

Bisulfite-Sequencing theory and Quality Control Felix Krueger [email protected] January...
41
-
Upload
james-wiggins -
Category
Documents
-
view
221 -
download
3
Transcript of Bisulfite-Sequencing theory and Quality Control Felix Krueger [email protected] January...

2
• 9:15-10:15 Bisulfite-Seq theory and QC• 10:15-10:30 coffee• 10:30-11:30 Mapping and QC practical• 11:30-12:30 Visualising and Exploring talk• 12:30-13:30 Lunch• 13:30-14:00 Methylation tools in SeqMonk• 14:00-15:30 Visualising and Exploring practical• 15:30-15:45 coffee• 15:45-16:35 Differential methylation talk & practical• 16:35-16:45 Introduction to other cytosine
modifications and oxBS

3
DNA Methylation
• Dynamic patterning• Correlation with gene expression• Reset during development• Suppression of repetitive elements • X-chromosome inactivation• Imprinting• Carcinogenesis
Cytosine 5-methyl Cytosine
DNA methyl-transferases
DNA-demethylase(s)?Hydroxymethyl cytosine? TETs?
Passive demethylation?

4
DNA methylation is sequence context dependent
sequenceCCAGTCGCTATACCAGTCGCTATACCAGTCGCTATA
contextCpGCHGCHH
mammalsYESno**no**
plantsYESYESYES
* H being anything but G** somewhat more relevant in certain cell types

5
Promoter methylation causes transcriptional silencing
Adapted from Stephen B BaylinNature Clinical Practice Oncology (2005)
Developmentally important gene or tumour suppressor gene

6
Imprinted Genes: mono-allelic expression
Imprinted Genes: Mono-allelic expression with parent-of-origin specificity. Have key roles in energy metabolism, placenta functions.
Differential allelicDNA methylation X
CGI (CpG island)
methylated CpGunmethylated CpG

7
Imprinted Genes: example
from Ferguson-Smith A. Nat. Rev. Genet. 2011
ICR / DMR
methylated CGI
unmethylated CGI
Rather unusual case where a non-methylated DMR causes silencing of a locus
ICR: Imprinting Control RegionDMR: Differentially Methylated Region

8
DNA methylation is maintained
from W. Reik & J. Walter, Nat. Rev. Genet. 2001

9
DNA methylation is reset during reprogramming

10
Measuring DNA methylation by Bisulfite-sequencing
Image by Illumina

11
Bisulfite Informatics
CCAGTCGCTATAGCGCGATATCGTAme me
TTAGTTGCTATAGTGCGATATTGTA
TTAGTTGCTATAGTGCGATATTGTA
...CCAGTCGCTATAGCGCGATATCGTA...|||||||||||||||||||||||||
Convert
Map

12
- 2 different PCR products and 4 possible different sequence strands from one genomic locus
Bisulfite conversion of a genomic locus
>>CCGGCATGTTTAAACGCT>><<GGCCGTACAAATTTGCGA<<
>>TCGGTATGTTTAAACGTT>><<AGCCATACAAATTTGCAA<<
>>CCAGCATGTTTAAACGCT>><<GGTCGTACAAATTTGCGA<<
Top strand Bottom strand
CTOTOT CTOB
OB
mC|
mC|
|mC
|mC
>>UCGGUATGTTTAAACGUT>><<GGUCGTACAAATTTGCGA<<
Bisulfite conversion
PCR amplification
|hmC
- each of these 4 sequence strands can theoretically exist in any possible conversion state

TTGGCATGTTTAAACGTT
cc..C.........Z.c.
…TTGGTATGTTTAAATGTT……AACCATACAAATTTACAA…
…CCAACATATTTAAACACT……GGTTGTATAAATTTGTGA…
align to bisulfite converted genomes
forward strand C -> T converted genome forward strand G -> A converted genome(equals reverse strand C -> T conversion)
5’…TTGGTATGTTTAAATGTT…3’ 5’…TTAACATATTTAAACATT…3’
(1)
(4)(3)
(2)
sequence of interest
(1) (2) (3) (4)
bisulfite convert read (treat sequence as bothforward and reverse strand)
read all 4 alignment outputs and extractthe unmodified genomic sequence if thesequence could be mapped uniquely
5’…CCGGCATGTTTAAACGCT…3’
CCGGCATGTTTAAACGCTTTGGCATGTTTAAACGTT
methylation call
read sequence
genomic sequence c unmethylated CC methylated Cz unmethylated C in CpG contextZ methylated C in CpG context
Mapping a Bisulfite-Seq read
methylation call
Bismark

14
Common sequencing protocols
>>CCGGCATGTTTAAACGCT>><<GGCCGTACAAATTTGCGA<<
Top strand
Bottom strand
>>TCGGTATGTTTAAACGTT>><<GGTCGTACAAATTTGCGA<<
OTOB
mC|
mC|
|mC
|mC
>>UCGGUATGTTTAAACGUT>>
<<GGUCGTACAAATTTGCGA<<
|hmC
1) Directional libraries (vast majority)
<<AGCCATACAAATTTGCAA<<>>CCAGCATGTTTAAACGCT>>
CTOTCTOB2) PBAT libraries
>>TCGGTATGTTTAAACGTT>><<AGCCATACAAATTTGCAA<<
>>CCAGCATGTTTAAACGCT>><<GGTCGTACAAATTTGCGA<<
CTOTOT
CTOBOB
3) Non-directional libraries

15
Validation
0
20
40
60
80
100
0
20
40
60
80
100
0 20 40 60 80 100
CpG CHG CHH Mapping efficiency
Bisulfite conversion rate (%)
Cyto
sine
sca
lled
unm
ethy
late
d(%
)
Mapping effi
ciency (%)
0
20
40
60
80
100
25 50 75 100 125 150
biasedunbiased
Read length (bp)
Map
ping
effi
cien
cy (%
)

16
BS-Seq Analysis Workflow
QC Trimming Mapping
Methylation extraction
Mapped QCAnalysis

17
Raw Sequence Data
...up to 1,000,000,000 lines per lane

18
Part I: Initial QC -What does QC tell you about your library?
• # of sequences• Basecall qualities• Base composition• Potential contaminants• Expected duplication rate

19
QC Raw data: Sequence Quality
10%
1%
0.1%
Error rate

QC: Base Composition
20
RRBSWGSBS

21
QC: Duplication rate

22
QC: Overrepresented sequences

23
Common problems in BS-SeqP
hred
sco
re
Position in read (bp)
Position in read (bp)
Bas
e co
nten
t (%
)
Not observed in ‘normal’ libraries, e.g. ChIP or RNA-Seq

24
Removing poor quality basecalls
before trimming after trimmingPh
red
scor
e

25
Removing adapter contamination
before trimming after trimming

26
Summary Adapter/Quality Trimming
Important to trim because failure to do so might result in:
Low mapping efficiency
Mis-alignments
Errors in methylation calls since adapters are methylated
Basecall errors tend toward 50% (C:mC)

27
Part II: Sequence alignment –Bismark primary alignment output (BAM file)
HISEQ2000-06:366:C3G4NACXX:3:1101:1316:2067_1:N:0: 99 16 71322125 255 100M = 71322232 207 NTTATTTAGTTTTTTAGGGTTTGTGTGTAGGAGTGTGGGAATTATGTTTTTTATGGTTGATATTTATTTAAAAGTGAGTATAAATTATATATATTTTTTT #1=DDDDDAAFFHIIIA:<FGHCCEFGHD?CFFBBBGEHHGHIII<FEHIIIII==DE??EHHFHEEEEEEEC>;>66;@CDEEEDCEEEEEEEDDDCBB NM:i:14 XX:Z:G8C2C7C21C13C6CC1C17CC3C4CC4 XM:Z:.........h..h.......x.....................h.............x......hh.h.................hh...h....hh.... XR:Z:CT XG:Z:CT XA:Z:1HISEQ2000-06:366:C3G4NACXX:3:1101:1316:2067_1:N:0: 147 16 71322232 255 100M = 71322125 -207 GGTTATTTTATTTAGGGTTATTGTTTTAGAGTTTTATTGTTGTGAACAGATATATGATTAAGGTAATTTTTATAAGGATAATATTTAATTGGAGTTGGTTCCCEEECADCFFFFHHGHGHIIGIHFIJJIJIHFGHGGGEHIJIIJGIGFJJJJJJJJJJIGJJJJGJJJJIIIJJIJIJJJJJJIJHHHHHFFFFFCCC NM:i:21 XX:Z:2G2CC1C1C1C11C11C2C10C1C4CC4C2C1C3C5C2C12C3C1 XM:Z:.....hh.h.h.x...........h...........x..x......X...h.h....hh....h..h.h...h.....h..h............x...h. XR:Z:GA XG:Z:CT XB:Z:1
methylation call
positionchromosome
Read 1
Read 2
sequence
quality

28
Sequence duplication
Complex/diverse library:
Duplicated library:
deduplication10055 17 100 100 71100percent methylation
33 50 100100 100 50 100percent methylation

29
Deduplication - considerations
RRBS
deduplication
Advisable for large genomes and moderate coverage
- unlikely to sequence several genuine copies of the same fragmentamongst >5bn possible fragments with different start sites- maximum coverage with duplication may still be (read length)-fold (even more with paired-end reads)
CCGG
NOT advisable for RRBS or other target enrichment methodswhere higher coverage is either desired or expected
CCGG
30
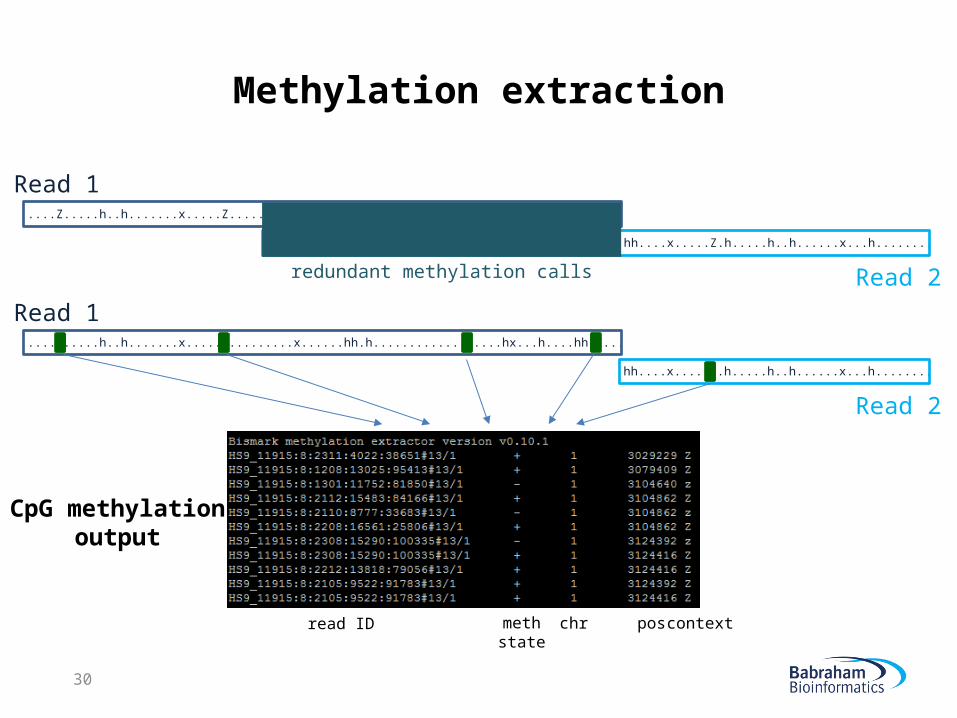
Methylation extraction
....Z.....h..h.......x.....Z.........x......hh.h.............z....hx...h....hh.Z...
...........hh.h.............z....hx...h....hh.Z...hh....x.....Z.h.....h..h......x...h........
Read 1
Read 2
....Z.....h..h.......x.....Z.........x......hh.h.............z....hx...h....hh.Z...
hh....x.....Z.h.....h..h......x...h........
Read 1
Read 2
CpG methylationoutput
read ID methstate
chr pos context
redundant methylation calls

31
Methylation extraction
CpG methylation output
bedGraph/coverage output
methylationpercentage
methposchr unmeth

32
Part III: Mapped QC -Methylation bias
good opportunity to look at conversion efficiency

33
Artificial methylation calls in paired-end libraries
5’- GGGNNNNNNNNNNNNNNNNNNNNNNNNNNNCCCA -3’3’- ACCCNNNNNNNNNNNNNNNNNNNNNNNNNNNGGG -5’
end repair + A-tailing

34
Specialist applications (I): Reduced representation BS-Seq (RRBS)
Sequence composition bias High duplication rate
35
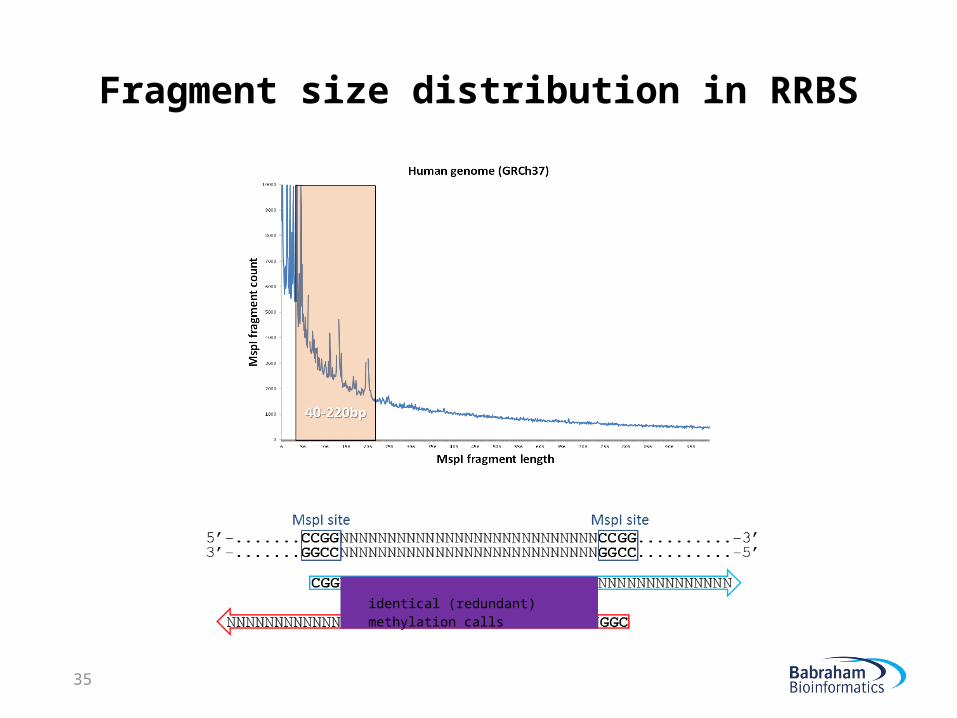
Fragment size distribution in RRBS
identical (redundant) methylation calls

36
Artificial methylation calls in RRBS libraries
C genomic cytosineC unmethylated cytosine

37
WGBS PBAT
Specialist application (II): Post-bisulfite adapter tagging (PBAT)
suitable for low input material

38
PBAT-Seq
trim off/ ignore first couple of basepairs

39
Bismark workflow
Pre AlignmentFastQC Initial quality controlTrim Galore Adapter/quality trimming using Cutadapt; handles RRBS
and paired-end reads; Trim Galore and RRBS User guideAlignmentBismark Output BAM
Post AlignmentDeduplication optionalMethylation extractor Output individual cytosine methylation calls; optionally
bedGraph or genome-wide cytosine reportM-bias analysis
bismark2report Graphical HTML report generation Example: http://www.bioinformatics.babraham.ac.uk/projects/bismark/PE_report.html
protocol: Quality Control, trimming and alignment of Bisulfite-Seq data

40
Useful links
• FastQC www.bioinformatics.babraham.ac.uk/projects/fastqc/
• Trim Galore www.bioinformatics.babraham.ac.uk/projects/trim_galore/
• Cutadapt https://code.google.com/p/cutadapt/
• Bismark www.bioinformatics.babraham.ac.uk/projects/bismark/
• Bowtie http://bowtie-bio.sourceforge.net/
• Bowtie 2 http://bowtie-bio.sourceforge.net/bowtie2/
• SeqMonk www.bioinformatics.babraham.ac.uk/projects/seqmonk/
• Cluster Flow www.bioinformatics.babraham.ac.uk/projects/clusterflow/
protocol: Quality control, trimming and alignment of Bisulfite-Seq datahttp://www.epigenesys.eu/en/protocols/bio-informatics/483-quality-control-trimming-and-alignment-of-bisulfite-seq-data-prot-57

41
FastQC: quality control for high throughput sequencing
FastQ Screen: organism andcontamination detection
Hi-C mapping
SeqMonk: Genome browser, quantitation and data analysis
Bismark: Bisulfite-sequencingalignments and methylation calls
Sierra: A web-based LIMS systemfor small sequencing facilities
ASAP: Allele-specific alignments
Trim Galore! Quality and adaptertrimming for (RRBS) libraries
www.bioinformatics.babraham.ac.uk


















