfatase
alcalina, para que pudéssemos localizar ambas as cadeias, leve e pesada (figura 37B).
Não foi possível observar nenhuma diferença no padrão de migração das amostras
submetida ou não a ação da enzima. Assim sendo, concluiu-se que, aparentemente, não está
ocorrendo N-glicosilação do scFab recombinante expresso pela levedura.
Figura 37. Análise por Western Blotting da deglicosilação do fragmento scFab recombinante. Logo após o tratamento com a enzina PNGase as amostras foram submetidas à eletroforese em gel SDS-PAGE 12% e este foi transferido para uma membrana de nitrocelulose. Procederam-se o bloqueio e a revelação com anticorpos diferentes para cada uma das membranas: (A) anti- Cκ humano como anticorpo primário e anti-IgG de cabra conjugado à fosfatase alcalina como secundário, (B) anti-human F(ab’)2 (H+L) feito em cabra conjugado a fosfatase alcalina.; IgG, IgG humana usada como controle da revelação; ( + ), amostra digerida com a enzima PNGase; ( - ), amostra intacta que foi incubada com os tampões da PNGase mas sem a enzima; ( P ), amostra processada sem o tratamento com os tampões da enzima. M - Marcador de massa molecular Bench Marker™ (Invitrogen®). Não foi possível observar diferença no padrão de migração entre as amostras de scFab deglicosilada e intacta (não deglicosilada).

92
Figura 38. Análise por SDS-PAGE da comparação da deglicosilação do fragmento scFab recombinante e IgG humana, coloração com prata. Logo após o tratamento com a enzina PNGase as amostras foram submetidas à eletroforese em gel SDS-PAGE 12% e este foi corado com prata. IgG, IgG humana usada como controle; ( + ), Amostra digerida com a enzima PNGase; ( - ), Amostra intacta que foi incubada com os tampões da PNGase mas sem a enzima; ( P ), Amostra processada sem o tratamento com os tampões da enzima. M - Marcador de massa molecular Bench Marker™ (Invitrogen®). Não foi possível observar diferença no padrão de migração entre as amostras de scFab deglicosilada e intacta (não deglicosilada), mas na IgG humana observou-se uma diferença no padrão de migração nas bandas próximas a 40 e 50 kDa correspondente a deglicosilação.
Para comparar os dados da deglicosilação foram feitas análises com base na seqüência
do scFab (descrita no Anexo 1), utilizando o programa NetNGlyc disponível na Internet, no
endereço www.cbs.dtu.dk/services/NetNGlyc/. Este programa prediz os sítios de N-
glicosilação. Também foi feita a análise de uma possível ocorrência de sítios para O-
glicosilação com o programa NetOGlyc disponível na Internet, no endereço
www.cbs.dtu.dk/services/NetOGlyc/.
Não foi detectado nenhum sitio de N-glicosilação, a partir dessas análises (figura 39).
Isso condiz com a verificação de que não houve diferença no padrão de migração entre as
amostras de scFab (figuras 37 e 38). Mas, a análise indicou dois possíveis sítios de O-
glicosilação na Treonina (posição 125) e na Serina (posição 129) como demonstrado na figura
40, o que indica que novas análises devem ser realizadas futuramente, para verificar a
ocorrência ou não desse processo de modificação pós-traducional.

93
Po
tenc
ial p
ara
N-G
licos
ilaçã
o
NetNGlyc 1.0: Predição de sítios de N-glicosilação na seqüência primária de aminoácidos
Posição na seqüência de aminoácidos
Pote
ncia
l par
a N
-Glic
osila
ção
NetNGlyc 1.0: Predição de sítios de N-glicosilação na seqüência primária de aminoácidos
Posição na seqüência de aminoácidos
Figura 39. Análise da seqüência do scFab pelo programa NetNGlyc para a averiguação de possíveis sítios de N-Glicosilação. A linha vermelha (Threshold) indica o limiar aceitável calculado pelo programa para indicar uma possível ocorrência de sítios para a N-glicosilação
Pote
ncia
l par
a O
-Glic
osila
ção
NetOGlyc 3.1: Predição de sítios de O-glicosilação na seqüência primária de aminoácidos
Posição na seqüência de aminoácidos
Pote
ncia
l par
a O
-Glic
osila
ção
NetOGlyc 3.1: Predição de sítios de O-glicosilação na seqüência primária de aminoácidos
Posição na seqüência de aminoácidos
Figura 40. Análise da seqüência do scFab pelo programa NetOGlyc para a averiguação de possíveis sítios de O-Glicosilação. A linha vermelha (Threshold) indica o limiar aceitável calculado pelo programa para indicar uma possível ocorrência de sítios para a O-glicosilação. As linhas azuis (Potential) indicam as posições dos possíveis sítios de O-glicosilação. foram detectados apenas dois pontos, nas posições 125 e 129 dos resíduos de aminoácidos, com potencial considerável para esse tipo de glicosilação.

94
6 – CONCLUSÃO e PERSPECTIVAS
Como resultado geral deste trabalho foram obtidas a expressão e caracterização de um
fragmento scFab recombinante, derivado do anticorpo anti Z-DNA mAbZ22, produzido na
levedura metilotrófica Pichia pastoris.
A partir dos dados relatados verificamos que é possível a expressão do Fab em gene
monocistrônico, pois a molécula mostrou-se ativa e funcional. Além de apresentar uma provável
dimerização mostrando montagem e processamento corretos. Apesar desses resultados serem
satisfatórios outros testes devem ser feitos para confirmar essa afirmativa. Entre eles, sugerimos
que a expectativa de massa (mass speck) por HPLC e sequenciamento, tanto da cadeia leve
quanto da pesada, seriam muito indicados. Isso porque os dois teste juntos podem confirmar o
processamento no sítio Kex2, no reticulo endoplasmático da levedura.
A quantidade de proteína total tanto em frasco (435 mg/L), quanto no sistema de
fermentação em biorreator (8,4 g/L), foram satisfatórias. Sendo que conseguimos aumentar
substancialmente a produção com o uso do biorreator. Foi possível estimar que, no biorreator,
aproximadamente 0,25 g/L do total de proteínas do sobrenadante correspondem ao fragmento
scFab recombinante. Uma vez que os parâmetros utilizados na fermentação em bioreator
foram determinados eles poderão ser novamente utilizados em outros sistemas de expressão.
Sugere-se que os próprios parâmetros determinados para o processo fermentativo (em
biorreator) do scFab devam ser utilizados para a produção deste nas condições ideais
estabelecidas. Assim como para outros fragmentos de anticorpos recombinantes utilizados
pelo grupo. Acreditamos, que estes parâmetros podem ser ainda melhor estabelecidos, a fim
de obtermos uma produção mais elevada. Entre os fatores a serem mais bem estabelecidos
encontram-se, por exemplo, a composição do meio e a forma de oxigenação do mesmo.
A purificação do scFab usando a coluna de troca iônica mostrou-se eficiente para
pequenas quantidades, sendo que, para uma purificação em larga escala o protocolo devera ser
devidamente ajustado.
Vencidas as etapas de otimização de produtividade e purificação, o cassete de expressão
desse vetor pode ser utilizado para a produção de fragmentos de anticorpos de interesse clínico.
Por exemplo, em diagnósticos utilizando citolocalização ou imunohistoquímica, ou até mesmo in
vivo, para tratamentos com anticorpos neutralizantes, mapeamento de tumores, desintoxicação de
fármacos ou mesmo Magneto Termo Citólise, desde que sua imunogenicidade sejam devidamente
testadas. Podendo em ultima instância está aumentando o leque de possibilidade de produção de
fragmentos de anticorpos em Pichia pastoris.

95
7 - REFERÊNCIAS BIBLIOGRÁFICAS
ABBAS A.; LICHTMAN A. e POBER J. Cellular and Molecular Immunology. (4. ed.) W. B. Sauders Company, 2000. AMORIN, H. V. e LEÃO, R. M. Fermentação Alcoólica: ciência e tecnologia. 1 ed., Piracicaba: Fermentec. 2005. ANDRADE, E. V.; FREITAS, S. M.; VENTURA, M. M.; MARANHÃO, A. Q. e BRÍGIDO, M. M. Thermodynamic basis for antibody binding to Z-DNA: Comparison of a monoclonal antibody and its recombinant derivatives. Biochimica et Biophysica Acta. 1726:293-301, 2005. ANDRADE, E.V.; ALBUQUERQUE, F.C.; MORAES, L.M.P.; BRIGIDO, M.M.; SANTOS- SILVA, M.A. Single-Chain Fv with Fc fragment of the Human IgG1 Tag: Construction, Pichia pastoris Expression and Antigen Binding Characterization. J. Biochem. 128: 891-895, 2000. BIRD, R. E.; HARDMAN, K. D.; JACOBSON, J. W.; JOHNSON, S.; KAUFMAN, B. M.; LEE, S. M.; LEE, T.; POPE, S. H.; RIORDAN, G. S. e WHITLOW, M. Single-chain antigen-binding proteins. Science. 242:423-6, 1988. Erratum in: Science. 244:409, 1989. BIRD, E. R. e WALKER, W. R. Single chain antibody variable regions. Trends in Biotechnology. 9:132-137, 1991. BOETTNER, M.; PRINZ, B.; HOLZ, C.; STAHL, U. e LANG C. High-throughput screening for expression of heterologous proteins in the yeast Pichia pastoris. Journal of Biotechnology 99:51-62, 2002. BROWN, T. A. Gemones. (2 ed.) Department of Biomolecular Sciences, UMIST, Manchester, UK. 2002. CEREGHINO, J. L. e CREGG, J. M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiology Reviews 24:45-66, 2000. CEREGHINO, G.; CEREGHINO, J.; ILGEN, C. e CREGG, J. Production of recombinant proteins in fermenter cultures of the yeast Pichia pastoris. Current Opinion in Biotechnology 13:329-32. 2002. CHENA, C. C.; WU, P. H.; HUANGA, C. T. e CHENG, K. J. A Pichia pastoris fermentation strategy for enhancing the heterologous expression of an Escherichia coli phytase. Enzyme and Microbial Technology 35:315–320, 2004. CLARE, J.; RAYMENT, F.; BALLANTINE, S.; SREEKRISHNA, K. e ROMANOS, M. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem integrations of the gene. Biotechnology 9:455-6. 1991.

96
CREGG J.M.; BARRINGER, K.J.; HESSLER, A.Y. e MADDEN, K. R. Pichia pastoris as a host system for transformation. Molecular and Cellular Biology. 5:3376-3385, 1985 CREGG J.M.; VEDVICK T.; RASCHKE W. Recent advances in the expression of foreign genes in Pichia pastoris. Biotechnology 11: 905-910, 1993. CREGG, J. M. Expression in the methilotrophic yeast Pichia pastoris. Expression in Pichia pastoris. Ed. Academic press. 157-191, 1999. CONTRERAS, R.; CARREZ, D.; KINGHORN, J.R.; VAN DEN HONDEL, C. A. M. J. J. e FIERS, W. Efficient Kex2-like processing of a Glucoamylase-Interleukin-6 fusion protein by Aspergillus nidulans and secretion of mature Interleukin-6. Biotechnology 9:378-381, 1991. CUNHA, A. E.; CLEMENTE, J. J.; GOMES, R.; PINTO, F.; THOMAZ, M.; MIRANDA, S.; PINTO, R. MOOSMAYER, D.; DONNER, P. e CARRONDO, M. J. Methanol induction optimization for scFv antibody fragment production in Pichia pastoris. Biotechnol Bioeng. 86:458-67 2004. DALY, R. E. e HEARN, M. T. W. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J. Mol. Recognit. 18: 119–138, 2005 DAMASCENO, L. M.; PLA, I.; CHANG, H-J.; COHEN, L.; RITTER, G.; OLD, L. J. e BATT, C. A. An optimized fermentation process for high-level production of a single-chain Fv antibody fragment in Pichia pastoris. Protein Expression and Purication 37:18–26, 2004. DE LORENZO, C.; COZZOLINO, R.; CARPENTIERI1, A.; PUCCI1, P.; LACCETTI, P. e D’ALESSIO, G. Biological properties of a human compact anti-ErbB2 antibody. Carcinogenesis. 26:1890-1895, 2005. DIGILIO, F.; MORRA, R.; PEDONE, E.; BARTOLUCCI, S. e ROSSI, M. High-level expression of Aliciclobacillus acidocaldarius thioredoxin in Pichia pastoris and Bacillus subtilis. Protein Expression and Purification 30:179-84, 2003. ELDIN P.; PAUZA M.; HIEDA Y.; LIN G.; MURTAUGH M.; PENTEL P. e PENNELL C. High-level secretion of two antibody single chain Fv fragments by Pichia pastoris. Journal of Immunological Methods 201:67-75, 1997. ELLIS S.B. et al. Isolation of alcohol oxidase and two other methanol regulatable genes from the yeast, Pichia pastoris. Molecular and Cellular Biology. 5:1111-1121, 1985. EMBERSON L.M.; TRIVETT A.J.; BLOWER P.J. e NICHOLLS P.J. Expression of an anti-CD33 single-chain antibody by Pichia pastoris. J Immunol Methods. 305:35-51, 2005. ENEVER, C.; TOMLINSON, I. M.; LUND, J.; LEVENS, M. e HOLLIGER, P. Engineering High Affinity Superantigens by Phage Display. J. Mol. Biol. 347:107–120, 2005.

97
FROMMER W.B. e NINNEMANN O. Heterologous expression of genes in bacterial, fungal, animal and plant cells. Annual Review of Plant Physiology and Plant Molecular Biology. 46:419-444, 1995. GOLOLOBOV, G. V.; CHERNOVA, E. A.; SCHOUROV, D. V.; SMIRNOV, I. V.; KUDELINA, I. A. e GABIBOV, A. G. Cleavage of supercoiled plasmid DNA by autoantibody Fab fragment: Application of the flow linear dichroism technique. Proc. Natl. Acad. Sci. 92:254-257, 1995. GOMORD V.; SOURROUILLE C.; FITCHETTE A.; BARDOR M.; PAGNY S.; LOUGE P.; FAYE L. Production and glycosylation of plant-made pharmaceuticals: the anticodies as a challenge. Plant Biotechnology Journal. 2:83-100,2004. HARLOW, E. e LANE, D. Antibodies: A laboratory Mannual. Cold Spring Harbor Lab. (2 ed.), 1988. HAYDEN, M.S. et al. Antibody engineering. Current Opinion in Immunology, 9:201-212, 1997. HELLWIG, S.; EMDE, F.; RAVEN, N. P. G.; HENKE, M. e FISCHER, R. Production of a single-chain Fv antibody fragment in 30-L fermentations of P. pastoris using on-line methanol control. Apresentado em: Jahrestagung der Biotechnologen, DECHEMA-Jahrestagung 99:2. 1999. HU, S.; LI, L.; QIAO, J.; GUO, Y.; CHENG, L. e LIU, J. Codon optimization, expression, and characterization of an internalizing anti-ErbB2 single-chain antibody in Pichia pastoris. Protein Expr Purif. In Press 2006 INGANAS, M. Comparison of mechanisms of interaction between protein A from Staphylococcus aureus and human monoclonal IgG, IgA and IgM in relation to the classical FC gamma and the alternative F(ab')2 epsilon protein A interactions. Scandinavo Journal of Immunology 13:343-52, 1981. KASTERN, W.; SJÖBRING, U. e BJÖRCK, L. Structure of Peptostreptococcal Protein L and Identification of a Repeated Immunoglobulin Light Chain-binding Domain. The Journal of Biological Chemistry. 267:12820-12825, 1992. KATO, K.; SAITO, T. e SEELAN, S. Reaction properties of catalytic antibodies encapsulated in organo substituted SiO2 Sol-Gel materials. Journal of Bioscience and Bioengineering. 100:478-480. 2005 KIPRIYANOV, S. M.; LITTLE, M. Genereration of Recombinat Antibodies. Molecular Biotechnology. 12:173-201, 1999. KONSTANTINOV, K. B. Monitoring and control of the physiological state of cell cultures. Biotechnol. Bioeng. 52:271-289, 1996. KUBY J. Immunology. W. H. Freeman and Company, USA, 5rd ed, 2002.

98
KUWAE, S.; OHYAMA, M.; OHYA, T.; OHI, H. e KOBAYASHI, K. Production of Recombinant Human Antithrobin by Pichia pastoris. Journal of Bioscience and Bioengineering. 99:264-271, 2005. LANGE, S.; SCHMITT, J. e SCHMID, R.D. High-yield expression of the recombinant, atrazine-specific Fab fragment K411B by the methylotrophic yeast Pichia pastoris. Journal of Immunological Methods 255:103–114. 2001 LEE, C. Y.; LEE, S. J.; JUNG, K. H.; KATOH, S. e LEE, E. K. High dissolved oxygen tension enhances heterologous protein expression by recombinant Pichia pastoris. Process Biochemistry. 38:1147-1154, 2003. LI, H.; SETHEURAMAN, N.; STADHEIM, T. A.; ZHA, D.; PRINZ, B.; BALLEW, N.; BOBROWICZ, P.; CHOI, B-K.; COOK, W. J.; CUKAN, M.; HOUSTOM-CUMMINGS, N. R.; DAVIDSON, R.; GONG, B.; HAMILTON, S. R.; HOOPES, J. P.; JIANG, Y.; KIM, N.; MANSFIELD, R.; NETT, J. H.; RIOS, S.; STRAWBRIDGE, R.; WILDT, S. e GERNGROSS, T. U. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nature Biotechnology. 24:210-215, 2006. LITTLE M; KIPRIYANOV S, GALL F, MOLDENHAUER G. Of mice and men: hybridoma and recombinant antibodies. Immunology Today 21(8): 364-370, 2000. LIU, H.; MULHOLLAND, N.; FU, H. e ZHAO, K. Cooperative Activity of BRG1 and Z-DNA Formation in Chromatin. Remodeling. Mol Cell Biol. 26:2550-2559. 2006 MACAULEY-PATRICK, S.; FAZENDA, M. L.; MCNEIL, B. e HARVEY, L. M. Heterologous protein production using the Pichia pastoris expression system. Yeast. 22:249–270, 2005. MAJEWSKI, J. e OTT, J. GT Repeats Are Associated with Recombination on Human Chromosome 22. Genome Res. 10:1108-1114. 2000 Manual do Kit de Expressão de proteínas recombinantes em Pichia pastoris. Invitrogen®. Número do Catálogo: K1710-01, Versão 3.0. MARANHÃO, A.; BRÍGIDO M. Anticorpos humanizados:Humanização de anticorpos de interesse clínico. Biotecnologia, Ciência e Desenvolvimento. 23:38-43, 2001. MARTY, C.; SCHEIDEGGER, P.; BALLMER-HOFER, K.; KLEMENZ, R. e SCHWENDENER, R.A. Production of functionalized single-chain Fv antibody fragments binding to the ED-B domain of the B-isoform of fibronectin in Pichia pastoris. Protein Expr Purif. 21:156-64. 2001. MOUNTAIN, A.; ADAIR, J. R. Engineering antibodies for therapy. Biotechnology and Genetic Engineering Reviews, 10:1-141, 1992. MURPHY, K.P.; GAGNE,; P., PAZMANY, C.; e MOODY, M.D. Expression of human interleukin-17 in Pichia pastoris: purification and characterization. Protein Expression and Purification 12:208-214. 1998.

99
NetNGlyc. Programa que prediz os sítios de N-glicosilação, encontra-se disponível na Internet, no endereço www.cbs.dtu.dk/services/NetNGlyc/. NetOGlyc. Programa que prediz os sítios de O-glicosilação, encontra-se disponível na Internet, no endereço www.cbs.dtu.dk/services/NetOGlyc/. NING, D.; JUNJIAN. X.; XUNZHANG. W.; WENYIN. C.; QING. Z.; KUANYUAN. S.; GUIRONG. R.; XIANGRONG. R.; QINGXIN. L, e ZHOUYAO. Y. Expression, purification, and characterization of humanized anti-HBs Fab fragment. J. Biochem. 134: 813–817. 2003 NING, D.; JUNJIAN, X.; QING, Z.; SHENG, X.; WENYIN, C.; GUIRONG, R. e XUNZHANG, W. Production of recombinant humanized anti-HBsAg Fab fragment from Pichia pastoris by fermentation. J Biochem Mol Biol. 38:294-299. 2005 OH, D-B.; KIM, Y-G. e RICH, A. Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. PNAS. 99: 16666–16671. 2002 ORLANDI, R.; GUSSOW, D. H.; JONES, P. T. e WINTER, G. Cloning immunoglobulin variable domains for expression by the polymerase chain reaction. Proc Natl Acad Sci U S A. 86:3833-7, 1989. OLEDZKA, G.; DABROWSKI, S. e KUR, J. High-level expression, secretion, and purification of the thermostable aqualysin I from Thermus aquaticus YT-1 in Pichia pastoris. Protein Expression and Purification. 29:223-9, 2003. OUYANG, J.; WANG, J.; DENG, R.; LONG, Q. e WANG, X. High-level expression, purification, and characterization of porcine somatotropin in Pichia pastoris. Protein Expression and Purification 32(1):28-34, 2003. PADLAN EA. Anatomy of the antibody molecule. Molecular Immunology, 31 (3): 169-217, 1994. PAUL, W. E. Fundamental Immunology. Lippincott Williams & Wilkins Publishers, 5th edition, 2003. PLÜNCKTHUN, A.; SKERRA, A.; Expression of functional antibody Fv and Fab fragments in Escherichia coli. Meth. Enzimol., 178:497-515, 1989. PRONGAY, A. J.; SMITH, T. J.: ROSSMANN, M. G.: EHRLICH, L. S.: CARTER, C. A. e MCCLURE, J. Preparation and crystallization of a human immunodeficiency virus p24-Fab complex. Proc. Natl. Acad. Sci. USA. 87:9980-9984, 1990.´ ProtParamTool. Programa de predição das características gerais das possíveis proteínas expressas que se encontra disponível na Internet, no endereço http://www.expasy.org/tools/. RUGGIERO, L. A. Clonagem e Expressão de Anticorpos Recombinantes em Células de Ovário de Hamster Chinês (CHO) em Cultura. Brasília: UnB, 2002. Dissertação (Mestrado em Biologia molecular). Instituto de Ciências Biológicas, Universidade de Brasília, 2002.

100
SAMBROOK R.; FRITSCH E. Maniatis T. Molecular Cloning: A Laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press, 1989.
STRATTON, J.; CHIRUVOLU, V. e MEAGHER. M. High Cell-Density Fermentation. In: Pichia Protocols. In: Methods in Molecular Biology (Editado por, Higgins, D. R. e Cregg, J. M.). Vol. 103, 1998. THIRY, M. E CINGOLANI, D. Optimizing scale-up fermentation processes. Trends in Biotechnology. 20:103-105, 2002. TONSO, A. Monitoramento e Operação de Cultivos de Células Animais em sistemas de Perfusão. Tese apresentada à escola Politécnica da Universidade de São Paulo para Obtenção do título de Doutor. São Paulo, 2000. TORMO, J.; FITA, I.; KANZLER, O. e BLAAS, D. Crystallization and Preliminary X-ray Diffraction Studies of the Fab Fragment of a Neutralizing Monoclonal Antibody Directed against Human Rhinovirus Serotype 2. The Journal of Biological Chemistry. 265:799-800, 1990. TORRES, F.; MORAES L.; Proteínas recombinantes produzidas em leveduras. Biotecnologia Ciência & Desenvolvimento. 12:20-22; 2000. UDA, T. e HIFUMI, E. Super Catalytic Antibody and Antigenase. Journal of Bioscience and Bioengineering. 97:143-152. 2004 VANAMALA, S.; SEETHARAM, S.; YAMMANI, R. e SEETHARAM, B. Human transcobalamin II receptor binds to Staphylococcus aureus protein A: implications as to its structure and function. Arch Biochemistry and Biophysics 411:204-14, 2003. VAZ, N. M. e FARIA, A. M. C. Guia Incompleto de Imunologia (Imunologia como se o organismo importasse). Belo Horizonte: Coopmed, 1993. VERMA R.; BOLETIA E.; GEORGE A. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. Journal of Immunological Methods. 216:165-181, 1998 VOET, D.; VOET, J. G. e PRATT, C. W. Fundamentos e Bioquímica. (1 ed) Porto Alegre: Artmed, 2000. WU T. e KABAT E. An analysis of the sequence of the variable regions of Bence-Jones proteins and myeloma light chains and their implication for antibody complementariy. Journal of Experimental Medicine 132:211-249,1970. ZHANG, W.; LI, Z. J. e Agblevor, F. A. Microbubble fermentation of recombinant Pichia pastoris for human serum albumin production. Process Biochemistry 40:2073–2078, 2005. ZHU, X.; DICKERSON, T. J.; ROGERS, C. J.; KAUFMANN, G. F.; MEE, J. M.; MCKENZIE, K. M.; JANDA, K. D. e WILSON, I. A. Complete reaction cycle of a cocaine catalytic antibody at atomic resolution. Structure. 14:205-16, 2006.

101
8 - ANEXO 1
Predição das características gerais das possíveis proteínas expressas pelo Sistema pPIg Fab feita pelo programa ProtParamTool que se encontra disponível na Internet, no endereço http://www.expasy.org/tools/. scFab sem qualquer processamento: Número de aminoácidos: 537 Peso Molecular: 58292.3 (58 kDa) pI Teórico: 6.94 scFab sem peptídeo sinal: Número de aminoácidos: 448 Peso Molecular: 48970.9 (48 kDa) pI Teórico: 8.88
Cadeias totalmente processadas: Cadeia leve Número de aminoácidos: 214 Peso Molecular: 23518.1 (23 kDa) pI Teórico: 6.35 Cadeia Pesada Número de aminoácidos: 227 Peso Molecular: 25470.8 (25 kDa) ou 24615.8 (24kDa) pI Teórico: 9.37
Encontram-se destacados: o pré-pró-peptídeo sinal (em negrito), o sítios de restrição
das endopeptidases Kex2 (quadrado preto) e Kex2-like (sublinhado), e os sítios de O-
glicosilação preditos (quadrados cinza).
> Cassete de expressão do scFab
MRFPSIFTAVLFAASSALAAPVNTTTEDETAQIPAEAVIGYSDLEGDFDVAVLPFSNSTNNGLLFINTTIASIAAKEEGVSLEKREAEARVQLVESGGGLVQPKGSLKLSCAASGFNFNTYAMNWVRQAPGKGLEWVARIRSKSNNYATYYADSMKDRFTISRDDSENMLYLQMINLKAEDTAMYYCVRQAYSNYGAMDYWGQGISVTVSSRSTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKRMDKRAPDLQMTQTTSSLSASLGDRVTISCSASQGISNYLNWYQQKPDGTVKLLIYYTSRLHSGVPSRFSGSGSGTDYSLTISNLEPEDIATYFCQQYSKFPFTFGSGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC

102
> scFab maduro RVQLVESGGGLVQPKGSLKLSCAASGFNFNTYAMNWVRQAPGKGLEWVARIRSKSNNYATYYADSMKDRFTISRDDSENMLYLQMINLKAEDTAMYYCVRQAYSNYGAMDYWGQGISVTVSSRSTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKRMDKRAPDLQMTQTTSSLSASLGDRVTISCSASQGISNYLNWYQQKPDGTVKLLIYYTSRLHSGVPSRFSGSGSGTDYSLTISNLEPEDIATYFCQQYSKFPFTFGSGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC > Cadeia leve: VLCK DLQMTQTTSSLSASLGDRVTISCSASQGISNYLNWYQQKPDGTVKLLIYYTSRLHSGVPSRFSGSGSGTDYSLTISNLEPEDIATYFCQQYSKFPFTFGSGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC > Cadeia pesada: VHCH1 RVQLVESGGGLVQPKGSLKLSCAASGFNFNTYAMNWVRQAPGKGLEWVARIRSKSNNYATYYADSMKDRFTISRDDSENMLYLQMINLKAEDTAMYYCVRQAYSNYGAMDYWGQGISVTVSSRSTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKRMDKR

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo



![[Factor Antihemofílico (Recombinante)]](https://static.fdocuments.net/doc/165x107/62e321857857632745738462/factor-antihemoflico-recombinante.jpg)














![Tecn DNA Recombinante[1]](https://static.fdocuments.net/doc/165x107/5571fcdc4979599169981392/tecn-dna-recombinante1.jpg)