

Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan.
70
Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan
-
date post
19-Dec-2015 -
Category
Documents
-
view
227 -
download
1
Transcript of Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan.

Biology Tutorial
Aarti BalasubramaniAnusha Bharadwaj
Massa ShouraStefan Giovan
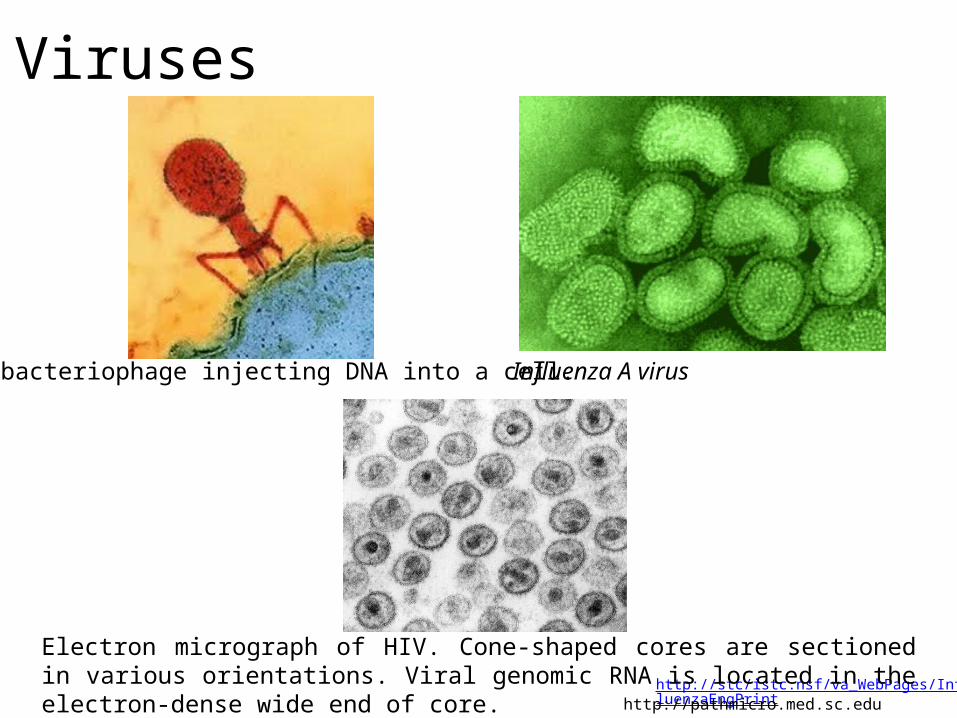
http://stc/istc.nsf/va_WebPages/InfluenzaEngPrint
Influenza A virus
Electron micrograph of HIV. Cone-shaped cores are sectioned in various orientations. Viral genomic RNA is located in the electron-dense wide end of core.
http://pathmicro.med.sc.edu
A T4 bacteriophage injecting DNA into a cell.
Viruses
Life Begins with Cells

http://course1.winona.edu/
All cells are Prokaryotic or Eukaryotic
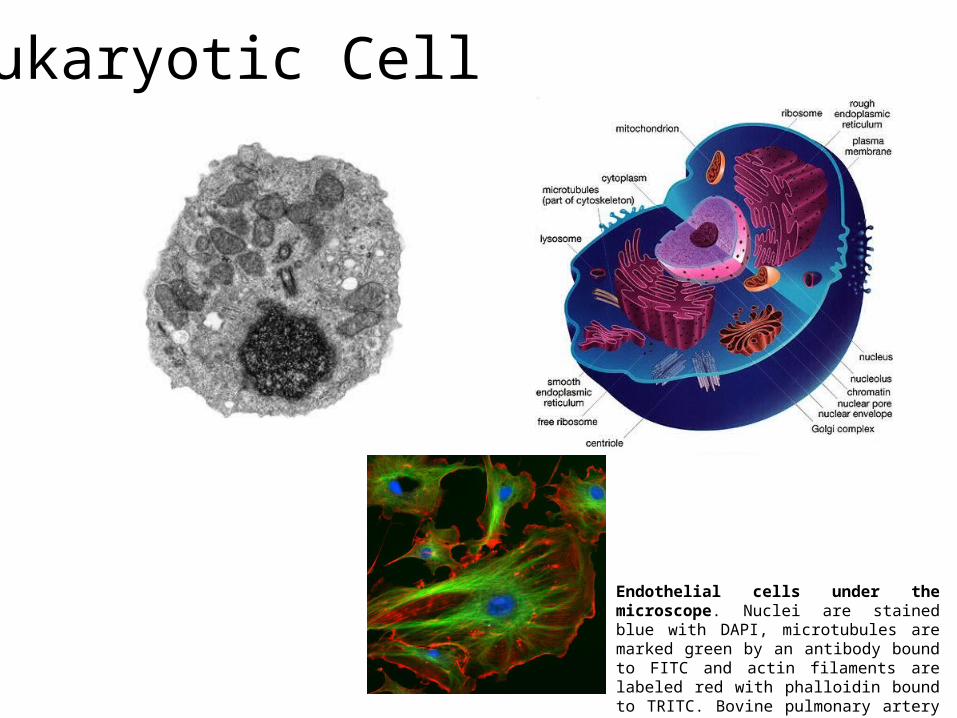
Eukaryotic Cell
Endothelial cells under the microscope. Nuclei are stained blue with DAPI, microtubules are marked green by an antibody bound to FITC and actin filaments are labeled red with phalloidin bound to TRITC. Bovine pulmonary artery endothelial cells

Nucleus= contains the genetic material
Cell Organelles
Mitochondrion= produces energy
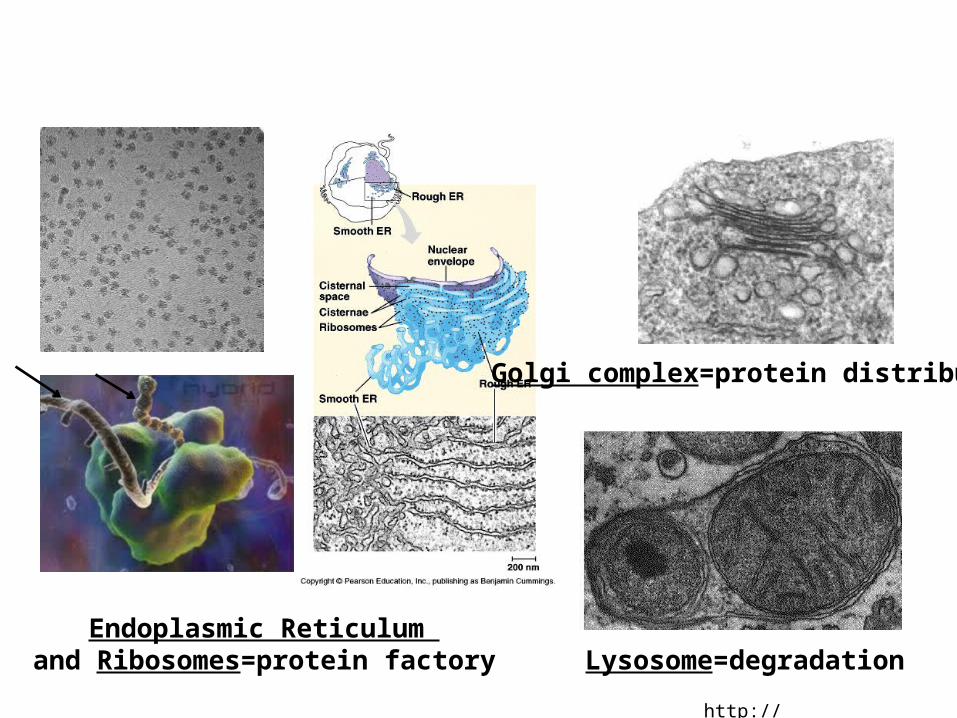
http://microbewiki.kenyon.edu/
Endoplasmic Reticulum and Ribosomes=protein factory
Golgi complex=protein distribution
Lysosome=degradation
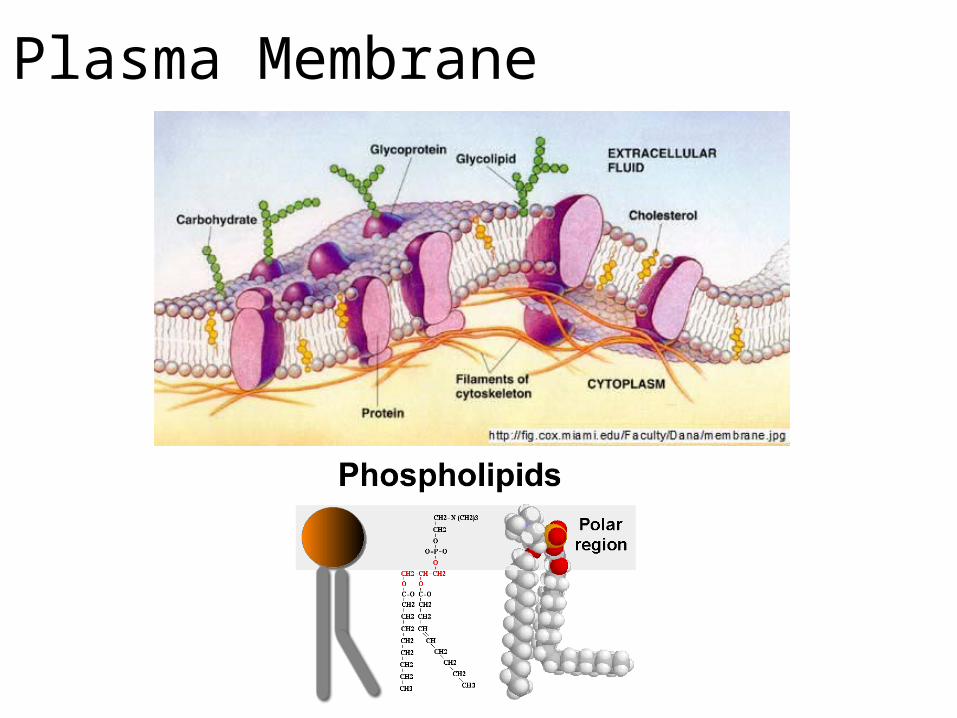
Plasma Membrane
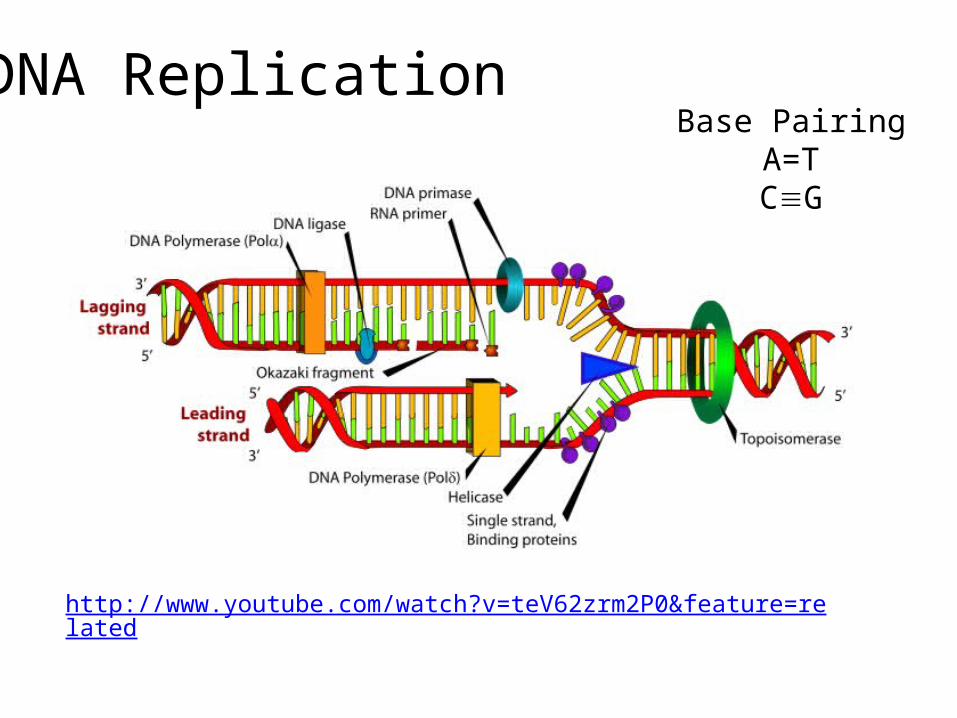
DNA Replication
http://www.youtube.com/watch?v=teV62zrm2P0&feature=related
Base PairingA=TCG
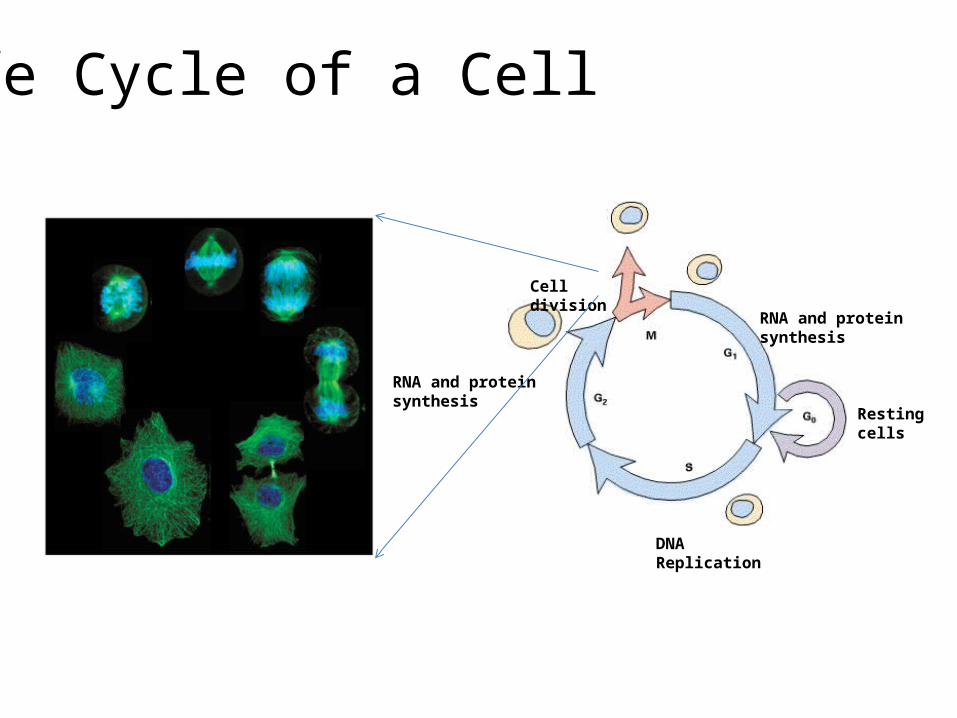
Life Cycle of a Cell
RNA and protein synthesis
DNA Replication
Resting cells
RNA and protein synthesis
Cell division
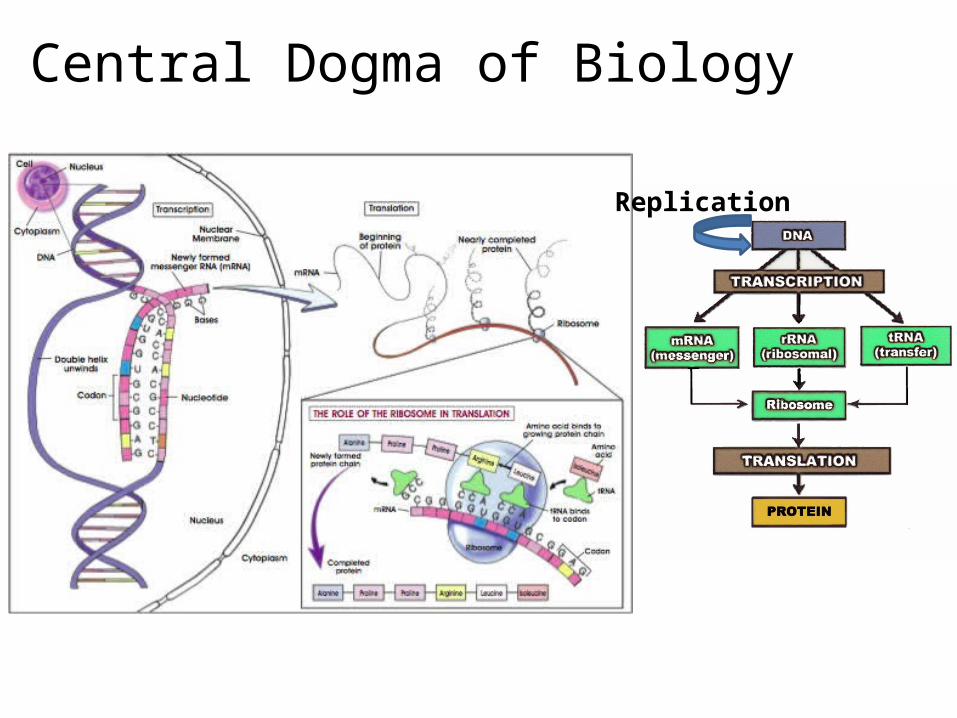
The Central Dogma of Biology
Replication

Transcription
http://www.youtube.com/watch?v=ztPkv7wc3yU
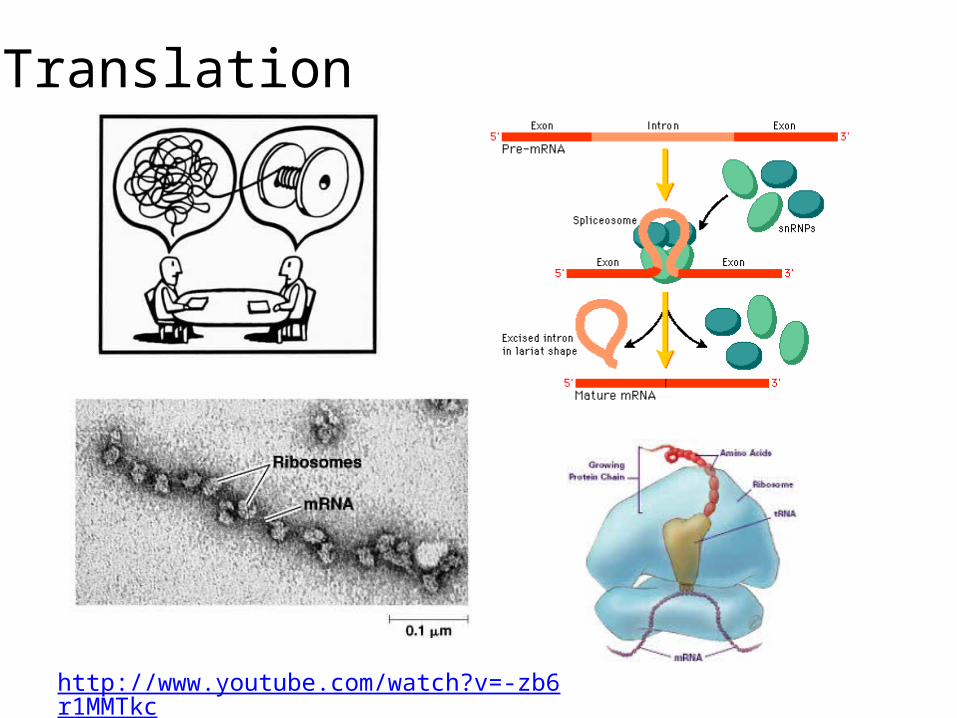
Translation
http://www.youtube.com/watch?v=-zb6r1MMTkc

Outline
• Cellular Biology– Organelle Structure/Function– Central Dogma
• Biochemistry– Energy Storage/Utilization– Macromolecules
• Bioinformatics – Sequences and Databases– Alignments, Tree Building, Modeling
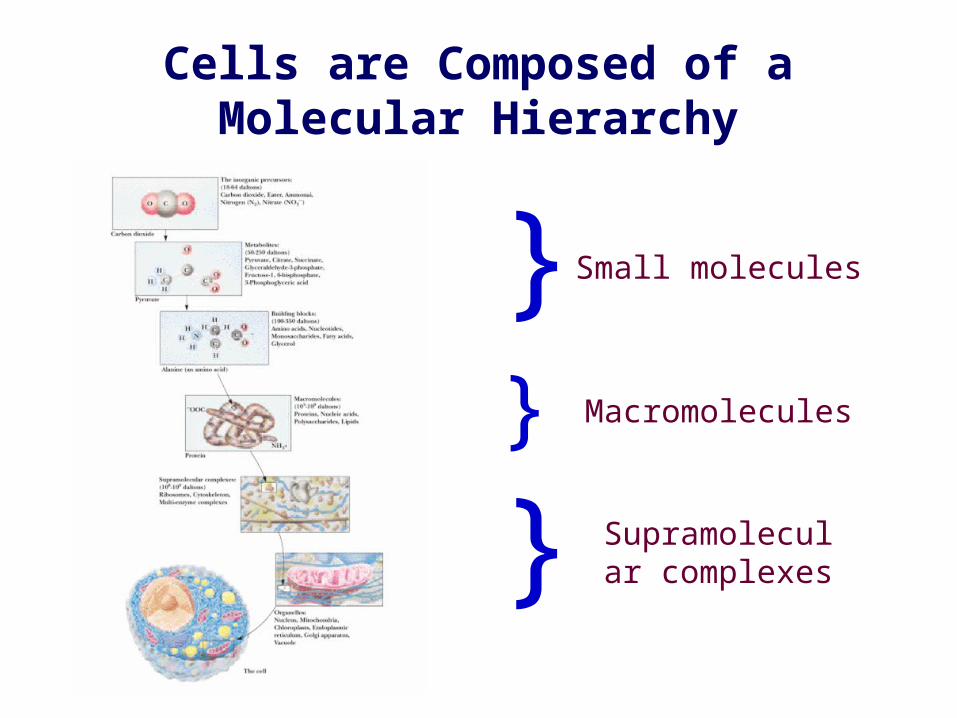
}
}}
Small molecules
Macromolecules
Supramolecular complexes
Cells are Composed of a Molecular Hierarchy
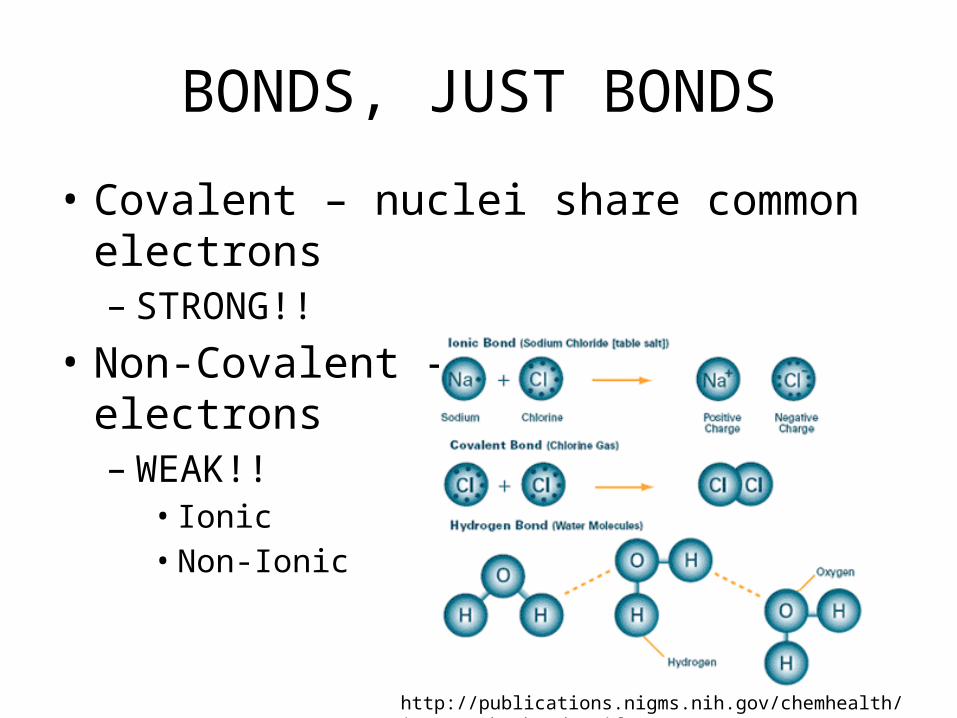
BONDS, JUST BONDS
• Covalent – nuclei share common electrons– STRONG!!
• Non-Covalent – No common electrons– WEAK!!
• Ionic• Non-Ionic
http://publications.nigms.nih.gov/chemhealth/images/ch1_bonds.gif
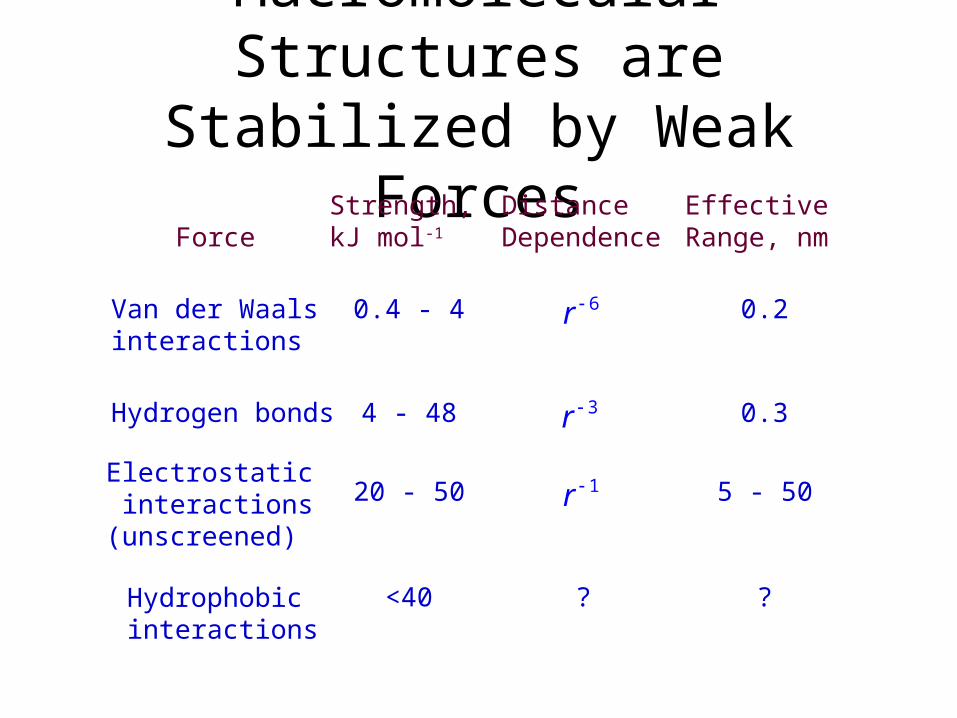
Macromolecular Structures are Stabilized by Weak Forces
ForceStrength, kJ mol-1
Effective Range, nm
Van der Waals interactions
Hydrogen bonds
Electrostatic interactions (unscreened)
Hydrophobic interactions
0.4 - 4
4 - 48
20 - 50
<40
0.2
0.3
5 - 50
?
DistanceDependence
6r
3r
1r
?
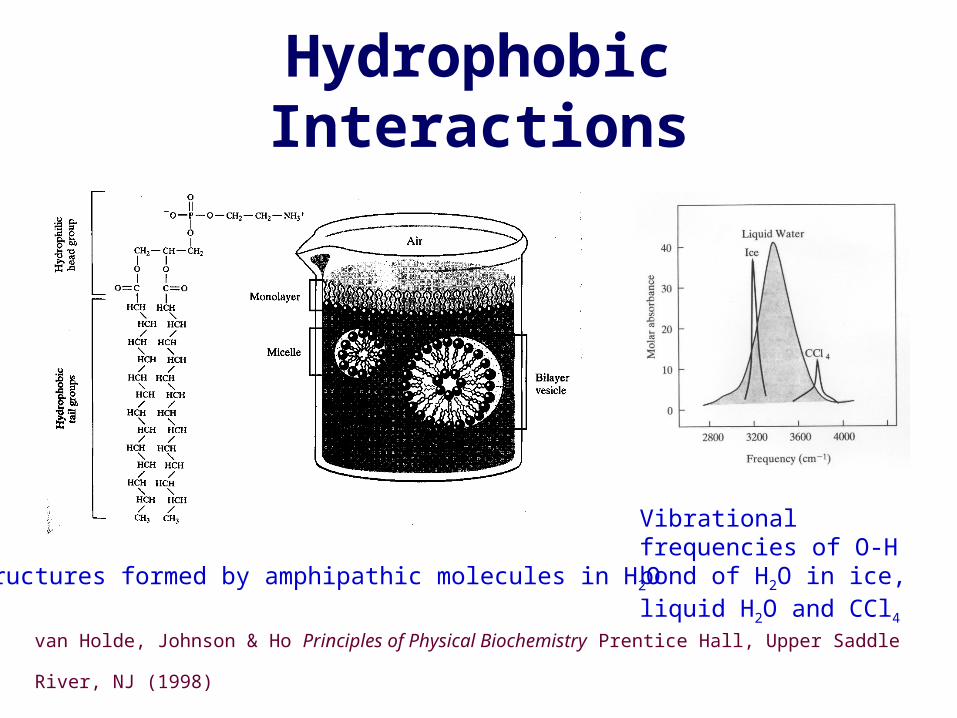
Hydrophobic Interactions
Structures formed by amphipathic molecules in H2O
van Holde, Johnson & Ho Principles of Physical Biochemistry Prentice Hall, Upper Saddle River,
NJ (1998)
Vibrational frequencies of O-H bond of H2O in ice, liquid H2O and CCl4
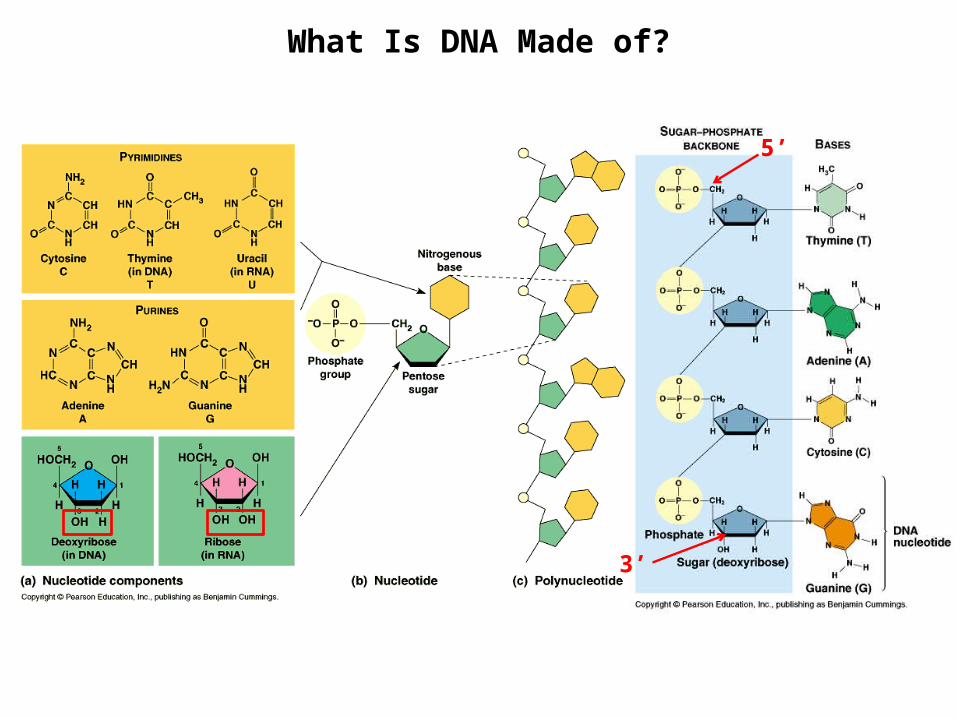
What Is DNA Made of?
3’
5’
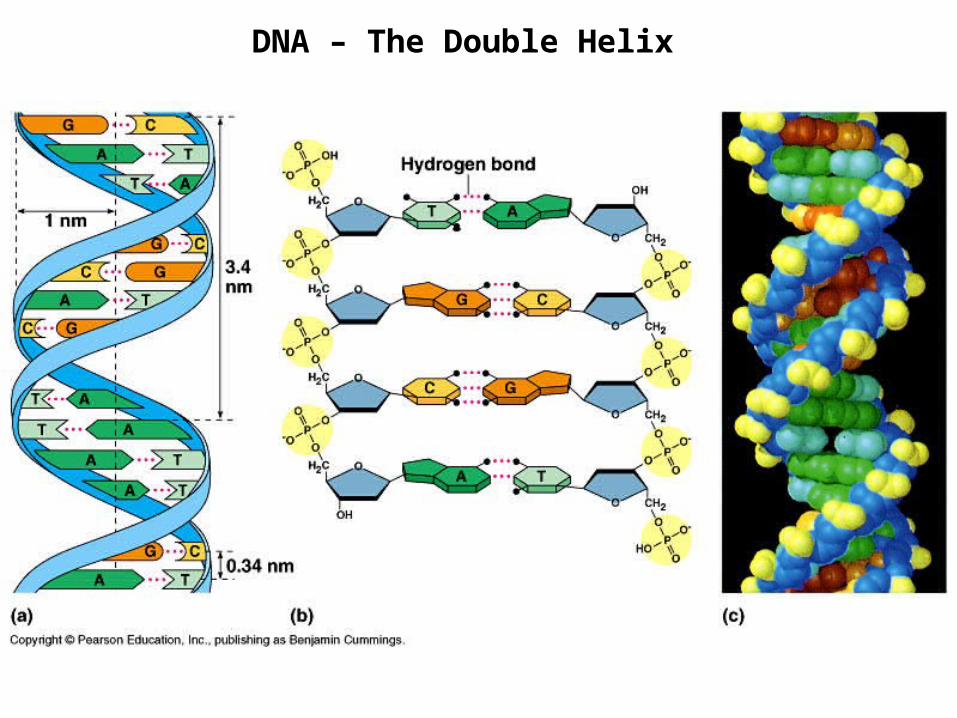
DNA – The Double Helix

Levels of Chromatin Packing
The Human Genome
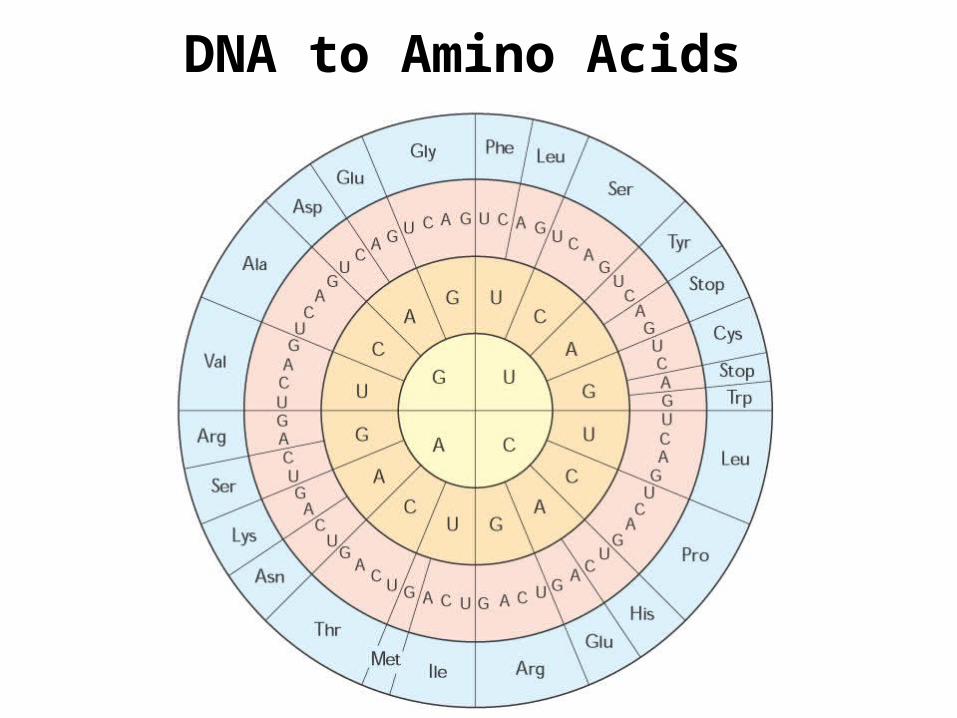
DNA to Amino Acids
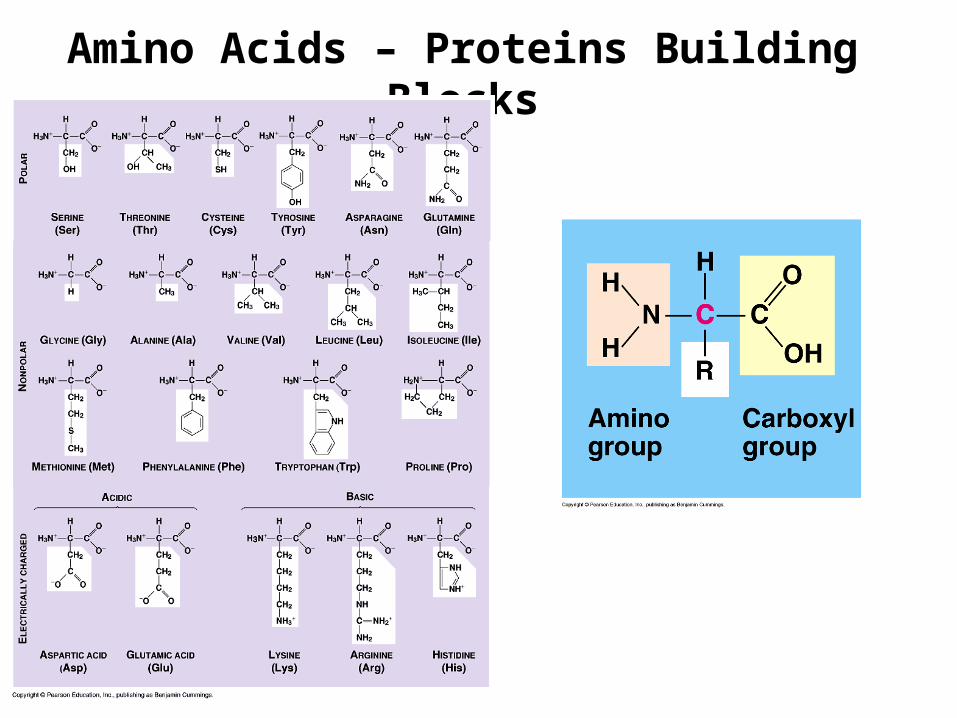
Amino Acids – Proteins Building Blocks
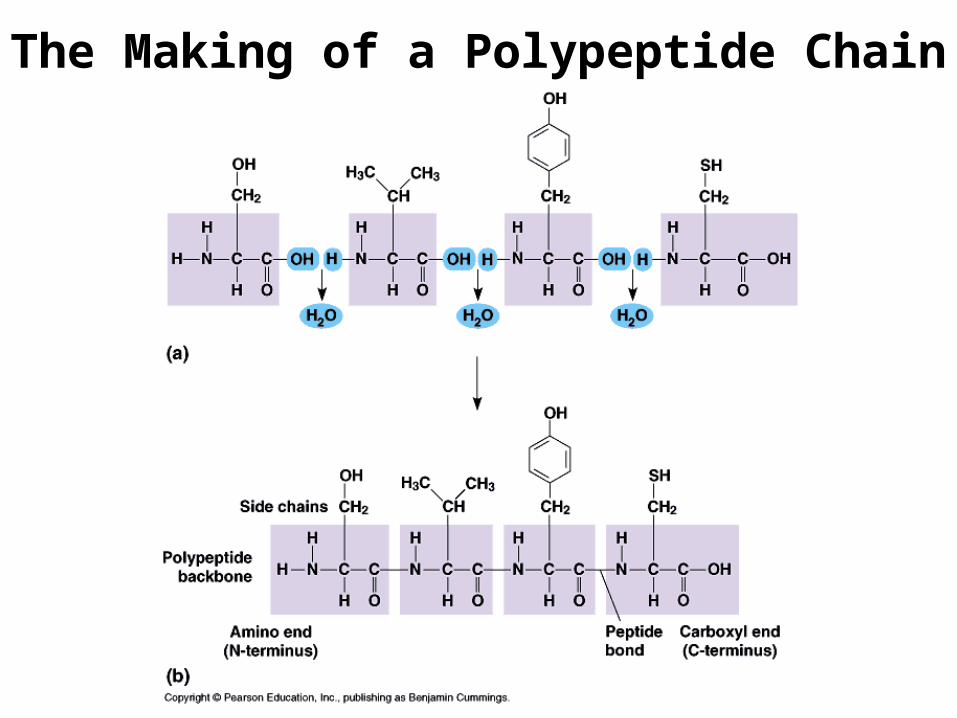
The Making of a Polypeptide Chain
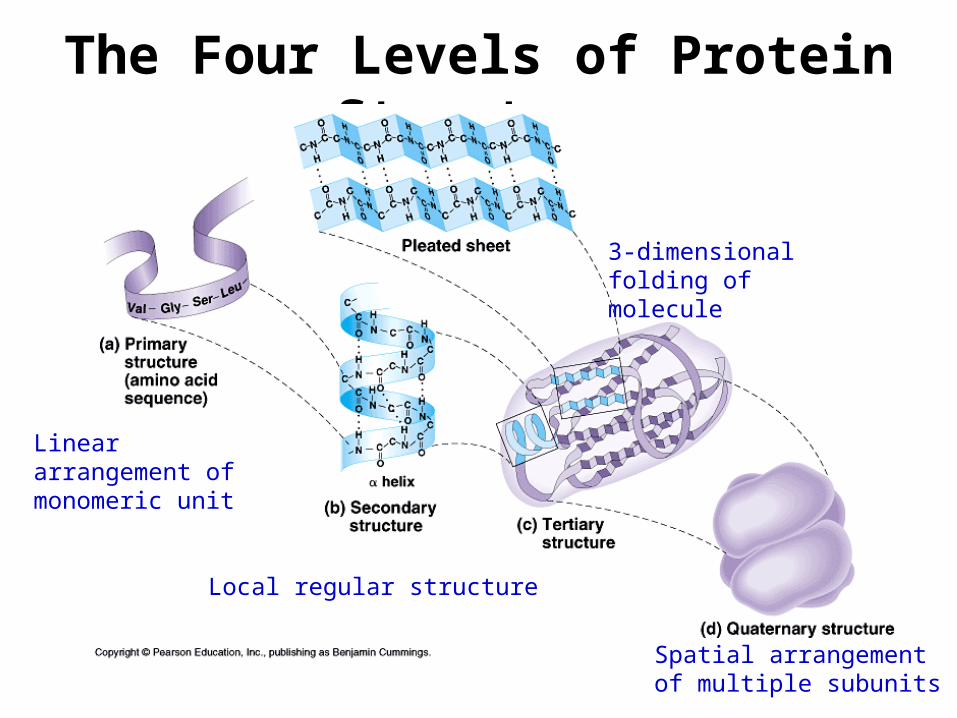
The Four Levels of Protein Structure
Linear arrangement of monomeric unit
Local regular structure
3-dimensional folding of molecule
Spatial arrangement of multiple subunits

Single Nucleotide Mutations

DNA Mutations

Experimental Techniques
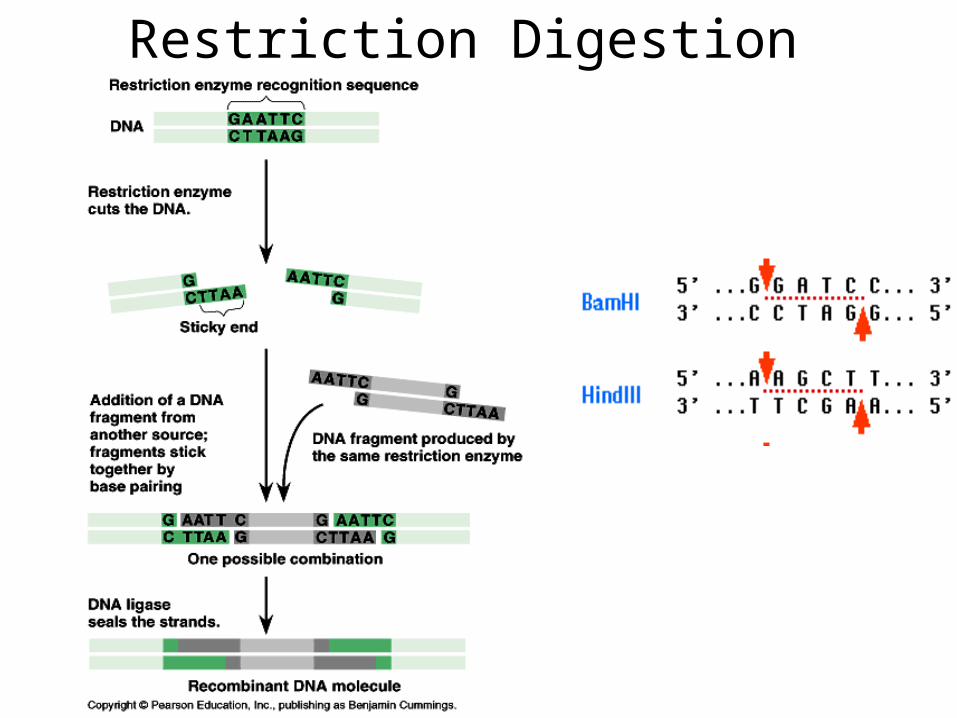
Restriction Digestion
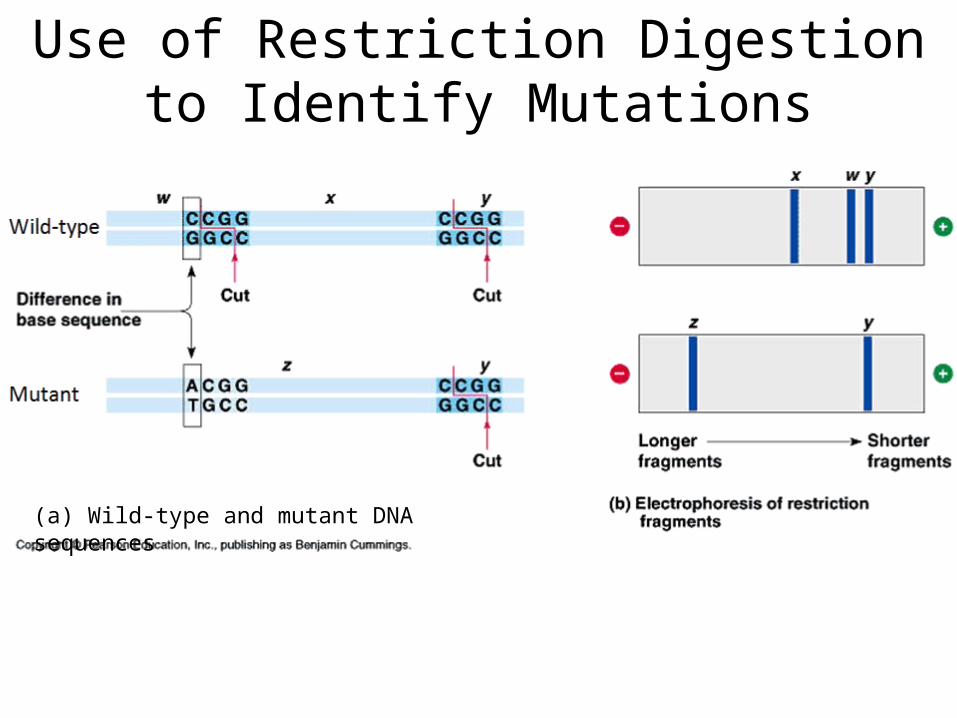
Use of Restriction Digestion to Identify Mutations
(a) Wild-type and mutant DNA sequences
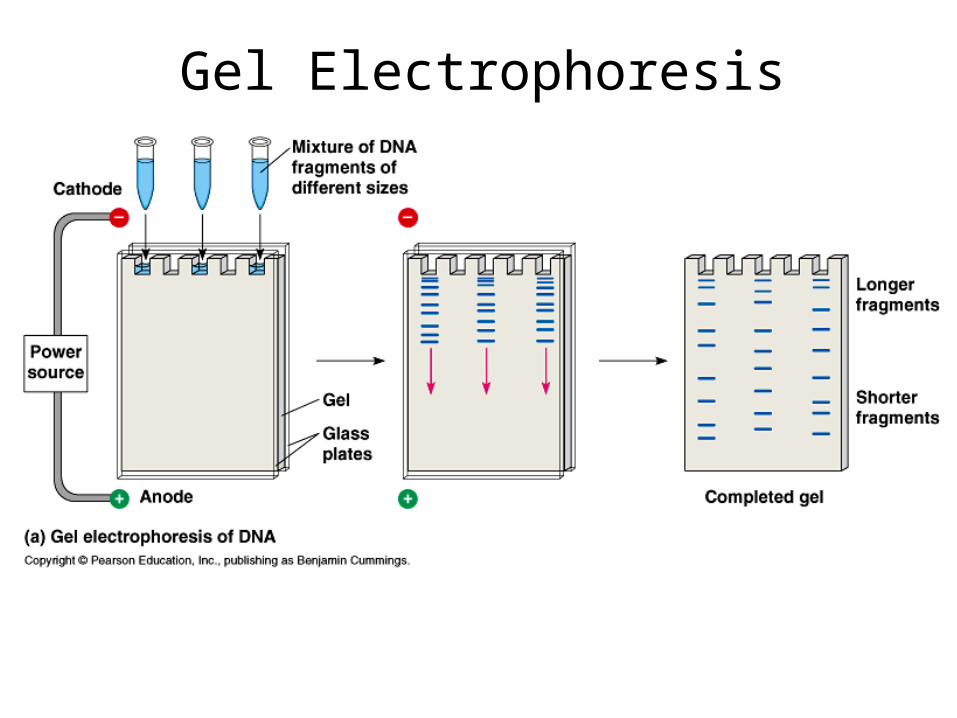
Gel Electrophoresis

Gel Electrophoresis-Visualizing DNA
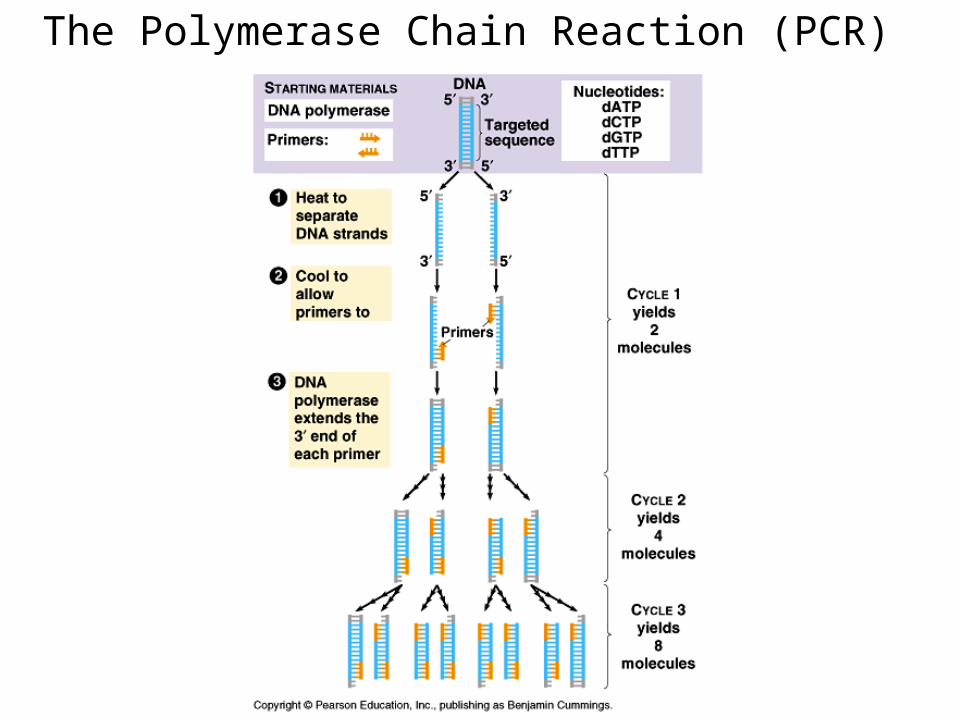
The Polymerase Chain Reaction (PCR)
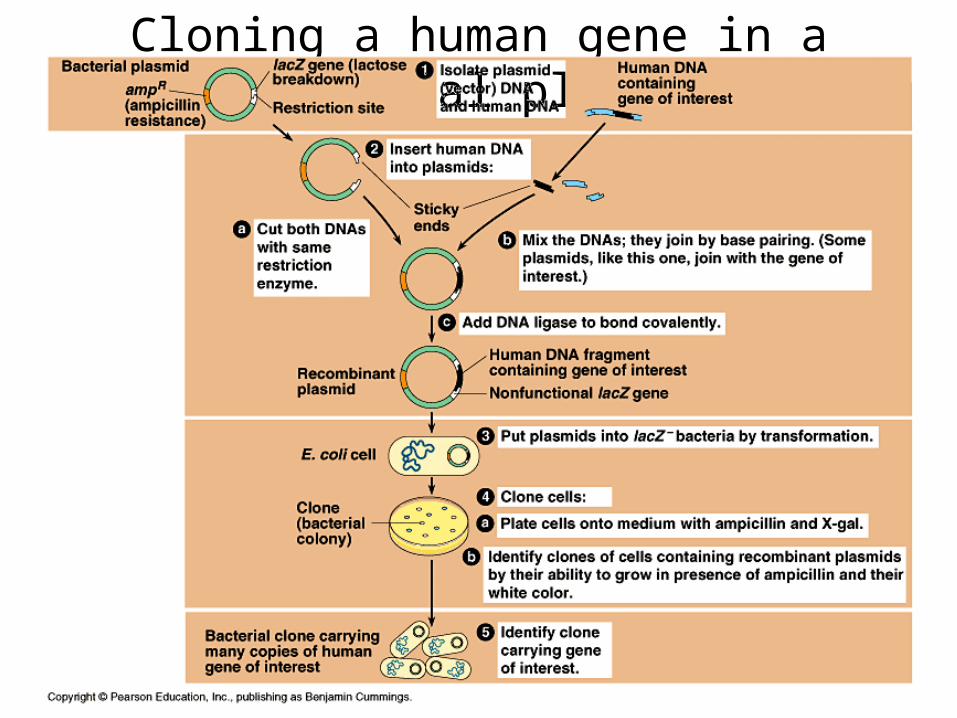
Cloning a human gene in a bacterial plasmid

Outline
• Cellular Biology– Organelle Structure/Function– Central Dogma
• Biochemistry– Energy Storage/Utilization– Macromolecules
• Bioinformatics– Sequences and Databases– Alignments, Tree Building, Modeling

Phenotype Tree BuildingHow Related are Organisms?
What do they eat? Where do they live? How do they divide? Move? Etc.Qualitative
http://nai.arc.nasa.gov/seminars/68_Rivera/tree.jpg

Genotype Tree BuildingHow Related are Organisms?
How similar is their genome? Proteome?MOLECULAR EVOLUTION
Quantitative
http://nai.arc.nasa.gov/seminars/68_Rivera/tree.jpg

Comparison of Genomes
• 1977- Φ-X174 genome sequenced – Only about 5.4 kbp
• 1997- E. coli K-12 genome sequenced– About 4.6x103 kbp
• 2007- Watson’s Genome sequenced! – About 3x106 kbp!
• About 0.1% difference between human genomes and 1% difference between humans and chimps!

Bioinformatics is…
• Highly Interdisciplinary– Proteomics and Genomics– Structural and Computational Biology– Systems Biology– Computer Science, Probabilistic Modeling
• Computational Sequence Analysis – What’s in a sequence?
STRUCTURE FUNCTIONSEQUENCE

Power of Prediction
• Can we …– predict structural and functional properties of
proteins given its sequence?– predict the consequences of a mutation?– design proteins or drugs with specific functions?
• Every thing we need to know is at our finger-tips, just need a better understanding of the natural world
STRUCTURE FUNCTIONSEQUENCE
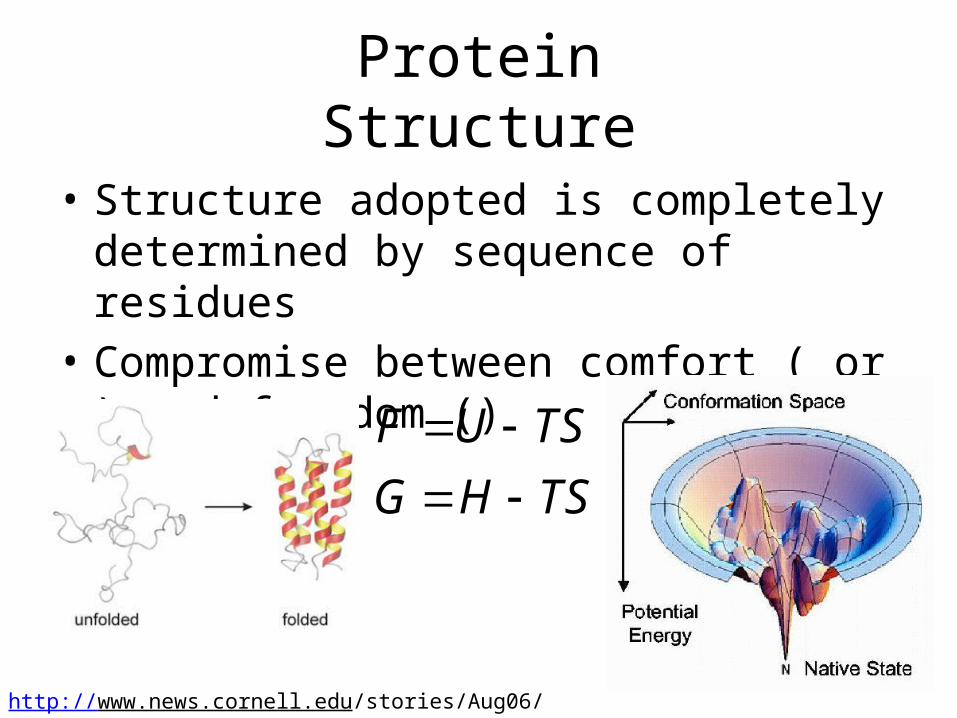
• Structure adopted is completely determined by sequence of residues
• Compromise between comfort ( or ) and freedom ()
Protein Structure
F U TS
G H TS
http://www.news.cornell.edu/stories/Aug06/protein_folding.jpg
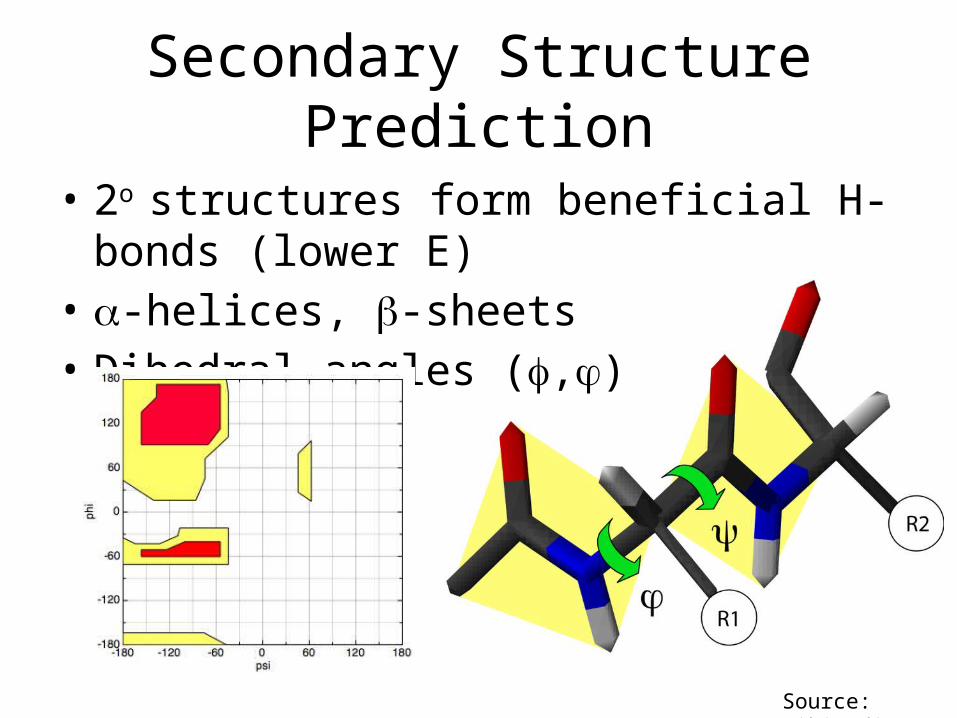
Secondary Structure Prediction
• 2o structures form beneficial H-bonds (lower E)• -helices, -sheets• Dihedral angles (,)
Source: Wikipedia

Tertiary Structure Prediction
• Homology/Comparative Modeling– BEST– Structure of very related protein is known
• Fold Recognition/Threading– OFTEN IS ENOUGH– Similar folds available but no close relative
• Knowledge Based or A Priori Predictions– ONLY POSSIBLE FOR VERY SHORT PROTEINS– Fold prediction but without experimental quality

Sequence Alignments
• FASTA Text Format >header – my sequence >header – my thesis THISISMYSEQ THESISTHYSTING• Alignment
T H I S I S – M Y S E – Q – T H E S I S T H Y S T I N G
• What can we learn from this?

Alignments
• Pairwise– Dot Plot– Global(N-W) or Local(S-W)
• Simple Database Searches– FASTA/BLAST
• Multiple Alignments– CLUSTAL
• Advanced Strategies– PSI/PHI-BLAST, HMM’s
Dot plot of two subunits inHuman Hemoglobin
Alpha Chain
Beta
Cha
in

Databases
• Nucleotide Sequence Database Collaboration– DDBJ, EMBL, GenBank at
NCBI• Amino Acid Databases
– UniProt, SWISS-PROT, TrEMBL
• Structural– PDB, MMDB, MSD
• Very Many Derivations! http://www.ncbi.nlm.nih.gov/Database/

Scoring Matrices
• PAM Matrix : Point Accepted Mutation– PAM1 estimates substitution rate if 1% of AA had
changed. Standards: PAM30 and PAM60• BLOSUM : BLOcks of Amino Acid SUbstitution
Matrix– BLOSUM80 “blocks” together sequences with
greater then 80% similarity.
More DivergentLess DivergentPAM1BLOSUM80
PAM250BLOSUM45

FASTA and BLAST
• FASTA - FAST All, Rapid AA or NT Alignments• BLAST – Basic Local Alignment Search Tool• Scoring Alignments
– Raw and Bit Scores;
– Significance of Local Alignment;
– Significance of Global Alignment; x uZ
'2 SE mn
ln'
ln 2
S KS

Nucleotide Sequence Distances
• Jukes-Cantor, single parameter
• Kimura, 2 parameter
3 4ln 14 3
d p
1 1 1 1ln ln2 1 2 4 1 2
dp q q
A C
G T
A C
G T

Distance Based Tree Building
• Tree Building => UPGMA– Smallest distance element -> nearest neighbors
1 2 120.5t t d 1- 2 0.1- 3 0.8 0.8- 4 0.8 1 0.3- 5 0.9 0.9 0.3 0.2-
1 20.050.05
1 2
345
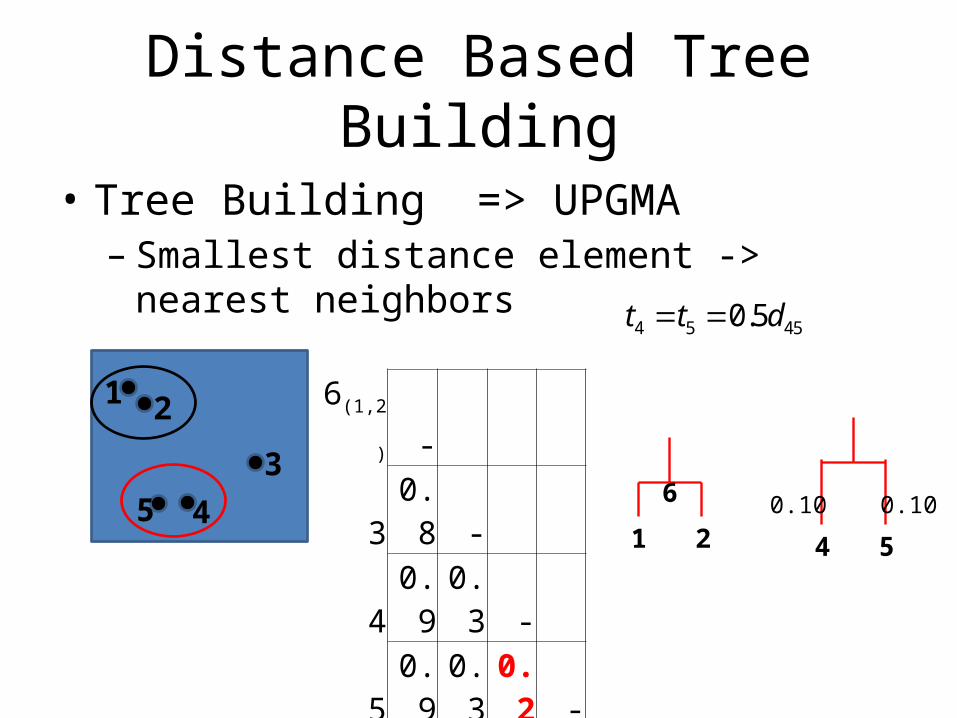
Distance Based Tree Building
• Tree Building => UPGMA– Smallest distance element -> nearest neighbors
6(1,2) - 3 0.8 - 4 0.9 0.3 -5 0.9 0.3 0.2 -
1 2
1 2
345
4 5
4 5 450.5t t d
0.10 0.106
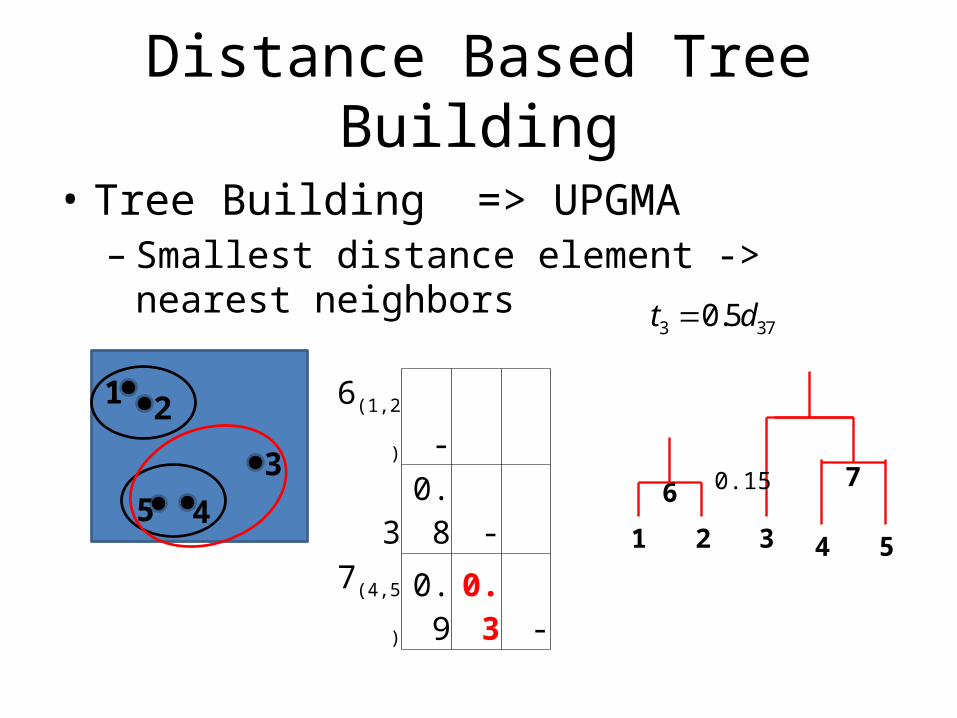
Distance Based Tree Building
• Tree Building => UPGMA– Smallest distance element -> nearest neighbors
6(1,2) - 3 0.8 -
7(4,5) 0.9 0.3 -
1 2 4 5
3 370.5t d
1 2
345 6
3
0.15 7

Distance Based Tree Building
• Tree Building => UPGMA– Smallest distance element -> nearest neighbors
6(1,2) - -8(3,4,5) 0.85 -
1 2 4 5
6 680.5t d
1 2
345 6
3
0.425
7
89

Distance Based Tree Building
• UPGMA is efficient but makes non-biological assumption that rate of substitution is constant for all branches– Useful in a variety of applications such as microarray
data processing• Neighbor-Joining does not make this assumption
and is still efficient– More accurate for use in phylogenetic analyses
• Also -> Maximum Parsimony, Maximum Likelihood, Minimum Evolution, and Bayesian methods

Energy Calculations
• Goal: Find Unique Arrangement of Atoms which Maximizes Stability
• Experimental (usually X-ray or NMR)• Monte Carlo
– Explore states – Let T->0 and discover low energy states
(Simulated Annealing)• Molecular Dynamics
– Newtonian mechanics to evolve the system

Molecular Mechanics
E K V
221 1
2 2i
i ii ii
pK mv
m
E : Total energyK : Kinetic energyV : Potential energy
iv: Velocity of particle i
ip: Momentum of particle i
i
i
i
Vx
Vi i y
Vz
F V
: Force acting on particle i (gradient of potential energy)iF
, ,i bonding i nonbondiV V V Sum of covalent and
noncovalent interactions

Pairwise Alignment
• Dot Plot– Visual and Qualitative
• Needleman-Wunsch Global Alignment– Alignment over entire
sequence• Smith-Waterman Local
Alignment– Alignment over sub-
sequenceshttp://lectures.molgen.mpg.de/Pairwise/DotPlots/
Dot plot of two subunits inHuman Hemoglobin
Alpha Chain
Beta
Cha
in

N-W Alignment
• Produces Optimal Global Alignment – Without exhaustive pairwise comparison
• Scoring Matrix, S• Simple scoring matrix for these
sequences• Matches get a score of +1• Mismatches (blank) get a score of -2
• One could also use BLOSUM or PAM scoring matrix for example
F M D T P L N EF 1KHM 1E 1D 1P 1L 1E 1

N-W Alignment
• Produces Optimal Global Alignment – Without exhaustive pairwise comparison
• Alignment Matrix, FF M D T P L N E
0 -2 -4 -6 -8 -10 -12 -14 -16F -2 +1K -4H -6M -8E -10D -12P -14L -16E -18
1, 1
1,
, 1
maxi j kl
ij i j
i j
F S
F F gap
F gap
Match always results in largest , else take the largest score from • mismatch,• gap in sequence 1 , or• gap in sequence 2 .

N-W Alignment
• Produces Optimal Global Alignment – Without exhaustive pairwise comparison
• Build Scoring Matrix, FF M D T P L N E
0 -2 -4 -6 -8 -10 -12 -14 -16F -2 +1 -1 -3 -5 -7 -9 -11 -13K -4 -1 -1H -6M -8E -10D -12P -14L -16E -18
1, 1
1,
, 1
maxi j kl
ij i j
i j
F S
F F gap
F gap

N-W Alignment
• Produces Optimal Global Alignment – Without exhaustive pairwise comparison
• Build Scoring Matrix, FF M D T P L N E
0 -2 -4 -6 -8 -10 -12 -14 -16F -2 +1 -1 -3 -5 -7 -9 -11 -13K -4 -1 -1 -3 -5 -7 -9 -11 -13H -6 -3 -3 -3 -5 -7 -9 -11 -13M -8 -5 -2 -4 -5 -7 -9 -11 -13E -10 -7 -4 -4 -6 -7 -9 -11 -10D -12 -9 -6 -3 -5 -7 -9 -11 -12P -14 -11 -8 -5 -5 -4 -6 -8 -10L -16 -13 -10 -7 -7 -6 -3 -5 -7E -18 -15 -12 -9 -9 -8 -5 -5 -4
Overall alignment score
1, 1
1,
, 1
maxi j kl
ij i j
i j
F S
F F gap
F gap

N-W Alignment
• Produces Optimal Global Alignment – Without exhaustive pairwise comparison
• Trace Back to Determine Optimum Alignment F M D T P L N E
0 -2 -4 -6 -8 -10 -12 -14 -16F -2 +1 -1 -3 -5 -7 -9 -11 -13K -4 -1 -1 -3 -5 -7 -9 -11 -13H -6 -3 -3 -3 -5 -7 -9 -11 -13M -8 -5 -2 -4 -5 -7 -9 -11 -13E -10 -7 -4 -4 -6 -7 -9 -11 -10D -12 -9 -6 -3 -5 -7 -9 -11 -12P -14 -11 -8 -5 -5 -4 -6 -8 -10L -16 -13 -10 -7 -7 -6 -3 -5 -7E -18 -15 -12 -9 -9 -8 -5 -5 -4
Seq1: F K HME D- P L - ESeq2: F - - M- DT P L NE
Match or MismatchGap in Sequence 1Gap in Sequence 2

Smith-Waterman Alignment
• Local alignment, Similar in Nature to N-W– S takes only non-negative values– Highest value in matrix corresponds to end of
alignment, need not be in corner– No penalty for gaps at ends
• Most rigorous method of aligning nucleotide or protein sequence domains

Database Searches
• Optimal pairwise alignment produced by S-W, but insufficient in scanning databases
• Scan for likely matches before performing more rigorous alignments– FASTA, BLAST
• Scan for words scoring higher than some threshold, extend alignment until score drops

Advanced Database Searches
• When BLAST falls short– Detecting homology between distantly related
proteins– Very long (>20kbp) genome sequences with highly
conserved regions and highly variable regions• PSI-BLAST (Position-Specific Iterated)
– BLAST generates Position Specific Scoring Matrix– PSSM used as query to re-search database
• Also, PHI-BLAST, HMMs…

Multiple Sequence Alignments
• Exact Approaches– e.g. N-W alignments– Prohibitive for many or long sequences
• Progressive Approaches– e.g. CLUSTAL
• Iterative Approaches• Consistency-Based Approaches• Structure-Based Methods

Distance Between Sequences
• Based on theory of molecular evolution
• Simplest method, Hamming distance, • Multiple substitutions at single site?• Poisson correction,
– Assume: Probability of observing a change is small, but constant across all sites
– Rate of mutation is constant over time– Mutations at different sites occur independently
100d p
ln 1d p
differences distances

James Watson, Francis Crick and Rosalind Franklin


















