
By Dr. Walaa Bayoumie El Gazzar
23
Biochemistry of muscle By Dr. Walaa Bayoumie El Gazzar
Transcript of By Dr. Walaa Bayoumie El Gazzar

Biochemistry of muscle
ByDr. Walaa Bayoumie El Gazzar

•Skeletal muscles are composed
of bundles of muscle fibers
•Muscle fibers are long cylindricalcells containing several nuclei andbounded by plasma membrane
called sarcolemma
•Muscle fibers are composed of
many myofibrils immersed ina cytosol that is rich in glycogen ,ATP, creatine phosphate andglycolytic enzymes .
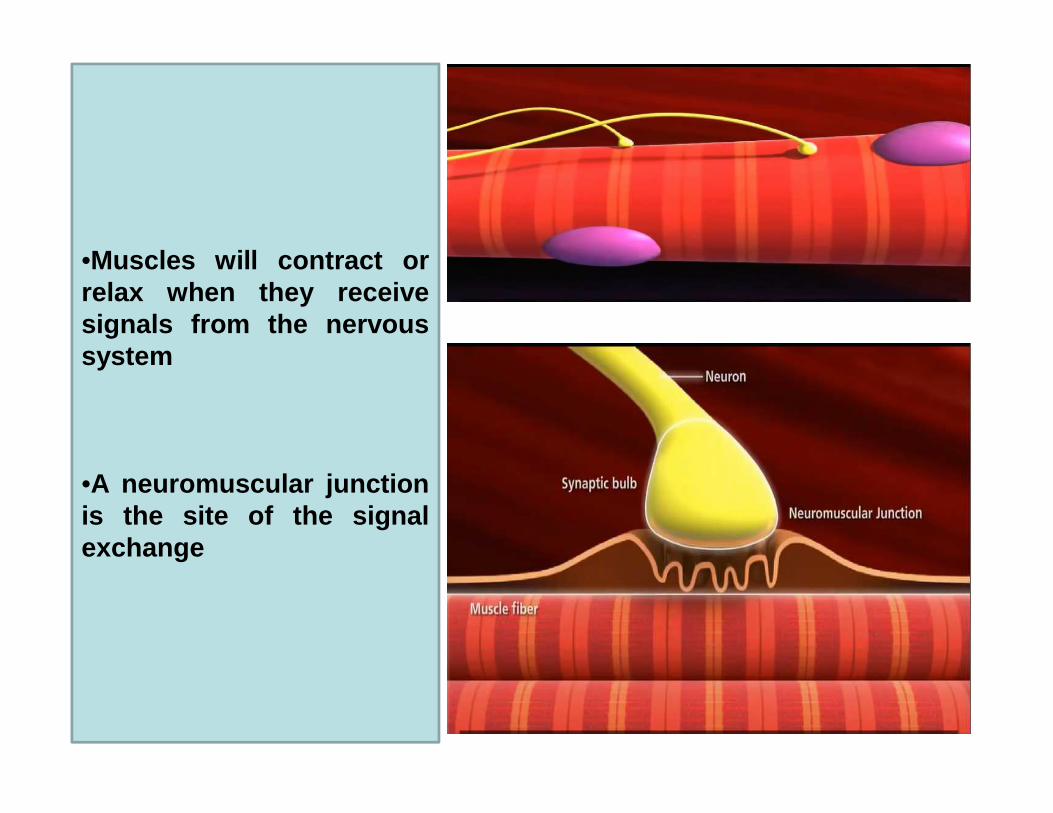
•Muscles will contract orrelax when they receivesignals from the nervoussystem
•A neuromuscular junctionis the site of the signalexchange

•The sarcoplasmic reticulum (SR) is smooth ER found in myocytes .Itstores Ca ions and pumps them out into the sarcoplasm when themuscle fiber is stimulated. The SR plays a major role in excitation –contraction coupling.
•T-tubules are deep invagination of the sarcolemma (ms. Fibe rplasma mem .) permit the conduction of electrical impulses.
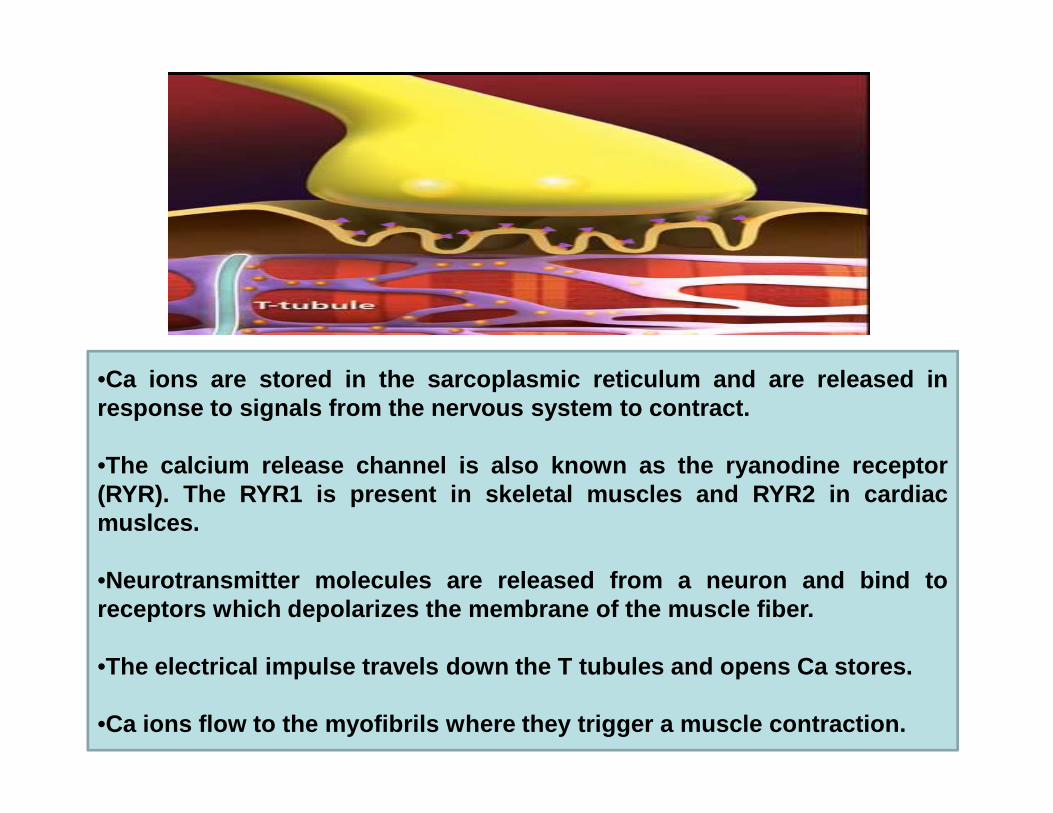
•Ca ions are stored in the sarcoplasmic reticulum and are rele ased inresponse to signals from the nervous system to contract .response to signals from the nervous system to contract .
•The calcium release channel is also known as the ryanodine re ceptor(RYR). The RYR1 is present in skeletal muscles and RYR2 in car diacmuslces.
•Neurotransmitter molecules are released from a neuron and b ind toreceptors which depolarizes the membrane of the muscle fiber .
•The electrical impulse travels down the T tubules and opens C a stores.
•Ca ions flow to the myofibrils where they trigger a muscle con traction.

Diagram of the relationships among the sarcolemma (p lasma membrane), a T tubule, and two cisternae of the SR of skeletal muscle (not to scale).
•The T tubule extends inward from the sarcolemma. A wave of dep olarization,initiated by a nerve impulse, is transmitted from the sarcol emma down the T tubule.It is then conveyed to the Ca 2+ release channel (RYR), perhaps by interactionbetween it and the dihydropyridine receptor (slow Ca 2+ voltage channel), which areshown in close proximity.
•Release of Ca 2+ from the Ca 2+ release channel into the cytosol initiates contraction.Subsequently, Ca 2+ is pumped back into the cisternae of the SR by the Ca 2+ ATPase(Ca2+ pump) and stored there, in part bound to calsequestrin.

Mutations in the Gene Encoding the Ca2+ Release Channel Are One Cause of Human Malignant
Hyperthermia
Some genetically predisposed patients experience a severe reaction,designated malignant hyperthermia (MH), on exposure to cer tainanesthetics (eg, halothane) and depolarizing skeletal musc le relaxants (eg,succinylcholine). The reaction consists primarily of rigi dity of skeletalmuscles, hypermetabolism, and high fever. A high cytosolic concentrationof Ca2+ in skeletal muscle is a major factor in its causation.
The mutation affects the function of the channel in that it op ens moreeasily and remains open longer; the net result is massive rel ease of Ca2+into the cytosol, ultimately causing sustained muscle cont raction.
Susceptibility is often inherited from a person's parents i n an autosomaldominant manner.

•Diagnosis is based on symptoms in the appropriatesituation. Family members may be tested to see ifthey are susceptible by muscle biopsy or genetictesting .
•Appropriate treatment is to stop the anesthetic andadminister the drug dantrolene intravenously.Dantrolene is a skeletal muscle relaxant that acts toDantrolene is a skeletal muscle relaxant that acts toinhibit release of Ca2+ from the SR into the cytosol,thus preventing the increase of cytosolic Ca2+ foundin MH.
•Males are more often affected than females. The riskof death with proper treatment is about 5% whilewithout it is around 75%.

THE DYSTROPHIN-GLYCOPROTEIN COMPLEX IN SKELETAL MUSCLE
•The dystrophin-glycoprotein complex (DGC) is a multi-subu nitcomplex within and across the membranes of cardiac and skele talmuscle cells as well as vascular smooth muscle cells.
•The complex mediates interactions between the cytoskeleto n,membrane, and the extracellular matrix. The complex is comp osedof up to 15 different proteins. The major intracellular prot ein is thelarge dystrophin protein.
•Dystrophin protein is integral to the structural stability of themyofiber. Without dystrophin, muscles are susceptible tomechanical injury and undergo repeated cycles of necrosis a ndregeneration. Ultimately, regenerative capabilities are exhaustedor inactivated.

•The dystrophin gene (one of the largest human genesdescribed to date) occupies almost 2% of the Xchromosome and nearly 0.05% of the entire genome .chromosome and nearly 0.05% of the entire genome .(Gene symbol: DMD).



The clinical significance of the dystrophinprotein relates to the fact that mutations inthe DMD gene are the cause of variousmuscular dystrophies: Duchenne musculardystrophy (DMD), Becker musculardystrophy (DMD), Becker musculardystrophy (BMD), and X-linked dilatedcardiomyopathy (XLDCM).

In DMD , a genetic mutation causes Dystrophin to be extremely short lackingthe dystroglycan binding end. Because of this every time the musclecontracts , small rips appear in the mem.

These rips allow diffusion of various molecules into and out of the myocyteas Ca ions.

Ca ions found outside the myocytes flow in through these smal l rips andactivate Ca dependent intracelluar enzymes that breakdown proteinscalled proteases


Normally with normal ca levels these proteases only breake d ownold and damaged proteins, however in DMD extremely high calevel activates too many of these proteases which breake dow nimportant functional proteins as well, this kills the myocy te

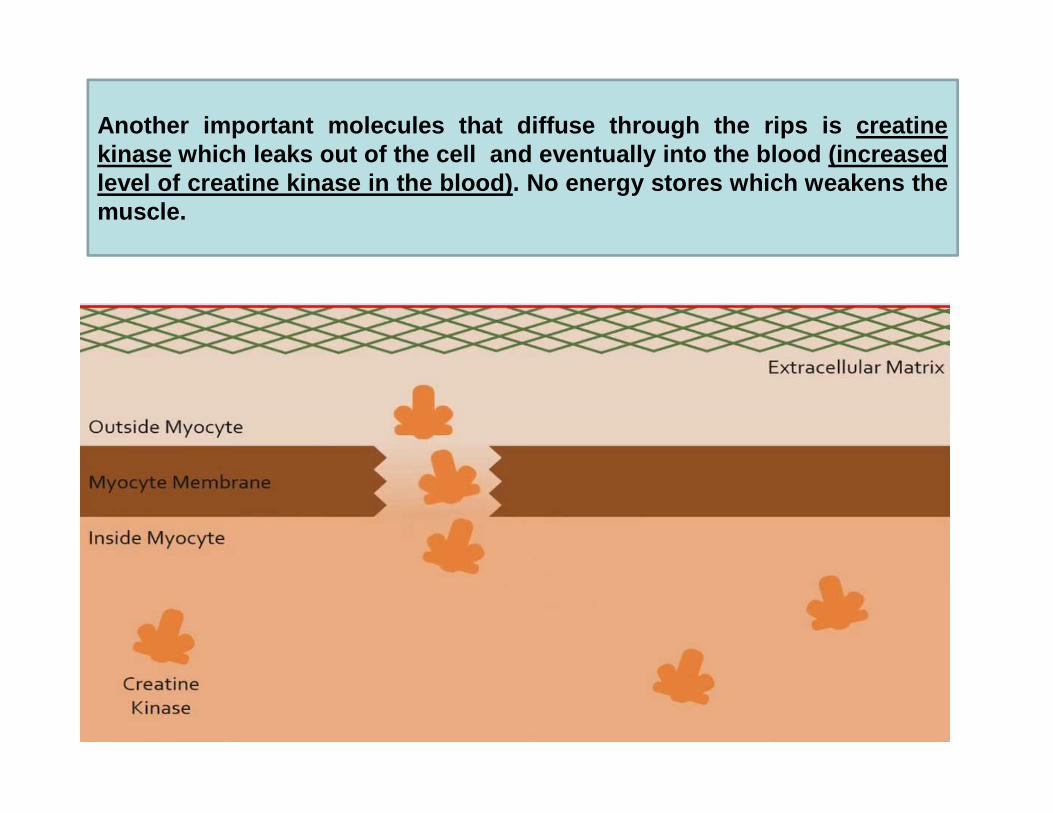
Another important molecules that diffuse through the rips i s creatinekinase which leaks out of the cell and eventually into the blood (inc reasedlevel of creatine kinase in the blood) . No energy stores which weakens themuscle.

•Duchenne muscular dystrophy (DMD) represents the most seve re formof all muscular dystrophies .
•DMD is a rapidly progressing, fatal form of muscular dystrop hy.Symptoms of DMD usually begin to appear within the first 6-mo nths oflife but can also be seen at birth in some afflicted infants. T hesymptoms of DMD are characterized by progressive muscledegeneration and weakness, eventually resulting in death. The averagelife span for DMD patients is around 25 years of age.
Becker muscular dystrophy (BMD) represents a milder form of musculardystrophy caused by mutations in the dystrophin gene. Whereas in thecase of Duchenne muscular dystrophy where no functional dys trophinprotein is made, BMD is associated with some functional prot ein . Forthis later reason the symptoms of BMD are much less severe, be ginmanifesting later in life than for DMD, and the disorder is no t lethal as inthe case of DMD.
The vast majority of the deletions found to cause D MDresult in frameshift mutations .

Make the cell skip the segment of the gene that contains an ear ly stopcodon , a technique known as exon skipping, resulting in a fun ctionaldystrophin ptn with a slightly shorter central rode



















