
BioSB meeting 2015
25
Scaffolding using long nanopore reads and more Hans Jansen Christiaan Henkel senior scientist
-
Upload
hansjansen9999 -
Category
Science
-
view
55 -
download
0
Transcript of BioSB meeting 2015

Scaffolding using long nanopore reads
and more
Hans Jansen
Christiaan Henkelsenior scientist

Dutch SME at Bioscience Park in Leiden , the Netherlands
• High throughput drug screens, and toxicity assays in zebrafish larvae
• Fish fertility (eel, pike perch, sole) to aid sustainable aquaculture
• Sequencing (genomes, transcriptomes)• Bioinformatics
ZF-screens B.V.

Genome projects
Common carp (Cyprinus carpio)High troughput screening modelGenome and transcriptomes
European and Japanese eel (Anguilla anguilla and Anguilla japonica)Completing the life cycle in aquacultureGenome and transcriptomes
King cobra (Ophiophagus hannah)Evolution and toxinsGenome and transcriptomes
But the quality of these genomes can be improved

But MAP is much more. It is about being a community and a playground to test new applications. As Gordon Sanghera (CEO of ONT) said "MAP will never end. There will always be a MAP“.
So if you think you're application can benefit from nanopore sensing then come join MAP and play with us.
Visible as a web portal with information from ONT and social media like system with blog possibilities, comment, likes, and a forum to ask advice.
MinION Access Program

We entered when MAP started.Our first MinION arrived in April 2014 and the first kits in June.Since then run 30 Flow Cells.
MAPpers competition Topped the leaderboard on read length and yield so we now have three MinION's.
MinION Access Program and ZF-genomics

Longest 2D read: 93.5 KbpLongest template read: 120 Kbp (231 Kbp)Highest yield: 1.32 Gevents
R7
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 280
50000000
100000000
150000000
200000000
250000000
300000000
350000000
template and 2D yield over the past year
template2D
Runs
Base pairs sequenced (Mbp)
R7.3R6

Scaffolding genomes using long readsor
How to untangle the assembly graph

Cheap short read sequencing technology has been used to generate many draft genomes
repeatunique sequence in unique sequence out
Draft genomes made with short read data suffer from a fundamental problem.Reads that are shorter than the length of a repeat can’t connect the unique sequence in with the unique sequence out
Genomic sequences
Short reads

repeatunique sequence in unique sequence out
Long reads can help to resolve repeat area’s in the assembly graph
And the resulting contigs will now look like this:
Untangle

1. Short read correction Quake (not for small genomes)2. Short read assembly Velvet3. MinION read alignment to Velvet contigs LAST4. Link filtering and contig tiling Untangle script5. Path detachment around repeats Untangle script6. Bubble popping Untangle script7. Delete unconfirmed connections Untangle script8. Contig extraction Untangle script
Assembly and scaffolding strategy
Task Software

Agrobacterium strain NCPPB 1771
Agrobacteria are the cause of crown gall disease, a tumorous growth of plant tissue.
Agrobacteria transfer part of their (plasmid) DNA to their host and this feature is used widely in plant research to genetically modify plants.
Agrobacteria have two chromosomes, and carry several plasmids. This strain also carries active transposons.
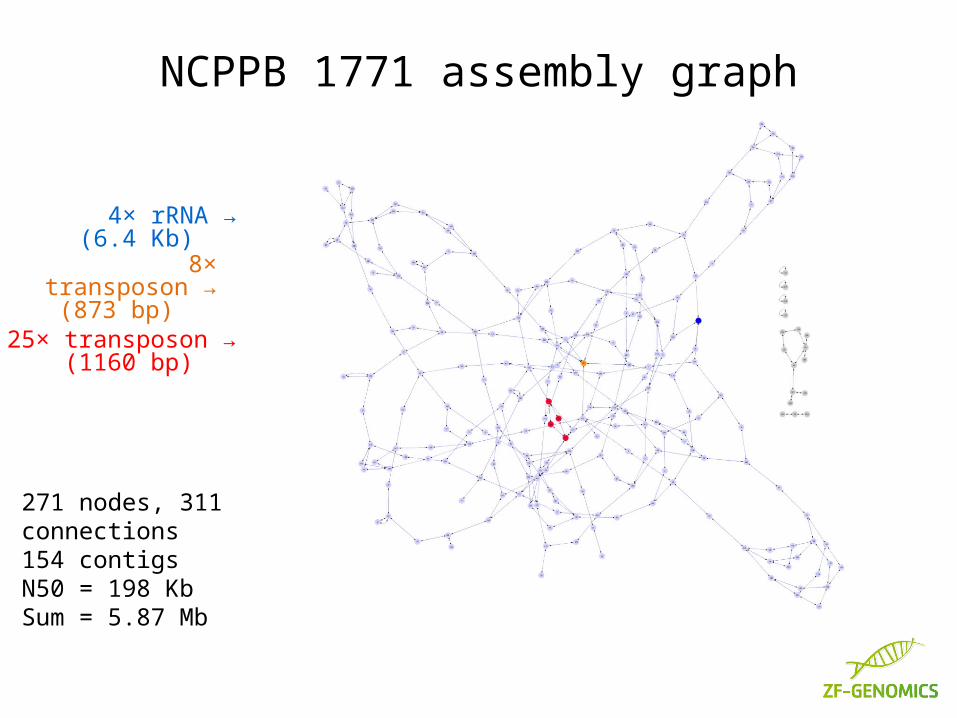
NCPPB 1771 assembly graph
25× transposon →(1160 bp)
8× transposon →(873 bp)
4× rRNA →(6.4 Kb)
271 nodes, 311 connections154 contigsN50 = 198 KbSum = 5.87 Mb

• Alignment: LAST with optimized settings
• Links: alignment filtering and contig tiling
• 7328 reads aligned to contigs
• 438 reads aligned to multiple contigs
• 585 links between contigs
• 13158 reads on R6 and R7 chemistry
• 73.8 Mb total yield (template and 2D)
• 5–85970 nt length, typical ~12 Kb
MinION sequencing and scaffolding
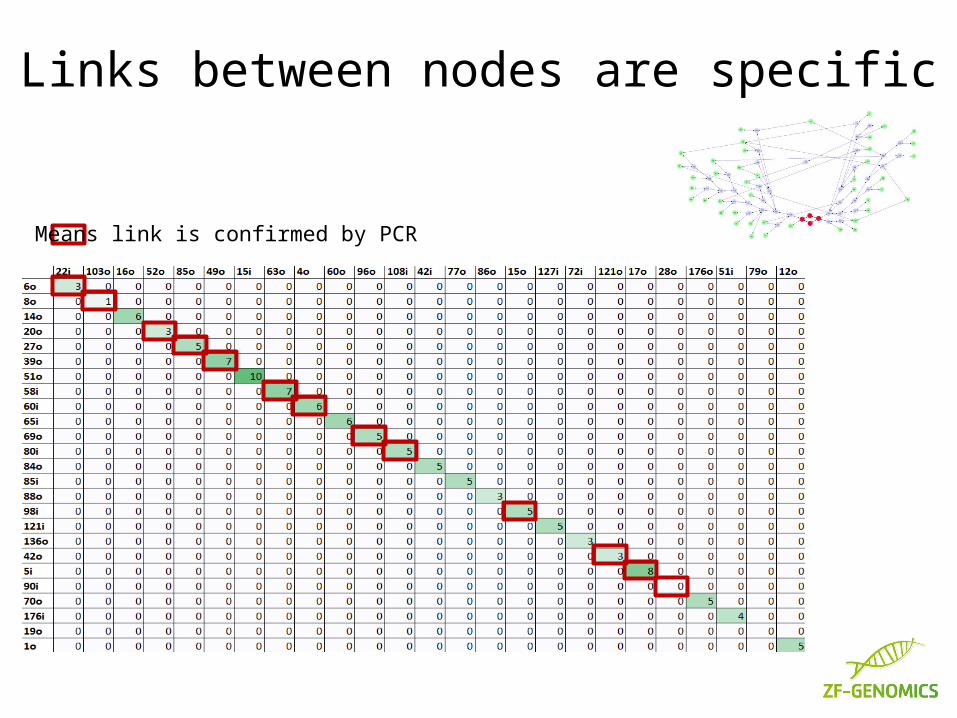
Links between nodes are specific
Means link is confirmed by PCR

Final assembly graph after scaffolding
• 271 nodes + 312 connections → 49 nodes + 5 connections• 154 contigs → ~8 contigs• Complete chromosome 2 (1.2 Mb), pTi (190 Kb), cryptic megaplasmid (746 Kb)• Slight residual fragmentation of chromosome 1

MinION Analysis and Reference Consortium
MARC is a consortium within MAP that seeks to establish sources of variation, optimize protocols and analysis.
It is open science. Data is shared in the consortium and will be made available through ENA.
~100 people have signed up. ~7 experimental groups and ~4 analysis groups are actively working.
Managed by weekly TC.

Different phases in MARC
Phase 1 is about being as standard as possible and establish variation in the system and between sites.This is done by 5 labs in the Netherlands, UK (2), USA ( east and west coast).
Phase 2 is all about tweaking the protocol. Things like DNA isolation, shearing (or not), running scripts, DNA modifications will be addressed in this phase.
Phase 3 is about examples of applications.
MinION Analysis and Reference Consortium

MinION Analysis and Reference Consortium
In phase 1 the 5 participating labs received Escherichia coli str. K-12 substr. MG1655.
Performed DNA isolation, library prep, and sequencing according to a detailed protocol.
Per lab a total of 4 libraries with 2 different kits were prepared and run.
This provides a excellent data set to understand sources of variance in ONT data.
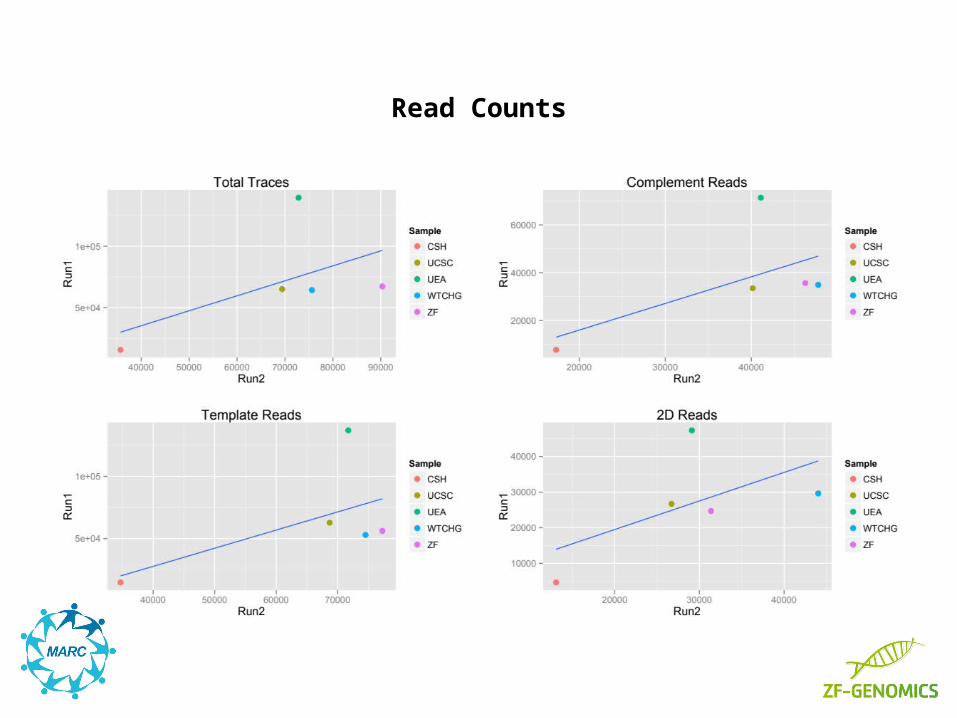
Read Counts

Read Length Statistics

Read Alignments

With the data of the first 10 runs analyzed we can already see that read length has a stronger lab effect than base pair identity to the reference.
Another set of 10 phase 1 runs is currently being analyzed and will give a clearer picture on variability.
Experiments for phase 2 will start shortly, while in parallel phase 3 experiments and analysis are being done.
Conclusions and perspectives

The king cobra genomeRapid expansion of the 3 FTx gen family in the king cobra

London Calling 2015
Highlights from Clive Brown’s talk
• Improvements to the basecaller . There’s still room for improvement.• Read until (and barcoding).• Fast mode on the MinION MkI (500 bp/sec instead of 30)• New 3000 channel ASIC with crumpet chip design to separate ASIC and fluidics part.• MinION MkII and PromethION will have this new ASIC.• Library prep on beads to reduce amounts of DNA needed (lower ng to pg).• Direct RNA sequencing.• Simplified sample preparation and VolTRAX.• Pricing will be “pay as you go”. Initial payment for hardware include some hrs sequencing.• MkI $270 and 3 hrs sequencing (~3 Gbp in fast mode).

Acknowledgements
Prof. Dr. Paul Hooykaas, Leiden University
Christiaan Henkelsenior scientist
Leiden University
Ron Dirks (CEO of ZF-screens B.V.)
All members of the MARC consortiumEwan Birney, EMBL-EBIJustin O’Grady, UEASara Goodwin, CSHL David Buck, WTCHG OxfordVadim Zalunin, EMBL-EBIMiten Jain, UCSCMatt Loose, NottinghamJared Simpson, OICR, Toronto


















