
Bypassing Cultivation To Identify Bacterial...
8
Bypassing Cultivation To Identify Bacterial Species Culture-independent genomic approaches identify credibly distinct clusters, avoid cultivation bias, and provide true insights into microbial species Luis M. Rodriguez-R and Konstantinos T. Konstantinidis Whether bacterial species exist as a natural unit remains an unresolved issue, one with important practical challenges, including that of correctly identifying microorganisms and diagnosing the causative agents of microbial diseases. The cur- rent bacterial species defınition is based on ge- netic and phenotypic distinctiveness of organ- isms grouped under the same name. However, the standard methods used to iden- tify bacterial species, including 16S rRNA gene sequence analysis, DNA-DNA hybridizations, and phenotypic tests under laboratory condi- tions, can lead to two ecologically and genetically distinct microorganisms being assigned to the same species. For example, Escherichia coli iso- lates can differ by as much as one-third of their genomes and represent important pathogens (e.g., O157:H7 lineage) or nonpathogenic, com- mensal organisms (e.g., MG1655 lineage). Another important limit to our ability to distinguish bacterial species is that current ap- proaches test bacterial isolates in the laboratory for phenotypic properties that may differ greatly from natural conditions. Such testing may not assign those isolates into clusters that are repre- sentative of natural populations. Instead, the isolates frequently fall along a continuum, a result that poses major challenges for any system that aims to assign organisms to distinct taxa. How- ever, whether that continuum reflects a natural pattern or, instead, an artifact of the methods used and/or the cultivation biases is diffıcult, if not impossible, to determine. Therefore, it is possible that organisms with distinct ecologies and preferred habitats and/or genotypes are be- ing grouped together incorrectly. In contrast, assessing organisms in their habi- tat (in-situ) enables one to observe credibly nat- ural diversity patterns. Thus, by assessing natural populations and bypassing cultivation bias, cul- ture-independent genomic approaches or, more simply, metagenomics can provide valuable in- sights into microbial species. Microbes Form Sequence-Discrete Populations From our review of fındings from large-scale metagenomic studies during the past fıve years, we came to realize that microbial communities are predominantly organized in sequence-dis- crete populations. These populations become evident after comparing the genome sequence of one member of the population against those of all co-occurring organisms in that same sample or habitat. Members within the same population share high sequence identity, ranging between 94 SUMMARY ➤ The current approach to defining bacterial species, based on genetic and phenotypic distinctiveness, is problematic. ➤ Bypassing cultivation to assess natural populations provides a valuable and perhaps more authoritative approach to identifying and defining bacterial species. ➤ Natural microbial communities are predominantly composed of sequence- discrete populations, with exceptions likely to be found within habitats that undergo frequent fluctuations or for organisms with unique ecologic characteristics. ➤ Sequence-discrete populations could be given candidate species names until appropriate isolates with ecologically relevant phenotypic properties are characterized. ➤ The mechanisms maintaining species, and perhaps more importantly, the relative importance of the mechanisms for different organisms and habi- tats, are not understood and demand further study. FEATURE ARTICLE Microbe—Volume 9, Number 3, 2014 • 111
Transcript of Bypassing Cultivation To Identify Bacterial...

Bypassing Cultivation ToIdentify Bacterial SpeciesCulture-independent genomic approaches identify credibly distinct clusters,avoid cultivation bias, and provide true insights into microbial species
Luis M. Rodriguez-R and Konstantinos T. Konstantinidis
Whether bacterial species exist as a natural unitremains an unresolved issue, one with importantpractical challenges, including that of correctlyidentifying microorganisms and diagnosing thecausative agents of microbial diseases. The cur-rent bacterial species defınition is based on ge-netic and phenotypic distinctiveness of organ-isms grouped under the same name.
However, the standard methods used to iden-tify bacterial species, including 16S rRNA genesequence analysis, DNA-DNA hybridizations,and phenotypic tests under laboratory condi-tions, can lead to two ecologically and geneticallydistinct microorganisms being assigned to thesame species. For example, Escherichia coli iso-lates can differ by as much as one-third of theirgenomes and represent important pathogens(e.g., O157:H7 lineage) or nonpathogenic, com-mensal organisms (e.g., MG1655 lineage).
Another important limit to our ability todistinguish bacterial species is that current ap-proaches test bacterial isolates in the laboratoryfor phenotypic properties that may differ greatlyfrom natural conditions. Such testing may notassign those isolates into clusters that are repre-sentative of natural populations. Instead, theisolates frequently fall along a continuum, a resultthat poses major challenges for any system thataims to assign organisms to distinct taxa. How-ever, whether that continuum reflects a naturalpattern or, instead, an artifact of the methodsused and/or the cultivation biases is diffıcult,if not impossible, to determine. Therefore, it ispossible that organisms with distinct ecologiesand preferred habitats and/or genotypes are be-ing grouped together incorrectly.
In contrast, assessing organisms in their habi-tat (in-situ) enables one to observe credibly nat-
ural diversity patterns. Thus, by assessing naturalpopulations and bypassing cultivation bias, cul-ture-independent genomic approaches or, moresimply, metagenomics can provide valuable in-sights into microbial species.
Microbes Form Sequence-DiscretePopulations
From our review of fındings from large-scalemetagenomic studies during the past fıve years,we came to realize that microbial communitiesare predominantly organized in sequence-dis-crete populations. These populations becomeevident after comparing the genome sequence ofone member of the population against those ofall co-occurring organisms in that same sampleor habitat. Members within the same populationshare high sequence identity, ranging between 94
SUMMARY
➤ The current approach to defining bacterial species, based on genetic andphenotypic distinctiveness, is problematic.
➤ Bypassing cultivation to assess natural populations provides a valuable andperhaps more authoritative approach to identifying and defining bacterialspecies.
➤ Natural microbial communities are predominantly composed of sequence-discrete populations, with exceptions likely to be found within habitatsthat undergo frequent fluctuations or for organisms with unique ecologiccharacteristics.
➤ Sequence-discrete populations could be given candidate species namesuntil appropriate isolates with ecologically relevant phenotypic propertiesare characterized.
➤ The mechanisms maintaining species, and perhaps more importantly, therelative importance of the mechanisms for different organisms and habi-tats, are not understood and demand further study.
FEATURE ARTICLE
Microbe—Volume 9, Number 3, 2014 • 111

and 100% genome average nucleotide identity(ANI).
This range depends on the age of the popula-tion, with younger populations showing lowersequence diversity. Those belonging to a partic-ular population show signifıcantly less geneticidentity to other co-occurring populations, typi-cally less than 80 – 85% ANI (genetic discontinu-ity; Fig. 1). Members of such a population alsotend to show similar abundances among them-selves, based on how many metagenomic readsmap on each genome sequence, indicating thatthey are ecologically homogeneous. In contrast,members of different populations, even closelyrelated ones, typically show different abun-dances, indicating that they are ecologically dif-ferentiated.
Within connected habitats having similar con-ditions, the same sequence-discrete populationsare found. In other words, when populationsare being dispersed between similar habitats,those populations are likely to remain indistin-guishable. These principles are based in part onour analyses of populations within fıve freshwaterlakes in the Southeastern United States thatconnect with one another via the ChattahoocheeRiver (Fig. 2).
Our fındings suggest that these microbial pop-ulations are not ephemeral, clonal amplifıcationsof one or a few cells. Instead, they are long-livedentities that may encompass substantial geneticdiversity. Moreover, non-discrete populationsare rare, at least for the abundant members ofnatural communities that can be robustly as-sessed by metagenomics, and typically ephem-eral, as they are associated with regular environ-mental perturbations such as the mixing ofdistinct populations that are adapted to living indifferent depths in the sea caused by ocean up-welling.
More generally, natural microbial communi-ties are predominantly composed of sequence-discrete populations, with exceptions likely to befound within habitats that undergo frequent fluc-tuations or for organisms with unique ecologiccharacteristics. Identifying such exceptions willhelp us to better understand the ecological andmolecular mechanisms that drive the diversitypatterns of populations described above.
The mechanisms may involve genetic ex-change among members of a bacterial popula-tion that keeps them consistent, analogous tosex in higher eukaryotes. On the other hand, eco-
logical coherence, in which different organismsoccupy the same niche, coupled with populationselective sweeps when a signifıcant genomic in-novation takes place, may drive population cohe-siveness. Further analysis of sequence-discretepopulations will lead to a fuller understandingof how discrete bacterial populations are main-tained, interact, and evolve within communities.In any case, such sequence-discrete populationsare important units within natural microbialcommunities.
Comparing Metagenome andConventional Species Definitions
The sequence-discrete populations identifıed bymetagenomics partly overlap with those encom-passed by the conventional species defınition.For instance, most— but not all—named bacte-rial species encompass organisms that show 95%or higher ANI among themselves (Fig. 3). Thislevel of relatedness contains the greatest diversityfor the oldest populations recovered in meta-genomes. Hence, the standard defınition usedfor defıning species encompasses the sequence-discrete populations. However, the latter tendto show lower genetic intradiversity than do con-ventionally named bacterial species.
Analysis of sequence-discrete populationshelps us to better recognize several limitationsin the conventional approach to defıning bac-terial species. Members of a population tend toshow smaller gene-content differences amongthemselves—typically, less than 5% of their totalgenes— compared to named species such as E.coli. This trend is consistent with the idea that theformer represent more ecologically and geneti-cally uniform clusters than those encompassed bythe current species defınition.
In contrast, several named species include or-ganisms isolated from, and hence adapted to liv-ing in, different habitats or hosts. For instance,the depth-stratifıed photosynthetic Procholoroc-cus marinus (Cyanobacteria) or the ammonia-oxidizing Marine Group I Thaumarchaeota sp.(Archaea) likely perform the same metabolismat every depth they inhabit, based on the genecontent recovered in the corresponding meta-genomes. However, their populations are se-quence-discrete, and hence not interchangeable,at different depths due to genomic adaptations tolight intensities and hydrostatic pressures thatthey encounter at their preferred depths.
FEATURE ARTICLE
112 • Microbe—Volume 9, Number 3, 2014
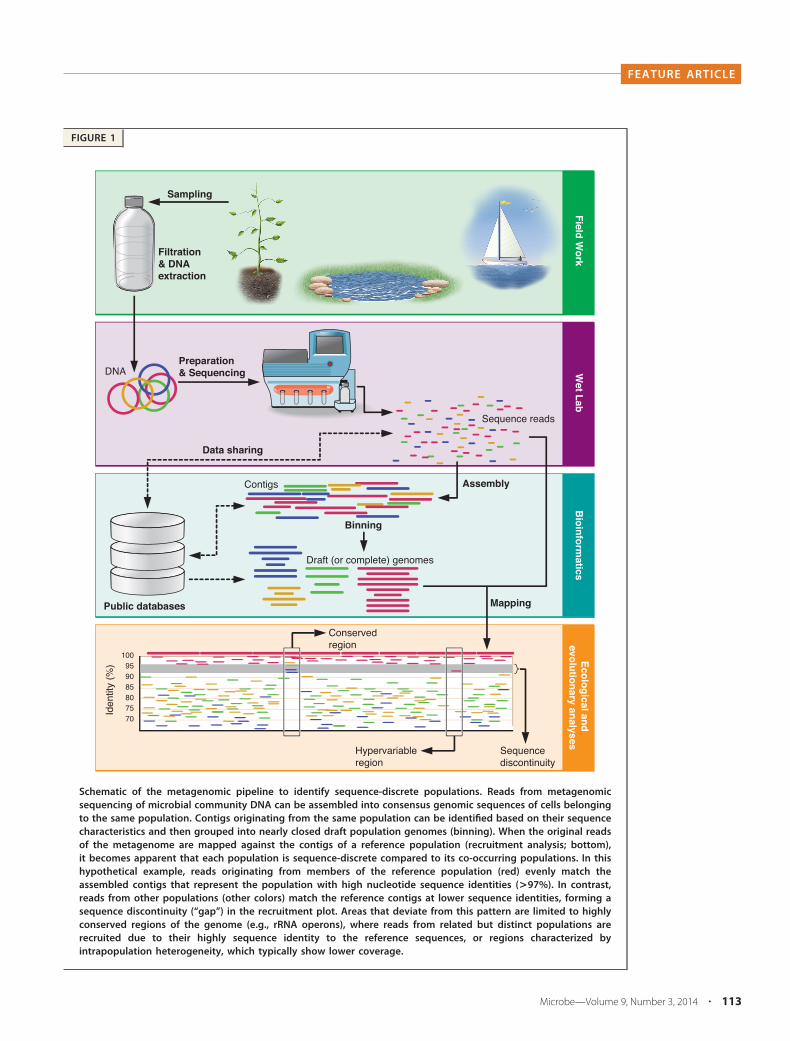
FIGURE 1
Schematic of the metagenomic pipeline to identify sequence-discrete populations. Reads from metagenomicsequencing of microbial community DNA can be assembled into consensus genomic sequences of cells belongingto the same population. Contigs originating from the same population can be identified based on their sequencecharacteristics and then grouped into nearly closed draft population genomes (binning). When the original readsof the metagenome are mapped against the contigs of a reference population (recruitment analysis; bottom),it becomes apparent that each population is sequence-discrete compared to its co-occurring populations. In thishypothetical example, reads originating from members of the reference population (red) evenly match theassembled contigs that represent the population with high nucleotide sequence identities (>97%). In contrast,reads from other populations (other colors) match the reference contigs at lower sequence identities, forming asequence discontinuity (“gap”) in the recruitment plot. Areas that deviate from this pattern are limited to highlyconserved regions of the genome (e.g., rRNA operons), where reads from related but distinct populations arerecruited due to their highly sequence identity to the reference sequences, or regions characterized byintrapopulation heterogeneity, which typically show lower coverage.
FEATURE ARTICLE
Microbe—Volume 9, Number 3, 2014 • 113
Similarly, several pathogens encode the samepathogenicity factors and cause similar symp-toms in humans or animals. However, thesefactors are encoded within different genomicbackgrounds, as is the case for several lineages ofE. coli.
From a taxonomic perspective, these sub-populations of the marine species, or the sub-populations of the animal pathogens, shouldbe assignable to the same species because they
are characterized by the same phenotypic ormetabolic properties that matter to us. From abacterial perspective, however, they are not inter-changeable and each occupies different ecologi-cal niches. These fındings argue for adopting amore ecological way of defıning bacterial speciesthan our current system allows, while also sug-gesting that modern culture-independent analyt-ical techniques may provide a better way of de-scribing species.
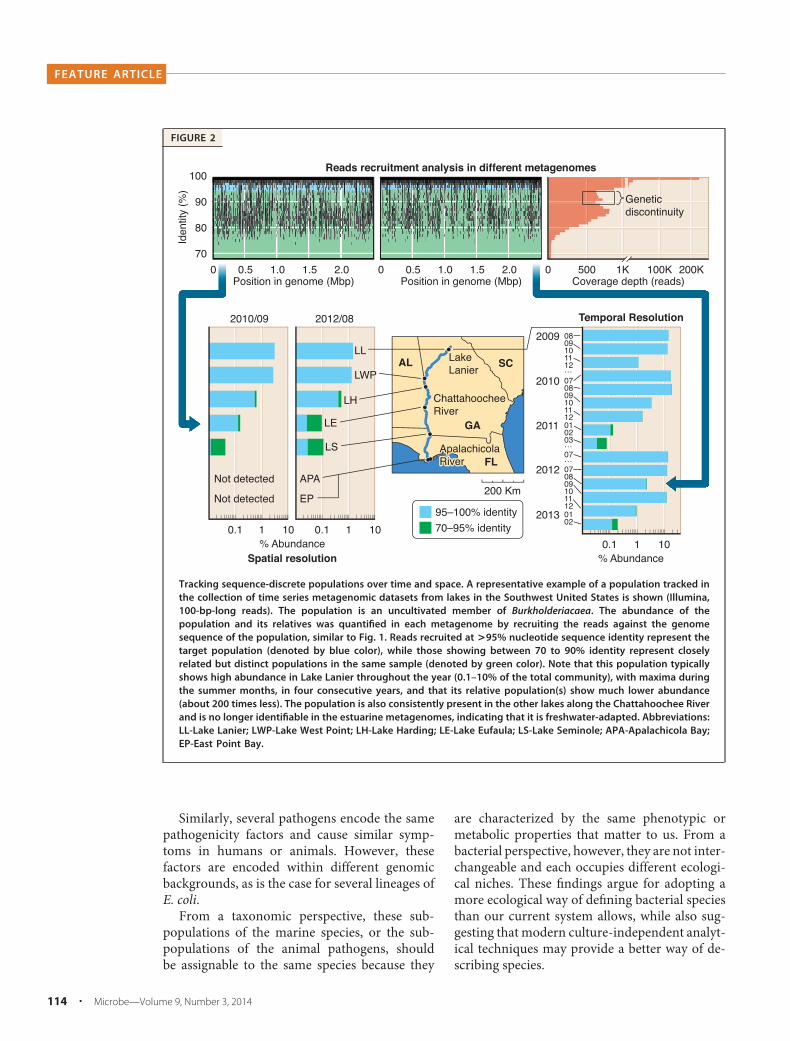
FIGURE 2
Tracking sequence-discrete populations over time and space. A representative example of a population tracked inthe collection of time series metagenomic datasets from lakes in the Southwest United States is shown (Illumina,100-bp-long reads). The population is an uncultivated member of Burkholderiacaea. The abundance of thepopulation and its relatives was quantified in each metagenome by recruiting the reads against the genomesequence of the population, similar to Fig. 1. Reads recruited at >95% nucleotide sequence identity represent thetarget population (denoted by blue color), while those showing between 70 to 90% identity represent closelyrelated but distinct populations in the same sample (denoted by green color). Note that this population typicallyshows high abundance in Lake Lanier throughout the year (0.1–10% of the total community), with maxima duringthe summer months, in four consecutive years, and that its relative population(s) show much lower abundance(about 200 times less). The population is also consistently present in the other lakes along the Chattahoochee Riverand is no longer identifiable in the estuarine metagenomes, indicating that it is freshwater-adapted. Abbreviations:LL-Lake Lanier; LWP-Lake West Point; LH-Lake Harding; LE-Lake Eufaula; LS-Lake Seminole; APA-Apalachicola Bay;EP-East Point Bay.
FEATURE ARTICLE
114 • Microbe—Volume 9, Number 3, 2014
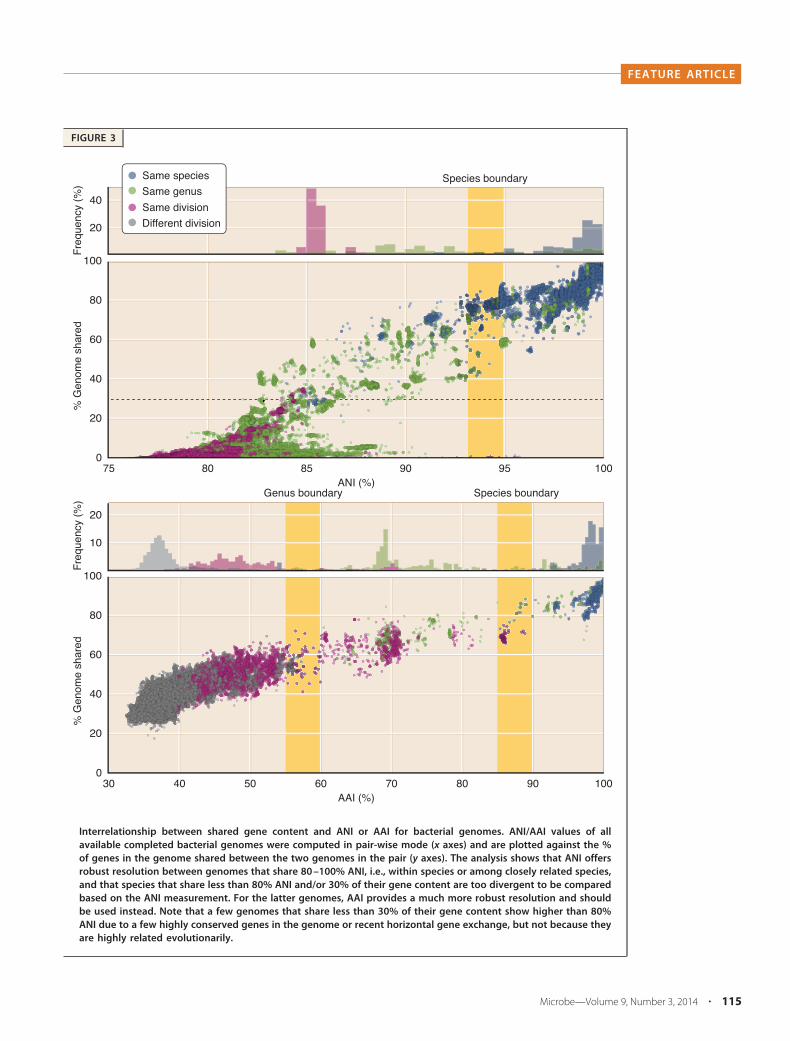
FIGURE 3
Interrelationship between shared gene content and ANI or AAI for bacterial genomes. ANI/AAI values of allavailable completed bacterial genomes were computed in pair-wise mode (x axes) and are plotted against the %of genes in the genome shared between the two genomes in the pair (y axes). The analysis shows that ANI offersrobust resolution between genomes that share 80 –100% ANI, i.e., within species or among closely related species,and that species that share less than 80% ANI and/or 30% of their gene content are too divergent to be comparedbased on the ANI measurement. For the latter genomes, AAI provides a much more robust resolution and shouldbe used instead. Note that a few genomes that share less than 30% of their gene content show higher than 80%ANI due to a few highly conserved genes in the genome or recent horizontal gene exchange, but not because theyare highly related evolutionarily.
FEATURE ARTICLE
Microbe—Volume 9, Number 3, 2014 • 115

Moving Forward: How To TaxonomicallyDescribe the Uncultivated Majority
Describing a new species depends in part on hav-ing a diagnostic phenotype based on traditionalbiochemical and physiological laboratory tech-niques as well as showing adequate geneticdistinctiveness. Such diagnostic phenotypes typ-ically are not available for sequence-discrete pop-ulations of uncultivated organisms.
The taxonomic status Candidatus provides away around this hurdle. Sequence-discrete popu-lations could be given candidate species namesuntil appropriate isolates with ecologically rele-vant phenotypic properties are characterized.Describing candidate species should be relativelyeasy for most of the sequence-discrete popula-tions because they can be identifıed and tracked
based on sequence data, which then can providemeans for developing probes with which to ana-lyze cell morphology and other characteristics.
Moreover, single-cell genomic techniquescould complement, perhaps even substitute for,shotgun metagenomic efforts because they canrecover the complete, or almost complete, ge-nome sequence of microorganisms under study.The Candidatus species is perhaps the only prag-matic approach for describing bacterial diversityin nature. If applied systematically to unculti-vated microorganisms, it also would greatly facil-itate communication among scientists.
Metagenomics data can reveal importantgene-content differences or genomic adaptationsamong sequence-discrete populations. Thesedifferences could account for important pheno-typic differences in situ. For instance, differential
AUTHOR PROFILE
Konstantinidis: Metagenomics and Environmental Microbiology amid Spear FishingKonstantinos (Kostas) Konstantinidis, 37, an assistant professorat Georgia Institute of Technology in Atlanta, studies thebehavior of environmental microorganisms, including in theocean, soil, or freshwater ecosystems, and the human gut. Amajor objective of his research involves developing culture-independent approaches to studying microbial communitiesas a means for distinguishing dangerous bacteria from theirinnocuous counterparts.
“Whether in soils, waters, deep subsurface environments,or in the atmosphere, microorganisms are affecting, if notcontrolling, the biogeochemical cycles that sustain life onEarth, but we don’t fully understand how,” he says. “Ourincomplete understanding of the microbial world is attrib-uted, at least in part, to the fact that the great majority ofmicroorganisms resists cultivation in the laboratory, andhence, cannot be studied efficiently.”
Konstantinidis grew up on the Greek island Andros, a2-hour ferry ride from Athens. He is the eldest of threechildren, with a younger brother and sister. His father, Theo-doros, an agronomist, retired three years ago and inspiredhis son to study agriculture in college. His mother, Sophia,a homemaker, died in 2002. “She taught me to strive forexcellence, to be persistent and patient in my work, and befair and honest to people, among many other things,” he says.“She influenced my character and work ethics more thananybody else.”
Konstantinidis left Andros for Thessaloniki, Greece’s secondlargest city, to attend Aristotle University. After completinghis undergraduate degree in agricultural sciences in 1999, he
moved to Michigan State University (MSU) in East Lansing tostudy crop and soil sciences, completing his Ph.D. in 2004. Hethen moved to the Massachusetts Institute of Technology inCambridge to do metagenomics research on deep-sea micro-bial communities. “Back in 2005, metagenomics was a verynew field and very few labs had engaged in it,” he says. “I amglad that I continued working on the environmental side ofmicrobiology, and I believe there is a big need for moreenvironmental microbiologists and biotechnologists.” After-wards, he went back to Greece to complete his militaryservice, before returning to the United States in 2007 andbeginning his tenure at Georgia Tech.
He is married to Kyriaki Kalaitzidou, a mechanical engineerwho is also at Georgia Tech. They met at MSU in 2001, he says,“So, Michigan State and East Lansing mean a lot to us.” They,including their three-year old son, Theodoros, “spend at leasta month each year in Greece so we get to enjoy the Greekfood and the more relaxing lifestyle there, as well as to see ourfamilies,” Konstantinidis says.
He devotes much of his free time to playing with Theo-doros. However, once his son is in bed, he scours the news“especially from my homeland, which is having some hardtimes recently, but will hopefully emerge stronger from therecession.” During his summer visits to Greece, he indulges hispassion for fishing in the “most ecologically friendly way,spear fishing with snorkel, no scuba,” he says.
Marlene CimonsMarlene Cimons lives and writes in Bethesda, Md.
FEATURE ARTICLE
116 • Microbe—Volume 9, Number 3, 2014

usage of amino acids in proteins to cope withhydrostatic pressure differences account, at leastin part, for the ability of distinct Marine Group IThaumarchaeota populations to occupy differentdepths in the oceans. Such differences are im-possible to reproduce in the laboratory based ontraditional methods or phenotypic assays.
Similarly, environmentally adapted E. colistrains possess ecologically important gene-content differences compared to their entericcounterparts. Yet, these organisms are indistin-guishable based on traditional phenotypic tests,apparently because those tests do not targetecologically appropriate genes and pathways. Insuch cases, the differences revealed by genomicsand metagenomics can be taken as adequate“phenotypic” differences for delineating speciesand guiding the design of discriminative pheno-typic tests.
As culture-independent transcriptomics andproteomics techniques are further refıned, theymay better help in assessing the activity of naturalmicrobial populations and, thus, in defıningspecies- or population-diagnostic signatures. Inour experience, transcriptomics and proteomicsdata typically corroborate population-specifıcsignatures revealed by metagenomics data.
Conclusions, Recommendations, Challenges
Natural microbial communities are predomi-nantly composed of sequence-discrete popula-tions that possess attributes expected for species,which contrasts with a genetic continuum ob-served between several named species. The dis-crepancy is presumably attributable to biases in-troduced by cultivation and human-centeredways of analyzing diversity. Therefore, Bacteriaand Archaea appear to form discrete biologicalunits, similar to eukaryotes, and these units arepartly encompassed by the current defınition ofspecies. Omics data can help to further refıne thespecies defınition.
Metagenomic fragment recruitment and ANIprovide a reliable means for assessing sequence-discrete populations and determining the levelof intrapopulation genetic diversity. For recruit-ment plots, it is important to use a genome se-quence or assembled contig from the same pop-ulation or a highly related population. ANI canalso discriminate between closely related popu-lations (sharing at least 70 –75% ANI), offershigher resolution than 16S rRNA gene or multi-
locus sequence analysis, and is less error-proneand more portable than is the DNA-DNA hy-bridization method. For more distantly relatedpopulations, the average amino acid identity(AAI) should be used because resolution is pro-gressively lost at the nucleotide level.
New species descriptions should be accompa-nied by their ANI and/or AAI relatedness values,and population relative abundance and persis-tence over time in situ, assessed by metagenomicsor other culture-independent technique. TheCandidatus species description provides a reli-able means to identify and characterize popula-tions with no sequenced representatives, and theANI values of such populations can be reliablycomputed based on assembled genome se-quences from metagenomics or single-cell tech-niques. To facilitate such efforts, we have devel-oped online implementations of the ANI andfragment recruitment tools (available throughhttp://enve-omics.gatech.edu/).
Meanwhile, the mechanisms maintaining se-quence-discrete populations (and species), andperhaps more importantly, the relative impor-tance of those mechanisms for different organ-isms and habitats, are not understood and de-mand further study. To this end, characterizingisolates of several sequence-discrete populationscould help to guide further use of omics data andhow to better interpret traditional phenotypicmeasurements.Luis M. Rodriguez-R is a Ph.D. candidate at the Center forBioinformatics and Computational Genomics, and School ofBiology, and Konstantinos T. Konstantinidis is the Carlton S.Wilder Assistant Professor at the Center for Bioinformatics andComputational Genomics, and School of Biology, and theSchool of Civil and Environmental Engineering, GeorgiaInstitute of Technology, Atlanta, Ga.
Acknowledgments
K.T.K. is indebted to Jim Tiedje for useful discussionsrelated to the species issue and Ed DeLong for exposinghim to the science of metagenomics. Our work is sup-ported in part by the U.S. DOE Offıce of Science, Biolog-ical and Environmental Research Division (BER),Genomic Science Program, Awards No. DE-SC0006662and DE-SC0004601, and by the U. S. National ScienceFoundation under Award No 1241046.
Suggested Reading
Fraser, C., W. P. Hanage, and B. G. Spratt. 2007. Recom-bination and the nature of bacterial speciation. Sci-ence. 315:476 – 480.
FEATURE ARTICLE
Microbe—Volume 9, Number 3, 2014 • 117

Gevers, D., F. M. Cohan, J. G. Lawrence, B. G. Spratt, T.Coenye, E. J. Feil, E. Stackebrandt, Y. Van de Peer,P. Vandamme, F. L. Thompson, and J. Swings. 2005.Opinion: re-evaluating prokaryotic species. NatureRev. Microbiol. 3:733–739.
Goris, J., K. T. Konstantinidis, T. Coenye, P. Van-damme, and J. M. Tiedje. 2007. DNA-DNA hybrid-ization values and their relation to whole genome se-quence. Int. J. Syst. Evol. Microbiol. 57:81–91.
Konstantinidis, K. T., J. Braff, D. M. Karl, and E. F.Delong. 2009. Comparative metagenomic analysisof a microbial community from 4000 m at StationALOHA in the North Pacifıc Subtropical Gyre. Appl.Environ. Microbiol. 75:5345–5355.
Konstantinidis, K. T., and E. F. DeLong. 2008. Genomicpatterns of recombination, clonal divergence and en-vironment in marine microbial populations. ISME J.10:1052–1065.
Konstantinidis, K. T., and E. Stackebrandt. 2013. Defın-ing taxonomic ranks, p. 29 –57. In The Prokaryotes.M. Dworkin et al. (ed.). Springer-Verlag. New York.
Luo, C., and K. T. Konstantinidis. 2011. Phosphorus-
related gene content is similar in Prochlorococcus pop-ulations from the North Pacifıc and North AtlanticOceans. Proc. Natl. Acad. Sci. USA 108:E62–3.
Luo, C., S. T. Walk, D. M. Gordon, M. Feldgarden, J. M.Tiedje, and K. T. Konstantinidis. 2011. Genome se-quencing of environmental Escherichia coli expandsunderstanding of the ecology and speciation of themodel bacterial species. Proc. Natl. Acad. Sci. USA108:7200 –7205.
Oh, S., A. Caro-Quintero, D. Tsementzi, N. Deleon-Rodriguez, C. Luo, R. Poretsky, and K. T. Konstan-tinidis. 2011. Metagenomic insights into the evolu-tion, function, and complexity of the planktonicmicrobial community of Lake Lanier, a temperatefreshwater ecosystem. Appl. Environ. Microbiol. 77:6000 – 6011.
Shapiro, B. J., J. Friedman, O. X. Cordero, S. P. Pre-heim, S. C. Timberlake, G. Szabo, M. F. Polz, andE. J. Alm. 2012. Population genomics of early eventsin the ecological differentiation of bacteria. Science.336:48 –51.
1752 N Street NW | Washington, DC 20036 | 202-942-9323 | [email protected]
Freely available: bit.ly/ASMFAQ
!"#$"%"&'%()&**+%,'#$%*--%$./&"%0."&%%1$''%2'#)&.3/%4'(*5$)'(6%Expert advice from the American Academy of
Microbiology brought to your classroom.
In!uenza
Topics include:
Adult Vaccina"on West Nile Virus Oil Spills
E.coli
FEATURE ARTICLE
118 • Microbe—Volume 9, Number 3, 2014


















