
Bioorthogonal PET probes -...
11
. Bioorthogonal PET probes … … serve as highly valuable tools in the field of pretargeted molecular imaging. In their Communication on page 9655 ff., H. Mikula, C. Kuntner, and co-workers describe the development, synthesis, and characterization of a low-molecular-weight 18 F-labeled tetrazine derivative that can be used for bioorthogonal PET imaging of dienophile- tagged (bio)molecules through the application of in vivo chemistry.
Transcript of Bioorthogonal PET probes -...

.Bioorthogonal PET probes …… serve as highly valuable tools in the field of pretargeted molecular imaging. In theirCommunication on page 9655 ff., H. Mikula, C. Kuntner, and co-workers describe thedevelopment, synthesis, and characterization of a low-molecular-weight 18F-labeledtetrazine derivative that can be used for bioorthogonal PET imaging of dienophile-tagged (bio)molecules through the application of in vivo chemistry.

Bioorthogonal ImagingDOI: 10.1002/anie.201404277
Development of a 18F-Labeled Tetrazine with FavorablePharmacokinetics for Bioorthogonal PET Imaging**Christoph Denk, Dennis Svatunek, Thomas Filip, Thomas Wanek, Daniel Lumpi,Johannes Frçhlich, Claudia Kuntner,* and Hannes Mikula*
Abstract: A low-molecular-weight 18F-labeled tetrazine deriv-ative was developed as a highly versatile tool for bioorthogonalPET imaging. Prosthetic groups and undesired carrying of 18Fthrough additional steps were evaded by direct 18F-fluorinationof an appropriate tetrazine precursor. Reaction kinetics of thecycloaddition with trans-cyclooctenes were investigated byapplying quantum chemical calculations and stopped-flowmeasurements in human plasma; the results indicated that thelabeled tetrazine is suitable as a bioorthogonal probe for theimaging of dienophile-tagged (bio)molecules. In vitro and invivo investigations revealed high stability and PET/MRI inmice showed fast homogeneous biodistribution of the 18F-labeled tetrazine that also passes the blood–brain barrier. An invivo click experiment confirmed the bioorthogonal behavior ofthis novel tetrazine probe. Due to favorable chemical andpharmacokinetic properties this bioorthogonal agent shouldfind application in bioimaging and biomedical research.
Bioorthogonal imaging applying in vivo chemistry hasemerged as a highly versatile tool in chemical biology andbiomedicine.[1] Since the development of copper-free clickchemistry for bioimaging using the strain-promoted azide–alkyne cycloaddition (SPAAC),[2] this bioorthogonal ligationhas been used for a wide variety of applications with the aimto examine the molecular details of biological processes.[3]
Although reaction kinetics were successively improvedduring the last years,[4] rate constants of SPAAC ligationsare still limited (< 5m!1 s!1, acetonitrile, 20 8C) and thereforethis chemistry is not fully suitable for bioconjugations in vivo.Nevertheless, in vivo SPAAC has recently been applied forthe development of a pretargeted imaging approach usingpositron emission tomography (PET).[5] To circumvent prob-
lems in terms of (relatively) low reaction rates of SPAACligations, Fox et al. and Weissleder et al. have independentlyintroduced the inverse-electron-demand Diels–Alder(IEDDA)-initiated conjugation between 1,2,4,5-tetrazines(Tz) and strained cycloalkenes as a highly efficient bioor-thogonal ligation.[6] Since then, this reaction has attractedbroad attention and has been extensively used in manybiomedical applications including the development of bio-orthogonal probes, live-cell imaging, protein labeling, and thesynthesis of PET imaging agents.[7] Furthermore, IEDDA wassuccessfully applied in combination with SPAAC for simulta-neous multitarget imaging.[8] Recently, Carlson et al. reportedthe development of advanced bioorthogonal turn-on probesthat apply through-bond energy transfer leading to fluores-cence quenching within boron dipyrromethene (BODIPY)-tetrazine derivatives.[9] Reaction kinetics of IEDDA ligationswere further improved by the development and application ofthe bicyclo[6.1.0]non-4-ene (s-TCO) moiety, an even morestrained trans-cyclooctene derivative, as a highly reactivedienophile leading to tetrazine ligations with rate constants ofup to approximately 20000m!1 s!1 (MeOH, 25 8C).[10] Inaddition to commonly used trans-cyclooctenes (TCO) and s-TCO,[11] cyclopropene (CP) tags have recently been reportedas useful dienophiles with lower steric demand and havesuccessfully been applied in live-cell imaging.[12]
Despite many applications of IEDDA ligations in the fieldof fluorescence imaging, the development of bioorthogonalPET approaches is still restricted due to a lack of efficientradiolabeled agents that distribute homogenously and showfast in vivo reaction kinetics, high stability, and adequateclearance. 18F-Labeling of dienophiles such as TCO andnorbornene was preferred in terms of pretargetingapproaches applying IEDDA bioconjugations so far due tothe previously observed and reported insufficient stability oftetrazines during direct fluorination.[13] Yet, this approach isstill cumbersome due to the fact that instead of readilyavailable cycloalkene tags the tetrazine moiety has to beattached to the pretargeting probe. Furthermore, a tetrazine-type radiolabeled agent would be capable of being applied forbioorthogonal imaging of various dienophiles and thusprovide high versatility. A 11C-labeled tetrazine was initiallydeveloped by Herth et al.[14] and metal radionuclides such as64Cu and 89Zr were attached to tetrazines using chelatingligands resulting in relatively large and complex bioorthog-onally imaging agents.[15] Furthermore, polymer-modifiedtetrazines were developed and used for pretargeted PETimaging.[16] However, the development of a readily accessibleradiolabeled low-molecular-weight tetrazine taking advant-age of the high specific radioactivity of 18F (allowing admin-
[*] C. Denk, D. Svatunek, Dr. D. Lumpi, Prof. J. Frçhlich, Dr. H. MikulaInstitut f!r Angewandte SynthesechemieTechnische Universit"t Wien (TUW) (Austria)E-mail: [email protected]
Dr. T. Filip, Dr. T. Wanek, Priv.-Doz. Dr. C. KuntnerHealth and Environment Department, Biomedical SystemsAustrian Institute of Technology (AIT)Seibersdorf (Austria)E-mail: [email protected]
[**] Financial support from the Vienna Anniversary Foundation forHigher Education (H-2500/2012) is gratefully acknowledged. Wethank Prof. Wolfgang Linert (VUT), Dr. Roland M!ller (SeibersdorfLaboratories), Dominik Matscheko (VUT), and Walter Kuba (VUT)for their kind support during this work. PET = positron emissiontomography.
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/anie.201404277.
AngewandteChemie
9655Angew. Chem. Int. Ed. 2014, 53, 9655 –9659 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

istration of low-mass dosages and shorter radiation expo-sures) is still significant, which inspired us to focus on thisparticular problem in the field of bioorthogonal PET imaging.
Herein we report our results on the synthesis andcharacterization of a low-molecular-weight 18F-labeled tetra-zine with favorable pharmacokinetics that is applicable asa versatile tool for pretargeted PET imaging using in vivoIEDDA chemistry.
During the development of improved procedures for thesynthesis of 1,2,4,5-tetrazines, the characterization of a varietyof these compounds revealed an inverse correlation betweentheir stability in fetal bovine serum and the reaction rates oftheir cycloaddition with dienophiles.[17] Therefore, wehypothesized that stability problems during direct tetrazinefluorination can be avoided or minimized by using lessreactive 3,6-dialkyltetrazines that still possess sufficientlyhigh reaction rates for pretargeted in vivo applications. Theavailability of physiologically stable radiolabeled tetrazines iscrucial for the development of reliable PET imagingapproaches. In addition to this trade-off (reactivity vs.stability), we intended to develop water-soluble low-molec-ular-weight compounds with the aim to obtain imaging agentswith favorable pharmacokinetic properties. Theseconsiderations led to the chemical structure of
tetrazine 1. The two alkyl substitu-ents were chosen to be as small asreasonable. A derivative witha methyl group was preferred
since it is more stable than an unsubstituted tetrazine,and a three-methylene spacer was consideredbetween the fluorine and the tetrazine moiety toavoid the undesired high reactivity of benzyl-type precursors(Tz-CH2-X) as well as possible elimination during nucleo-philic fluorination (Tz-CH2CH2-X!Tz-CH=CH2). Quantumchemical calculations on the basis of a recently reportedcomputational approach (DFT, M06-2X/6-31G(d,p), Gaus-sian 09)[18] were applied to estimate the reaction kinetics(Table 1) of 1 in comparison to that of phenyl-substitutedtetrazines (e.g. 2),[19] which are commonly used in combina-tion with TCO tags for various bioorthogonal ligationapplications.[20] As expected, modeling of IEDDA reactionswith trans-cyclooctene (3) showed an increased activationfree energy for 1 (18.8 kcalmol!1) relative to that for 2(15.0 kcal mol!1), but this difference is compensated when s-TCO 4 is used for cycloaddition with 1 (15.3 kcalmol!1). Thus,applying the bioorthogonal reaction pair 1/4 should result inreasonable rate constants similar to those of the frequentlyapplied ligation between 2 and 3. This assumption wasverified by stopped-flow measurements after preparation of1 starting from 4-hydroxypropanenitrile (6) applying metal-catalyzed tetrazine synthesis[21] followed by DAST-mediatedfluorination of the intermediate 7 (Scheme 1). Furthermore,a significantly increased reaction rate was observed for thecycloaddition of 1 with the water-soluble s-TCO derivative 5in human plasma (k2> 8000m!1 s!1, 37 8C), which is in agree-ment with previously reported studies on the effect of wateron IEDDA reaction kinetics (Table 1).[22]
Regarding the preparation of compound 7, we havestudied the impact of different reaction parameters on
tetrazine synthesis and have mainly focused on the reagentgrade of hydrazine. Finally, we did not observe significantlydecreased yields of 7 when the monohydrate (18 %) was usedinstead of anhydrous hydrazine (21 %) that was originallyused for the metal-catalyzed preparation of 3,6-dialkyltetra-zines.[21] This is of particular interest, since the availability ofanhydrous hydrazine has been restricted in Europe since 2011due to the REACH regulation of the European ChemicalsAgency.[23] Although commercially available hydrazine cya-nurate can be used as a reagent for the safe preparation ofanhydrous hydrazine of high purity,[24] reliable syntheticmethods using the monohydrate or other forms (e.g. hydra-zine salts) should be preferred.
The in vitro stability of 1 in human plasma was assessed byspectrophotometric measurements and HPLC. Almost nodegradation was observed after incubation at 37 8C for 12 h(99 % recovery) further revealing the capability of 1 asa promising scaffold for the development of a radiolabeledagent for pretargeted PET imaging. Although the excellentstability of 4 (no degradation in human serum, 30 mmbutylamine in MeOH or 5 mm ethanethiol in MeOH) wasshown by Taylor et al. ,[10] they also observed isomerization(58 % after 3.5 h) to the unreactive cis isomer at a highconcentration of ethanethiol (30 mm in MeOH). Further-more, Rossin et al. have recently reported that trans-cyclo-octenes are probably isomerized by interactions with copper-containing proteins; they also presented strategies to circum-vent this problem by increasing the steric bulk of the tag.[25]
Table 1: Reaction kinetics of IEDDA cycloadditions obtained by compu-tational methods (M06-2X/6-31G(d,p), Gaussian 09) and stopped-flowmeasurements.
Tetrazine DP[a] Solvent DG!calcd
[b] DG! [c] k2 [m!1 s!1][d]
1 3 1,4-dioxane 18.7 17.5 1.49"0.011 4 1,4-dioxane 15.3 15.0 85.5"2.31 5 human plasma n.d.[e] n.d. 8200"3002 3 1,4-dioxane 15.0 15.1 79.2"1.3
[a] DP= dienophile. [b] Calculated activation free energies (kcalmol!1;25 8C). [c] Activation free energies determined by stopped-flow meas-urements (kcalmol!1; 25 8C). [d] Second-order rate constants (meas-urements at 37 8C). [e] n.d.= not determined.
Scheme 1. Synthesis of tetrazine 1 (DAST=diethylaminosulfur trifluoride).
.AngewandteCommunications
9656 www.angewandte.org ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2014, 53, 9655 –9659

The stability of compound 5 was assessed by incubation inhuman plasma at 37 8C for 24 h leading to a recovery of> 80%, a significantly higher value than those previouslyreported for reactive tetrazines.[17] Hence, the combination ofradiolabeled tetrazines as pull-down reagents and (s-)TCOlabeling should be advantageous for bioorthogonal in vivoimaging in contrast to inverse strategies involving tetrazinetags and radiolabeled dienophiles.
Radiosynthesis affording [18F]-1 was successfully carriedout by direct fluorination of the tosylated intermediate 8 ina radiochemical yield of up to 18 % (Figure 1). Cycloadditionof [18F]-1 with s-TCO 4 occurred rapidly forming theconjugate [18F]-9 that slowly isomerized to [18F]-10 (bothproducts as a mixture of regio- and stereoisomers).
In vivo studies were performed by administration of [18F]-1 to female BALB/c mice followed by dynamic PET (n = 4)and PET/MR (n = 2) imaging (MR = magnetic resonancetomography). A very homogenous biodistribution of [18F]-1 was observed in all regions in the body including the brain,which clearly indicates the ability of [18F]-1 to pass the blood–brain barrier (Figure 2). Furthermore, PET/MR revealed nosignificant extent of in vivo defluorination of [18F]-1, since noenhanced radioactivity uptake in bone was observed.[26]
Time–activity curves were similar for all analyzed organs
(Figure 3a). After the initial perfusion directly after intra-venous administration of [18F]-1 a very uniform uptakesteadily decreasing from 2 to 1.5 SUV (standardized uptakevalue) was observed in all organs (except the urinary bladder)during the onset of the PET scan. Conversely, SUV in theurinary bladder increased from 2 to 10 SUV, suggesting
Figure 1. a) Radiosynthesis of [18F]-1 and IEDDA cycloaddition withs-TCO 4 (n.c.a. =no carrier added; only one isomer shown for [18F]-9and [18F]-10). b) HPLC of 1 (UV/Vis, 540 nm), radio-HPLC of purified[18F]-1 and ligation products after click reaction with s-TCO 4. HPLCconditions: Zorbax SB-Aq reversed phase, H2O/MeCN gradient elu-tion.
Figure 2. Combined PET/MRI summation images: 0–5 min (a) and 5–120 min (b) after injection of [18F]-1.
Figure 3. a) Mean organ time–activity curves obtained in BALB/c mice(n = 6; for bone n= 2). b) Ex vivo biodistribution of [18F]-1 in indicatedorgans 2 h after intravenous administration in mice (n= 6). Data arepresented as mean SUV ! standard deviation. All organs (apart fromthe bladder) displayed similar radioactivity concentration values.
AngewandteChemie
9657Angew. Chem. Int. Ed. 2014, 53, 9655 –9659 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org

a rapid renal clearance of [18F]-1. Evaluation of theex vivo biodistribution determined 120 min after[18F]-1 injection gave values ranging from 1 SUV to1.5 SUV with the exception of the urinary bladder(Figure 3b), which is in good agreement with thePET results. The in vivo stability of [18F]-1 wasfurther examined by radio-TLC of mouse plasmaobtained immediately after the imaging experiments(120 min after administration). We observed onlya low degree of metabolic degradation (approxi-mately 15%) as shown by the formation of morepolar metabolites, which is assumed to occur viaenzyme-catalyzed oxidation of the methyl group of[18F]-1 thus leading to xenobiotic metabolizationsimilar to toluene and related compounds.[27]
Furthermore, the bioconjugation of [18F]-1 andthe water-soluble dienophile 5 was performed in vivoto verify the bioorthogonal behavior of this noveltetrazine-type PET probe.[18F]-1 was administered tofemale BALB/c mice (n = 4) followed by 5 after aninitial period of 20 min. In this experiment an inverseapproach was used to allow for the homogenousbiodistribution of the radiolabeled tetrazine prior toin vivo reaction. After 5, 15, and 30 min retroorbitalblood was collected and the click reaction wasquenched by addition of the more reactive tetrazine 2.Murine plasma was obtained and analyzed using radio-TLC(Figure 4). Finally, we observed > 90 % conversion after5 min and complete in vivo reaction 30 min after adminis-tration of 5 (see the Supporting Information, Figure S4).These results clearly indicate and confirm the capability of[18F]-1 as a highly valuable and versatile bioorthogonal probefor pretargeted PET imaging.
In summary, we have developed a fast and reliableprocedure for the synthesis of a low-molecular-weight andwater-soluble radiolabeled tetrazine; direct [18F]-fluorinationwas applied to avoid additional steps and the need forprosthetic groups. Furthermore, [18F]-1 was prepared usingreadily accessible and/or commercially available reagentswithout the use of anhydrous hydrazine and [18F]-1 was shownto possess highly favorable pharmacokinetic properties.Although high stability of [18F]-1 was observed, its metabolicbehavior has still to be verified in further studies. In vivoinvestigations clearly indicate the suitability of this radiola-beled tetrazine as a pull-down reagent for pretargeted PETimaging. Hence, we expect this highly versatile and readilyaccessible compound, which enables in vivo detection ofdienophile-tagged (bio)molecules, to find many applicationsand lead to further investigations in the fields of bioorthog-onal imaging and biomedical research.
Received: April 13, 2014Revised: May 9, 2014Published online: July 2, 2014
.Keywords: bioorthogonal chemistry · click chemistry ·imaging agents · kinetics · tetrazines
[1] a) L. Carroll, H. L. Evans, E. O. Aboagye, A. C. Spivey, Org.Biomol. Chem. 2013, 11, 5772 – 5781; b) N. K. Devaraj, R.Weissleder, Acc. Chem. Res. 2011, 44, 816 – 827; c) E. M. Sletten,C. R. Bertozzi, Acc. Chem. Res. 2011, 44, 666 – 676.
[2] J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V.Chang, I. A. Miller, A. Lo, J. A. Codelli, C. R. Bertozzi, Proc.Natl. Acad. Sci. USA 2007, 104, 16793 – 16797.
[3] a) S. T. Laughlin, J. M. Baskin, S. L. Amacher, C. R. Bertozzi,Science 2008, 320, 664 – 667; b) A. B. Neef, C. Schultz, Angew.Chem. 2009, 121, 1526 – 1529; Angew. Chem. Int. Ed. 2009, 48,1498 – 1500; c) T. Plass, S. Milles, C. Koehler, C. Schultz, E. A.Lemke, Angew. Chem. 2011, 123, 3964 – 3967; Angew. Chem. Int.Ed. 2011, 50, 3878 – 3881; d) R. Xie, S. Hong, X. Chen, Curr.Opin. Chem. Biol. 2013, 17, 747 – 752; e) J. C. Jewett, C. R.Bertozzi, Chem. Soc. Rev. 2010, 39, 1272 – 1279; f) J. C. Jewett,E. M. Sletten, C. R. Bertozzi, J. Am. Chem. Soc. 2010, 132, 3688 –3690.
[4] G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan,C. R. Bertozzi, Angew. Chem. 2012, 124, 2493 – 2497; Angew.Chem. Int. Ed. 2012, 51, 2443 – 2447.
[5] S. B. Lee, H. L. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn, D. W.Kim, Angew. Chem. 2013, 125, 10743 – 10746; Angew. Chem. Int.Ed. 2013, 52, 10549 – 10552.
[6] a) M. L. Blackman, M. Royzen, J. M. Fox, J. Am. Chem. Soc.2008, 130, 13518 – 13519; b) N. K. Devaraj, R. Weissleder, S. A.Hilderbrand, Bioconjugate Chem. 2008, 19, 2297 – 2299.
[7] a) G. Budin, K. S. Yang, T. Reiner, R. Weissleder, Angew. Chem.2011, 123, 9550 – 9553; Angew. Chem. Int. Ed. 2011, 50, 9378 –9381; b) K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters,J. W. Chin, Nat. Chem. 2012, 4, 298 – 304; c) K. S. Yang, G. Budin,T. Reiner, C. Vinegoni, R. Weissleder, Angew. Chem. 2012, 124,6702 – 6707; Angew. Chem. Int. Ed. 2012, 51, 6598 – 6603; d) D. S.Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox,A. Y. Ting, J. Am. Chem. Soc. 2011, 134, 792 – 795; e) J. L.Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W.Rhoads, R. B. Cooley, C. Refakis, J. M. Fox, R. A. Mehl, J.Am. Chem. Soc. 2012, 134, 2898 – 2901; f) E. Kim, K. S. Yang, R.Weissleder, PLoS One 2013, 8, e81275; g) T. Reiner, E. J.
Figure 4. a) Bioorthogonal in vivo reaction of [18F]-1 and dienophile 5 leading tothe formation of [18F]-11 and [18F]-12 (one isomer shown). b) Radio-TLC (SiO2) ofplasma obtained after 5 min of reaction time (left) and blank obtained 35 minafter administration of [18F]-1 (right).
.AngewandteCommunications
9658 www.angewandte.org ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2014, 53, 9655 –9659

Keliher, S. Earley, B. Marinelli, R. Weissleder, Angew. Chem.2011, 123, 1963 – 1966; Angew. Chem. Int. Ed. 2011, 50, 1922 –1925.
[8] M. R. Karver, R. Weissleder, S. A. Hilderbrand, Angew. Chem.2012, 124, 944 – 946; Angew. Chem. Int. Ed. 2012, 51, 920 – 922.
[9] J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand, R. Weis-sleder, Angew. Chem. 2013, 125, 7055 – 7058; Angew. Chem. Int.Ed. 2013, 52, 6917 – 6920.
[10] M. T. Taylor, M. L. Blackman, O. Dmitrenko, J. M. Fox, J. Am.Chem. Soc. 2011, 133, 9646 – 9649.
[11] R. Selvaraj, J. M. Fox, Curr. Opin. Chem. Biol. 2013, 17, 753 –760.
[12] J. Yang, J. Seckute, C. M. Cole, N. K. Devaraj, Angew. Chem.2012, 124, 7594 – 7597; Angew. Chem. Int. Ed. 2012, 51, 7476 –7479.
[13] a) J. Seckute, N. K. Devaraj, Curr. Opin. Chem. Biol. 2013, 17,761 – 767; b) Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D.Brown, P. S. Conti, J. M. Fox, Chem. Commun. 2010, 46, 8043 –8045; c) J. C. Knight, S. Richter, M. Wuest, J. D. Way, F. Wuest,Org. Biomol. Chem. 2013, 11, 3817 – 3825.
[14] M. M. Herth, V. L. Andersen, S. Lehel, J. Madsen, G. M.Knudsen, J. L. Kristensen, Chem. Commun. 2013, 49, 3805 –3807.
[15] a) R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. M.Vulders, I. Verel, J. Lub, M. S. Robillard, Angew. Chem. 2010,122, 3447 – 3450; Angew. Chem. Int. Ed. 2010, 49, 3375 – 3378;b) B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D.Carlin, P. Zanzonico, R. Weissleder, J. S. Lewis, J. Nucl. Med.2013, 54, 1389 – 1396.
[16] N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli, R.Weissleder, Proc. Natl. Acad. Sci. USA 2012, 109, 4762 – 4767.
[17] M. R. Karver, R. Weissleder, S. A. Hilderbrand, BioconjugateChem. 2011, 22, 2263 – 2270.
[18] F. Liu, R. S. Paton, S. Kim, Y. Liang, K. N. Houk, J. Am. Chem.Soc. 2013, 135, 15642 – 15649.
[19] S. A. Lang, B. D. Johnson, E. Cohen, J. Heterocycl. Chem. 1975,12, 1143 – 1153.
[20] a) E. J. Keliher, T. Reiner, G. M. Thurber, R. Upadhyay, R.Weissleder, ChemistryOpen 2012, 1, 177 – 183; b) E. J. Keliher, T.Reiner, A. Turetsky, S. A. Hilderbrand, R. Weissleder, Chem-MedChem 2011, 6, 424 – 427; c) B. M. Zeglis, P. Mohindra, G. I.Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder, J. S.Lewis, Bioconjugate Chem. 2011, 22, 2048 – 2059; d) H.-S. Han,N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder, M. G.Bawendi, J. Am. Chem. Soc. 2010, 132, 7838 – 7839; e) J. B.Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee, R. Weissleder,Nat. Nanotechnol. 2010, 5, 660 – 665.
[21] J. Yang, M. R. Karver, W. Li, S. Sahu, N. K. Devaraj, Angew.Chem. 2012, 124, 5312 – 5315; Angew. Chem. Int. Ed. 2012, 51,5222 – 5225.
[22] J. W. Wijnen, S. Zavarise, J. B. F. N. Engberts, M. Charton, J.Org. Chem. 1996, 61, 2001 – 2005.
[23] Inclusion of Substances of Very High Concern in the CandidateList (2011). ED/31/2011 published in accordance with Article59(10) of the Regulation No 1907/2006 of the EuropeanParliament and of the Council of 18 December 2006 concerningthe Registration, Evaluation, Authorisation and Restriction ofChemicals (REACH).
[24] E. Nachbaur, G. Leiseder, Monatsh. Chem. 1971, 102, 1718 –1723.
[25] R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli,R. M. Versteegen, J. Lub, M. S. Robillard, Bioconjugate Chem.2013, 24, 1210 – 1217.
[26] a) R. A. Hawkins, Y. Choi, S.-C. Huang, C. K. Hoh, M.Dahlbom, C. Schiepers, N. Satyamurthy, J. R. Barrio, M. E.Phelps, J. Nucl. Med. 1992, 33, 633 – 642; b) E. Even-Sapir, E.Mishani, G. Flusser, U. Metser, Semin. Nucl. Med. 2007, 37, 462 –469.
[27] D. E. Chapman, T. J. Moore, S. R. Michener, G. Powis, DrugMetab. Dispos. 1990, 18, 929 – 936.
AngewandteChemie
9659Angew. Chem. Int. Ed. 2014, 53, 9655 –9659 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org

Bioorthogonale BildgebungDOI: 10.1002/ange.201404277
Entwicklung eines 18F-markierten Tetrazins mit vorteilhaften pharma-kokinetischen Eigenschaften f!r die bioorthogonale Positronenemissi-onstomographie**Christoph Denk, Dennis Svatunek, Thomas Filip, Thomas Wanek, Daniel Lumpi,Johannes Frçhlich, Claudia Kuntner* und Hannes Mikula*
Abstract: Es wurde ein niedermolekulares 18F-markiertes Te-trazin entwickelt, das im Bereich der bioorthogonalen Bild-gebung mithilfe von PET verwendet werden kann. Durch 18F-Fluorierung einer geeigneten Vorstufe konnten die Verwen-dung von prosthetischen Gruppen und die Notwendigkeitmehrerer radiochemischer Syntheseschritte vermieden werden.Die Reaktionskinetik der Cycloaddition mit trans-Cyclooc-tenen wurde mithilfe quantenchemischer Rechungen undStopped-Flow-Messungen in humanem Plasma untersucht.Eine hohe Stabilit!t konnte in vitro und in vivo nachgewiesenwerden, w!hrend PET/MR-Untersuchungen an M!usen eineschnelle homogene Biodistribution offenbarten. Anhand einesIn-vivo-Experiments konnte die Bioorthogonalit!t der neuar-tigen Tetrazinsonde best!tigt werden. Die hervorragenden Ei-genschaften dieser bioorthogonalen Sonde zur Lokalisierungund Visualisierung von Dienophil-markierten Molek"lenbilden die Grundlage f"r viele Anwendungsmçglichkeiten imBereich der molekularen Bildgebung.
Die bioorthogonale Bildgebung unter Anwendung von „In-vivo-Klick-Chemie“ hat sich im letzten Jahrzehnt als vielsei-tige und vorteilhafte Technologie im Bereich der chemischenBiologie und der Biomedizin etabliert.[1] Seit der Entwicklungder kupferfreien Klick-Chemie unter Anwendung der durcherhçhte Ringspannung beg!nstigten (strain-promoted) Azid-Alkin-Cycloaddition (SPAAC)[2] wurde diese bioorthogonaleLigation f!r eine Vielzahl von Anwendungen herangezogen,mit dem Ziel, biologische Prozesse auf molekularer Ebenebesser untersuchen und beschreiben zu kçnnen.[3] Obwohl dieReaktionsgeschwindigkeit dieser Ligation durch die Ent-wicklung optimierter Cyclooctine in den letzten Jahren suk-
zessiv verbessert wurde,[4] ist diese nach wie vor limitiert(< 5m!1 s!1, Acetonitril, 20 8C) und daher f!r schnelle In-vivo-Biokonjugationen nur unzureichend geeignet. Dennochwurde In-vivo-SPAAC erst k!rzlich f!r die Entwicklung eineszweistufigen (pretargeted) Positronenemissionstomographie-(PET)-Verfahrens angewendet (erste Stufe: Verabreichungeiner sich im Zielgewebe anreichernden Verbindung; zweiteStufe: Injektion einer radiomarkierten Sonde, die durch In-vivo-Ligation die Detektion und Visualisierung der erstenVerbindung ermçglicht).[5] Um Probleme in Verbindung mitden relativ geringen Reaktionsgeschwindigkeiten derSPAAC-Ligation zu umgehen, wurde unabh"ngig von Foxet al. und Weissleder et al. die durch eine Diels-Alder-Reak-tion mit inversem Elektronenbedarf (inverse electrondemand Diels–Alder, IEDDA) initiierte Konjugation von1,2,4,5-Tetrazinen (Tz) und gespannten Cycloalkenen (z. B.trans-Cyclooctenen (TCO)) als effiziente bioorthogonale Li-gation entwickelt.[6] Diese Reaktion wurde seither im Zugeeiner Vielzahl von biomedizinischen Applikationen verwen-det, darunter die Entwicklung von bioorthogonalen Sondenf!r die molekulare Bildgebung, die effiziente Modifikationvon Proteinen sowie die schnelle Synthese von radioaktivmarkierten Substanzen.[7] Zudem konnte IEDDA in Kombi-nation mit SPAAC f!r die simultane Bildgebung zweierZielsubstanzen (Targets) verwendet werden.[8] Die Verkn!p-fung eines Tetrazins mit einem Bodipy-Fluorophor ermçg-lichte die Entwicklung von hçchst effektiven fluorogenenbioorthogonalen Sonden, wie erst k!rzlich von Carlson et al.beschrieben.[9] Die Reaktionsgeschwindigkeit der IEDDA-Ligation konnte durch die Anwendung von substituiertenBicyclo[6.1.0]non-4-enen als deutlich reaktivere Dienophile(s-TCO, zus"tzlich gespannte (strained) trans-Cyclooctene))signifikant gesteigert werden, wodurch sich Reaktionsge-schwindigkeiten > 20 000m!1 s!1 (MeOH, 25 8C) erreichenlassen.[10] Um sterische Einfl!sse auf das Biomolek!l zu mi-nimieren, wurden zus"tzlich zu den etablierten Dienophilen(TCO und s-TCO)[11] neuartige, stabilisierte Cyclopropene(CP) mit deutlich geringerem Raumbedarf entwickelt, diebereits f!r die molekulare Bildgebung in lebenden Zellenangewendet wurden.[12]
Trotz einer Vielzahl von Anwendungen der IEDDA-Li-gation im Bereich der Fluoreszenzmikroskopie ist die Ent-wicklung von bioorthogonalen PET-Verfahren wegen desMangels an radiomarkierten bioorthogonalen Sonden mitgeeigneten Eigenschaften (homogene Biodistribution,schnelle In-vivo-Reaktion, hohe Stabilit"t, ad"quate Aus-scheidung) immer noch limitiert. Da die Radiosynthese von
[*] C. Denk, D. Svatunek, Dr. D. Lumpi, Prof. J. Frçhlich, Dr. H. MikulaInstitut f!r Angewandte SynthesechemieTechnische Universit"t Wien (TUW) (#sterreich)E-Mail : [email protected]
Dr. T. Filip, Dr. T. Wanek, Priv.-Doz. Dr. C. KuntnerHealth and Environment Department, Biomedical SystemsAustrian Institute of Technology (AIT)Seibersdorf (#sterreich)E-Mail : [email protected]
[**] Wir danken der Hochschuljubil"umsstiftung der Stadt Wien (H-2500/2012) f!r die Finanzierung und zudem Prof. Wolfgang Linert(TUW), Dr. Roland M!ller (Seibersdorf Laboratories), DominikMatscheko und Walter Kuba (beide TUW) f!r ihre Unterst!tzungdieser Arbeit.
Hintergrundinformationen zu diesem Beitrag sind im WWW unterhttp://dx.doi.org/10.1002/ange.201404277 zu finden.
.AngewandteZuschriften
9810 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2014, 126, 9810 –9814

18F-Tetrazinen wegen deren geringer Stabilit!t unter dennotwendigen Reaktionsbedingungen der direkten Fluorie-rung nicht realisiert werden konnte, wurde die 18F-Markie-rung von Dienophilen, wie TCO oder Norbornen, bislangbevorzugt.[13] Der Nachteil bei diesem Verfahren ist jedoch,dass anstelle der leicht verf"gbaren Cycloalkene eine Tetra-zinmodifikation am Zielmolek"l angebracht werden muss.Zudem kçnnte ein radiomarkiertes Tetrazin f"r die bioor-thogonale Bildgebung unterschiedlicher Dienophile einge-setzt werden. Die erstmalige Radiosynthese eines 11C-mar-kierten Tetrazins wurde von Herth et al. vorgestellt,[14] undmetallische Radionuklide wie 64Cu oder 89Zr wurden mit Te-trazinen unter Verwendung von Chelatliganden verkn"pft.[15]
Des Weiteren wurden Polymer-modifizierte Tetrazine entwi-ckelt und bereits f"r zweistufige PET-Verfahren angewen-det.[16] In all diesen F!llen wurden jedoch relativ große undkomplexe Sonden hergestellt und verwendet. Die Entwick-lung von einfach verf"gbaren oder herzustellenden radio-markierten Tetrazinsonden unter Verwendung des Radioiso-tops 18F ist daher von unver!nderter Wichtigkeit, auch umwegen der hohen spezifischen Radioaktivit!t dieses Nuklidseine Verabreichung geringerer Mengen zu ermçglichen. Ausdiesem Grund konzentrierten wir uns auf dieses spezielleProblem im Bereich der bioorthogonalen Bildgebung.
Hier berichten wir "ber unsere Resultate der Syntheseund Charakterisierung eines niedermolekularen 18F-mar-kierten Tetrazins mit vorteilhaften pharmakokinetischen Ei-genschaften, das als vielseitige Sonde f"r die Entwicklung vonzweistufigen bioorthogonalen PET-Verfahren unter Verwen-dung der IEDDA-Reaktion eingesetzt werden kann.
Im Zuge der Entwicklung optimierter Syntheseverfahrenf"r die Herstellung von unterschiedlichen 1,2,4,5-Tetrazinenergaben weitere Untersuchungen ein indirekt proportionalesVerh!ltnis zwischen der Reaktivit!t und der Stabilit!t (infetalem K!lberserum) dieser Verbindungen.[17] Aufgrunddieser Befunde vermuteten wir, dass das Problem der gerin-gen Stabilit!t bei der direkten Radiofluorierung durch dieVerwendung von weniger reaktiven 3,6-Dialkyltetrazinenvermieden oder minimiert werden kann. Die Verf"gbarkeitvon physiologisch stabilen radiomarkierten Tetrazinen istausschlaggebend f"r die Entwicklung zuverl!ssiger PET-Verfahren. Zus!tzlich zu diesem Kompromiss (Reaktivit!tgegen"ber Stabilit!t) fokussierten wir uns zudem auf was-serlçsliche und niedermolekulare Verbindungen, mit demZiel, optimale pharmakokinetische Eigenschaften zu errei-
chen. Diese #berlegungen f"hrten schließ-lich zu der chemischen Struktur des Tetra-zins 1, wobei beide Substituenten so kleinwie mçglich gew!hlt wurden. Ein Derivatmit Methylgruppe wurde einem unsubsti-
tuierten Tetrazin wegen der hçheren Stabilit!t vorgezogen,und drei Methylengruppen zwischen dem Tetrazinring unddem Fluoratom wurden in Betracht gezogen, um einerseitseine hohe Reaktivit!t und dadurch geringere Stabi-lit!t einer entsprechenden Benzylvorstufe (Tz-CH2-X) sowie mçgliche Eliminierungsreaktionen (Tz-CH2CH2-X!Tz-CH=CH2) vermeiden zu kçnnen. Eswurden quantenchemische Rechnungen unter An-wendung von bereits beschriebenen Methoden
(DFT, M06-2X/6-31G(d,p), Gaussian 09)[18] angewendet, umeine erste Absch!tzung der Reaktionsgeschwindigkeit von1 im Vergleich zu jener von Phenyl-substituierten Tetrazinen
(z. B. 2),[19] die h!ufig in Kombination mit TCO verwendetwerden,[20] zu erhalten (Tabelle 1). Entsprechend den Erwar-tungen ergab die Modellierung der IEDDA-Reaktionen mittrans-Cycloocten (3) eine erhçhte freie Aktivierungsenthal-
pie f"r 1 (18.8 kcal mol!1) gegen"ber 2 (15.0 kcal mol!1). Al-lerdings wird dieser Unterschied durch die Verwendung vons-TCO 4 f"r die Cycloaddition mit 1 (15.3 kcalmol!1) kom-pensiert. Die Anwendung des bioorthogonalen Reaktanten-paares 1/4 sollte daher zu ausreichenden Reaktionsge-schwindigkeiten !hnlich jener der h!ufig angewendeten Li-gation zwischen 2 und 3 f"hren. Diese Annahme wurde inweiterer Folge durch Stopped-Flow-Messungen best!tigt,wobei 1 zuvor ausgehend von 4-Hydroxypropannitril (6)hergestellt wurde. Eine metallkatalysierte Tetrazinsynthese,gefolgt von einer direkten Fluorierung des Intermediates 7durch Reaktion mit DAST, f"hrte zur Zielverbindung(Schema 1). Des Weiteren konnten wir eine deutlich gestei-gerte Reaktionsgeschwindigkeit der Ligation von 1 mit demwasserlçslichen s-TCO-Derivat 5 in humanem Plasma fest-stellen (k2> 8000m!1 s!1, 37 8C), und zwar in #bereinstim-mung mit fr"heren Berichten "ber den Effekt von Wasser aufdie IEDDA-Reaktionskinetik (Tabelle 1).[22]
Tabelle 1: Untersuchung der Reaktionskinetik der IEDDA-Cycloadditionunter Anwendung von quantenchemischen Rechungen (M06-2X/6-31G-(d,p), Gaussian 09) und Stopped-Flow-Messungen.
Tetrazin DP[a] Lçsungsmittel DG!ber.
[b] DG![c] k2 [m!1 s!1][d]
1 3 1,4-Dioxan 18.7 17.5 1.49"0.011 4 1,4-Dioxan 15.3 15.0 85.5"2.31 5 Humanplasma n.b.[e] n.b. 8200"3002 3 1,4-Dioxan 15.0 15.1 79.2"1.3
[a] DP= Dienophil. [b] Berechnete freie Aktivierungsenthalpien (kcalmol!1; 25 8C). [c] Freie Aktivierungsenthalpien, ermittelt durch Stopped-Flow-Messungen (kcalmol!1; 25 8C). [d] Geschwindigkeitskonstanten(Messungen bei 37 8C). [e] n.b.= nicht berechnet.
Schema 1. Synthese des Tetrazins 1 (DAST= Diethylaminoschwefeltrifluorid).
AngewandteChemie
9811Angew. Chem. 2014, 126, 9810 –9814 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
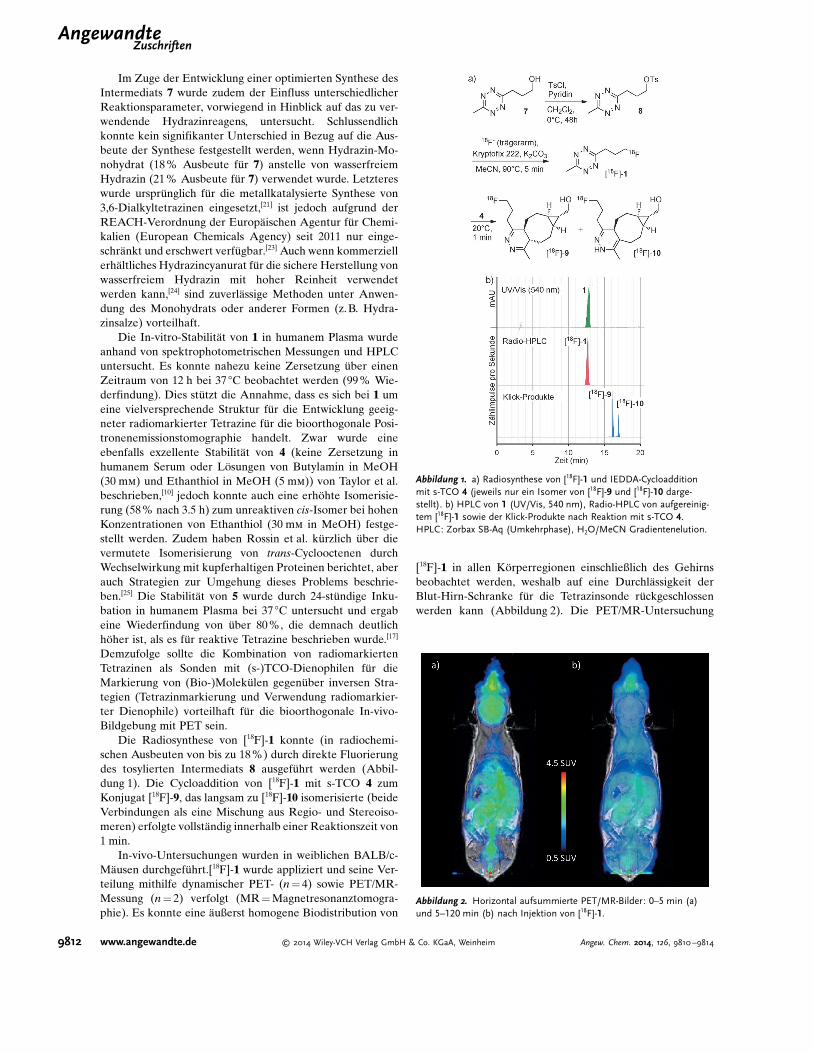
Im Zuge der Entwicklung einer optimierten Synthese desIntermediats 7 wurde zudem der Einfluss unterschiedlicherReaktionsparameter, vorwiegend in Hinblick auf das zu ver-wendende Hydrazinreagens, untersucht. Schlussendlichkonnte kein signifikanter Unterschied in Bezug auf die Aus-beute der Synthese festgestellt werden, wenn Hydrazin-Mo-nohydrat (18 % Ausbeute f!r 7) anstelle von wasserfreiemHydrazin (21 % Ausbeute f!r 7) verwendet wurde. Letztereswurde urspr!nglich f!r die metallkatalysierte Synthese von3,6-Dialkyltetrazinen eingesetzt,[21] ist jedoch aufgrund derREACH-Verordnung der Europ"ischen Agentur f!r Chemi-kalien (European Chemicals Agency) seit 2011 nur einge-schr"nkt und erschwert verf!gbar.[23] Auch wenn kommerziellerh"ltliches Hydrazincyanurat f!r die sichere Herstellung vonwasserfreiem Hydrazin mit hoher Reinheit verwendetwerden kann,[24] sind zuverl"ssige Methoden unter Anwen-dung des Monohydrats oder anderer Formen (z. B. Hydra-zinsalze) vorteilhaft.
Die In-vitro-Stabilit"t von 1 in humanem Plasma wurdeanhand von spektrophotometrischen Messungen und HPLCuntersucht. Es konnte nahezu keine Zersetzung !ber einenZeitraum von 12 h bei 37 8C beobachtet werden (99 % Wie-derfindung). Dies st!tzt die Annahme, dass es sich bei 1 umeine vielversprechende Struktur f!r die Entwicklung geeig-neter radiomarkierter Tetrazine f!r die bioorthogonale Posi-tronenemissionstomographie handelt. Zwar wurde eineebenfalls exzellente Stabilit"t von 4 (keine Zersetzung inhumanem Serum oder Lçsungen von Butylamin in MeOH(30 mm) und Ethanthiol in MeOH (5 mm)) von Taylor et al.beschrieben,[10] jedoch konnte auch eine erhçhte Isomerisie-rung (58% nach 3.5 h) zum unreaktiven cis-Isomer bei hohenKonzentrationen von Ethanthiol (30 mm in MeOH) festge-stellt werden. Zudem haben Rossin et al. k!rzlich !ber dievermutete Isomerisierung von trans-Cyclooctenen durchWechselwirkung mit kupferhaltigen Proteinen berichtet, aberauch Strategien zur Umgehung dieses Problems beschrie-ben.[25] Die Stabilit"t von 5 wurde durch 24-st!ndige Inku-bation in humanem Plasma bei 37 8C untersucht und ergabeine Wiederfindung von !ber 80 %, die demnach deutlichhçher ist, als es f!r reaktive Tetrazine beschrieben wurde.[17]
Demzufolge sollte die Kombination von radiomarkiertenTetrazinen als Sonden mit (s-)TCO-Dienophilen f!r dieMarkierung von (Bio-)Molek!len gegen!ber inversen Stra-tegien (Tetrazinmarkierung und Verwendung radiomarkier-ter Dienophile) vorteilhaft f!r die bioorthogonale In-vivo-Bildgebung mit PET sein.
Die Radiosynthese von [18F]-1 konnte (in radiochemi-schen Ausbeuten von bis zu 18%) durch direkte Fluorierungdes tosylierten Intermediats 8 ausgef!hrt werden (Abbil-dung 1). Die Cycloaddition von [18F]-1 mit s-TCO 4 zumKonjugat [18F]-9, das langsam zu [18F]-10 isomerisierte (beideVerbindungen als eine Mischung aus Regio- und Stereoiso-meren) erfolgte vollst"ndig innerhalb einer Reaktionszeit von1 min.
In-vivo-Untersuchungen wurden in weiblichen BALB/c-M"usen durchgef!hrt.[18F]-1 wurde appliziert und seine Ver-teilung mithilfe dynamischer PET- (n = 4) sowie PET/MR-Messung (n = 2) verfolgt (MR = Magnetresonanztomogra-phie). Es konnte eine "ußerst homogene Biodistribution von
[18F]-1 in allen Kçrperregionen einschließlich des Gehirnsbeobachtet werden, weshalb auf eine Durchl"ssigkeit derBlut-Hirn-Schranke f!r die Tetrazinsonde r!ckgeschlossenwerden kann (Abbildung 2). Die PET/MR-Untersuchung
Abbildung 1. a) Radiosynthese von [18F]-1 und IEDDA-Cycloadditionmit s-TCO 4 (jeweils nur ein Isomer von [18F]-9 und [18F]-10 darge-stellt). b) HPLC von 1 (UV/Vis, 540 nm), Radio-HPLC von aufgereinig-tem [18F]-1 sowie der Klick-Produkte nach Reaktion mit s-TCO 4.HPLC: Zorbax SB-Aq (Umkehrphase), H2O/MeCN Gradientenelution.
Abbildung 2. Horizontal aufsummierte PET/MR-Bilder: 0–5 min (a)und 5–120 min (b) nach Injektion von [18F]-1.
.AngewandteZuschriften
9812 www.angewandte.de ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2014, 126, 9810 –9814

lieferte zudem keine signifikanten Hinweise auf eine In-vivo-Defluorierung von [18F]-1, da keine verst!rkte Aufnahme vonRadioaktivit!t im Knochengewebe beobachtet werdenkonnte.[26] Zeit-Aktivit!ts-Kurven zeigten einen sehr !hnli-chen Verlauf f"r alle untersuchten Organe mit Ausnahme derHarnblase (Abbildung 3a). Nach einer anf!nglichen Perfu-sion direkt nach der intravençsen Injektion von [18F]-1 wurdeim gesamten Verlauf der PET-Messung eine einheitlicheAbnahme der Aktivit!tskonzentration von 2 auf 1.5 SUV(standardized uptake value) in allen Organen mit Ausnahmeder Harnblase beobachtet. Bei Letzterer stieg die Aktivi-t!tskonzentration von 2 auf 10 SUV, weshalb von einerschnellen renalen Ausscheidung von [18F]-1 ausgegangenwerden kann. Die Resultate der Ex-vivo-Untersuchungen,die direkt im Anschluss (120 min nach der Injektion von [18F]-1) durchgef"hrt wurden, zeigten Aktivit!tskonzentrationenim Bereich von 1 bis 1.5 SUV (erneut mit Ausnahme derHarnblase; Abbildung 3b) und stimmen daher gut mit denDaten der PET-Messungen "berein. Die In-vivo-Stabilit!tvon [18F]-1 wurde in weiterer Folge per Radio-D"nnschicht-chromatographie (Radio-DC) von Mausplasma, das direktnach den PET-Experimenten erhalten wurde (120 min nachInjektion), untersucht. Es konnte lediglich ein geringesAusmaß an metabolischem Abbau festgestellt werden (ca.15%), wobei die Bildung polarer Metaboliten beobachtetwurde. Auf Grundlage dieser Resultate wird eine enzymka-talysierte Oxidation der Methylgruppe von [18F]-1 !hnlichzum xenobiotischen Metabolismus von Toluol und verwand-ten Verbindungen vermutet.[27]
Die Biokonjugation von [18F]-1 und dem wasserlçslichenDienophil 5 wurde in vivo durchgef"hrt, um die Bioortho-gonalit!t dieser Reaktion und somit des 18F-Tetrazins verifi-zieren zu kçnnen.[18F]-1 wurde weiblichen BALB/c-M!usen verabreicht (n = 4). Nach einer anf!nglichenPeriode von 20 min, um eine homogene Verteilungvon [18F]-1 vor der In-vivo-Reaktion gew!hrleistenzu kçnnen, wurde 5 injiziert (inverse Vorgehens-weise). Nach weiteren 5, 15 und 30 min wurde Blutdurch Punktion des retroorbitalen Plexus entnom-men und die IEDDA-Reaktion von [18F]-1 durchZugabe des reaktiveren Tetrazins 2 gestoppt. Muri-nes Plasma wurde anschließend mithilfe von Radio-DC untersucht (Abbildung 4), wobei bereits nach5 min ein Reaktionsumsatz von "ber 90 % sowie einevollst!ndige In-vivo-Reaktion 30 min nach Verab-reichung von 5 festgestellt wurden (siehe Hinter-grundinformationen, Abbildung S4). Diese Befundebest!tigen eindeutig das große Potenzial von [18F]-1 als vielseitige Sonde f"r zweistufige PET-Verfah-ren.
Zusammenfassend konnte eine schnelle und zu-verl!ssige Methode f"r die Synthese eines nieder-molekularen und wasserlçslichen radiomarkiertenTetrazins unter Anwendung direkter [18F]-Fluorie-rung entwickelt und dadurch die Nowendigkeitprosthetischer Gruppen sowie weiterer Synthese-schritte vermieden werden. [18F]-1 wurde zudemunter Verwendung von kommerziell erh!ltlichenund/oder leicht zug!nglichen Reagentien hergestellt,
Abbildung 3. a) Durchschnittliche Aktivit!tskonzentration in unter-schiedlichen Organen von BALB/c-M!usen (n= 6; f"r Knochen n = 2).b) Ex-vivo-Biodistribution von [18F]-1 in den angef"hrten Organen 2 hnach der intravençsen Injektion (n =6). Durchschnittliche Aktivit!ts-konzentrationen sind jeweils als SUV !Standardabweichung darge-stellt. Es wurde eine !hnliche Aktivit!t in allen Organen (mit Ausnah-me der Harnblase) festgestellt.
Abbildung 4. a) Bioorthogonale In-vivo-Reaktion von [18F]-1 und Dienophil 5 unterBildung der Ligationsprodukte [18F]-11 und [18F]-12 (ein Isomer dargestellt).b) Radio-D"nnschichtchromatogramm (SiO2) einer Plasmaprobe nach einer Reak-tionszeit von 5 min (links) sowie einer Blindprobe, die 35 min nach der Injektionvon [18F]-1 erhalten wurde (rechts).
AngewandteChemie
9813Angew. Chem. 2014, 126, 9810 –9814 ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

wobei der Gebrauch von wasserfreiem Hydrazin umgangenwerden konnte. Die neuartige Tetrazinsonde zeigte zudemhervorragende pharmakokinetische Eigenschaften und einehohe Stabilit!t, wobei der genaue Metabolismus dieser Ver-bindung im Zuge weiterer Untersuchungen noch verifiziertwerden muss. In-vivo-Versuche belegten eindeutig die Eig-nung von [18F]-1 als bioorthogonale Sonde f"r zweistufigePET-Verfahren. Die vielseitigen und vorteilhaften Eigen-schaften dieses Tetrazins, das die In-vivo-Detektion vonDienophil-markierten (Bio-)Molek"len ermçglicht, sprechenf"r eine Vielzahl von Anwendungsmçglichkeiten im Bereichder bioorthogonalen Bildgebung sowie der biomedizinischenForschung.
Eingegangen am 13. April 2014,ver!nderte Fassung am 9. Mai 2014Online verçffentlicht am 2. Juli 2014
.Stichwçrter: Bildgebende Verfahren · Bioorthogonale Chemie ·Klick-Chemie · Reaktionskinetik · Tetrazine
[1] a) L. Carroll, H. L. Evans, E. O. Aboagye, A. C. Spivey, Org.Biomol. Chem. 2013, 11, 5772 – 5781; b) N. K. Devaraj, R.Weissleder, Acc. Chem. Res. 2011, 44, 816 – 827; c) E. M. Sletten,C. R. Bertozzi, Acc. Chem. Res. 2011, 44, 666 – 676.
[2] J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V.Chang, I. A. Miller, A. Lo, J. A. Codelli, C. R. Bertozzi, Proc.Natl. Acad. Sci. USA 2007, 104, 16793 – 16797.
[3] a) S. T. Laughlin, J. M. Baskin, S. L. Amacher, C. R. Bertozzi,Science 2008, 320, 664 – 667; b) A. B. Neef, C. Schultz, Angew.Chem. 2009, 121, 1526 – 1529; Angew. Chem. Int. Ed. 2009, 48,1498 – 1500; c) T. Plass, S. Milles, C. Koehler, C. Schultz, E. A.Lemke, Angew. Chem. 2011, 123, 3964 – 3967; Angew. Chem. Int.Ed. 2011, 50, 3878 – 3881; d) R. Xie, S. Hong, X. Chen, Curr.Opin. Chem. Biol. 2013, 17, 747 – 752; e) J. C. Jewett, C. R.Bertozzi, Chem. Soc. Rev. 2010, 39, 1272 – 1279; f) J. C. Jewett,E. M. Sletten, C. R. Bertozzi, J. Am. Chem. Soc. 2010, 132, 3688 –3690.
[4] G. deAlmeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan,C. R. Bertozzi, Angew. Chem. 2012, 124, 2493 – 2497; Angew.Chem. Int. Ed. 2012, 51, 2443 – 2447.
[5] S. B. Lee, H. L. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn, D. W.Kim, Angew. Chem. 2013, 125, 10743 – 10746; Angew. Chem. Int.Ed. 2013, 52, 10549 – 10552.
[6] a) M. L. Blackman, M. Royzen, J. M. Fox, J. Am. Chem. Soc.2008, 130, 13518 – 13519; b) N. K. Devaraj, R. Weissleder, S. A.Hilderbrand, Bioconjugate Chem. 2008, 19, 2297 – 2299.
[7] a) G. Budin, K. S. Yang, T. Reiner, R. Weissleder, Angew. Chem.2011, 123, 9550 – 9553; Angew. Chem. Int. Ed. 2011, 50, 9378 –9381; b) K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters,J. W. Chin, Nat. Chem. 2012, 4, 298 – 304; c) K. S. Yang, G. Budin,T. Reiner, C. Vinegoni, R. Weissleder, Angew. Chem. 2012, 124,6702 – 6707; Angew. Chem. Int. Ed. 2012, 51, 6598 – 6603; d) D. S.Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox,A. Y. Ting, J. Am. Chem. Soc. 2012, 134, 792 – 795; e) J. L. Seit-chik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads,R. B. Cooley, C. Refakis, J. M. Fox, R. A. Mehl, J. Am. Chem.Soc. 2012, 134, 2898 – 2901; f) E. Kim, K. S. Yang, R. Weissleder,PLoS One 2013, 8, e81275; g) T. Reiner, E. J. Keliher, S. Earley,B. Marinelli, R. Weissleder, Angew. Chem. 2011, 123, 1963 –1966; Angew. Chem. Int. Ed. 2011, 50, 1922 – 1925.
[8] M. R. Karver, R. Weissleder, S. A. Hilderbrand, Angew. Chem.2012, 124, 944 – 946; Angew. Chem. Int. Ed. 2012, 51, 920 – 922.
[9] J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand, R. Weissle-der, Angew. Chem. 2013, 125, 7055 – 7058; Angew. Chem. Int. Ed.2013, 52, 6917 – 6920.
[10] M. T. Taylor, M. L. Blackman, O. Dmitrenko, J. M. Fox, J. Am.Chem. Soc. 2011, 133, 9646 – 9649.
[11] R. Selvaraj, J. M. Fox, Curr. Opin. Chem. Biol. 2013, 17, 753 –760.
[12] J. Yang, J. Seckute, C. M. Cole, N. K. Devaraj, Angew. Chem.2012, 124, 7594 – 7597; Angew. Chem. Int. Ed. 2012, 51, 7476 –7479.
[13] a) J. Seckute, N. K. Devaraj, Curr. Opin. Chem. Biol. 2013, 17,761 – 767; b) Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D.Brown, P. S. Conti, J. M. Fox, Chem. Commun. 2010, 46, 8043 –8045; c) J. C. Knight, S. Richter, M. Wuest, J. D. Way, F. Wuest,Org. Biomol. Chem. 2013, 11, 3817 – 3825.
[14] M. M. Herth, V. L. Andersen, S. Lehel, J. Madsen, G. M.Knudsen, J. L. Kristensen, Chem. Commun. 2013, 49, 3805 –3807.
[15] a) R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. M. Vul-ders, I. Verel, J. Lub, M. S. Robillard, Angew. Chem. 2010, 122,3447 – 3450; Angew. Chem. Int. Ed. 2010, 49, 3375 – 3378;b) B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D.Carlin, P. Zanzonico, R. Weissleder, J. S. Lewis, J. Nucl. Med.2013, 54, 1389 – 1396.
[16] N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli, R.Weissleder, Proc. Natl. Acad. Sci. USA 2012, 109, 4762 – 4767.
[17] M. R. Karver, R. Weissleder, S. A. Hilderbrand, BioconjugateChem. 2011, 22, 2263 – 2270.
[18] F. Liu, R. S. Paton, S. Kim, Y. Liang, K. N. Houk, J. Am. Chem.Soc. 2013, 135, 15642 – 15649.
[19] S. A. Lang, B. D. Johnson, E. Cohen, J. Heterocycl. Chem. 1975,12, 1143 – 1153.
[20] a) E. J. Keliher, T. Reiner, G. M. Thurber, R. Upadhyay, R.Weissleder, ChemistryOpen 2012, 1, 177 – 183; b) E. J. Keliher, T.Reiner, A. Turetsky, S. A. Hilderbrand, R. Weissleder, Chem-MedChem 2011, 6, 424 – 427; c) B. M. Zeglis, P. Mohindra, G. I.Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder, J. S.Lewis, Bioconjugate Chem. 2011, 22, 2048 – 2059; d) H.-S. Han,N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder, M. G.Bawendi, J. Am. Chem. Soc. 2010, 132, 7838 – 7839; e) J. B.Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee, R. Weissleder,Nat. Nanotechnol. 2010, 5, 660 – 665.
[21] J. Yang, M. R. Karver, W. Li, S. Sahu, N. K. Devaraj, Angew.Chem. 2012, 124, 5312 – 5315; Angew. Chem. Int. Ed. 2012, 51,5222 – 5225.
[22] J. W. Wijnen, S. Zavarise, J. B. F. N. Engberts, M. Charton, J.Org. Chem. 1996, 61, 2001 – 2005.
[23] Aufnahme von Substanzen mit hoher Gefahrenstufe in dieKandidatenliste (2011). ED/31/2011 verçffentlicht in #berein-stimmung mit Artikel 59(10) der Verordnung Nr. 1907/2006 desEurop!ischen Parlaments und Rates vom 18. Dezember 2006betreffend die Registrierung, Einsch!tzung, Autorisierung undBeschr!nkung von Chemikalien (REACH).
[24] E. Nachbaur, G. Leiseder, Monatsh. Chem. 1971, 102, 1718 –1723.
[25] R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli,R. M. Versteegen, J. Lub, M. S. Robillard, Bioconjugate Chem.2013, 24, 1210 – 1217.
[26] a) R. A. Hawkins, Y. Choi, S.-C. Huang, C. K. Hoh, M. Dahl-bom, C. Schiepers, N. Satyamurthy, J. R. Barrio, M. E. Phelps, J.Nucl. Med. 1992, 33, 633 – 642; b) E. Even-Sapir, E. Mishani, G.Flusser, U. Metser, Semin. Nucl. Med. 2007, 37, 462 – 469.
[27] D. E. Chapman, T. J. Moore, S. R. Michener, G. Powis, DrugMetab. Dispos. 1990, 18, 929 – 936.
.AngewandteZuschriften
9814 www.angewandte.de ! 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2014, 126, 9810 –9814


















