BRAF/NRAS Wild-Type Melanomas Have a High Mutation ... · In one case, matched blood was not...
11
Human Cancer Biology BRAF/NRAS Wild-Type Melanomas Have a High Mutation Load Correlating with Histologic and Molecular Signatures of UV Damage Victoria J. Mar 1,10,13 , Stephen Q. Wong 2 , Jason Li 4 , Richard A. Scolyer 14,15,16 , Catriona McLean 10,11 , Anthony T. Papenfuss 7,8 , Richard W. Tothill 4 , Hojabr Kakavand 14,15,16 , Graham J. Mann 15 , John F. Thompson 14,15,16 , Andreas Behren 12 , Jonathan S. Cebon 12 , Rory Wolfe 13 , John W. Kelly 10 , Alexander Dobrovic 2,6 , and Grant A. McArthur 1,3,5,6,9 Abstract Purpose: The mutation load in melanoma is generally high compared with other tumor types due to extensive UV damage. Translation of exome sequencing data into clinically relevant information is therefore challenging. This study sought to characterize mutations identified in primary cutaneous melanomas and correlate these with clinicopathologic features. Experimental Design: DNA was extracted from 34 fresh-frozen primary cutaneous melanomas and matched peripheral blood. Tumor histopathology was reviewed by two dermatopathologists. Exome sequencing was conducted and mutation rates were correlated with age, sex, tumor site, and histopathologic variables. Differences in mutations between categories of solar elastosis, pigmentation, and BRAF/NRAS mutational status were investigated. Results: The average mutation rate was 12 per megabase, similar to published results in metastases. The average mutation rate in severely sun damaged (SSD) skin was 21 per Mb compared with 3.8 per Mb in non- SSD skin (P ¼ 0.001). BRAF/NRAS wild-type (WT) tumors had a higher average mutation rate compared with BRAF/NRAS–mutant tumors (27 vs. 5.6 mutations per Mb; P ¼ 0.0001). Tandem CC>TT/GG>AA mutations comprised 70% of all dinucleotide substitutions and were more common in tumors arising in SSD skin (P ¼ 0.0008) and in BRAF/NRAS WT tumors (P ¼ 0.0007). Targetable and potentially targetable mutations in WT tumors, including NF1, KIT, and NOTCH1, were spread over various signaling pathways. Conclusion: Melanomas arising in SSD skin have higher mutation loads and contain a spectrum of molecular subtypes compared with BRAF- and NRAS-mutant tumors indicating multigene screening approaches and combination therapies may be required for management of these patients. Clin Cancer Res; 19(17); 4589–98. Ó2013 AACR. Introduction The development of BRAF inhibitors, vemurafenib and dabrafenib, marked a turning point in the treatment and prognosis of patients with advanced stage metastatic mel- anoma. The BRAF oncogene is mutated in up to 50% of cutaneous melanomas and its clinical and histologic asso- ciations have been well characterized (1, 2). BRAF V600E mutations occur more commonly in patients under the age of 50 years with higher nevus counts, and are more common in melanomas arising on the trunk in intermittently sun- exposed skin (1, 3). They are also more prevalent in super- ficial spreading melanomas (SSM) as compared with other melanoma subtypes and have characteristic histopatholog- ic features (1, 4, 5). In contrast, BRAF V600K mutations are Authors' Affiliations: 1 Molecular Oncology Laboratory, Oncogenic Sig- naling and Growth Control Program; 2 Molecular Pathology Research and Development Laboratory; 3 Translational Research Laboratory, Can- cer Therapeutics Program; 4 Bioinformatics Core Facility, Peter Mac- Callum Cancer Centre, East Melbourne; 5 Sir Peter MacCallum Depart- ment of Oncology, Departments of 6 Pathology and 7 Mathematics and Statistics, The University of Melbourne; 8 Bioinformatics Division, The Walter and Eliza Hall Institute for Medical Research, Parkville; 9 Depart- ment of Medicine, St. Vincent's Hospital, The University of Melbourne, Fitzroy; 10 Victorian Melanoma Service, 11 Department of Anatomical Pathology, Alfred Hospital, Prahran; 12 Ludwig Institute for Cancer Research, Austin Hospital, Heidelberg; 13 Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, Victoria; 14 Royal Prince Alfred Hospital, Camperdown; 15 Melanoma Institute Australia; and 16 The University of Sydney, Sydney, New South Wales, Australia Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). V. Mar and S.Q. Wong are co-first authors. Corresponding Author: Grant McArthur, Research Division, Peter Mac- Callum Cancer Centre, Locked Bag 1, A'Beckett St, East Melbourne, VIC 8006, Australia. Phone: 61-3-9656-1954; Fax: 61-3-9656-3717; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-13-0398 Ó2013 American Association for Cancer Research. Clinical Cancer Research www.aacrjournals.org 4589 on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398
Transcript of BRAF/NRAS Wild-Type Melanomas Have a High Mutation ... · In one case, matched blood was not...

Human Cancer Biology
BRAF/NRAS Wild-Type Melanomas Have a High MutationLoadCorrelatingwith Histologic andMolecular Signatures ofUV Damage
Victoria J. Mar1,10,13, Stephen Q. Wong2, Jason Li4, Richard A. Scolyer14,15,16, Catriona McLean10,11,Anthony T. Papenfuss7,8, Richard W. Tothill4, Hojabr Kakavand14,15,16, Graham J. Mann15,John F. Thompson14,15,16, Andreas Behren12, Jonathan S. Cebon12, Rory Wolfe13, John W. Kelly10,Alexander Dobrovic2,6, and Grant A. McArthur1,3,5,6,9
AbstractPurpose: The mutation load in melanoma is generally high compared with other tumor types due to
extensiveUVdamage. Translation of exome sequencing data into clinically relevant information is therefore
challenging. This study sought to characterize mutations identified in primary cutaneous melanomas and
correlate these with clinicopathologic features.
Experimental Design: DNA was extracted from 34 fresh-frozen primary cutaneous melanomas and
matched peripheral blood. Tumor histopathology was reviewed by two dermatopathologists. Exome
sequencingwas conducted andmutation rateswere correlatedwith age, sex, tumor site, and histopathologic
variables. Differences in mutations between categories of solar elastosis, pigmentation, and BRAF/NRAS
mutational status were investigated.
Results: The average mutation rate was 12 per megabase, similar to published results in metastases. The
averagemutation rate in severely sun damaged (SSD) skinwas 21 perMb comparedwith 3.8 perMb in non-
SSD skin (P ¼ 0.001). BRAF/NRAS wild-type (WT) tumors had a higher average mutation rate compared
with BRAF/NRAS–mutant tumors (27 vs. 5.6 mutations per Mb; P ¼ 0.0001). Tandem CC>TT/GG>AAmutations comprised 70% of all dinucleotide substitutions and were more common in tumors arising in
SSD skin (P ¼ 0.0008) and in BRAF/NRASWT tumors (P ¼ 0.0007). Targetable and potentially targetable
mutations in WT tumors, includingNF1, KIT, andNOTCH1, were spread over various signaling pathways.
Conclusion: Melanomas arising in SSD skin have higher mutation loads and contain a spectrum of
molecular subtypes compared with BRAF- and NRAS-mutant tumors indicating multigene screening
approaches and combination therapies may be required for management of these patients. Clin Cancer
Res; 19(17); 4589–98. �2013 AACR.
IntroductionThe development of BRAF inhibitors, vemurafenib and
dabrafenib, marked a turning point in the treatment andprognosis of patients with advanced stage metastatic mel-anoma. The BRAF oncogene is mutated in up to 50% ofcutaneous melanomas and its clinical and histologic asso-ciations have been well characterized (1, 2). BRAF V600E
mutations occur more commonly in patients under the ageof 50yearswithhigher nevus counts, andaremore commonin melanomas arising on the trunk in intermittently sun-exposed skin (1, 3). They are also more prevalent in super-ficial spreading melanomas (SSM) as compared with othermelanoma subtypes and have characteristic histopatholog-ic features (1, 4, 5). In contrast, BRAF V600K mutations are
Authors' Affiliations: 1Molecular Oncology Laboratory, Oncogenic Sig-naling and Growth Control Program; 2Molecular Pathology Researchand Development Laboratory; 3Translational Research Laboratory, Can-cer Therapeutics Program; 4Bioinformatics Core Facility, Peter Mac-Callum Cancer Centre, East Melbourne; 5Sir Peter MacCallum Depart-ment of Oncology, Departments of 6Pathology and 7Mathematics andStatistics, The University of Melbourne; 8Bioinformatics Division, TheWalter and Eliza Hall Institute for Medical Research, Parkville; 9Depart-ment of Medicine, St. Vincent's Hospital, The University of Melbourne,Fitzroy; 10Victorian Melanoma Service, 11Department of AnatomicalPathology, Alfred Hospital, Prahran; 12Ludwig Institute for CancerResearch, Austin Hospital, Heidelberg; 13Department of Epidemiologyand Preventive Medicine, Monash University, Melbourne, Victoria;14Royal Prince Alfred Hospital, Camperdown; 15Melanoma Institute
Australia; and 16The University of Sydney, Sydney, New South Wales,Australia
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
V. Mar and S.Q. Wong are co-first authors.
Corresponding Author: Grant McArthur, Research Division, Peter Mac-Callum Cancer Centre, Locked Bag 1, A'Beckett St, East Melbourne, VIC8006, Australia. Phone: 61-3-9656-1954; Fax: 61-3-9656-3717; E-mail:[email protected]
doi: 10.1158/1078-0432.CCR-13-0398
�2013 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 4589
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

more common with increasing age and in melanomas onthe head and neck (6). BRAF-mutant melanomas are morelikely to metastasize to regional lymph nodes, comparedwith BRAFwild-type (WT) tumors, which more commonlymetastasize to non-nodal sites (4). Mutually exclusive ofBRAF, mutations in NRAS are found in approximately15% of cutaneous melanomas, which tend to be thicker,with a higher mitotic rate (7). Therapeutic targeting ofNRAS has to date been less successful than targeting ofBRAF (2, 8), though inhibitors targeting downstream ofNRAS show promise and are currently undergoing clinicaltrials (8). Tumors that are WT for BRAF and NRAS areless well characterized and are likely to be more hetero-geneous (4).
In recent years, exome sequencing of melanoma hasprovided valuable insights into the complexity of thistumor. The mutation load in melanoma is far greater thanother tumor types, mostly due to UV damage (9). Conse-quently, uncovering new therapeutic targets by sifting outdriver mutations responsible for tumor development andprogression from inconsequential passenger mutations is amajor challenge. Themajority of exome studies to date havefocused on sequencing data from cell lines derived frommetastatic melanomas (10–16). Few studies have includedsamples from primary melanomas (14, 15, 17) and nonehave correlated exome data with clinical and histologicvariables of the primary tumor.
Integration of molecular changes with clinical and path-ologic characteristics of the primary tumor may assist thetranslation of exome data to a clinically relevant under-standing of tumor biology. The aim of this study was toidentify mutations associated with primary cutaneous mel-anomas and correlate these with clinicopathologic features.
Materials and MethodsSelection of samples for discovery phase
Fresh-frozen tissue from primary cutaneous melanomaswas collected prospectively from melanoma clinics at theVictorianMelanoma Service, AlfredHospital (Prahran, VIC,Australia) and Peter MacCallum Cancer Centre (East Mel-bourne, VIC, Australia; n¼ 5) aswell as retrospectively fromthe Victorian Cancer Biobank (n ¼ 8) and MelanomaInstitute Australia Biospecimen Bank (n ¼ 21). Matchedpatient blood was also collected to distinguish somaticmutations from germline mutations. In one case, matchedblood was not available, therefore normal DNA wasextracted from dissected adjacent normal tissue (caseNM002). All patients gave informed consent and ethicsapproval was obtained from the Peter MacCallum CancerCentre Ethics Committee (11 of 25) for all human tissuesand clinicopathologic data used in this study. The histopa-thology of all cases was reviewed by two dermatopatholo-gists (C. McLean and R.A. Scolyer) and various histopath-ologic features were scored (detailed later). Thirty-twotumors were verified on hematoxylin and eosin stain ascontaining more than 80% of tumor. The remaining twosamples (NM002 and NM019) contained 50% to 60%tumor respectively and were included as the sensitivity ofmutation detection by next-generation sequencing remainshigh even for impure samples (18). Tumor and matchednormal DNA was extracted using the Qiagen Gentra Pure-gene Kit (Qiagen) according to themanufacturer’s protocol.Extracted DNA was quantified using a Qubit fluorometer(Invitrogen) and a Quant-iT dsDNA HS kit (Invitrogen).
Assessment of clinical and histologic variablesPatient information including date of birth, date of
surgery, gender, and site of tumor was collected. Histologicvariables such as Breslow thickness (mm), ulceration,mitotic rate (n/mm2), melanoma subtype, presence ofregression, lymphovascular invasion, neurotropism, andmicrosatellite lesionswere assessed.Melanoma subtypewasclassified according to the World Health Organization(WHO) criteria into nodular melanoma (NM), SSM, andlentigo maligna melanoma (LMM; ref. 19). Solar elastosiswas graded (0, none; 1, mild; 2, moderate; 3, severe)according to the amount of elastotic fibers in normal skinadjacent to the melanoma in the excision specimen (3).Histologic evidenceof solar damagewas alsodichotomized;tumors arising on non–severely sun damaged (non-SSD;solar elastosis scores of 0 or 1) and tumors arising on SSDskin (solar elastosis scores of 2 or 3). Pigmentation wasgraded according to the amount of pigment within constit-uent melanocytes (3). Tumor lymphocytic infiltration wasgraded 0–3 according to the density and distribution of thelymphocytic infiltrate within the dermal component of thetumor (20). Features of nesting, scatter, epidermal contour,and circumscription were also assessed (3).
Exome sequencingA schematic summary of how samples were sequenced
and analyzed is shown in Supplementary Fig. S1. One
Translational RelevanceTargeted therapy directed at the oncogene, BRAF, has
improved melanoma patient outcome in recent years.Targeted strategies forNRAS-mutant melanoma are cur-rently under investigation. Here, we show that BRAF/NRASwild-type (WT) tumors are a complex group,morecommonly arising in sun-exposed sites associated withsevere solar elastosis. Consistent with this, they have ahigh mutation load with a large proportion of C>Ttransitions as well as dinucleotide CC>TT transitionsspecific forUV-induceddamage. It is likely that anumberof genomic insults are required cumulatively for mela-noma progression in this group. Potentially targetablemutations in BRAF/NRAS WT melanomas are spreadover several different signaling pathways and thismay inthe future have implications for therapeutic approachesto patients harboring such tumors. Classification ofmelanoma into BRAF-mutant, NRAS-mutant, andhigh-mutation load groups may assist in identificationof patients more likely to respond to particular com-bined drug therapies.
Mar et al.
Clin Cancer Res; 19(17) September 1, 2013 Clinical Cancer Research4590
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

microgramofDNAwas sheared to approximately 200bp bysonication (Covaris). Exome enrichment was conductedusing the NimbleGen EZ Exome Library v2.0 Kit (n ¼25) or the Agilent SureSelect Human All Exon version 2Kit (n ¼ 9) according to the recommended protocols.Sequencing was carried out on an Illumina HiSeq2000instrument. Samples were loaded in an indexed pool of 3samples per lane, and an average coverage of 141� wasachieved across all samples. We did not observe any signif-icant differences in performance of the different exomecapture platforms. Library preparation and sequencinginformation for each sample is provided in SupplementaryTable S1.
Sequencing alignment and variant callingSequence readswere aligned to the humangenome (hg19
assembly) using the Burrows–Wheeler Aligner (BWA) pro-gram (21). Local realignment around indels and basequality score recalibration were conducted using theGenome Analysis Tool Kit (GATK; ref. 22) software, andduplicate reads removed using Picard (23). Single-nucleo-tide variants (SNV) and indels were identified using theGATKUnifiedGenotyper, Somatic Indel Detector (24), andMuTect (Broad Institute, Cambridge, MA; ref. 25). Variantswere annotated with information from Ensembl (26)Release 64 using the Ensembl Perl Application ProgramInterface including SNP Effect Predictor. Data with SNVsidentified in this cohort are provided in SupplementaryTable S2.
Candidate variant identificationVariants were first filtered for confident calls using a
quality score cutoff of�30 and a read depth of �20. Next,variants were filtered to include only somatic mutations,located in canonical transcripts [the most prevalent tran-script as detailed by the UniProt Knowledgebase (27)],with bidirectional read support, and mutations predictedto be potentially deleterious (mutations which potential-ly change the coding of a protein, i.e., nonsynonymous,splice site, indels, stop codon lost, and stop codon gainmutations). A list of potentially "actionable" mutations,predicted to have diagnostic, prognostic, or therapeuticimplications, was generated by comparing our list offiltered mutations to potential targets listed by Catalogueof Somatic Mutations in Cancer (COSMIC; ref. 28) andthe Genomics of Drug Sensitivity in Cancer Project(GDSC), Sanger Institute (Cambridgeshire, United King-dom; ref. 29).To assess the frequency of commonly mutated genes in
this cohort, a literature search was conducted to identifymutations validated as occurring in more than 10% ofmelanomas.
Pathway analysisFor each tumor, the number of mutations in eight
pathways, well described in melanoma (30), was assessed.Gene(s) listed in each pathway are described in Supple-mentary Table S3.
Validation of mutationsThe BRAF and NRAS mutation status of each tumor was
determined using high-resolution melting analysis (HRM)and Sanger sequencing as previously described (31).
Actionable mutations shown in Supplementary Table S4were validated using Sanger sequencing. PCR amplificationwas conducted on a Rotor-Gene Q (Qiagen) or a Light-Cycler 480 (Roche Diagnostics). Primer sequences andspecific PCR reaction conditions are available upon request.Following PCR amplification, a 1:10 dilution PCR productwas sequenced using the BigDye Terminator v3.1 CycleSequencing Kit (Applied Biosystems). The sequencing pro-ducts were purified with Agencourt CleanSEQ beads (Beck-man Coulter), followed by capillary electrophoresis on anABI 3730 DNA Sequencing instrument (Applied Biosys-tems). Data analysis was conducted with Sequencher soft-ware, version 4.6 (Gene Codes).
Validation of mutation load in BRAF/NRASWT tumorsTo validate our concept of a high-mutation load group of
melanomas, we retrospectively examined exome data fromHodis and colleagues (14) and Krauthammer and collea-gues (15), which sequenced independent cohorts with atotal of 198 cutaneousmelanomas and described themuta-tion rate together with BRAF andNRAS status. Acral, muco-sal, and uveal melanomas were excluded.
Statistical analysisNonparametric Spearman correlation was used to inves-
tigate associations between mutation rate and other con-tinuous variables (thickness and mitotic rate) to avoid anyundue influence of outliers. The Mann–Whitney U andKruskal–Wallis tests were used to assess associationsbetween mutation rate and respectively dichotomous vari-ables and variables with three or more categories. Orderedlogistic regression was used to assess associations betweensolar elastosis scores and continuous variables. Multinomi-al logistic regression was used to assess for associationsbetweenBRAFV600E,BRAFV600K,NRAS, andWT tumors.All analyses were conducted using Stata12 statistical soft-ware (Stata Corporation).
ResultsA total of 34 primary cutaneous melanomas were
sequenced from 34 patients (59% female) with a medianage at diagnosis of 67 years.Melanomaswere from the lowerlimb (47%), head and neck (26%), trunk (18%), and upperlimb(9%). Fifty-threepercentof cases arose innon-SSDskin.Notably, none of the cases were from acral or mucosal sites.Seventeen cases were NM, 16 were SSM, and 1 was LMM. Ofthe16SSM,5hadaprominentdermalnodule. Thirtypercentof melanomas were amelanotic. The median thickness was6.2 mm and median mitotic rate 10 per mm2 (Table 1).
Landscape of mutations in primarymelanoma samplesA total of 139,962 somatic mutations located in canon-
ical transcripts were identified (seeMaterials andMethods),
Mutation Load in BRAF/NRAS Wild-Type Melanomas
www.aacrjournals.org Clin Cancer Res; 19(17) September 1, 2013 4591
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

with an average of 4,117 (range, 105–28,507) mutationsper tumor (or on average 33 mutations/Mb). Of these,37,981 mutations (12 mutations/Mb) were predicted to bedeleterious (Fig. 1), consistent with rates of nonsynon-ymous changes reported in other studies (14, 15).
Signatures of UV-induced damageThe ratio of nonsynonymous to synonymous changes
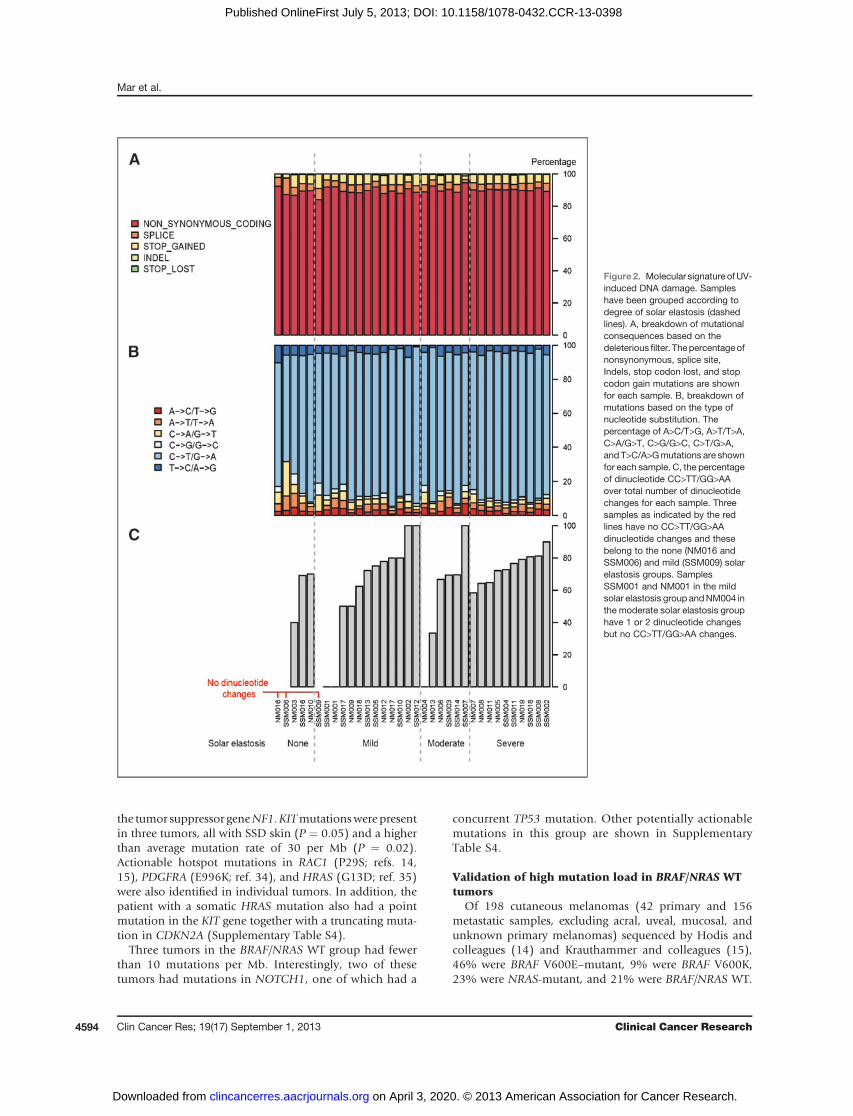
was 1.7, consistent with other melanoma exome studies(11, 32). A breakdown of mutational consequences basedon the deleterious filter is shown in Fig. 2A. Eighty-fivepercent of SNVs were composed of C>T or G>A transitionsconsistent withUV-inducedDNAdamage (Fig. 2B).Ninety-nine percent of all SNVs were at a dipyrimidine site and ofthose 86%wereC>T transitions, similar to results publishedelsewhere (10, 15). The UV signature of DNA damage wasalso confirmed by investigating dinucleotide CC>TT/GG>AA transitions, which is a more specific signature ofUV damage as the likelihood of obtaining two tandemtransitions is very low compared with SNV C>T changes,which may not be due to UV damage. All but six tumorsshowed tandemCC>TT changes, whichmade up 73%of alldinucleotide substitutions (Fig. 2C). Tumors arising in SSDskin had an average of 26 CC>TT transitions, comparedwith an average of four in tumors arising in non-SSD skin(P ¼ 0.0008). The number of CC>TT transitions as a
percentage of total dinucleotide transitions in tumors withno solar elastosis was 36% compared with 74% in tumorswith severe solar elastosis (P ¼ 0.04; Fig. 2C). BRAF/NRASWT tumors had an average of 34 CC>TT transitions com-pared with an average of six in both NRAS- and BRAF-mutant tumors (P ¼ 0.0007).
Tumors which showed no CC>TT/GG>AA changes (n ¼6)were fromrelatively sun-protected sites (such as the lowerlimb and trunk)with low solar elastosis scores (Fig. 2C) andwere either BRAF- or NRAS-mutant. Only one of thesetumors arose in SSD skin (solar elastosis score of 2) andwas BRAF V600K–mutant.
Correlation of mutation rate with clinical andhistopathologic variables
Melanoma arising on sun-exposed anatomic sites (head,neck, and upper limb) had highermutation rates comparedwith those arising on the lower limb and trunk as would beexpected, although this difference did not reach statisticalsignificance (P ¼ 0.07). Mutation rates were more than5-fold in tumors arising in skin with severe solar elastosiscomparedwith tumors arising in skinwith no solar elastosis(P ¼ 0.001; Fig. 3).
Interestingly there was an inverse correlation betweenthickness and mutation rate (r ¼ �0.4; P ¼ 0.02; Fig. 4).There was no statistically significant association betweenmutation rate and other clinicopathologic features such asage, sex,mitotic rate, tumor subtype, pigmentation, featuresof regression, or tumor-infiltrating lymphocytes.
Mutation rate in tumors with classical melanomamutations
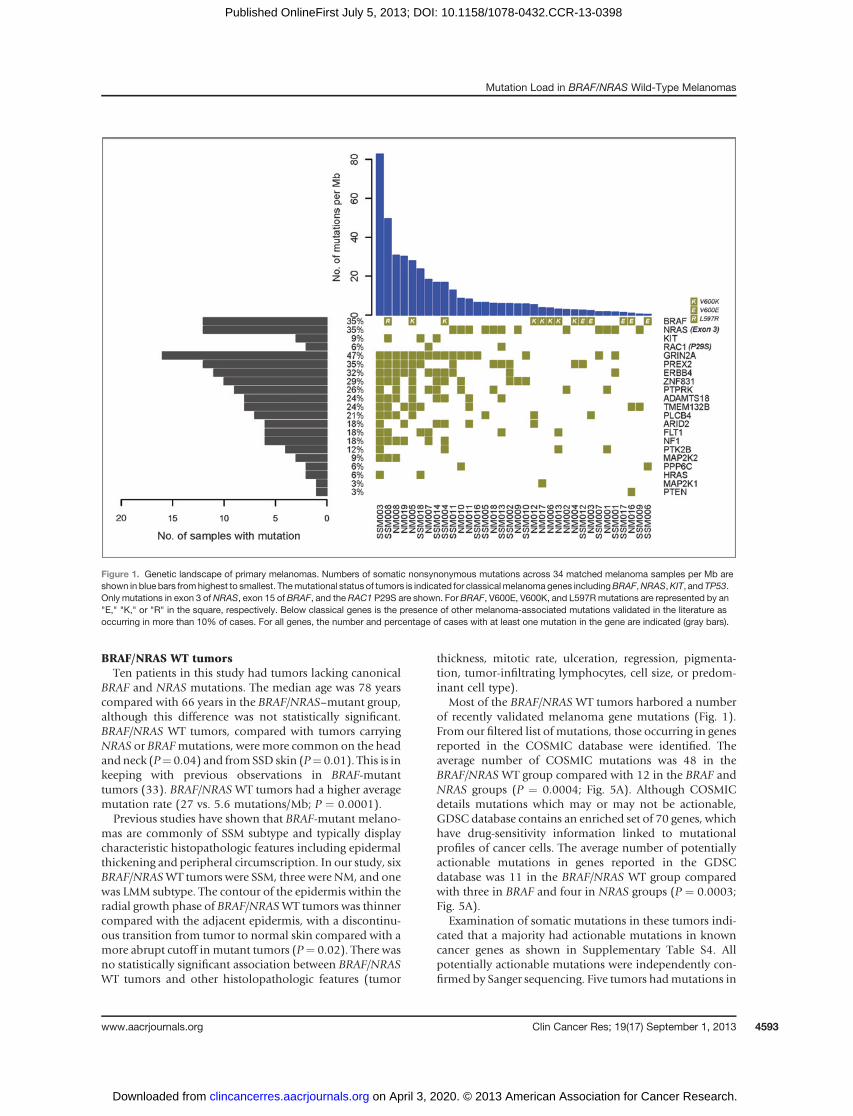
Thirty-fivepercent of tumors in thediscovery cohort had acanonical BRAF V600E or V600K mutation and 35% wereNRAS-mutant in exon 3 (Fig. 1). Interestingly, the propor-tion of BRAF V600K (7 of 34) mutations was greater thanBRAF V600E mutations (5/34). All BRAF and NRAS muta-tions were independently confirmed by Sanger sequencing.Consistent with previous reports (6), most BRAF V600Kmutations were in tumors arising in SSD skin, whereasBRAF V600E mutations were more common in tumorsfrom sun-protected sites with low solar elastosis scores(P ¼ 0.02).
The presence of a BRAF orNRASmutationwas associatedwith a lower mutation load within the tumor comparedwithWT tumors (P<0.0001). This associationwas strongestfor BRAFV600E–mutant tumors [relative risk ratio (RRR)¼0.38/mutation; 95% confidence interval (CI), 0.16–0.91],and less strong for BRAF V600K- andNRAS-mutant tumors(RRR ¼ 0.91/mutation; 95% CI, 0.82–1.02 and RRR ¼0.82/mutation; 95% CI, 0.69–0.97, respectively).
This cohort of melanomas exhibited a range of nonsy-nonymous mutations in genes classically associated withmelanoma including c-KIT (n ¼ 3), RAC1 (n ¼ 2), andCDKN2A (n ¼ 3). Other melanoma-associated mutationsvalidated in the literature as occurring in more than 10% ofcases are shown in Fig. 1. The majority of these mutationsclustered in tumors with high mutation loads.
Table 1. Clinical and histopathologiccharacteristics of patient cohort
Continuous variables Mean Median Range
Age, y 69 67 40–90Thickness, mm 7.4 6.2 2.5–25.0Mitotic rate, n/mm2 12 10 2–33
Categorical variables n %
Sex Male 14 41Female 20 59
Site HN 9 26UL 3 9T 6 18LL 16 47
Ulceration No 7 21Yes 27 79
Melanoma subtype SSM 11 32SSMþN 5 15NM 17 50LMM 1 3
Solar elastosis None 5 15Mild 13 38Moderate 6 18Severe 10 29
Abbreviations: HN, head and neck; LL, lower limb; SSMþN,SSM with a prominent dermal nodule; T, trunk; UL, upperlimb.
Mar et al.
Clin Cancer Res; 19(17) September 1, 2013 Clinical Cancer Research4592
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398
BRAF/NRAS WT tumorsTen patients in this study had tumors lacking canonical
BRAF and NRAS mutations. The median age was 78 yearscompared with 66 years in the BRAF/NRAS–mutant group,although this difference was not statistically significant.BRAF/NRAS WT tumors, compared with tumors carryingNRAS or BRAFmutations, weremore common on the headandneck (P¼ 0.04) and fromSSD skin (P¼ 0.01). This is inkeeping with previous observations in BRAF-mutanttumors (33). BRAF/NRAS WT tumors had a higher averagemutation rate (27 vs. 5.6 mutations/Mb; P ¼ 0.0001).Previous studies have shown that BRAF-mutant melano-
mas are commonly of SSM subtype and typically displaycharacteristic histopathologic features including epidermalthickening and peripheral circumscription. In our study, sixBRAF/NRASWT tumors were SSM, three were NM, and onewas LMM subtype. The contour of the epidermis within theradial growth phase of BRAF/NRASWT tumors was thinnercompared with the adjacent epidermis, with a discontinu-ous transition from tumor to normal skin compared with amore abrupt cutoff in mutant tumors (P¼ 0.02). There wasno statistically significant association between BRAF/NRASWT tumors and other histolopathologic features (tumor
thickness, mitotic rate, ulceration, regression, pigmenta-tion, tumor-infiltrating lymphocytes, cell size, or predom-inant cell type).
Most of the BRAF/NRAS WT tumors harbored a numberof recently validated melanoma gene mutations (Fig. 1).From our filtered list of mutations, those occurring in genesreported in the COSMIC database were identified. Theaverage number of COSMIC mutations was 48 in theBRAF/NRAS WT group compared with 12 in the BRAF andNRAS groups (P ¼ 0.0004; Fig. 5A). Although COSMICdetails mutations which may or may not be actionable,GDSC database contains an enriched set of 70 genes, whichhave drug-sensitivity information linked to mutationalprofiles of cancer cells. The average number of potentiallyactionable mutations in genes reported in the GDSCdatabase was 11 in the BRAF/NRAS WT group comparedwith three in BRAF and four in NRAS groups (P ¼ 0.0003;Fig. 5A).
Examination of somatic mutations in these tumors indi-cated that a majority had actionable mutations in knowncancer genes as shown in Supplementary Table S4. Allpotentially actionable mutations were independently con-firmed by Sanger sequencing. Five tumors hadmutations in
Figure 1. Genetic landscape of primary melanomas. Numbers of somatic nonsynonymous mutations across 34 matched melanoma samples per Mb areshown in blue bars fromhighest to smallest. Themutational status of tumors is indicated for classicalmelanoma genes includingBRAF,NRAS,KIT, and TP53.Only mutations in exon 3 ofNRAS, exon 15 of BRAF, and the RAC1 P29S are shown. For BRAF, V600E, V600K, and L597Rmutations are represented by an"E," "K," or "R" in the square, respectively. Below classical genes is the presence of other melanoma-associated mutations validated in the literature asoccurring in more than 10% of cases. For all genes, the number and percentage of cases with at least one mutation in the gene are indicated (gray bars).
Mutation Load in BRAF/NRAS Wild-Type Melanomas
www.aacrjournals.org Clin Cancer Res; 19(17) September 1, 2013 4593
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398
the tumor suppressor geneNF1.KITmutationswere presentin three tumors, all with SSD skin (P ¼ 0.05) and a higherthan average mutation rate of 30 per Mb (P ¼ 0.02).Actionable hotspot mutations in RAC1 (P29S; refs. 14,15), PDGFRA (E996K; ref. 34), and HRAS (G13D; ref. 35)were also identified in individual tumors. In addition, thepatient with a somatic HRAS mutation also had a pointmutation in the KIT gene together with a truncating muta-tion in CDKN2A (Supplementary Table S4).
Three tumors in the BRAF/NRAS WT group had fewerthan 10 mutations per Mb. Interestingly, two of thesetumors had mutations in NOTCH1, one of which had a
concurrent TP53 mutation. Other potentially actionablemutations in this group are shown in SupplementaryTable S4.
Validation of high mutation load in BRAF/NRAS WTtumors
Of 198 cutaneous melanomas (42 primary and 156metastatic samples, excluding acral, uveal, mucosal, andunknown primary melanomas) sequenced by Hodis andcolleagues (14) and Krauthammer and colleagues (15),46% were BRAF V600E–mutant, 9% were BRAF V600K,23% were NRAS-mutant, and 21% were BRAF/NRAS WT.
Figure2. Molecular signature ofUV-induced DNA damage. Sampleshave been grouped according todegree of solar elastosis (dashedlines). A, breakdown of mutationalconsequences based on thedeleterious filter. Thepercentageofnonsynonymous, splice site,Indels, stop codon lost, and stopcodon gain mutations are shownfor each sample. B, breakdown ofmutations based on the type ofnucleotide substitution. Thepercentage of A>C/T>G, A>T/T>A,C>A/G>T, C>G/G>C, C>T/G>A,and T>C/A>Gmutations are shownfor each sample. C, the percentageof dinucleotide CC>TT/GG>AAover total number of dinucleotidechanges for each sample. Threesamples as indicated by the redlines have no CC>TT/GG>AAdinucleotide changes and thesebelong to the none (NM016 andSSM006) and mild (SSM009) solarelastosis groups. SamplesSSM001 and NM001 in the mildsolar elastosis group andNM004 inthe moderate solar elastosis grouphave 1 or 2 dinucleotide changesbut no CC>TT/GG>AA changes.
Mar et al.
Clin Cancer Res; 19(17) September 1, 2013 Clinical Cancer Research4594
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

The mean number of somatic substitutions was 397, 577,495, and 940 for BRAF V600E, V600K, NRAS, and WTtumors, respectively (P ¼ 0.006).
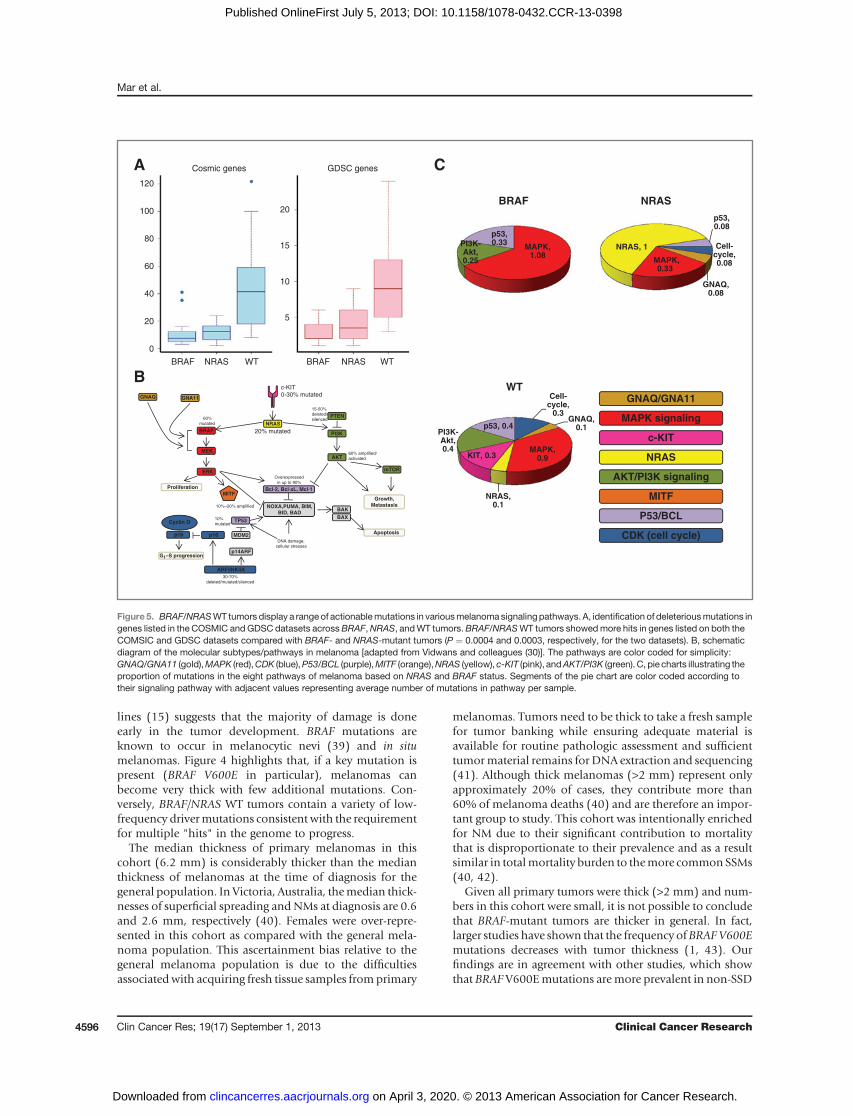
Pathway analysisAn analysis of the number ofmutations across eight well-
describedmelanoma pathways in BRAF- andNRAS-mutanttumors compared with BRAF/NRAS WT tumors is shownin Fig. 5B and C. Although theMAPK pathway contains themajority of mutations for all groups, BRAF/NRAS WTtumors have multiple mutations spread across seven of
these pathways, in particular the PI3K-Akt and p53 path-ways. NRAS mutants on the other hand, had a slightpreponderance to other pathways but NRAS remains thedominantly mutated gene.
DiscussionThis study shows that BRAF and NRAS WT melanomas
are a complex group with a high mutation load due toextensive UV damage. This finding was validated in a largercohort of 198 primary and metastatic melanomas. BRAF/NRAS WT melanomas are strongly associated with UVdamage as evidenced clinically by the higher degree of solarelastosis, and on a molecular level, with a high proportionof C>T transitions at a dipyrimidine, and more specifically,more frequent tandem CC>TT transitions. It is likely thatdifferent treatment strategies will be required when treatingpatients with high mutation load melanomas, which har-bor an array of potentially targetable mutations. Classifica-tion of melanoma into BRAF-mutant, NRAS-mutant, andhigh-mutation load groups may be helpful for identifica-tion of patients suitable for particular combined drugtherapies.
To our knowledge, this is the first study to sequenceprimary cutaneous melanomas and correlate moleculardata with clinical characteristics. Although previous studieshave shown an association betweenmolecular signatures ofUV damage and cutaneous melanomas (14, 15), none havequantified the degree of solar elastosis adjacent to theprimary tumor. In fact, melanomas arising in nonglabrous,nonmucosal sites are often universally classified as "sun-exposed." Krauthammer and colleagues also found thatBRAF/NRAS WT cutaneous melanomas had a high muta-tion burden, but reported BRAF/NRAS–mutantmelanomasto have mutation loads in the mid range. That cohort wasskewed by uveal, acral, and mucosal tumors with lowmutation burdens. Although acral melanomas can haveBRAF or NRAS mutations and perhaps be associated withUV damage (36), mucosal [which may be NRAS- or BRAF-mutant (14, 15, 37)], and uveal melanomas are not UVrelated. With histologic assessment of solar damage, wehavebeen able to showdistinctmolecular signatures amongcommon histologic subtypes of cutaneous melanoma,which may have important implications for treatment.
The inverse correlation between mutation load and pri-mary tumor thickness shown in this study was surprising.The clonal evolution model rests on the notion that cancerprogresses from a low metastatic potential (thin early-stagemelanomas) to a strong metastatic state (thicker morelocally advanced melanomas) through the accumulationof molecular alterations such as mutations which increasethe invasive and proliferative potential of cancer cells astumor burden increases (38). Our data suggest that inmelanomas there is not a simple relationship betweentumor burden and mutation load. Rather, it suggests thatmelanomas do not necessarily accumulate mutations asthey get thicker. In support of this, a similar range ofmutation rates published for metastatic disease and cell
Figure3. Highmutation load tumors are associatedwithmore severe solarelastosis of the surrounding skin. Box-and-whisker plot of mutation rate(perMb) vs. the degree of solar elastosis ranking frommild (score¼ 0; n¼5),mild (score¼ 1; n¼ 13), moderate (score¼ 2; n¼ 6), and severe (score¼ 3; n ¼ 10). There was a significant association between higher solarelastosis scores and increasing mutation rate (P ¼ 0.001).
Figure 4. BRAF-mutant melanomas have lower mutations ratescompared with WT tumors. Mutation rate (per Mb) vs. Breslow thickness(mm) of each tumor. The mutational status of each tumor is describedwith BRAF V600E (green triangle), BRAF V600K (orange square), NRASexon 3–positive (red diamond), and non-BRAF/NRAS–positive or WT(blue cross) marked for each tumor on the graph. There was an inversecorrelation between thickness and mutation rate (Spearman correlation,r ¼ �0.4; P ¼ 0.02).
Mutation Load in BRAF/NRAS Wild-Type Melanomas
www.aacrjournals.org Clin Cancer Res; 19(17) September 1, 2013 4595
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398
lines (15) suggests that the majority of damage is doneearly in the tumor development. BRAF mutations areknown to occur in melanocytic nevi (39) and in situmelanomas. Figure 4 highlights that, if a key mutation ispresent (BRAF V600E in particular), melanomas canbecome very thick with few additional mutations. Con-versely, BRAF/NRAS WT tumors contain a variety of low-frequency drivermutations consistent with the requirementfor multiple "hits" in the genome to progress.
The median thickness of primary melanomas in thiscohort (6.2 mm) is considerably thicker than the medianthickness of melanomas at the time of diagnosis for thegeneral population. In Victoria, Australia, themedian thick-nesses of superficial spreading and NMs at diagnosis are 0.6and 2.6 mm, respectively (40). Females were over-repre-sented in this cohort as compared with the general mela-noma population. This ascertainment bias relative to thegeneral melanoma population is due to the difficultiesassociated with acquiring fresh tissue samples fromprimary
melanomas. Tumors need to be thick to take a fresh samplefor tumor banking while ensuring adequate material isavailable for routine pathologic assessment and sufficienttumormaterial remains for DNA extraction and sequencing(41). Although thick melanomas (>2 mm) represent onlyapproximately 20% of cases, they contribute more than60% of melanoma deaths (40) and are therefore an impor-tant group to study. This cohort was intentionally enrichedfor NM due to their significant contribution to mortalitythat is disproportionate to their prevalence and as a resultsimilar in totalmortality burden to themore commonSSMs(40, 42).
Given all primary tumors were thick (>2 mm) and num-bers in this cohort were small, it is not possible to concludethat BRAF-mutant tumors are thicker in general. In fact,larger studies have shown that the frequency ofBRAFV600Emutations decreases with tumor thickness (1, 43). Ourfindings are in agreement with other studies, which showthat BRAF V600Emutations aremore prevalent in non-SSD
BRAF
BRAF
NRAS
NRAS
WT
WT BRAF NRAS WT
MAPK,1.08
PI3K-Akt,0.25
p53,0.33
GNAQ/GNA11
MAPK signaling
c-KIT
NRAS
AKT/PI3K signaling
MITF
P53/BCL
CDK (cell cycle)
Cell-cycle,0.08
GNAQ,0.08
MAPK,0.33
NRAS, 1
p53,0.08
Cell-cycle,
0.3GNAQ,
0.1
MAPK,0.9
NRAS,0.1
KIT, 0.3
PI3K-Akt,0.4
p53, 0.4
c-KIT0-30% mutated
NRAS
20% mutated
15-50%
deleted/mutated/
silencedPTEN
PI3K
AKT60% amplified/
activated
BRAF
60%
mutated
MEK
ERK
ProliferationMITF
10%–20% amplified
Bcl-2, Bcl-xL, Mcl-1
NOXA,PUMA, BIM,BID, BAD
Overexpressed
in up to 90%
Growth,Metastasis
mTOR
DNA damage,
cellular stresses
BAK
BAX
Apoptosis
TP5310%
mutated
ARF/INK4A30-70%
deleted/mutated/silenced
MDM2
p14ARF
p16p16
Cyclin D
G1–S progression
GNAQ GNA11
A
B
CCosmic genes
20
15
10
5
120
100
80
60
40
20
0
GDSC genes
Figure5. BRAF/NRASWT tumors display a range of actionablemutations in variousmelanomasignalingpathways. A, identification of deleteriousmutations ingenes listed in the COSMIC andGDSC datasets acrossBRAF,NRAS, andWT tumors.BRAF/NRASWT tumors showedmore hits in genes listed on both theCOMSIC and GDSC datasets compared with BRAF- and NRAS-mutant tumors (P ¼ 0.0004 and 0.0003, respectively, for the two datasets). B, schematicdiagram of the molecular subtypes/pathways in melanoma [adapted from Vidwans and colleagues (30)]. The pathways are color coded for simplicity:GNAQ/GNA11 (gold),MAPK (red),CDK (blue),P53/BCL (purple),MITF (orange),NRAS (yellow), c-KIT (pink), andAKT/PI3K (green). C, pie charts illustrating theproportion of mutations in the eight pathways of melanoma based on NRAS and BRAF status. Segments of the pie chart are color coded according totheir signaling pathway with adjacent values representing average number of mutations in pathway per sample.
Mar et al.
Clin Cancer Res; 19(17) September 1, 2013 Clinical Cancer Research4596
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

skin (1), and V600K and KITmutations are more prevalentin SSD skin (6, 44). Furthermore, our findings that BRAFand NRAS WT tumors tend to have a thinner epidermis incontrast to adjacent normal skin and a tendency for thetransition from tumor to normal skin to be discontinuousare also supported by larger studies (3).The correlation between BRAF status and mutation rate
suggests that where a predominant driver mutation ispresent, the mutation rate will often remain low. AlthoughKIT mutations occurred in three BRAF/NRAS WT tumors,these all had a high mutation burden. There were threetumors in the BRAF/NRAS WT group with mutation ratesless than 10 perMb. Two of these tumors had amutation inNOTCH1, which has recently been implicated in growthand invasion of uveal melanoma (45) and is potentiallytargetable (46, 47).Inactivatingmutations in the tumor suppressor geneNF1
were present in 50% of WT tumors compared with just 4%of BRAF/NRAS–mutant tumors. Hodis and colleaguesreported a similarly high frequency of NF1 mutationsamong BRAF/NRAS WT compared with mutant tumors(25% vs. 2%; ref. 14). Loss of NF1 tumor suppression canlead to constitutive MAPK pathway activation through Ras(ref. 48; Fig. 5B). This highlights the potential for NF1 tobecome an important therapeutic target in WT tumors.It is important to note that, given the current knowledge
on the sensitivity and resistance of therapeutics to specificmutations, classifying mutations into driver and passengermutations aswell as actionable and/or druggable is difficult.An "actionable mutation" as defined earlier, is a geneticalteration which may have significant diagnostic, prognos-tic, or therapeutic implications for a patient. A subset ofthesemay be "druggable," i.e., it would predict sensitivity orresistance to a specific drug (49). Therapeutic targeting ofmutations in tumor suppressor genes is particularly chal-lenging, as research into restoring normal gene function inpatients (i.e., gene therapy) is currently ineffective for mosttumor suppressor genes.Although systematic identification of actionable muta-
tions in this cohort is particularly challenging due to thelarge number of mutations present, we have attempted toidentify actionable mutations by comparing variants with anumber of cancer mutation databases. BRAF/NRAS WTtumors contain a number of low-frequency driver muta-tions and therefore require a larger number of UV-inducedinsults to the genome to progress. Importantly, they docontain a number of potentially targetable mutations,though these are spread over different pathways. LikeBRAF-mutant tumors, theMAPK pathway is most frequent-ly involved in BRAF/NRAS WT tumors, however, this is
accompanied by greater burden of mutations in this groupoverall. Although there is significant cross-talk betweenpathways, results from this study suggest that therapeutictargeting ofmultiple pathwaysmay be necessary rather thanfocused targeting of a single pathway in BRAF/NRAS WTtumors with a high mutation burden.
Disclosure of Potential Conflicts of InterestJ.F. Thompson has other commercial research support from GlaxoS-
mithKline and Provectus, has honoraria from Speakers Bureau of GlaxoS-mithKline, and is a consultant/advisory board member of GlaxoSmithKline.G.A. McArthur has a commercial research grant from Novartis, Millennium,and Pfizer and is a consultant/advisory board member of Roche Genentech,GlaxoSmithKline, Novartis, Millennium, Amgen, Ventana, and Bristol-Myers Squibb. No potential conflicts of interest were disclosed by the otherauthors.
Authors' ContributionsConception and design: V. Mar, S.Q. Wong, A.T. Papenfuss, J. Cebon, A.Dobrovic, G.A. McArthurDevelopment of methodology: V. Mar, S.Q. Wong, J. Li, R.A. Scolyer, R.Tothill, A. DobrovicAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): V. Mar, S.Q. Wong, R.A. Scolyer, C. McLean, G.J.Mann, J.F. Thompson, J. Cebon, J.W. Kelly, A. Dobrovic, G.A. McArthurAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): V. Mar, S.Q. Wong, J. Li, R.A. Scolyer, C.McLean, A.T. Papenfuss, J. Cebon, R. Wolfe, A. Dobrovic, G.A. McArthurWriting, review, and/or revision of themanuscript: V.Mar, S.Q.Wong, J.Li, R.A. Scolyer, A.T. Papenfuss, H. Kakavand,G.J.Mann, A. Behren, J. Cebon,R. Wolfe, J.W. Kelly, A. Dobrovic, G.A. McArthurAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): S.Q. Wong, J. Li, H. Kakavand, A.Behren, J. CebonStudy supervision: S.Q.Wong, R.A. Scolyer, A.T. Papenfuss,G.J.Mann,G.A.McArthur
AcknowledgmentsThe authors thank the contributions from staff at the Victorian Cancer
Biobank, Melbourne Melanoma Project, and Melanoma Institute Australiafor the collection of samples.
Grant SupportThis project was enabled by the Melbourne Melanoma Project funded by
the Victorian Government through the Victorian Cancer Agency Transla-tional Research ProgramGrant (EOI09_27) and established through supportof the Victor Smorgon Charitable Fund. This work was also supported byProgramGrant 633004of theNationalHealth andMedical ResearchCouncilof Australia (NHMRC) and Translational Research Program Grant 10/TPG/1-02 of the Cancer Institute New South Wales. V. Mar was supported by aNational Health and Medical Research Council of Australia (NHMRC) PhDScholarship. R.A. Scolyer is supported by the Cancer Institute New SouthWales Fellowship program. A.T. Papenfuss was supported by an NHMRCCareer Development Fellowship with contributions also made possiblethrough Victorian State Government Operational Infrastructure Supportand NHMRC IRIISS.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received February 13, 2013; revisedMay 31, 2013; accepted June 15, 2013;published OnlineFirst July 5, 2013.
References1. Liu W, Kelly JW, Trivett M, Murray WK, Dowling JP, Wolfe R, et al.
Distinct clinical and pathological features are associated with theBRAF(T1799A(V600E)) mutation in primary melanoma. J Invest Der-matol 2007;127:900–5.
2. Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthe-sis of epidemiological, clinical, histopathological, genetic, and
biological aspects, supporting distinct subtypes, causal path-ways, and cells of origin. Pigment Cell Melanoma Res 2011;24:879–97.
3. Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, et al.Improving melanoma classification by integrating genetic and mor-phologic features. PLoS Med 2008;5:e120.
Mutation Load in BRAF/NRAS Wild-Type Melanomas
www.aacrjournals.org Clin Cancer Res; 19(17) September 1, 2013 4597
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

4. Broekaert SM, Roy R, Okamoto I, van den Oord J, Bauer J, Garbe C,et al. Genetic and morphologic features for melanoma classification.Pigment Cell Melanoma Res 2010;23:763–70.
5. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H,et al. Distinct sets of genetic alterations in melanoma. N Engl J Med2005;353:2135–47.
6. Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, ThompsonJF, et al. Distinguishing clinicopathologic features of patients withV600E and V600K BRAF-mutant metastatic melanoma. Clin CancerRes 2012;18:3242–9.
7. Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, et al. Clinicaloutcome and pathological features associated with NRASmutation incutaneous melanoma. Pigment Cell Melanoma Res 2011;24:666–72.
8. Kelleher FC, McArthur GA. Targeting NRAS in melanoma. CancerJ 2012;18:132–6.
9. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G,et al. Patterns of somatic mutation in human cancer genomes. Nature2007;446:153–8.
10. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al.Exome sequencing identifies recurrent somaticMAP2K1 andMAP2K2mutations in melanoma. Nat Genet 2012;44:133–9.
11. Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exomesequencing identifiesGRIN2A as frequentlymutated inmelanoma.NatGenet 2011;43:442–6.
12. Palavalli LH, Prickett TD, Wunderlich JR, Wei X, Burrell AS, Porter-GillP, et al. Analysis of the matrix metalloproteinase family reveals thatMMP8 is often mutated in melanoma. Nat Genet 2009;41:518–20.
13. Prickett TD, Agrawal NS,Wei X, Yates KE, Lin JC,Wunderlich JR, et al.Analysis of the tyrosine kinome in melanoma reveals recurrent muta-tions in ERBB4. Nat Genet 2009;41:1127–32.
14. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP,et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
15. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCuskerJP, et al. Exome sequencing identifies recurrent somatic RAC1 muta-tions in melanoma. Nat Genet 2012;44:1006–14.
16. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Proto-popovA, et al.Melanomagenome sequencing reveals frequentPREX2mutations. Nature 2012;485:502–6.
17. Turajlic S, Furney SJ, Lambros MB, Mitsopoulos C, Kozarewa I, GeyerFC, et al. Whole genome sequencing of matched primary and meta-static acral melanomas. Genome Res 2012;22:196–207.
18. Cibulskis K, LawrenceMS, Carter SL, Sivachenko A, Jaffe D, SougnezC, et al. Sensitive detection of somatic point mutations in impure andheterogeneous cancer samples. Nat Biotechnol 2013;31:213–9.
19. LeBoit P, Burg G, Weedon D, Sarasin A. Skin Tumors. Pathology andgenetics. Lyon, France: IARC Press; 2006.
20. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthySW, et al. Tumor-infiltrating lymphocyte grade is an independentpredictor of sentinel lymph node status and survival in patients withcutaneous melanoma. J Clin Oncol 2012;30:2678–83.
21. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60.
22. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, KernytskyA, et al. The Genome Analysis Toolkit: a MapReduce framework foranalyzing next-generation DNA sequencing data. Genome Res2010;20:1297–303.
23. Picard [Internet]. [updated 2009May 18; cited Feb 13, 2013]. Availablefrom: http://picard.sourceforge.net/.
24. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C,et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8.
25. MuTect—cancer gene analysis tools [Internet]. Cambridge,MA: BroadInstitute 2012 [cited Feb 13, 2013]. Available from:https://confluence.broadinstitute.org/display/CGATools/MuTect.
26. Flicek P, AmodeMR, Barrell D, Beal K, Brent S, Carvalho-Silva D, et al.Ensembl 2012. Nucleic Acids Res 2012;40:D84–90.
27. Universal protein resource (UniProt) knowledgebase [Internet] 2002–2013 [citedFeb13, 2013].Available from:http://www.uniprot.org/faq/30.
28. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, et al.COSMIC (the Catalogue of Somatic Mutations in Cancer): a resourceto investigate acquiredmutations in human cancer. Nucleic Acids Res2010;38:D652–7.
29. Genomics of drug sensitivity in cancer [Internet]. Cambridgeshire, UK:Wellcome Trust Sanger Institute, 2012 [cited Feb 13, 2013]. Availablefrom: http://www.cancerrxgene.org/.
30. Vidwans SJ, Flaherty KT, Fisher DE, Tenenbaum JM, Travers MD,Shrager J. A melanoma molecular disease model. PLoS ONE 2011;6:e18257.
31. Blombery PA,Wong SQ, Hewitt CA, Dobrovic A,Maxwell EL, Juneja S,et al. Detection of BRAF mutations in patients with hairy cell leukemiaand related lymphoproliferative disorders. Haematologica 2012;97:780–3.
32. Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K,Aoude LG, et al. Frequent somaticmutations inMAP3K5 andMAP3K9in metastatic melanoma identified by exome sequencing. Nat Genet2012;44:165–9.
33. Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T,et al. Determinants of BRAF mutations in primary melanomas. J NatlCancer Inst 2003;95:1878–90.
34. Dutton-Regester K, Irwin D, Hunt P, Aoude LG, Tembe V, Pupo GM,et al. A high-throughput panel for identifying clinically relevant muta-tion profiles in melanoma. Mol Cancer Ther 2012;11:888–97.
35. Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X,et al. Personalized oncology through integrative high-throughputsequencing: a pilot study. Sci Transl Med 2011;3:111ra21.
36. Saldanha G, Potter L, Daforno P, Pringle JH. Cutaneous melanomasubtypes show different BRAF and NRAS mutation frequencies. ClinCancer Res 2006;12:4499–505.
37. Si L, Kong Y, Xu X, Flaherty KT, Sheng X, Cui C, et al. Prevalence ofBRAF V600E mutation in Chinese melanoma patients: large scaleanalysis of BRAF and NRAS mutations in a 432-case cohort. Eur JCancer 2012;48:94–100.
38. Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med 2006;355:51–65.39. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM,
et al. High frequency of BRAF mutations in nevi. Nat Genet 2003;33:19–20.
40. Mar V, Roberts H, Wolfe R, English DR, Kelly JW. Nodular mela-noma: a distinct clinical entity and the largest contributor to mel-anoma deaths in Victoria, Australia. J Am Acad Dermatol 2013;68:568–75.
41. Scolyer RA, Thompson JF. Biospecimen banking: the pathway topersonalized medicine for patients with cancer. J Surg Oncol 2013;107:681–2.
42. Shaikh WR, Xiong M, Weinstock MA. The contribution of nodularsubtype to melanoma mortality in the United States, 1978 to 2007.Arch Dermatol 2012;148:30–6.
43. Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, JohnsonMM, Cooke CP, et al. Clinical correlates of NRAS and BRAFmutations in primary human melanoma. Clin Cancer Res 2011;17:229–35.
44. BeadlingC, Jacobson-DunlopE,Hodi FS, LeC,WarrickA,Patterson J,et al. KITgenemutations andcopy number inmelanoma subtypes.ClinCancer Res 2008;14:6821–8.
45. Asnaghi L, Ebrahimi KB, Schreck KC, Bar EE, Coonfield ML, Bell WR,et al. Notch signaling promotes growth and invasion in uveal mela-noma. Clin Cancer Res 2012;18:654–65.
46. Huang X,Wang L, ZhangH,WangH, Zhao X,QianG, et al. Therapeuticefficacy by targeting correction of Notch1-induced aberrants in uvealtumors. PLoS ONE 2012;7:e44301.
47. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, LauKW, et al. Systematic identification of genomic markers of drugsensitivity in cancer cells. Nature 2012;483:570–5.
48. Basu TN,GutmannDH, Fletcher JA, Glover TW, Collins FS, DownwardJ. Aberrant regulation of ras proteins in malignant tumour cells fromtype 1 neurofibromatosis patients. Nature 1992;356:713–5.
49. Dancey JE, Bedard PL, Onetto N, Hudson TJ. The genetic basis forcancer treatment decisions. Cell 2012;148:409–20.
Mar et al.
Clin Cancer Res; 19(17) September 1, 2013 Clinical Cancer Research4598
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398

2013;19:4589-4598. Published OnlineFirst July 5, 2013.Clin Cancer Res Victoria J. Mar, Stephen Q. Wong, Jason Li, et al.
DamageCorrelating with Histologic and Molecular Signatures of UV Wild-Type Melanomas Have a High Mutation LoadBRAF/NRAS
Updated version
10.1158/1078-0432.CCR-13-0398doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2013/07/03/1078-0432.CCR-13-0398.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/19/17/4589.full#ref-list-1
This article cites 44 articles, 10 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/19/17/4589.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/19/17/4589To request permission to re-use all or part of this article, use this link
on April 3, 2020. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 5, 2013; DOI: 10.1158/1078-0432.CCR-13-0398