B Cells Limit Repair after Ischemic Acute Kidney...
12
B Cells Limit Repair after Ischemic Acute Kidney Injury Hye Ryoun Jang,* † Maria Teresa Gandolfo, ‡ Gang Jee Ko,* Shailesh R. Satpute,* Lorraine Racusen, ‡ and Hamid Rabb* *Nephrology Division, Department of Medicine, and ‡ Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, Maryland; and † Nephrology Division, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea ABSTRACT There is no established modality to repair kidney damage resulting from ischemia-reperfusion injury (IRI). Early responses to IRI involve lymphocytes, but the role of B cells in tissue repair after IRI is unknown. Here, we examined B cell trafficking into postischemic mouse kidneys and compared the repair response between control (wild-type) and MT (B cell-deficient) mice with and without adoptive transfer of B cells. B cells infiltrated postischemic kidneys and subsequently activated and differentiated to plasma cells during the repair phase. Plasma cells expressing CD126 increased and B-1 B cells trafficked into postischemic kidneys with distinct kinetics. An increase in B lymphocyte chemoattractant in the kidney preceded B cell trafficking. Postischemic kidneys of MT mice expressed higher IL-10 and vascular endothelial growth factor and exhibited more tubular proliferation and less tubular atrophy. Adoptive transfer of B cells into MT mice reduced tubular proliferation and increased tubular atrophy. Treatment with anti-CD126 antibody increased tubular proliferation and reduced tubular atrophy in the late repair phase. These results demonstrate that B cells may limit the repair process after kidney IRI. Targeting B cells could have therapeutic potential to improve repair after IRI. J Am Soc Nephrol 21: 654 –665, 2010. doi: 10.1681/ASN.2009020182 Ischemia is a leading cause of acute kidney injury (AKI) in both native kidneys and allografts. In allo- grafts, ischemic AKI frequently results in delayed graft function. 1 Many studies have demonstrated that both innate and adaptive immune responses are involved in the pathogenesis of renal injury after renal isch- emia-reperfusion injury (IRI). 2,3 On the basis of tradi- tional concepts of adaptive immunity, lymphocytes were not expected to play an important role in the early renal injury after IRI; however, T cells were found to mediate the early phase of IRI in kidney and in other organs, both directly and indirectly. 4–6 B cells also seem to participate in the early injury response of renal IRI, 7 and B cell products are also important in early IRI response in skeletal muscle. 8 B cells have been identified as important mediators of various autoimmune diseases, such as experimental allergic encephalomyelitis (EAE), collagen-induced arthritis, and inflammatory bowel disease. 9 –11 In EAE, B cells seem to function as antigen-presenting cells during the initiation phase. 12,13 In a recent report, B cells were involved in both initiation and progression of EAE. 14 Clinical trials using mAb to CD20 expressed on B cells have suggested beneficial effects in autoim- mune diseases such as rheumatoid arthritis, lupus er- ythematosus, and multiple sclerosis. 15–18 Although ischemic AKI and autoimmune disease are tradition- ally viewed as different disease categories, they share a crucial feature: A prominent immune/inflammatory response. It was previously shown that B cells traffic into chronically inflamed organs, activate and form ec- topic germinal centers, and locally differentiate to Received February 14, 2009. Accepted November 25, 2009. Published online ahead of print. Publication date available at www.jasn.org. Correspondence: Dr. Hamid Rabb, Division of Nephrology, Johns Hopkins University School of Medicine, Ross Building, Room 965, 720 Rutland Avenue, Baltimore, MD 21205. Phone: 410-502-1555; Fax: 410-614-5129; E-mail: [email protected] Copyright 2010 by the American Society of Nephrology BASIC RESEARCH www.jasn.org 654 ISSN : 1046-6673/2104-654 J Am Soc Nephrol 21: 654–665, 2010
Transcript of B Cells Limit Repair after Ischemic Acute Kidney...

B Cells Limit Repair after Ischemic Acute Kidney Injury
Hye Ryoun Jang,*† Maria Teresa Gandolfo,‡ Gang Jee Ko,* Shailesh R. Satpute,*Lorraine Racusen,‡ and Hamid Rabb*
*Nephrology Division, Department of Medicine, and ‡Department of Pathology, Johns Hopkins University School ofMedicine, Baltimore, Maryland; and †Nephrology Division, Department of Medicine, Samsung Medical Center,Sungkyunkwan University School of Medicine, Seoul, Korea
ABSTRACTThere is no established modality to repair kidney damage resulting from ischemia-reperfusion injury (IRI).Early responses to IRI involve lymphocytes, but the role of B cells in tissue repair after IRI is unknown.Here, we examined B cell trafficking into postischemic mouse kidneys and compared the repair responsebetween control (wild-type) and �MT (B cell-deficient) mice with and without adoptive transfer of B cells.B cells infiltrated postischemic kidneys and subsequently activated and differentiated to plasma cellsduring the repair phase. Plasma cells expressing CD126 increased and B-1 B cells trafficked intopostischemic kidneys with distinct kinetics. An increase in B lymphocyte chemoattractant in the kidneypreceded B cell trafficking. Postischemic kidneys of �MT mice expressed higher IL-10 and vascularendothelial growth factor and exhibited more tubular proliferation and less tubular atrophy. Adoptivetransfer of B cells into �MT mice reduced tubular proliferation and increased tubular atrophy. Treatmentwith anti-CD126 antibody increased tubular proliferation and reduced tubular atrophy in the late repairphase. These results demonstrate that B cells may limit the repair process after kidney IRI. Targeting Bcells could have therapeutic potential to improve repair after IRI.
J Am Soc Nephrol 21: 654–665, 2010. doi: 10.1681/ASN.2009020182
Ischemia is a leading cause of acute kidney injury(AKI) in both native kidneys and allografts. In allo-grafts, ischemic AKI frequently results in delayed graftfunction.1 Many studies have demonstrated that bothinnate and adaptive immune responses are involvedin the pathogenesis of renal injury after renal isch-emia-reperfusion injury (IRI).2,3 On the basis of tradi-tional concepts of adaptive immunity, lymphocyteswere not expected to play an important role in theearly renal injury after IRI; however, T cells werefound to mediate the early phase of IRI in kidney andin other organs, both directly and indirectly.4–6 B cellsalso seem to participate in the early injury response ofrenal IRI,7 and B cell products are also important inearly IRI response in skeletal muscle.8
B cells have been identified as important mediatorsof various autoimmune diseases, such as experimentalallergic encephalomyelitis (EAE), collagen-inducedarthritis, and inflammatory bowel disease.9–11 In EAE,B cells seem to function as antigen-presenting cellsduring the initiation phase.12,13 In a recent report, B
cells were involved in both initiation and progressionof EAE.14 Clinical trials using mAb to CD20 expressedon B cells have suggested beneficial effects in autoim-mune diseases such as rheumatoid arthritis, lupus er-ythematosus, and multiple sclerosis.15–18 Althoughischemic AKI and autoimmune disease are tradition-ally viewed as different disease categories, they share acrucial feature: A prominent immune/inflammatoryresponse.
It was previously shown that B cells traffic intochronically inflamed organs, activate and form ec-topic germinal centers, and locally differentiate to
Received February 14, 2009. Accepted November 25, 2009.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Hamid Rabb, Division of Nephrology,Johns Hopkins University School of Medicine, Ross Building,Room 965, 720 Rutland Avenue, Baltimore, MD 21205. Phone:410-502-1555; Fax: 410-614-5129; E-mail: [email protected]
Copyright � 2010 by the American Society of Nephrology
BASIC RESEARCH www.jasn.org
654 ISSN : 1046-6673/2104-654 J Am Soc Nephrol 21: 654–665, 2010

plasma cells.19,20 A number of studies have demonstrated that Bcells infiltrate into renal allografts and contribute to rejection21,22;however, the exact role of B cells that have infiltrated into renalallografts is still unclear. Some studies reported that B cells couldcause transplant acute cellular rejection as well as humoral rejec-tion and increase the risk for graft failure independent of C4dperitubular deposition,23,24 whereas other studies have not shownthis clinical correlation.25,26 One recent article characterized intra-graft B cells during renal allograft rejection: Both mature B cellsand interstitial plasmablasts correlated with circulating donor-specific antibody concentration and poor response to steroidtherapy during rejection.27 The presence of mature B cells wasassociated with reduced graft survival.
On the basis of recent advances in studies of B cells in auto-and alloimmune diseases, the increasingly recognized patho-genic role for lymphocytes in IRI, and lack of treatment toaugment repair, we tested the hypothesis that B cells modulatethe repair process after kidney IRI. We analyzed the numbersand phenotypes of kidney-infiltrating B cells and the expres-sion of B lymphocyte chemoattractant (BLC) during the repairphase. We found marked trafficking of B cells into the post-ischemic kidney during repair, with a distinct phenotype atdifferent time points, along with increased BLC expression.We then evaluated the renal repair status of control (wild-type)mice, mature B cell– deficient (�MT) mice, �MT mice withadoptive B cell transfer, and �MT mice with serum transfer.We found that B cells modify tubular repair and proliferation.Finally, we targeted CD126-expressing plasma cells with ananti-CD126 antibody and found a significant improvement intissue repair after IRI.
RESULTS
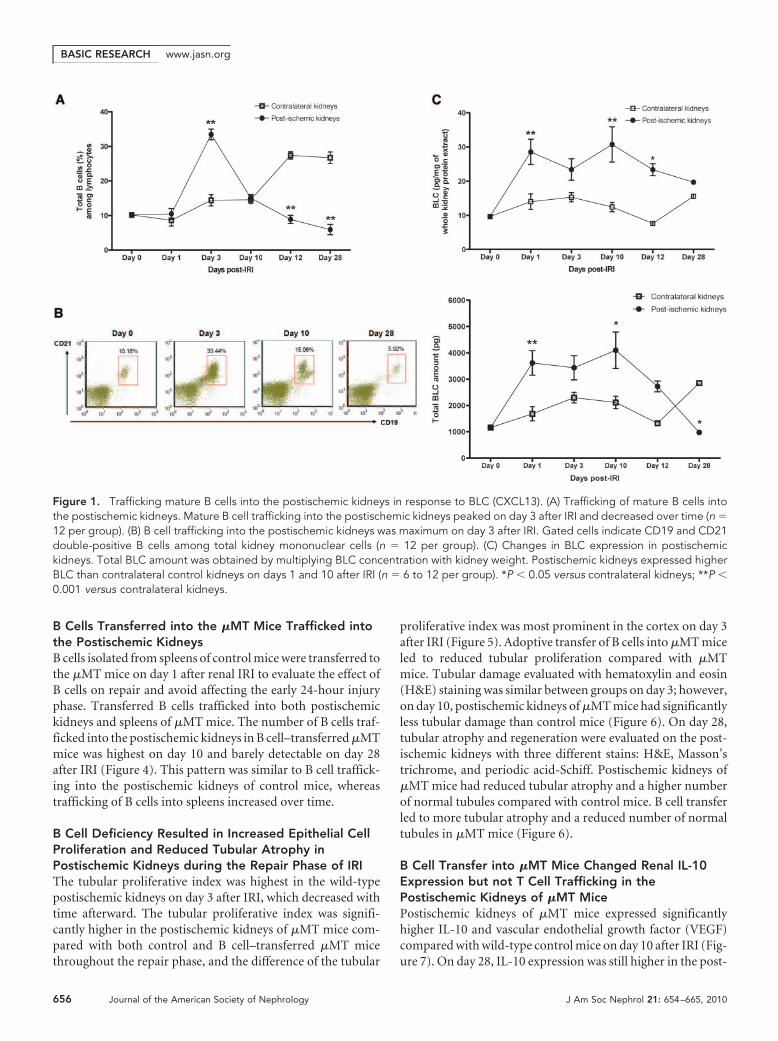
B Cells Trafficked into the Postischemic Kidneys andDifferentiated into Plasma CellsThe number of total kidney mononuclear cells (KMNCs) washighest in the postischemic kidneys on day 3 after IRI (con-
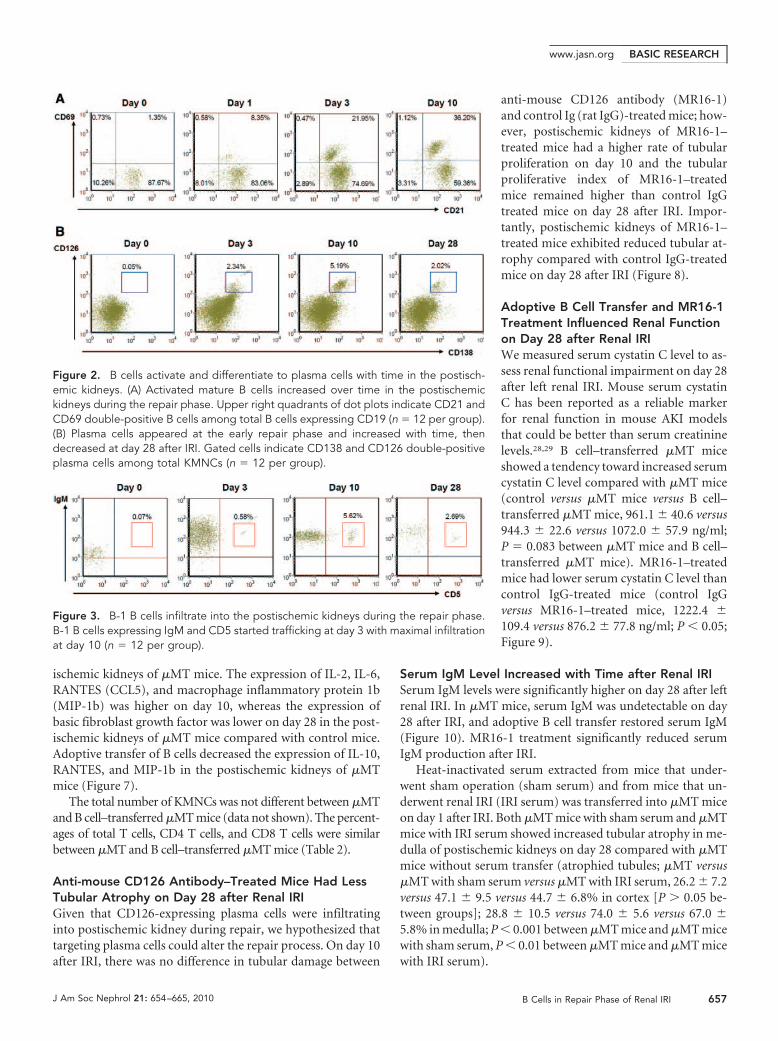
tralateral versus postischemic kidneys, 0.59 � 0.04 versus1.01 � 0.05 � 106 per pair of kidneys; P � 0.05). The numberof total KMNCs did not differ between contralateral and post-ischemic kidneys on days 1, 10, 12, and 28 after IRI (data notshown); however, an increased number of mature B cells (ex-pressing both CD19 and CD21) trafficked into the postisch-emic kidneys during the repair phase (Table 1). The percentageof B cells in the postischemic kidneys was the highest on day 3after IRI and decreased over time (Figure 1). BLC, measured inkidney protein extract, was increased on day 1 after IRI. Post-ischemic kidneys expressed higher BLC protein than contralat-eral kidneys on days 1 and 10 after IRI (Figure 1). Both matu-ration and activation status of infiltrating B cells wereevaluated with several surface markers including IgM, MHCclass II, CD21, CD38, CD40, CD69, CD126, and CD138. MHCclass II expression on B cells trafficked into the postischemickidneys was highest on day 3 after IRI, whereas CD69 (activa-tion marker) expression on B cells remained elevated through-out the repair phase (Figure 2, Table 1). We used anti-CD138 antibody and anti-CD126 antibody to identifyplasma cells. Plasma cells appeared on day 3 and reachedpeak numbers on day 10 after IRI, then decreased over time(Figure 2). There was no difference in the expression ofCD38 (surface marker for plasmablasts) and CD40 (surfacemarker reacting with CD40L on T cells) on B cells traffick-ing into postischemic kidneys at various time points of therepair phase (data not shown). The percentage of total Tcells among KMNCs markedly increased over time in thepostischemic kidneys (Table 1).
B-1 B Cells Trafficked into the Postischemic KidneysB-1 B cells, a minor subset of B cells abundant in the peritonealcavity, were examined with anti-CD5 antibody and anti-IgMantibody. The percentage of B-1 B cells among total B cells inthe kidneys was very low before IRI and remained low until day3 after IRI; however, the percentage of B-1 B cells markedlyincreased in the postischemic kidneys on day 10, then de-creased over time (Figure 3, Table 1).
Table 1. Postischemic kidney B cell and T cell populations in the repair phase of renal IRI in wild-type control mice
% of CellsDay 3 after IRI Day 10 after IRI Day 28 after IRI
CT IRI CT IRI CT IRI
Total B cells 14.41 � 1.63 33.44 � 1.54a 14.57 � 1.12 15.06 � 0.98b 26.71 � 1.64 5.92 � 1.49a,c
MHCII� B cells 9.42 � 1.31 23.60 � 1.76d 10.39 � 0.64 14.71 � 0.76b,d 13.58 � 2.65 4.65 � 0.42c,d
CD69� B cells 4.68 � 0.54 8.67 � 1.25d 4.46 � 0.53 17.32 � 3.03b,d 4.05 � 0.21 9.92 � 1.15d,e
B-1 B cells 0.54 � 0.11 0.58 � 0.11 0.65 � 0.14 5.62 � 1.16a,b 0.11 � 0.02 2.69 � 0.36a,c
CD126� plasma cells 0.07 � 0.02 2.34 � 0.33a 0.04 � 0.01 5.19 � 0.69a,b 0.07 � 0.01 2.02 � 0.19a,e
Total T cells 25.80 � 0.14 29.38 � 0.69 22.78 � 1.01 48.60 � 5.27a,b 27.35 � 0.74 66.61 � 2.65a,c
Data are means � SEM (n � 12 per group). CT, contralateral kidneys; IRI, postischemic kidneys; total B cells, mature B cells expressing CD19 and CD21 amongtotal KMNCs; MHCII� B cells, MHC class II expressing B cells among total KMNCs; CD69� B cells, CD69 (early activation marker) expressing B cells amongtotal KMNCs; B-1 B cells, B-1 B cell subset expressing CD5 and IgM among total B cells; plasma cells, plasma cells expressing CD138 and CD126 among totalKMNCs; total T cells, T cells expressing T cell receptor �.aP � 0.001 versus contralateral kidneys at the same time point.bP � 0.05 versus postischemic kidneys on day 3 after IRI.cP � 0.05 versus postischemic kidneys on day 3 and day 10 after IRI.dP � 0.05 versus contralateral kidneys at the same time point.eP � 0.05 versus postischemic kidneys on day 10 after IRI.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 655
B Cells Transferred into the �MT Mice Trafficked intothe Postischemic KidneysB cells isolated from spleens of control mice were transferred tothe �MT mice on day 1 after renal IRI to evaluate the effect ofB cells on repair and avoid affecting the early 24-hour injuryphase. Transferred B cells trafficked into both postischemickidneys and spleens of �MT mice. The number of B cells traf-ficked into the postischemic kidneys in B cell–transferred �MTmice was highest on day 10 and barely detectable on day 28after IRI (Figure 4). This pattern was similar to B cell traffick-ing into the postischemic kidneys of control mice, whereastrafficking of B cells into spleens increased over time.
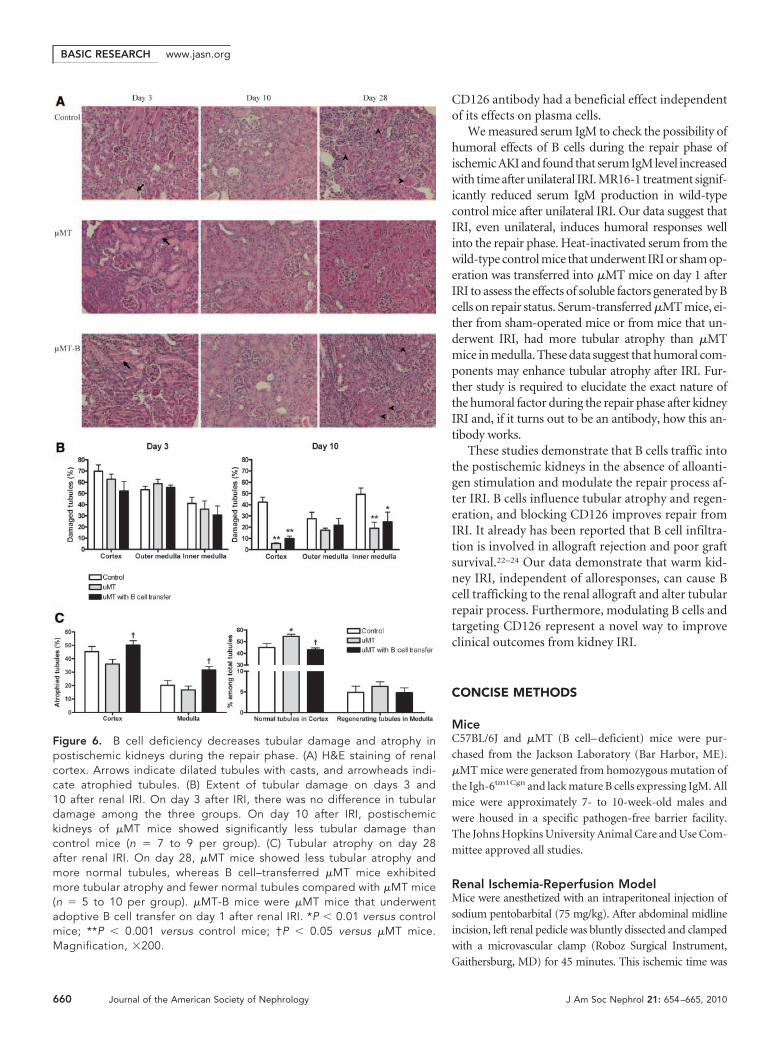
B Cell Deficiency Resulted in Increased Epithelial CellProliferation and Reduced Tubular Atrophy inPostischemic Kidneys during the Repair Phase of IRIThe tubular proliferative index was highest in the wild-typepostischemic kidneys on day 3 after IRI, which decreased withtime afterward. The tubular proliferative index was signifi-cantly higher in the postischemic kidneys of �MT mice com-pared with both control and B cell–transferred �MT micethroughout the repair phase, and the difference of the tubular
proliferative index was most prominent in the cortex on day 3after IRI (Figure 5). Adoptive transfer of B cells into �MT miceled to reduced tubular proliferation compared with �MTmice. Tubular damage evaluated with hematoxylin and eosin(H&E) staining was similar between groups on day 3; however,on day 10, postischemic kidneys of �MT mice had significantlyless tubular damage than control mice (Figure 6). On day 28,tubular atrophy and regeneration were evaluated on the post-ischemic kidneys with three different stains: H&E, Masson’strichrome, and periodic acid-Schiff. Postischemic kidneys of�MT mice had reduced tubular atrophy and a higher numberof normal tubules compared with control mice. B cell transferled to more tubular atrophy and a reduced number of normaltubules in �MT mice (Figure 6).
B Cell Transfer into �MT Mice Changed Renal IL-10Expression but not T Cell Trafficking in thePostischemic Kidneys of �MT MicePostischemic kidneys of �MT mice expressed significantlyhigher IL-10 and vascular endothelial growth factor (VEGF)compared with wild-type control mice on day 10 after IRI (Fig-ure 7). On day 28, IL-10 expression was still higher in the post-
Figure 1. Trafficking mature B cells into the postischemic kidneys in response to BLC (CXCL13). (A) Trafficking of mature B cells intothe postischemic kidneys. Mature B cell trafficking into the postischemic kidneys peaked on day 3 after IRI and decreased over time (n �12 per group). (B) B cell trafficking into the postischemic kidneys was maximum on day 3 after IRI. Gated cells indicate CD19 and CD21double-positive B cells among total kidney mononuclear cells (n � 12 per group). (C) Changes in BLC expression in postischemickidneys. Total BLC amount was obtained by multiplying BLC concentration with kidney weight. Postischemic kidneys expressed higherBLC than contralateral control kidneys on days 1 and 10 after IRI (n � 6 to 12 per group). *P � 0.05 versus contralateral kidneys; **P �0.001 versus contralateral kidneys.
BASIC RESEARCH www.jasn.org
656 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 654–665, 2010
ischemic kidneys of �MT mice. The expression of IL-2, IL-6,RANTES (CCL5), and macrophage inflammatory protein 1b(MIP-1b) was higher on day 10, whereas the expression ofbasic fibroblast growth factor was lower on day 28 in the post-ischemic kidneys of �MT mice compared with control mice.Adoptive transfer of B cells decreased the expression of IL-10,RANTES, and MIP-1b in the postischemic kidneys of �MTmice (Figure 7).
The total number of KMNCs was not different between �MTand B cell–transferred �MT mice (data not shown). The percent-ages of total T cells, CD4 T cells, and CD8 T cells were similarbetween �MT and B cell–transferred �MT mice (Table 2).
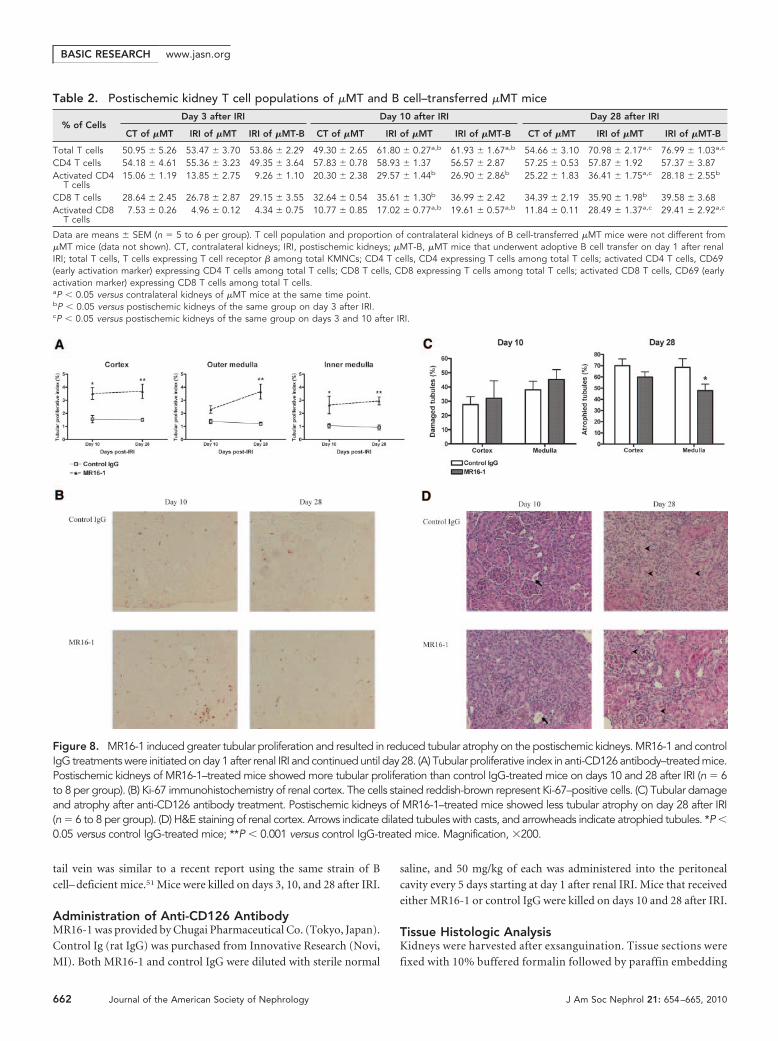
Anti-mouse CD126 Antibody–Treated Mice Had LessTubular Atrophy on Day 28 after Renal IRIGiven that CD126-expressing plasma cells were infiltratinginto postischemic kidney during repair, we hypothesized thattargeting plasma cells could alter the repair process. On day 10after IRI, there was no difference in tubular damage between
anti-mouse CD126 antibody (MR16-1)and control Ig (rat IgG)-treated mice; how-ever, postischemic kidneys of MR16-1–treated mice had a higher rate of tubularproliferation on day 10 and the tubularproliferative index of MR16-1–treatedmice remained higher than control IgGtreated mice on day 28 after IRI. Impor-tantly, postischemic kidneys of MR16-1–treated mice exhibited reduced tubular at-rophy compared with control IgG-treatedmice on day 28 after IRI (Figure 8).
Adoptive B Cell Transfer and MR16-1Treatment Influenced Renal Functionon Day 28 after Renal IRIWe measured serum cystatin C level to as-sess renal functional impairment on day 28after left renal IRI. Mouse serum cystatinC has been reported as a reliable markerfor renal function in mouse AKI modelsthat could be better than serum creatininelevels.28,29 B cell–transferred �MT miceshowed a tendency toward increased serumcystatin C level compared with �MT mice(control versus �MT mice versus B cell–transferred �MT mice, 961.1 � 40.6 versus944.3 � 22.6 versus 1072.0 � 57.9 ng/ml;P � 0.083 between �MT mice and B cell–transferred �MT mice). MR16-1–treatedmice had lower serum cystatin C level thancontrol IgG-treated mice (control IgGversus MR16-1–treated mice, 1222.4 �109.4 versus 876.2 � 77.8 ng/ml; P � 0.05;Figure 9).
Serum IgM Level Increased with Time after Renal IRISerum IgM levels were significantly higher on day 28 after leftrenal IRI. In �MT mice, serum IgM was undetectable on day28 after IRI, and adoptive B cell transfer restored serum IgM(Figure 10). MR16-1 treatment significantly reduced serumIgM production after IRI.
Heat-inactivated serum extracted from mice that under-went sham operation (sham serum) and from mice that un-derwent renal IRI (IRI serum) was transferred into �MT miceon day 1 after IRI. Both �MT mice with sham serum and �MTmice with IRI serum showed increased tubular atrophy in me-dulla of postischemic kidneys on day 28 compared with �MTmice without serum transfer (atrophied tubules; �MT versus�MT with sham serum versus �MT with IRI serum, 26.2 � 7.2versus 47.1 � 9.5 versus 44.7 � 6.8% in cortex [P � 0.05 be-tween groups]; 28.8 � 10.5 versus 74.0 � 5.6 versus 67.0 �5.8% in medulla; P � 0.001 between �MT mice and �MT micewith sham serum, P � 0.01 between �MT mice and �MT micewith IRI serum).
Figure 2. B cells activate and differentiate to plasma cells with time in the postisch-emic kidneys. (A) Activated mature B cells increased over time in the postischemickidneys during the repair phase. Upper right quadrants of dot plots indicate CD21 andCD69 double-positive B cells among total B cells expressing CD19 (n � 12 per group).(B) Plasma cells appeared at the early repair phase and increased with time, thendecreased at day 28 after IRI. Gated cells indicate CD138 and CD126 double-positiveplasma cells among total KMNCs (n � 12 per group).
Figure 3. B-1 B cells infiltrate into the postischemic kidneys during the repair phase.B-1 B cells expressing IgM and CD5 started trafficking at day 3 with maximal infiltrationat day 10 (n � 12 per group).
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 657

DISCUSSION
Our study demonstrates that B cells modulate kidney repairafter IRI. B cells migrate into the postischemic kidney duringthe repair phase, with specific changes in populations and ac-tivation status with time. B cell deficiency reduces renal tubularatrophy by enhancing tubular proliferation. Adoptive transferof B cells into �MT mice increases renal tubular atrophy andreduces tubular proliferation in �MT mice, demonstratingthat the altered tubular atrophy and proliferation in �MT micewere indeed due to B cell deficiency. Targeting infiltratingCD126-expressing plasma cells using anti-CD126 antibody ledto reduced tubular atrophy with enhanced tubular prolifera-tion and reduced functional impairment. These data are thefirst demonstration of a role for B cells in repair from kidneywarm IRI and reveal the promise of targeting B cells to improveclinical outcomes in this common condition.
We initially studied B cell infiltration into the post-ischemic kidneys to look for evidence supporting a rolefor B cells in the repair process. The maximum increasein mature infiltrating B cells occurred at day 3 and re-turned to control levels by day 10. T cell trafficking intothe postischemic kidneys was markedly increased at day10 and remained increased during the late repair phaseup to day 28. That B cell trafficking preceded T celltrafficking during repair suggests that B cells could beinvolved in regulating T cell infiltration and expansionin postischemic kidney. An important role of B cells asantigen-presenting or co-stimulatory cells was also re-ported in animal models of autoimmune disease30;however, adoptive B cell transfer into �MT mice did notaffect T cell trafficking into the postischemic kidneys,suggesting that B cells could be acting on tubular repairindependent of T cell trafficking.
On day 10, there was a peak in plasma cells and B-1 Bcells in the postischemic kidneys. Intragraft plasma cellswere found to correlate with circulating donor-specific an-tibodies in a recent clinical study27; however, our IRImodel was alloantigen independent. There is evidence thatB-1 B cells are involved in initiation of early injury in amurine intestinal IRI model but not in repair.31 A recentclinical report also suggested an important role of B cellsexpressing both CD19 and CD5 (expressed on B-1 B cells)in the pathogenesis of primary IgA nephropathy.32
After finding B cell trafficking in postischemic kid-ney during repair, we examined the expression of a BLCthat could be involved in recruiting the B cells. BLC is a16- to 18-kD member of the �- or CXC family of che-mokine and induces migration of both naive B cells andB-1 B cells.33,34 The important role of BLC as a B cellchemoattractant is supported by a report demonstrat-ing that overexpression of CXCR5, the receptor forBLC, was sufficient to overcome antigen-induced B cellmovement to the T cell zone.33 BLC expression in thepostischemic kidneys was significantly increased after
renal IRI, remained elevated during the repair phase, and couldhave mediated the B cell trafficking.
Given that B cells were trafficking into postischemic kidney,with a distinct phenotypic change with time and an upregulationof BLC, we hypothesized that B cells directly modulate the repairprocess from kidney IRI. We therefore examined repair from kid-ney IRI in �MT mice. Ki-67 immunostaining was used to evaluatetubular proliferation; this staining was previously reported as auseful and accurate tool in assessing renal tubular prolifera-tion.35,36 At day 3, there was no difference in the tubular injury ofpostischemic kidneys between wild-type control and �MT mice;however, at day 10, there was greater tubular proliferation andreduced tubular injury in �MT mice. To test whether thesechanges in the �MT mice were due to the absence of B cells, weadoptively transferred B cells into the �MT mice. With adoptivetransfer of B cells, the �MT mice regained tubular proliferationand tubular injury similar to wild-type control mice, demonstrat-
Figure 4. B cells adoptively transfer into �MT mice. B cells were isolatedfrom spleens of control mice and transferred into �MT mice on day 1 afterIRI. (A) B cells transferred into �MT mice trafficked into both spleens andpostischemic kidneys (n � 5 to 6 per group). (B) Postischemic kidneys ofB cell–transferred �MT mice showed the highest number of B cells on day10 after IRI. Gated cells indicate CD19� B cells (n � 5 to 6 per group). (C)B cells trafficked into the postischemic kidneys of B cell–transferred �MTmice differentiated into plasma cells. Gated cells indicate plasma cellsexpressing CD138 and CD126 (n � 5 to 6 per group). *P � 0.05 versus�MT mice; **P � 0.001 versus �MT mice.
BASIC RESEARCH www.jasn.org
658 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 654–665, 2010

ing that it was indeed the B cells that were mediating this effect onpostischemic kidney repair. Renal tubular cells in the cortex ofpostischemic kidneys after blood flow has returned to near nor-mal levels have been suggested as an early site of repair processesduring AKI.37 In �MT mice, the tubular proliferative index ofpostischemic kidneys was significantly higher than control miceon day 3, and this may have led to less tubular damage of post-ischemic kidneys compared with control mice on day 10 after IRI.On day 28 after renal IRI, postischemic kidneys of �MT mice hadreduced tubular atrophy and more normal tubules than control
mice, which may have resulted from higher tubularproliferation of �MT mice on day 10. B cell defi-ciency has also been reported to attenuate liver fi-brosis in a murine CCl4-induced liver fibrosis mod-el.38 Adoptive transfer of B cells into �MT mice inour study resulted in a marked decrease of the tubu-lar proliferative index of postischemic kidneys, andtubular damage and atrophy were comparable tocontrol mice. Adoptively transferred B cells traf-ficked into the postischemic kidneys and differenti-ated into plasma cells. B cell transfer also affectedintrarenal cytokine expression. Postischemic kid-neys of �MT mice expressed higher IL-10 on days10 and 28 and higher VEGF on day 10 after IRI. Aprotective role of IL-10 in renal IRI has been sug-gested in several studies,39 – 42 and a beneficial ef-fect of VEGF on renal IRI has also been reported.43,44
After adoptive B cell transfer into �MT mice, re-nal expression of these cytokines was reducedto a level comparable to control mice. We alsoinvestigated the degree of fibrosis using Mas-son’s trichrome staining on day 28 and founddiscordant effects of B cells on fibrosis com-pared with tubular atrophy. Future studies thatexamine fibrosis more carefully at various and latertime points would be useful in evaluating the influ-ence of B cell manipulation on fibrosis after AKI.
One of the unexpected findings was the in-crease in kidney-infiltrating plasma cells express-ing CD138 and CD126 (IL-6 receptor) during re-pair, particularly at day 10. Intrarenal plasma cellswere recently implicated in humoral rejection.27
We tested the functional significance of plasmacells during repair from IRI using MR16-1 givenat day 1 after IRI (rather than before or at the timeof ischemia so as not to interfere with early injurymechanisms). MR16-1 is known to bind andblock both soluble and membrane-bound typesof IL-6 receptor.45 The postischemic kidneys ofMR16-1–treated mice did not show any differ-ence in tubular injury on day 10; however, therewas less tubular atrophy on day 28 after renal IRIwith MR16-1 treatment compared with controlmice. The tubular proliferative index of postisch-emic kidneys of MR16-1–treated mice was al-
ready higher than control mice on day 10 and remained so onday 28 after IRI. The beneficial effect of MR16-1 has been re-ported in other disease models in which B cells contribute tothe pathogenesis, such as collagen-induced arthritis andCastleman disease, and in a murine autoimmune kidney dis-ease model.46 – 48 Although CD126 is not entirely specific forplasma cells, CD126 is strongly expressed on plasma cells andrelatively weakly expressed on other leukocytes.49 Plasma cellsmay function in renal IRI in a manner distinct from their rolein secreting Igs; however, we cannot exclude the possibility that
Figure 5. B cell deficiency increases tubular proliferation in postischemickidneys during the repair phase. (A) Tubular proliferative index in postischemickidneys. The tubular proliferative index of postischemic kidneys was highest onday 3 after IRI in all groups and decreased over time (n � 5 to 8 per group). (B)Ki-67 immunohistochemistry of renal cortex. The tubular proliferative index of�MT mice was higher than both control and B cell–transferred �MT mice in thecortex throughout the repair phase and also in the medulla on days 3 and 10after IRI. The cells stained reddish-brown represent Ki-67–positive cells, whichreflect the proliferation process. �MT-B mice were �MT mice that underwentadoptive B cell transfer on day 1 after renal IRI. *P � 0.05 versus control mice;**P � 0.001 versus control mice. Magnification, �200.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 659
CD126 antibody had a beneficial effect independentof its effects on plasma cells.
We measured serum IgM to check the possibility ofhumoral effects of B cells during the repair phase ofischemic AKI and found that serum IgM level increasedwith time after unilateral IRI. MR16-1 treatment signif-icantly reduced serum IgM production in wild-typecontrol mice after unilateral IRI. Our data suggest thatIRI, even unilateral, induces humoral responses wellinto the repair phase. Heat-inactivated serum from thewild-type control mice that underwent IRI or sham op-eration was transferred into �MT mice on day 1 afterIRI to assess the effects of soluble factors generated by Bcells on repair status. Serum-transferred �MT mice, ei-ther from sham-operated mice or from mice that un-derwent IRI, had more tubular atrophy than �MTmice in medulla. These data suggest that humoral com-ponents may enhance tubular atrophy after IRI. Fur-ther study is required to elucidate the exact nature ofthe humoral factor during the repair phase after kidneyIRI and, if it turns out to be an antibody, how this an-tibody works.
These studies demonstrate that B cells traffic intothe postischemic kidneys in the absence of alloanti-gen stimulation and modulate the repair process af-ter IRI. B cells influence tubular atrophy and regen-eration, and blocking CD126 improves repair fromIRI. It already has been reported that B cell infiltra-tion is involved in allograft rejection and poor graftsurvival.22–24 Our data demonstrate that warm kid-ney IRI, independent of alloresponses, can cause Bcell trafficking to the renal allograft and alter tubularrepair process. Furthermore, modulating B cells andtargeting CD126 represent a novel way to improveclinical outcomes from kidney IRI.
CONCISE METHODS
MiceC57BL/6J and �MT (B cell– deficient) mice were pur-
chased from the Jackson Laboratory (Bar Harbor, ME).
�MT mice were generated from homozygous mutation of
the Igh-6tm1Cgn and lack mature B cells expressing IgM. All
mice were approximately 7- to 10-week-old males and
were housed in a specific pathogen-free barrier facility.
The Johns Hopkins University Animal Care and Use Com-
mittee approved all studies.
Renal Ischemia-Reperfusion ModelMice were anesthetized with an intraperitoneal injection of
sodium pentobarbital (75 mg/kg). After abdominal midline
incision, left renal pedicle was bluntly dissected and clamped
with a microvascular clamp (Roboz Surgical Instrument,
Gaithersburg, MD) for 45 minutes. This ischemic time was
Figure 6. B cell deficiency decreases tubular damage and atrophy inpostischemic kidneys during the repair phase. (A) H&E staining of renalcortex. Arrows indicate dilated tubules with casts, and arrowheads indi-cate atrophied tubules. (B) Extent of tubular damage on days 3 and10 after renal IRI. On day 3 after IRI, there was no difference in tubulardamage among the three groups. On day 10 after IRI, postischemickidneys of �MT mice showed significantly less tubular damage thancontrol mice (n � 7 to 9 per group). (C) Tubular atrophy on day 28after renal IRI. On day 28, �MT mice showed less tubular atrophy andmore normal tubules, whereas B cell–transferred �MT mice exhibitedmore tubular atrophy and fewer normal tubules compared with �MT mice(n � 5 to 10 per group). �MT-B mice were �MT mice that underwentadoptive B cell transfer on day 1 after renal IRI. *P � 0.01 versus controlmice; **P � 0.001 versus control mice; †P � 0.05 versus �MT mice.Magnification, �200.
BASIC RESEARCH www.jasn.org
660 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 654–665, 2010

chosen after preliminary experiments, in which 30 minutes of ischemia
led to too mild histologic response on days 10 and 28 and 60 minutes of
ischemia led to too severe histologic injury. During the procedures, mice
were kept well hydrated with warm sterile saline at a constant tempera-
ture (37°C). After the clamps were removed, the wounds were sutured
and the mice were allowed to recover with free access to food and water.
Cohorts of mice were killed on days 1, 3, 10, 12, and 28 after surgery. Both
postischemic kidneys and contralateral (right, untouched) kidneys were
collected and compared.
Flow Cytometry Analysis of KMNCsKMNCs were isolated according to the method previously de-
scribed.50 Briefly, decapsulated kidneys were immersed in RPMI
buffer (Mediatech, Manassas, VA) containing 5% FBS and disrupted
mechanically using Stomacher 80 Biomaster (Seward, Worthing,
West Sussex, UK). Samples were strained, washed, and resuspended
in 36% Percoll (Amersham Pharmacia Biotech, Piscataway, NJ) fol-
lowed by gentle overlaying onto 72% Percoll. After centrifugation at
1000 � g for 30 minutes at room temperature, KMNCs were collected
from the Percoll interface, washed twice, and counted on a hemocy-
tometer using trypan blue exclusion.
Isolated KMNCs were preincubated with anti-CD16/CD32 Fc re-
ceptor blocking antibody for 10 minutes to minimize nonspecific an-
tibody binding. Cells were then incubated with anti-mouse anti-
CD19, CD21, CD38, CD40, CD69, CD126, CD138, IgM, and MHC
class II (all from BD Biosciences except for anti-mouse FITC-conju-
gated anti-IgM antibody from eBioscience, San Diego, CA) for 25
minutes at 4°C, washed with FACS buffer, and fixed in 1% parafor-
maldehyde solution. Four-color immunofluorescence staining was
acquired and analyzed using a FACSCalibur instrument (BD Bio-
sciences, San Jose, CA) and FCS Express V3 (De Novo Software, Los
Angeles, CA). Each assay included at least 10,000 gated events.
Mouse BLC (CXCL13) ImmunoassayMouse BLC (CXCL13) was measured on kidney protein samples
extracted from whole-kidney tissues using mouse BLC immuno-
assay kit (R&D Systems, Minneapolis, MN) according to the man-
ufacturer’s protocol. BLC concentration (pg/ml) of all samples was
divided by protein concentration (mg/ml) for normalization.
Isolation of B Cells and Adoptive Transfer into �MTMiceMature B cells were isolated from spleens of C57BL/6J mice using a
mouse B cell enrichment kit (StemCell Technologies, Vancouver,
British Columbia, Canada). Briefly, spleens were macerated in RPMI
buffer containing 5% FBS and centrifuged, followed by red blood cell
lysis using 1� red blood cell lysis buffer (eBioscience). The splenocyte
pellet was resuspended in PBS with 2% normal rat serum. The cell
suspension was incubated with SpinSep mouse B cell enrichment
cocktail, biotin selection cocktail, and dense particles sequentially.
Then samples were diluted and layered on top of the SpinSep density
medium. After centrifugation at 1200 � g for 10 minutes at room
temperature, B cells were collected from the top of the density me-
dium. B cells were resuspended in sterile normal saline and injected
into �MT mice on day 1 after renal IRI. We used a B cell isolation
method based on negative selection to avoid any disruption of B cell
integrity. Mean purity of CD19-expressing mature B cells analyzed
with flow cytometry was 95%. Approximately 1.5 � 107 cells were
injected into the tail vein and 4.5 � 107 cells were injected into peri-
toneal cavity in each mouse. The amount of B cells injected into the
Figure 7. Cytokines are expressed in postischemic kidneys. (A)Postischemic kidneys of �MT mice expressed higher IL-10 andVEGF on day 10 after IRI than wild-type controls. B cell transferreduced the expression of IL-10 and VEGF in the postischemickidneys of �MT mice (n � 5 per group). (B) Postischemic kidneysof �MT mice expressed higher IL-2, IL-6, RANTES, and MIP-1b onday 10 but lower basic fibroblast growth factor (FGF) on day 28compared with control mice. B cell transfer reduced the expres-sion of RANTES and MIP-1b in the postischemic kidneys of �MTmice (n � 5 per group). *P � 0.05 versus control mice; **P �0.001 versus control mice; †P � 0.05 versus �MT mice.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 661
tail vein was similar to a recent report using the same strain of B
cell– deficient mice.51 Mice were killed on days 3, 10, and 28 after IRI.
Administration of Anti-CD126 AntibodyMR16-1 was provided by Chugai Pharmaceutical Co. (Tokyo, Japan).
Control Ig (rat IgG) was purchased from Innovative Research (Novi,
MI). Both MR16-1 and control IgG were diluted with sterile normal
saline, and 50 mg/kg of each was administered into the peritoneal
cavity every 5 days starting at day 1 after renal IRI. Mice that received
either MR16-1 or control IgG were killed on days 10 and 28 after IRI.
Tissue Histologic AnalysisKidneys were harvested after exsanguination. Tissue sections were
fixed with 10% buffered formalin followed by paraffin embedding
Table 2. Postischemic kidney T cell populations of �MT and B cell–transferred �MT mice
% of CellsDay 3 after IRI Day 10 after IRI Day 28 after IRI
CT of �MT IRI of �MT IRI of �MT-B CT of �MT IRI of �MT IRI of �MT-B CT of �MT IRI of �MT IRI of �MT-B
Total T cells 50.95 � 5.26 53.47 � 3.70 53.86 � 2.29 49.30 � 2.65 61.80 � 0.27a,b 61.93 � 1.67a,b 54.66 � 3.10 70.98 � 2.17a,c 76.99 � 1.03a,c
CD4 T cells 54.18 � 4.61 55.36 � 3.23 49.35 � 3.64 57.83 � 0.78 58.93 � 1.37 56.57 � 2.87 57.25 � 0.53 57.87 � 1.92 57.37 � 3.87Activated CD4
T cells15.06 � 1.19 13.85 � 2.75 9.26 � 1.10 20.30 � 2.38 29.57 � 1.44b 26.90 � 2.86b 25.22 � 1.83 36.41 � 1.75a,c 28.18 � 2.55b
CD8 T cells 28.64 � 2.45 26.78 � 2.87 29.15 � 3.55 32.64 � 0.54 35.61 � 1.30b 36.99 � 2.42 34.39 � 2.19 35.90 � 1.98b 39.58 � 3.68Activated CD8
T cells7.53 � 0.26 4.96 � 0.12 4.34 � 0.75 10.77 � 0.85 17.02 � 0.77a,b 19.61 � 0.57a,b 11.84 � 0.11 28.49 � 1.37a,c 29.41 � 2.92a,c
Data are means � SEM (n � 5 to 6 per group). T cell population and proportion of contralateral kidneys of B cell-transferred �MT mice were not different from�MT mice (data not shown). CT, contralateral kidneys; IRI, postischemic kidneys; �MT-B, �MT mice that underwent adoptive B cell transfer on day 1 after renalIRI; total T cells, T cells expressing T cell receptor � among total KMNCs; CD4 T cells, CD4 expressing T cells among total T cells; activated CD4 T cells, CD69(early activation marker) expressing CD4 T cells among total T cells; CD8 T cells, CD8 expressing T cells among total T cells; activated CD8 T cells, CD69 (earlyactivation marker) expressing CD8 T cells among total T cells.aP � 0.05 versus contralateral kidneys of �MT mice at the same time point.bP � 0.05 versus postischemic kidneys of the same group on day 3 after IRI.cP � 0.05 versus postischemic kidneys of the same group on days 3 and 10 after IRI.
Figure 8. MR16-1 induced greater tubular proliferation and resulted in reduced tubular atrophy on the postischemic kidneys. MR16-1 and controlIgG treatments were initiated on day 1 after renal IRI and continued until day 28. (A) Tubular proliferative index in anti-CD126 antibody–treated mice.Postischemic kidneys of MR16-1–treated mice showed more tubular proliferation than control IgG-treated mice on days 10 and 28 after IRI (n � 6to 8 per group). (B) Ki-67 immunohistochemistry of renal cortex. The cells stained reddish-brown represent Ki-67–positive cells. (C) Tubular damageand atrophy after anti-CD126 antibody treatment. Postischemic kidneys of MR16-1–treated mice showed less tubular atrophy on day 28 after IRI(n � 6 to 8 per group). (D) H&E staining of renal cortex. Arrows indicate dilated tubules with casts, and arrowheads indicate atrophied tubules. *P �0.05 versus control IgG-treated mice; **P � 0.001 versus control IgG-treated mice. Magnification, �200.
BASIC RESEARCH www.jasn.org
662 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 654–665, 2010

and stained with H&E, periodic acid-Schiff, or Masson’s
trichrome. Renal tubular damage, atrophy, and regeneration were
scored by a renal pathologist who was blinded to the experimental
groups.
Immunohistochemistry of Postischemic Kidneys withKi-67Immunohistochemistry staining with Ki-67 was performed on for-
malin-fixed kidney tissues. Sections (4 �m) were deparaffinized
with xylene and rehydrated in a graded alcohol series, then placed
in citrate buffer solution (pH 6.0). Slides were placed in a pressure
cooker and heated with microwave for 10 minutes to enhance
antigen retrieval. After cooling, sections were immersed in 3%
hydrogen peroxide for 10 minutes to block endogenous peroxi-
dase, then treated with avidin/biotin block and protein block se-
rum-free (DAKO, Carpinteria, CA) sequentially and incubated
overnight at 4°C with 1:50 diluted monoclonal rat anti-mouse an-
tibody to Ki-67 (clone TEC-3; DAKO), a 360-kD nuclear protein,
expressed by proliferating cells in all phases of the active cell cycle
but absent in resting cells. The next day, the slides were incubated
with a biotin-conjugated goat anti-rat IgG secondary antibody
(Jackson ImmunoResearch, West Grove, PA) for 30 minutes at
room temperature, followed by an avidin-biotin horseradish per-
oxidase complex (ABC peroxidase; Vector Laboratories, Burlin-
game, CA). 3,3�-Diaminobenzidene tetrahydrochloride (Vector
Laboratories) was applied to the slides for developing brown color.
Counterstaining was done with eosin.
For calculation of the tubular proliferative index, Ki-67–incu-
bated slides were also incubated with Hoechst Dye solution (In-
vitrogen, Carlsbad, CA) for 5 minutes in the dark to counterstain
total nuclei. Light and fluorescence microscopy of the same field
were performed at �200 magnification (Eclipse E600 microscope;
Nikon, Melville, NY). Three different areas of cortex, outer me-
dulla, and inner medulla were evaluated for each slide. Images
were captured and digitized using Digital Still Camera DXM1200
and ACT-1 software (Nikon) and then analyzed for cell counting
using ImageJ software (National Institutes of Health, Bethesda,
MD). For each animal, the tubular proliferative index in cortex,
outer medulla, and inner medulla was expressed as ratio between
the tubular Ki-67–positive cells and total tubular cell nuclei of the
examined fields.36
Bioplex Protein ArrayA panel of cytokines were measured in whole-kidney protein extracts
with Bioplex Protein Array system (Bio-Rad, Hercules, CA) according
to the manufacturer’s method. This is a multiplexed, particle-based,
flow cytometric assay that uses anti-cytokine mAbs linked to micro-
spheres incorporating distinct properties of two fluorescence dyes.
Our assay was designed to detect and quantify IL-2, IL-6, IL-10, RAN-
TES (CCL5), VEGF, MIP-1b, and basic fibroblast growth factor. Each
cytokine value was normalized by dividing the raw cytokine concen-
tration (pg/ml) with kidney protein concentration (mg/ml) measured
by the Bradford method.
Figure 10. Serum IgM levels varied after IRI. (A) In the control(wild-type) mice that underwent renal IRI, serum samples ob-tained on day 28 after renal IRI contained significantly higher IgMcompared with serum samples obtained on days 0 and 10 (n � 5to 12 per group). *P � 0.05 versus both days 0 and 10. (B) On day28 after renal IRI, serum IgM was not detected in �MT mice. Bcell–transferred �MT mice had significantly higher IgM in theirserum than �MT mice but a comparable level of IgM to controlmice (n � 7 to 12 per group). *P � 0.001 versus control; †P �0.001 versus �MT. (C) On day 28 after renal IRI, MR16-1–treatedmice revealed a significantly reduced level of serum IgM com-pared with control IgG-treated mice (n � 6 per group). *P � 0.01versus control IgG.
Figure 9. Serum cystatin C levels varied on day 28 after IRI. (A)Serum cystatin C level tended to be higher in B cell–transferred�MT mice compared with �MT mice on day 28 after left renal IRI(n � 4 per group). (B) MR16-1–treated mice revealed lower serumcystatin C than control IgG-treated mice on day 28 after left renalIRI (n � 6 per group). �MT-B mice were �MT mice that underwentadoptive B cell transfer on day 1 after renal IRI. *P � 0.05 versuscontrol IgG-treated mice.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 663

Measurement of Serum Cystatin C LevelCystatin C level was measured with serum samples obtained on day 28
after left renal IRI using ELISA kit (Biovendor, Candler, NC) accord-
ing to the manufacturer’s recommended method.
Measurement of Serum IgM LevelIgM level was measured with serum samples obtained on days 0, 10,
and 28 after left renal IRI using ELISA kit (Immunology Consultants
Laboratory, Inc., Newberg, OR) according to the manufacturer’s rec-
ommended method.
Serum Transfer into �MT MiceSerum was extracted from the control (C57BL/6J) mice that under-
went left renal IRI or sham operation on day 10. All serum was heated
at 56°C for 30 minutes before being transferred to inactivate comple-
ment. A total of 0.5 ml of heat-inactivated serum (half into the tail
vein and half into the peritoneal cavity) was injected into each �MT
mice on day 1 after renal IRI. Mice were killed on day 28 after IRI.
Statistical AnalysisAll data are expressed as mean � SEM. Group means were compared
using Mann-Whitney test using SPSS 12.0K or ANOVA followed by
Newman-Keuls post hoc analysis using GraphPad Prism 4. Statistical
significance was determined at P � 0.05.
ACKNOWLEDGMENTS
This study was supported by National Institute of Diabetes and Di-
gestive and Kidney Diseases grant R01DK054770 to H.R. H.R.J. was
supported by the Korea Research Foundation.
We thank Chaitali Sarkar and Priya Kesari for technical assistance
and Dr. Yanfei Huang for helpful advice.
DISCLOSURESNone.
REFERENCES
1. Perico N, Cattaneo D, Sayegh MH, Remuzzi G: Delayed graft functionin kidney transplantation. Lancet 364: 1814–1827, 2004
2. Jang HR, Rabb H: The innate immune response in ischemic acutekidney injury. Clin Immunol 130: 41–50, 2009
3. Okusa MD: The inflammatory cascade in acute ischemic renal failure.Nephron 90: 133–138, 2002
4. Rabb H, Daniels F, O’Donnell M, Haq M, Saba SR, Keane W, TangWW: Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 279: F525–F531,2000
5. Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB,O’Donnell MP, Rabb H: Identification of the CD4(�) T cell as a majorpathogenic factor in ischemic acute renal failure. J Clin Invest 108:1283–1290, 2001
6. Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD: Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protec-
tion: The role of CD4� T cells and IFN-gamma. J Immunol 176:3108–3114, 2006
7. Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H:B cell deficiency confers protection from renal ischemia reperfusioninjury. J Immunol 171: 3210–3215, 2003
8. Weiser MR, Williams JP, Moore FD Jr, Kobzik L, Ma M, Hechtman HB,Carroll MC: Reperfusion injury of ischemic skeletal muscle is mediated bynatural antibody and complement. J Exp Med 183: 2343–2348, 1996
9. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM: B cellsregulate autoimmunity by provision of IL-10. Nat Immunol 3: 944–950,2002
10. Mauri C, Gray D, Mushtaq N, Londei M: Prevention of arthritis byinterleukin 10-producing B cells. J Exp Med 197: 489–501, 2003
11. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK:Chronic intestinal inflammatory condition generates IL-10-producingregulatory B cell subset characterized by CD1d upregulation. Immu-nity 16: 219–230, 2002
12. Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK: Myelin oligo-dendrocyte glycoprotein-specific T and B cells cooperate to induce aDevic-like disease in mice. J Clin Invest 116: 2393–2402, 2006
13. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A: Spontaneousopticospinal encephalomyelitis in a double-transgenic mouse modelof autoimmune T cell/B cell cooperation. J Clin Invest 116: 2385–2392, 2006
14. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF: Regula-tory B cells inhibit EAE initiation in mice while other B cells promotedisease progression. J Clin Invest 118: 3420–3430, 2008
15. Edwards JC, Cambridge G: Sustained improvement in rheumatoidarthritis following a protocol designed to deplete B lymphocytes.Rheumatology (Oxford) 40: 205–211, 2001
16. Anolik JH, Barnard J, Cappione A, Pugh-Bernard AE, Felgar RE,Looney RJ, Sanz I: Rituximab improves peripheral B cell abnormalitiesin human systemic lupus erythematosus. Arthritis Rheum 50: 3580–3590, 2004
17. Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ,Sloand JA, Rosenblatt J, Sanz I: B cell depletion as a novel treatmentfor systemic lupus erythematosus: A phase I/II dose-escalation trial ofrituximab. Arthritis Rheum 50: 2580–2589, 2004
18. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-OrA, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH: B-celldepletion with rituximab in relapsing-remitting multiple sclerosis.N Engl J Med 358: 676–688, 2008
19. Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G,Berek C, Hiepe F, Krenn V, Radbruch A, Manz RA: Inflamed kidneys ofNZB/W mice are a major site for the homeostasis of plasma cells. EurJ Immunol 31: 2726–2732, 2001
20. Magalhaes R, Stiehl P, Morawietz L, Berek C, Krenn V: Morphologicaland molecular pathology of the B cell response in synovitis of rheu-matoid arthritis. Virchows Arch 441: 415–427, 2002
21. Sarwal M, Chua MS, Kambham N, Hsieh SC, Satterwhite T, Masek M,Salvatierra O Jr: Molecular heterogeneity in acute renal allograft re-jection identified by DNA microarray profiling. N Engl J Med 349:125–138, 2003
22. Martins HL, Silva C, Martini D, Noronha IL: Detection of B lymphocytes(CD20�) in renal allograft biopsy specimens. Transplant Proc 39:432–434, 2007
23. Hippen BE, DeMattos A, Cook WJ, Kew CE 2nd, Gaston RS: Associ-ation of CD20� infiltrates with poorer clinical outcomes in acutecellular rejection of renal allografts. Am J Transplant 5: 2248–2252,2005
24. Tsai EW, Rianthavorn P, Gjertson DW, Wallace WD, Reed EF, EttengerRB: CD20� lymphocytes in renal allografts are associated with poorgraft survival in pediatric patients. Transplantation 82: 1769–1773,2006
25. Bagnasco SM, Tsai W, Rahman MH, Kraus ES, Barisoni L, Vega R,Racusen LC, Haas M, Mohammed BS, Zachary AA, Montgomery RA:
BASIC RESEARCH www.jasn.org
664 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 654–665, 2010

CD20-positive infiltrates in renal allograft biopsies with acute cellularrejection are not associated with worse graft survival. Am J Transplant7: 1968–1973, 2007
26. Doria C, di Francesco F, Ramirez CB, Frank A, Iaria M, Francos G,Marino IR, Farber JL: The presence of B-cell nodules does not neces-sarily portend a less favorable outcome to therapy in patients withacute cellular rejection of a renal allograft. Transplant Proc 38: 3441–3444, 2006
27. Zarkhin V, Kambham N, Li L, Kwok S, Hsieh SC, Salvatierra O, SarwalMM: Characterization of intra-graft B cells during renal allograft rejec-tion. Kidney Int 74: 664–673, 2008
28. Song S, Meyer M, Turk TR, Wilde B, Feldkamp T, Assert R, Wu K,Kribben A, Witzke O: Serum cystatin C in mouse models: A reliableand precise marker for renal function and superior to serum creatinine.Nephrol Dial Transplant 24: 1157–1161, 2009
29. Sahsivar MO, Narin C, Kiyici A, Toy H, Ege E, Sarigul A: The effect ofiloprost on renal dysfunction following renal ischemia reperfusionusing cystatin C and beta2-microglobulin monitoring. Shock 32: 498–502, 2009
30. Bouaziz JD, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, TedderTF: Therapeutic B cell depletion impairs adaptive and autoreactiveCD4� T cell activation in mice. Proc Natl Acad Sci U S A 104: 20878–20883, 2007
31. Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, WaldschmidtTJ, Gipson MG, Tsokos GC, Holers VM: Mice deficient in complementreceptors 1 and 2 lack a tissue injury-inducing subset of the naturalantibody repertoire. J Immunol 169: 2126–2133, 2002
32. Yuling H, Ruijing X, Xiang J, Yanping J, Lang C, Li L, Dingping Y,Xinti T, Jingyi L, Zhiqing T, Yongyi B, Bing X, Xinxing W, Youxin J,Fox DA, Lundy SK, Guohua D, Jinquan T: CD19�CD5� B Cells inprimary IgA nephropathy. J Am Soc Nephrol 19: 2130 –2139, 2008
33. Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, Cyster JG:Balanced responsiveness to chemoattractants from adjacent zonesdetermines B-cell position. Nature 416: 94–99, 2002
34. Brandes M, Legler DF, Spoerri B, Schaerli P, Moser B: Activation-dependent modulation of B lymphocyte migration to chemokines. IntImmunol 12: 1285–1292, 2000
35. Hall PA, Greenwood RA, d’Ardenne AJ, Levison DA: In situ demon-stration of renal tubular regeneration using the monoclonal antibodyKi67. Nephron 49: 122–125, 1988
36. Nadasdy T, Laszik Z, Blick KE, Johnson LD, Silva FG: Proliferativeactivity of intrinsic cell populations in the normal human kidney. J AmSoc Nephrol 4: 2032–2039, 1994
37. Molitoris BA, Sutton TA: Endothelial injury and dysfunction: Role inthe extension phase of acute renal failure. Kidney Int 66: 496 – 499,2004
38. Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, CasolaS, Shlomchik MJ, Koteliansky V, Hochman PS, Ibraghimov A: Attenu-ated liver fibrosis in the absence of B cells. J Clin Invest 115: 3072–3082, 2005
39. Daemen MA, van de Ven MW, Heineman E, Buurman WA: Involve-
ment of endogenous interleukin-10 and tumor necrosis factor-alpha in renal ischemia-reperfusion injury. Transplantation 67: 792–800, 1999
40. Deng J, Kohda Y, Chiao H, Wang Y, Hu X, Hewitt SM, Miyaji T,McLeroy P, Nibhanupudy B, Li S, Star RA: Interleukin-10 inhibitsischemic and cisplatin-induced acute renal injury. Kidney Int 60: 2118–2128, 2001
41. Koken T, Serteser M, Kahraman A, Akbulut G, Dilek ON: Which ismore effective in the prevention of renal ischemia-reperfusion-in-duced oxidative injury in the early period in mice: Interleukin (IL)-10 oranti-IL-12? Clin Biochem 37: 50–55, 2004
42. Godet C, Goujon JM, Petit I, Lecron JC, Hauet T, Mauco G, CarretierM, Robert R: Endotoxin tolerance enhances interleukin-10 renal ex-pression and decreases ischemia-reperfusion renal injury in rats. Shock25: 384–388, 2006
43. Lemos FB, Ijzermans JN, Zondervan PE, Peeters AM, van den Engel S,Mol WM, Weimar W, Baan CC: Differential expression of heme oxy-genase-1 and vascular endothelial growth factor in cadaveric andliving donor kidneys after ischemia-reperfusion. J Am Soc Nephrol 14:3278–3287, 2003
44. Faleo G, Neto JS, Kohmoto J, Tomiyama K, Shimizu H, Takahashi T,Wang Y, Sugimoto R, Choi AM, Stolz DB, Carrieri G, McCurry KR,Murase N, Nakao A: Carbon monoxide ameliorates renal cold isch-emia-reperfusion injury with an upregulation of vascular endothelialgrowth factor by activation of hypoxia-inducible factor. Transplanta-tion 85: 1833–1840, 2008
45. Okazaki M, Yamada Y, Nishimoto N, Yoshizaki K, Mihara M: Charac-terization of anti-mouse interleukin-6 receptor antibody. Immunol Lett84: 231–240, 2002
46. Takagi N, Mihara M, Moriya Y, Nishimoto N, Yoshizaki K, Kishimoto T,Takeda Y, Ohsugi Y: Blockage of interleukin-6 receptor amelioratesjoint disease in murine collagen-induced arthritis. Arthritis Rheum 41:2117–2121, 1998
47. Katsume A, Saito H, Yamada Y, Yorozu K, Ueda O, Akamatsu K,Nishimoto N, Kishimoto T, Yoshizaki K, Ohsugi Y: Anti-interleukin 6(IL-6) receptor antibody suppresses Castleman’s disease likesymptoms emerged in IL-6 transgenic mice. Cytokine 20: 304 –311,2002
48. Mihara M, Takagi N, Takeda Y, Ohsugi Y: IL-6 receptor blockageinhibits the onset of autoimmune kidney disease in NZB/W F1 mice.Clin Exp Immunol 112: 397–402, 1998
49. Janeway C: CD antigens. In: Immunobiology, 6th Ed., New York,Garland Science Publishing, 2005, pp 731–746
50. Ascon DB, Lopez-Briones S, Liu M, Ascon M, Savransky V, Colvin RB,Soloski MJ, Rabb H: Phenotypic and functional characterization ofkidney-infiltrating lymphocytes in renal ischemia reperfusion injury.J Immunol 177: 3380–3387, 2006
51. Sun JB, Flach CF, Czerkinsky C, Holmgren J: B lymphocytes promoteexpansion of regulatory T cells in oral tolerance: Powerful induction byantigen coupled to cholera toxin B subunit. J Immunol 181: 8278–8287, 2008
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 21: 654–665, 2010 B Cells in Repair Phase of Renal IRI 665