
BIPN140 Lecture 11: Synaptic Plasticity (I) - Class...
14
BIPN140 Lecture 11: Synaptic Plasticity (I) Su (FA16) 1. Short-Term Synaptic Plasticity 2. Long-Term Synaptic Plasticity 3. Molecular Mechanisms of Long-Term Potentiation
Transcript of BIPN140 Lecture 11: Synaptic Plasticity (I) - Class...
BIPN140 Lecture 11: Synaptic Plasticity (I)
Su (FA16)
1. Short-Term Synaptic Plasticity
2. Long-Term Synaptic Plasticity
3. Molecular Mechanisms of Long-Term Potentiation

Synaptic Plasticity
Synaptic plasticity: the connection between neurons are subject to modification (development, activity, physiological state, experience, disease, drugs, etc.).
Changes in synaptic efficacy are widely believed as the basis for:(1) certain forms of learning(2) storage of memories(3) refinement of connections during development
How to modify the strength/nature of connection between neurons? (1) changes in the number of synapses(2) changes in the probability of NT release (presynaptic mechanism)(3) changes in the number and/or property of NT receptors (postsynaptic
mechanism)
Synaptic plasticity varies in time scale from milliseconds to years.
Short-term Synaptic Plasticity Most short-term synaptic plasticity (milliseconds to minutes) affect the
amount the neurotransmitter released from presynaptic terminals. Key regulators: frequency of presynaptic APs, presynaptic [Ca2+] /sensitivity to Ca2+, availability of readily releasable SVs.
Four major types: (1) Synaptic facilitation: ms, Ca2+ build up due to quick succession of APs(2) Synaptic depression: ms, transient depletion of readily releasable SVs.(3) Synaptic augmentation: seconds, more effective use of Ca2+.(4) Post-tetanic potentiation (PTP): seconds to minutes, better mobilization
of SV pools (more phosphorylation of synapsin to mobilize SVs)
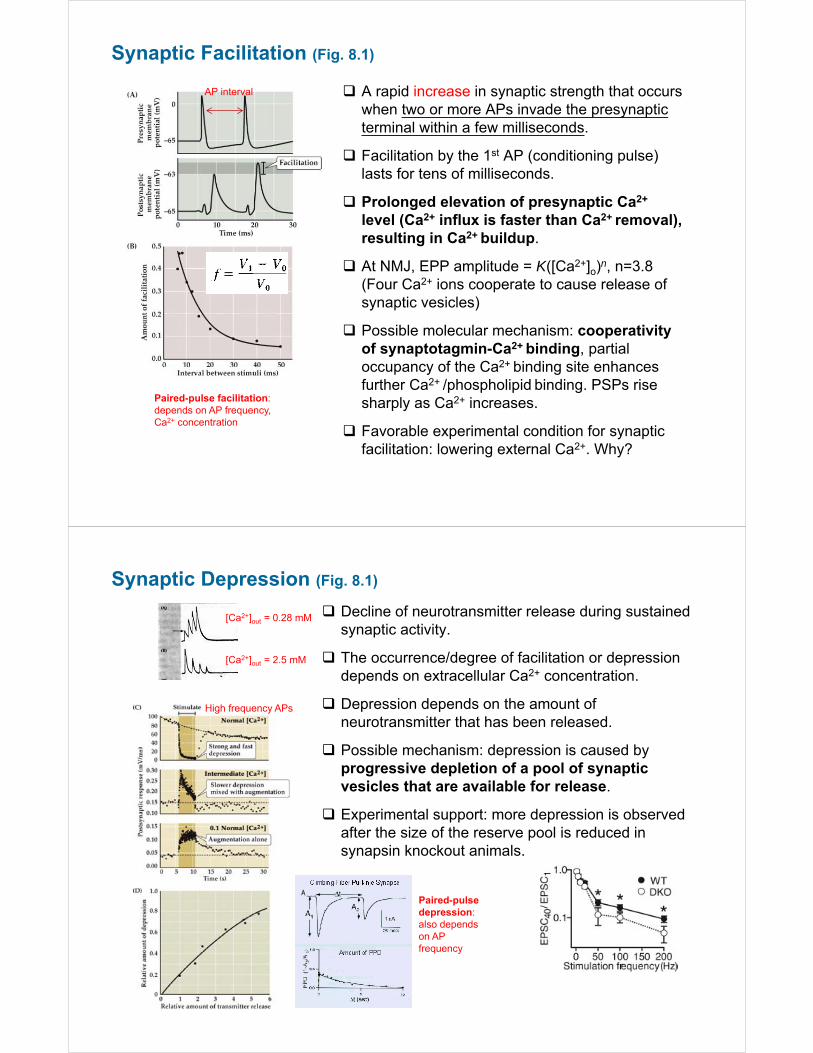
Synaptic Facilitation (Fig. 8.1)
A rapid increase in synaptic strength that occurs when two or more APs invade the presynaptic terminal within a few milliseconds.
Facilitation by the 1st AP (conditioning pulse) lasts for tens of milliseconds.
Prolonged elevation of presynaptic Ca2+
level (Ca2+ influx is faster than Ca2+ removal), resulting in Ca2+ buildup.
At NMJ, EPP amplitude = K([Ca2+]o)n, n=3.8 (Four Ca2+ ions cooperate to cause release of synaptic vesicles)
Possible molecular mechanism: cooperativity of synaptotagmin-Ca2+ binding, partial occupancy of the Ca2+ binding site enhances further Ca2+ /phospholipid binding. PSPs rise sharply as Ca2+ increases.
Favorable experimental condition for synaptic facilitation: lowering external Ca2+. Why?
AP interval
Paired-pulse facilitation: depends on AP frequency, Ca2+ concentration
Synaptic Depression (Fig. 8.1)
Decline of neurotransmitter release during sustained synaptic activity.
The occurrence/degree of facilitation or depression depends on extracellular Ca2+ concentration.
Depression depends on the amount of neurotransmitter that has been released.
Possible mechanism: depression is caused by progressive depletion of a pool of synaptic vesicles that are available for release.
Experimental support: more depression is observed after the size of the reserve pool is reduced in synapsin knockout animals.
[Ca2+]out = 0.28 mM
[Ca2+]out = 2.5 mM
High frequency APs
Paired-pulse depression: also depends on AP frequency
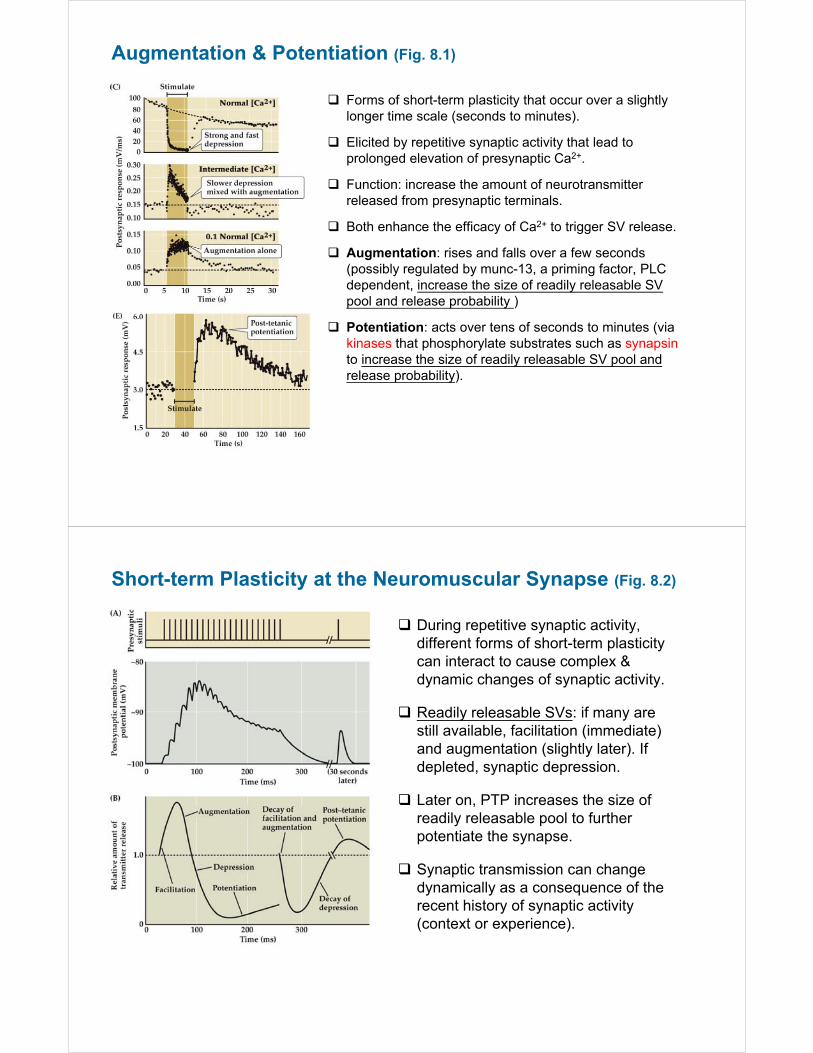
Augmentation & Potentiation (Fig. 8.1)
Forms of short-term plasticity that occur over a slightly longer time scale (seconds to minutes).
Elicited by repetitive synaptic activity that lead to prolonged elevation of presynaptic Ca2+.
Function: increase the amount of neurotransmitter released from presynaptic terminals.
Both enhance the efficacy of Ca2+ to trigger SV release.
Augmentation: rises and falls over a few seconds (possibly regulated by munc-13, a priming factor, PLC dependent, increase the size of readily releasable SV pool and release probability )
Potentiation: acts over tens of seconds to minutes (via kinases that phosphorylate substrates such as synapsinto increase the size of readily releasable SV pool and release probability).
Short-term Plasticity at the Neuromuscular Synapse (Fig. 8.2)
During repetitive synaptic activity, different forms of short-term plasticity can interact to cause complex & dynamic changes of synaptic activity.
Readily releasable SVs: if many are still available, facilitation (immediate) and augmentation (slightly later). If depleted, synaptic depression.
Later on, PTP increases the size of readily releasable pool to further potentiate the synapse.
Synaptic transmission can change dynamically as a consequence of the recent history of synaptic activity (context or experience).

Long-term Synaptic Plasticity
Long-term synaptic plasticity alter synaptic transmission over time scales of 30 minutes or longer.
Arises from molecular mechanisms that vary over time (short to long):(1) post-translational modifications (e.g. phosphorylation) of existing proteins
(changes in AMPA receptor trafficking)(2) changes in translations (local translation)(3) changes in gene transcription + translation (producing enduring changes
in synaptic transmission).
Two major forms: (1) long-term potentiation (LTP): long-lasting increase in synaptic strength(2) long-term depression (LTD): long-lasting decrease in synaptic strength
Changes in synaptic efficacy that are likely the basis for:(1) certain forms of learning(2) storage of memories(3) refinement of synaptic connections during development
Hippocampus (Fig. 8.6)
A cortical structure in the medial portion of the temporal lobe (medial temporal lobe in primates).
Critical for the consolidation of information from short-term memory to long-term memory and spatial navigation & spatial memory.
Different types of neurons and collaterals (axon bundles) are organized in an orderly and relatively planar manner that is conducive to slice preparation electrophysiology. Most of the relevant circuitry is left intact in a hippocampus slice.

The Tri-synaptic Circuit of the Hippocampus (Fig. 8.6)
Tri-synaptic circuit: 3 axon bundles (perforant path, mossy fibers & Schaffer collaterals) + 3 main neuronal types (granule cells, CA3 & CA1 pyramidal neurons)
Input from entorhinal cortex (via perforantpath) => granule cell (via mossy fibers) => CA3 pyramidal cell (via Schaffer collaterals) => CA1 pyramidal cell.
Excitatory glutamatergic connections.
Clusters of monosynaptic connections.CA: cornu ammonis (ram’s horn)
Long-term Potentiation (LTP)
Granule cells
Perforant path
Extracellular recording
High frequency stimulation
A few seconds of high-frequency electrical stimulation can enhance synaptic transmission in the rabbit hippocampus for days.
Inducing LTP: a tetanus (high-frequency electrical stimulus, e.g. 100 Hz).
Recording: extracellular recording to measure amplitude or the slope of field potential (collective EPSPs) or intracellular patch-clamp recording to measure the amplitude of EPSPs.
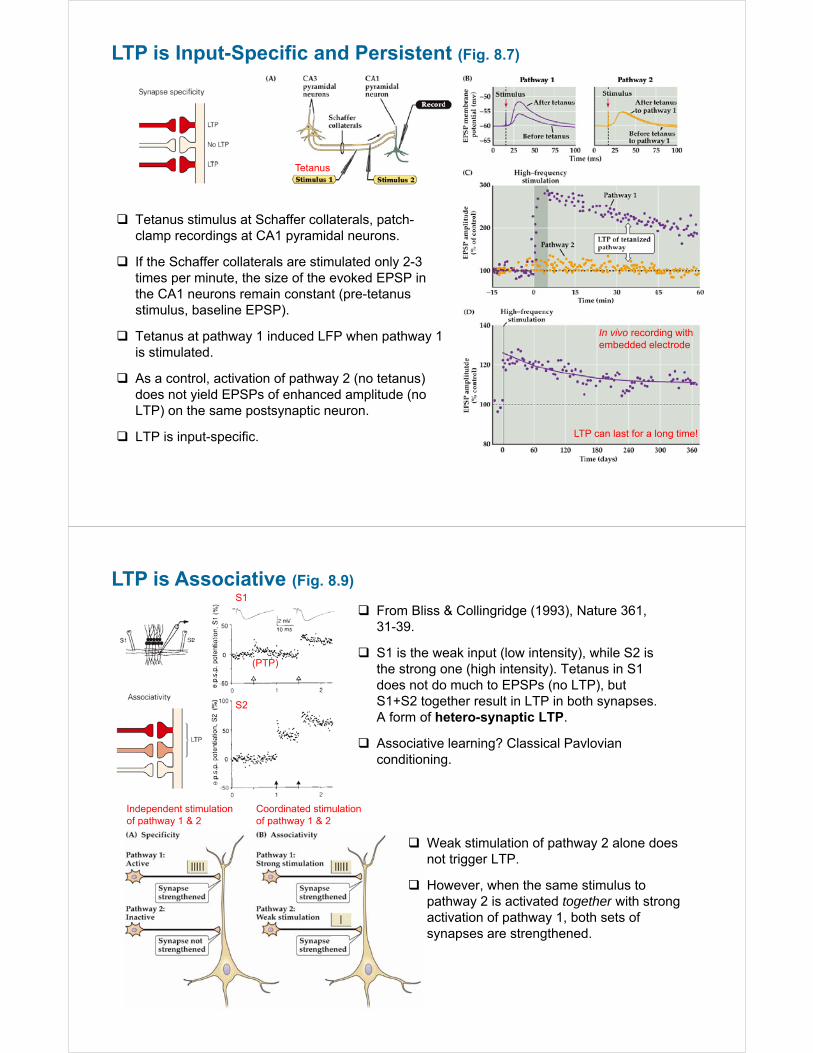
LTP is Input-Specific and Persistent (Fig. 8.7)
Tetanus stimulus at Schaffer collaterals, patch-clamp recordings at CA1 pyramidal neurons.
If the Schaffer collaterals are stimulated only 2-3 times per minute, the size of the evoked EPSP in the CA1 neurons remain constant (pre-tetanus stimulus, baseline EPSP).
Tetanus at pathway 1 induced LFP when pathway 1 is stimulated.
As a control, activation of pathway 2 (no tetanus) does not yield EPSPs of enhanced amplitude (no LTP) on the same postsynaptic neuron.
LTP is input-specific.
Tetanus
In vivo recording with embedded electrode
LTP can last for a long time!
LTP is Associative (Fig. 8.9)
Independent stimulation of pathway 1 & 2
Coordinated stimulation of pathway 1 & 2
From Bliss & Collingridge (1993), Nature 361, 31-39.
S1 is the weak input (low intensity), while S2 is the strong one (high intensity). Tetanus in S1 does not do much to EPSPs (no LTP), but S1+S2 together result in LTP in both synapses. A form of hetero-synaptic LTP.
Associative learning? Classical Pavlovian conditioning.
Weak stimulation of pathway 2 alone does not trigger LTP.
However, when the same stimulus to pathway 2 is activated together with strong activation of pathway 1, both sets of synapses are strengthened.
(PTP)
S1
S2

LTP involves Coincidence Detection (Fig. 8.8)
(in the CA1 neurons)
Associative LTP suggests that LTP occurs when both presynaptic and postsynaptic neurons are active. That is, the presynaptic neurons fires action potentials and the postsynaptic membrane is depolarized.
To test, low intensity tetanus at Schaffer collateral => no LTP in CA1 neurons.
Paired the low-intensity stimulus with strong depolarization of the CA1 neurons, within 100 ms of neurotransmitter release from the Schaffer collaterals => result in LTP.
LTP occurs only when presynaptic action potentials and postsynaptic depolarization are tightly linked in time => a coincidence detector.
LTP: Potential for Information Storage
Input-specific: when LTP is induced by activation of one synapse, it does not occur in other, inactive synapses that contact the same neuron. Selective storage of information likely takes place at synapses, not neurons.
State-dependent: pairing of pre- and postsynaptic events, coincidence detector.
Hebbian Principle: Donald Hebb, a Canadian psychologist, in 1949 stated “when an axon of cell A is near enough to excite a cell B and repeatedly or persistently takes part in firing it, some growth process or metabolic change takes place in one or both cells such that A ' s efficiency, as one of the cells firing B, is increased”. In other words, coordinated activity of a presynaptic terminal and postsynaptic neuron would strengthen the synaptic connection between them, as observed in LTP much later.
A simplified version of Hebb’s rule: neurons that fire together wire together.
LTP at synapses shows many properties for learning.
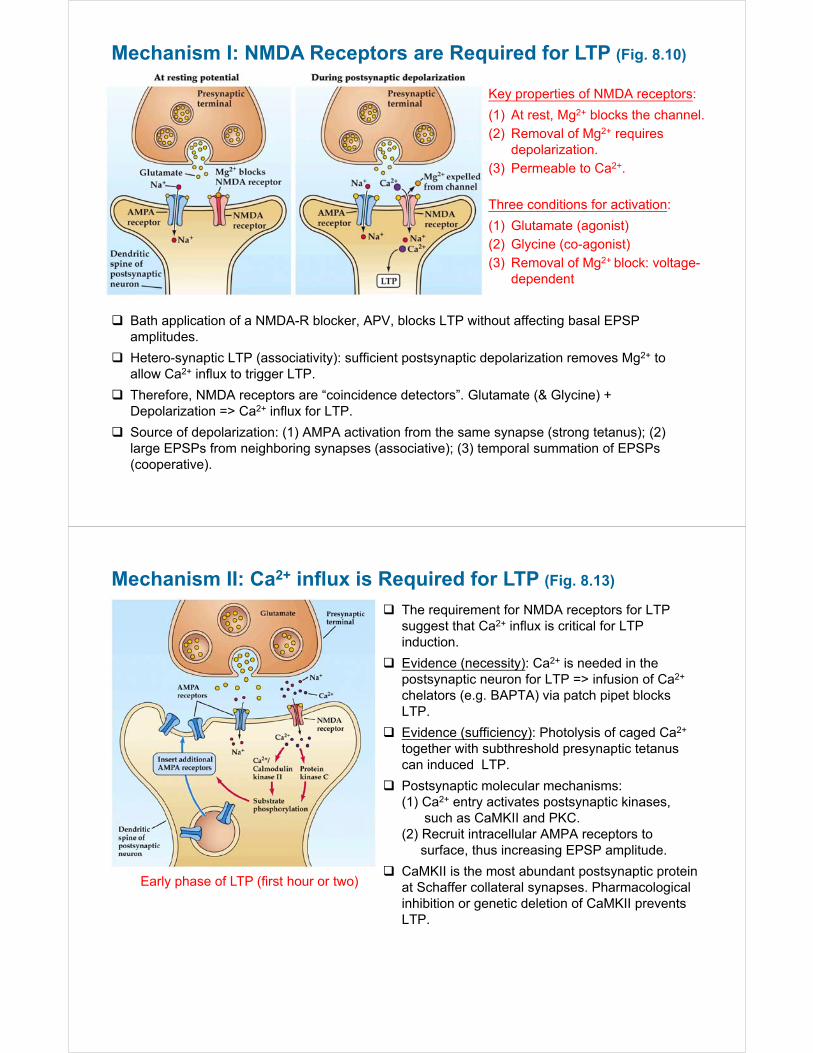
Mechanism I: NMDA Receptors are Required for LTP (Fig. 8.10)
Bath application of a NMDA-R blocker, APV, blocks LTP without affecting basal EPSP amplitudes.
Hetero-synaptic LTP (associativity): sufficient postsynaptic depolarization removes Mg2+ to allow Ca2+ influx to trigger LTP.
Therefore, NMDA receptors are “coincidence detectors”. Glutamate (& Glycine) + Depolarization => Ca2+ influx for LTP.
Source of depolarization: (1) AMPA activation from the same synapse (strong tetanus); (2) large EPSPs from neighboring synapses (associative); (3) temporal summation of EPSPs (cooperative).
Key properties of NMDA receptors:
(1) At rest, Mg2+ blocks the channel.(2) Removal of Mg2+ requires
depolarization.(3) Permeable to Ca2+.
Three conditions for activation:
(1) Glutamate (agonist)(2) Glycine (co-agonist)(3) Removal of Mg2+ block: voltage-
dependent
Mechanism II: Ca2+ influx is Required for LTP (Fig. 8.13)
The requirement for NMDA receptors for LTP suggest that Ca2+ influx is critical for LTP induction.
Evidence (necessity): Ca2+ is needed in the postsynaptic neuron for LTP => infusion of Ca2+
chelators (e.g. BAPTA) via patch pipet blocks LTP.
Evidence (sufficiency): Photolysis of caged Ca2+
together with subthreshold presynaptic tetanus can induced LTP.
Postsynaptic molecular mechanisms:(1) Ca2+ entry activates postsynaptic kinases,
such as CaMKII and PKC.(2) Recruit intracellular AMPA receptors to
surface, thus increasing EPSP amplitude.
CaMKII is the most abundant postsynaptic protein at Schaffer collateral synapses. Pharmacological inhibition or genetic deletion of CaMKII prevents LTP.
Early phase of LTP (first hour or two)
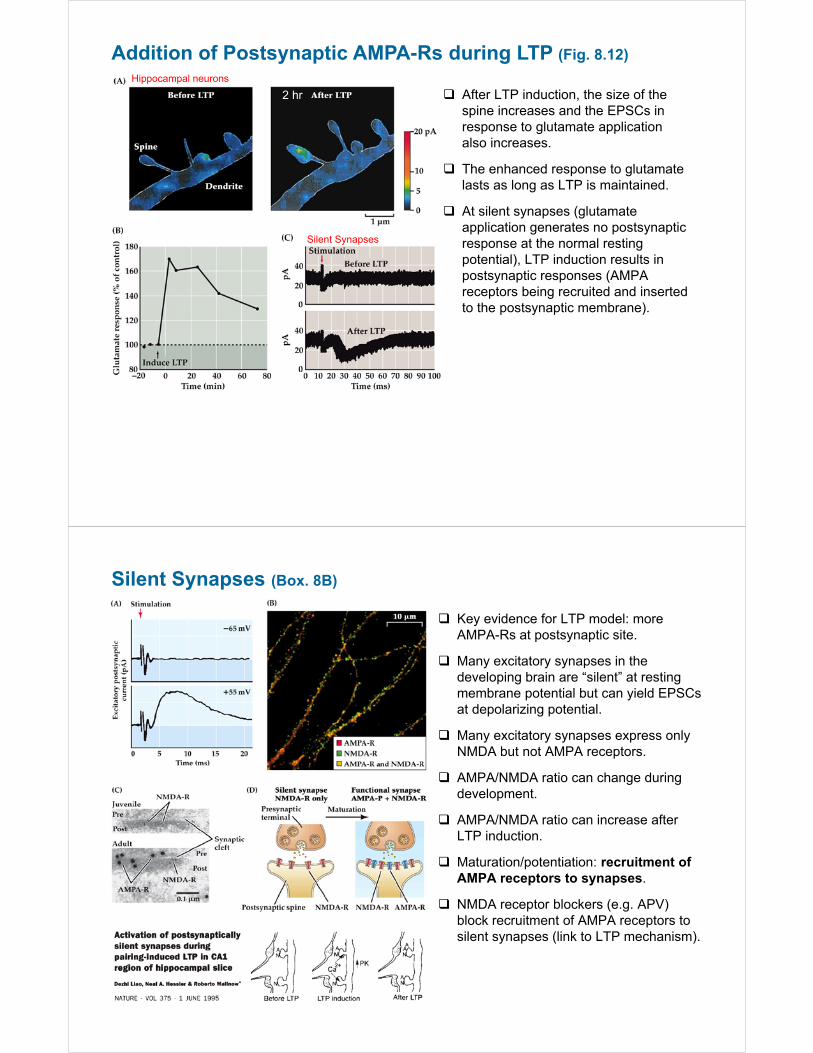
Addition of Postsynaptic AMPA-Rs during LTP (Fig. 8.12)
2 hr
Hippocampal neurons
After LTP induction, the size of the spine increases and the EPSCs in response to glutamate application also increases.
The enhanced response to glutamate lasts as long as LTP is maintained.
At silent synapses (glutamate application generates no postsynaptic response at the normal resting potential), LTP induction results in postsynaptic responses (AMPA receptors being recruited and inserted to the postsynaptic membrane).
Silent Synapses
Silent Synapses (Box. 8B)
Key evidence for LTP model: more AMPA-Rs at postsynaptic site.
Many excitatory synapses in the developing brain are “silent” at resting membrane potential but can yield EPSCs at depolarizing potential.
Many excitatory synapses express only NMDA but not AMPA receptors.
AMPA/NMDA ratio can change during development.
AMPA/NMDA ratio can increase after LTP induction.
Maturation/potentiation: recruitment of AMPA receptors to synapses.
NMDA receptor blockers (e.g. APV) block recruitment of AMPA receptors to silent synapses (link to LTP mechanism).
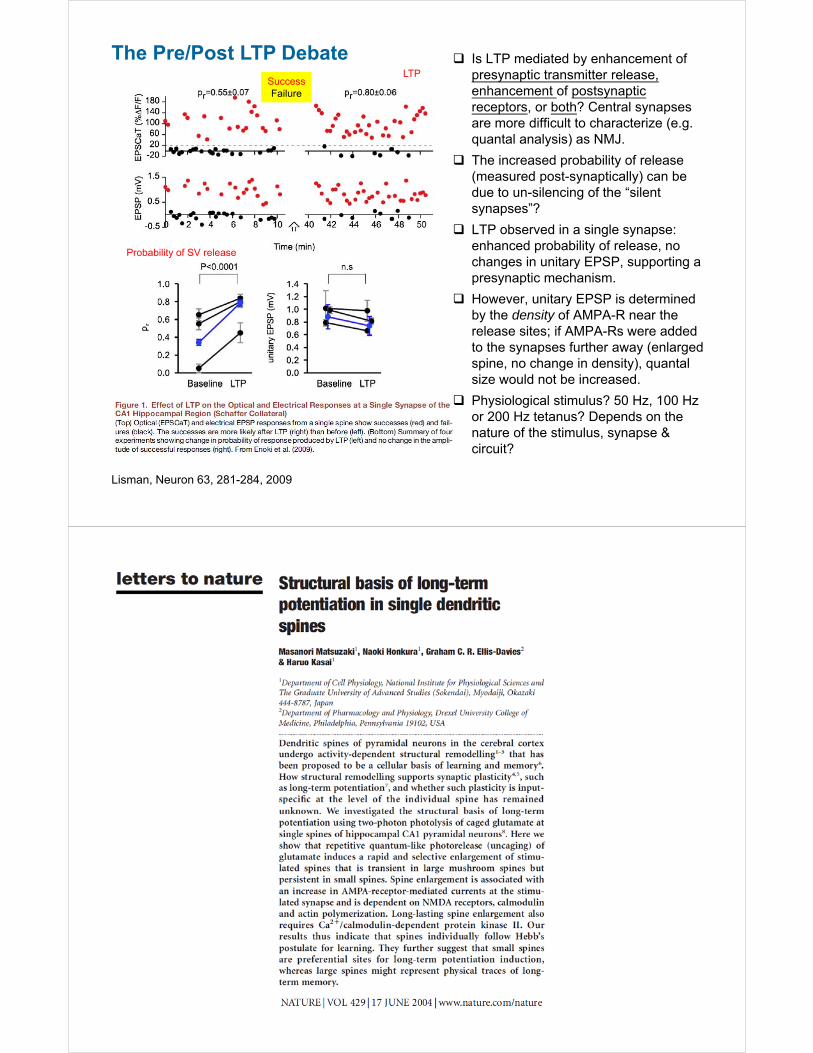
The Pre/Post LTP Debate
Lisman, Neuron 63, 281-284, 2009
Is LTP mediated by enhancement of presynaptic transmitter release, enhancement of postsynaptic receptors, or both? Central synapses are more difficult to characterize (e.g. quantal analysis) as NMJ.
The increased probability of release (measured post-synaptically) can be due to un-silencing of the “silent synapses”?
LTP observed in a single synapse: enhanced probability of release, no changes in unitary EPSP, supporting a presynaptic mechanism.
However, unitary EPSP is determined by the density of AMPA-R near the release sites; if AMPA-Rs were added to the synapses further away (enlarged spine, no change in density), quantal size would not be increased.
Physiological stimulus? 50 Hz, 100 Hz or 200 Hz tetanus? Depends on the nature of the stimulus, synapse & circuit?
SuccessFailure
LTP
Probability of SV release

Background: In the cortex, dendritic spines undergo activity-dependent structural remodeling (structural plasticity). What is the link between spine structural changes and LTP? Is it input specific at the level of individual spines? Entirely regulated postsynaptically?
Experiments: Using two-photon photolysis (two-photon uncaging) of caged glutamate to stimulate a single spine at hippocampal CA1 pyramidal neuron (instead of electrical stimulation of Schaffer collaterals). This stimulus protocol completely bypass presynaptic requirement. Test the effect of repetitive uncaging on spine morphology and AMPA/NMDA currents. Test the requirement of CaMKII in activity-dependent spine morphological and electrophysiological changes.
Fig. 1. Spine-head enlargement induced by repetitive uncaging of MNI-glutamate.
Hippocampal Slice
Sparse labeling of CA1 neurons with GFP
Caged glutamate (MNI-glutamate)
Two-photon laser beam: ~1 m (single spine)stimulatedneighboring spineUncaging
Electrical Stimulation (Schaffer collaterals)
Rapid structural change (within ~60s)
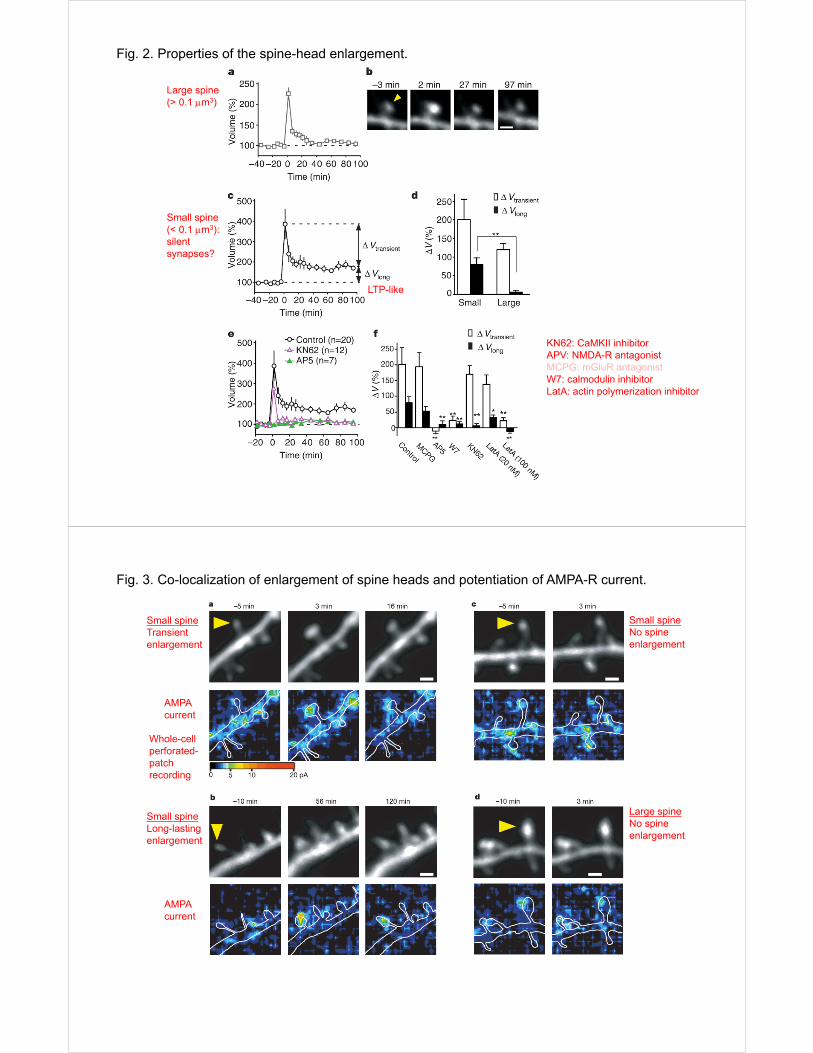
Fig. 2. Properties of the spine-head enlargement.
Large spine(> 0.1 m3)
Small spine(< 0.1 m3): silent synapses?
KN62: CaMKII inhibitorAPV: NMDA-R antagonistMCPG: mGluR antagonistW7: calmodulin inhibitorLatA: actin polymerization inhibitor
LTP-like
Fig. 3. Co-localization of enlargement of spine heads and potentiation of AMPA-R current.
Small spineTransient enlargement
Small spineLong-lasting enlargement
AMPA current
AMPA current
Small spineNo spine enlargement
Large spineNo spine enlargement
Whole-cellperforated-patch recording
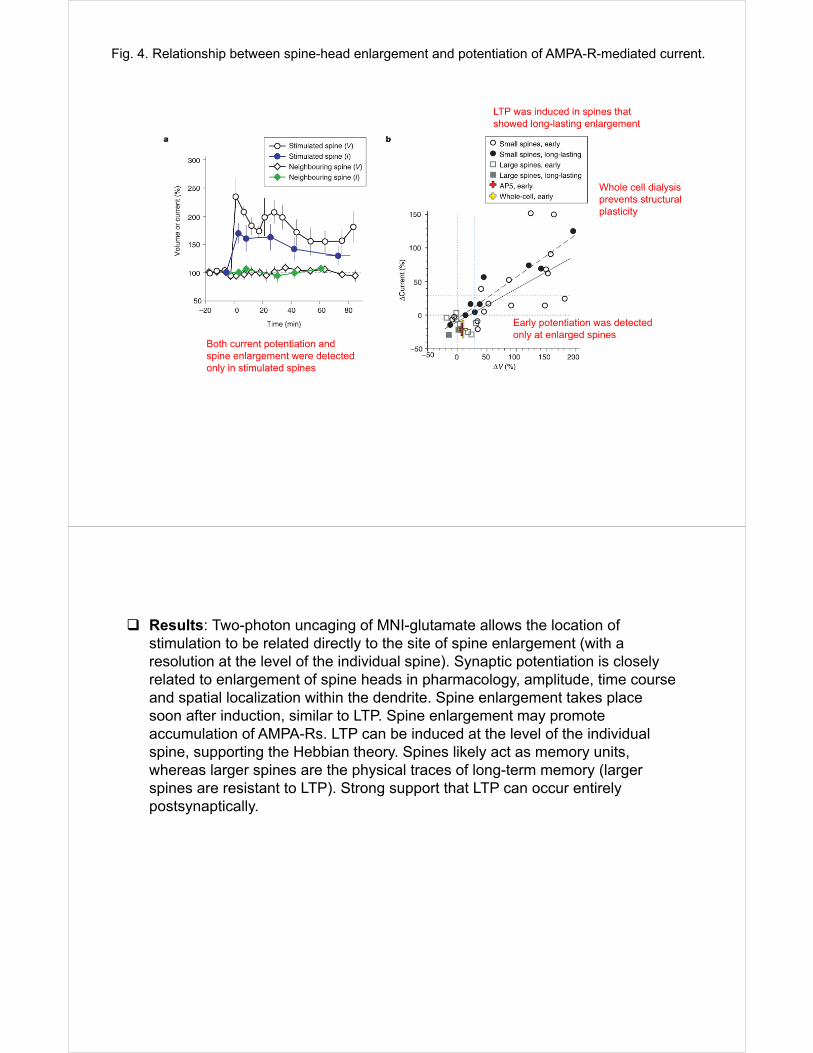
Fig. 4. Relationship between spine-head enlargement and potentiation of AMPA-R-mediated current.
Early potentiation was detected only at enlarged spines
LTP was induced in spines that showed long-lasting enlargement
Both current potentiation and spine enlargement were detected only in stimulated spines
Whole cell dialysis prevents structural plasticity
Results: Two-photon uncaging of MNI-glutamate allows the location of stimulation to be related directly to the site of spine enlargement (with a resolution at the level of the individual spine). Synaptic potentiation is closely related to enlargement of spine heads in pharmacology, amplitude, time course and spatial localization within the dendrite. Spine enlargement takes place soon after induction, similar to LTP. Spine enlargement may promote accumulation of AMPA-Rs. LTP can be induced at the level of the individual spine, supporting the Hebbian theory. Spines likely act as memory units, whereas larger spines are the physical traces of long-term memory (larger spines are resistant to LTP). Strong support that LTP can occur entirely postsynaptically.


















