
Autoimmune Gastritis Mediated by CD4þ T Cells Promotes the...
11
Microenvironment and Immunology Autoimmune Gastritis Mediated by CD4þ T Cells Promotes the Development of Gastric Cancer Thanh-Long M. Nguyen 1 , Shradha S. Khurana 3 , Clifford J. Bellone 1 , Benjamin J. Capoccia 3 , John E. Sagartz 2 , Russell A. Kesman Jr 1 , Jason C. Mills 3 , and Richard J. DiPaolo 1 Abstract Chronic inflammation is a major risk factor for cancer, including gastric cancers and other gastrointestinal cancers. For example, chronic inflammation caused by autoimmune gastritis (AIG) is associated with an increased risk of gastric polyps, gastric carcinoid tumors, and possibly adenocarcinomas. In this study, we characterized the progression of gastric cancer in a novel mouse model of AIG. In this model, disease was caused by CD4 þ T cells expressing a transgenic T-cell receptor specific for a peptide from the H þ /K þ ATPase proton pump, a protein expressed by parietal cells in the stomach. AIG caused epithelial cell aberrations that mimicked most of those seen in progression of human gastric cancers, including chronic gastritis followed by oxyntic atrophy, mucous neck cell hyperplasia, spasmolytic polypeptide-expressing metaplasia, dysplasia, and ultimately gastric intraepithelial neoplasias. Our work provides the first direct evidence that AIG supports the develop- ment of gastric neoplasia and provides a useful model to study how inflammation drives gastric cancer. Cancer Res; 73(7); 2117–26. Ó2013 AACR. Introduction Autoimmune gastritis (AIG) is one of the most common autoimmune conditions in humans and is caused when the adaptive immune system (T and B cells) targets self-anti- gens expressed by parietal cells and chief cells in the gastric mucosa. AIG may persist in an asymptomatic form for many years. A subset of individuals will eventually develop pernicious anemia (PA). Pernicious anemia is the major cause of vitamin B12 deficiency. AIG and pernicious anemia have respective prevalence of 2% and 0.15% to 1% in the general population (1, 2), which is increased 3- to 5-fold in individuals with other, concomitant autoimmune diseases, such as type 1 diabetes (3, 4) and autoimmune thyroid disease (5, 6). Gastric carcinoid tumors, evolving from enterochromaffine-like (ECL) cell hyper/dysplasia induced by hypergastrinemia, develop in 4% to 9% of patients with AIG/PA (7–9). Gastric carcinoid tumors are relatively benign lesions, metastasizing in less than 10% of cases (10). Several studies have examined whether individuals with AIG/PA also have a higher risk of developing gastric adenocarcinomas, which is the second leading cause of cancer related deaths in the world. Two recent studies, one with 4.5 million retired male veterans in the United States and the other included 9 million individuals from Sweden, reported that individuals with pernicious anemia had an increased risk of developing not only gastrointes- tinal carcinoids but also stomach adenocarcinomas, small intestinal adenocarcinomas, squamous cell carcinomas (SCC), and esophageal SCCs (11, 12). Gastric cancer is the fourth most common cancer and the second most deadly malignant neoplasia in the world. A model, referred to as the Correa pathway, describes the development of gastric adenocarcinomas in humans from a histologic perspective (13). This model details the progression of gastric cancer through a series of pathologic steps the epithelium undergoes starting with chronic inflammation (gastritis), fol- lowed by atrophy (especially loss of parietal cells), metaplasia, dysplasia, and eventually neoplasia. A better understanding of how inflammation induces gastric epithelial cell changes could provide potential therapeutic strategies for diagnosing and preventing gastric cancer (14). To gain a better understanding of the progression of gastric cancer from a cellular and molecular perspective, numerous groups have developed ani- mal models, mouse models in particular, to study gastric carcinogenesis. Such strategies have included chronic infec- tion with Helicobacter (15), chemical depletion of parietal cells (16, 17), and several different lines of genetically modified mice. While these models have increased our understanding of the roles of infection, parietal cell loss, and genes involved in regulating epithelial cell biology, none have directly examined the role of chronic inflammation as the primary inducer of epithelial cell change, which would be useful for understanding the roles of cytokines and immune cells in promoting gastric cancer and for addressing the potential link between AIG and gastric cancer. Authors' Affiliations: Departments of 1 Molecular Microbiology and Immu- nology and 2 Comparative Medicine, Saint Louis University School of Medicine; and 3 Division of Gastroenterology, Washington University School of Medicine, St. Louis, Missouri Corresponding Author: Richard J. DiPaolo, Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, Saint Louis, MO 63104. Phone: 314-977-8860; Fax: 314-977-8717; E-mail: [email protected] doi: 10.1158/0008-5472.CAN-12-3957 Ó2013 American Association for Cancer Research. Cancer Research www.aacrjournals.org 2117 on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957
Transcript of Autoimmune Gastritis Mediated by CD4þ T Cells Promotes the...

Microenvironment and Immunology
Autoimmune Gastritis Mediated by CD4þ T Cells Promotesthe Development of Gastric Cancer
Thanh-LongM. Nguyen1, Shradha S. Khurana3, Clifford J. Bellone1, Benjamin J. Capoccia3, John E. Sagartz2,Russell A. Kesman Jr1, Jason C. Mills3, and Richard J. DiPaolo1
AbstractChronic inflammation is a major risk factor for cancer, including gastric cancers and other gastrointestinal
cancers. For example, chronic inflammation caused by autoimmune gastritis (AIG) is associated with anincreased risk of gastric polyps, gastric carcinoid tumors, and possibly adenocarcinomas. In this study, wecharacterized the progression of gastric cancer in a novel mouse model of AIG. In this model, disease was causedby CD4þ T cells expressing a transgenic T-cell receptor specific for a peptide from the Hþ/Kþ ATPase protonpump, a protein expressed by parietal cells in the stomach. AIG caused epithelial cell aberrations that mimickedmost of those seen in progression of human gastric cancers, including chronic gastritis followed by oxynticatrophy, mucous neck cell hyperplasia, spasmolytic polypeptide-expressing metaplasia, dysplasia, and ultimatelygastric intraepithelial neoplasias. Our work provides the first direct evidence that AIG supports the develop-ment of gastric neoplasia and provides a useful model to study how inflammation drives gastric cancer. CancerRes; 73(7); 2117–26. �2013 AACR.
IntroductionAutoimmune gastritis (AIG) is one of the most common
autoimmune conditions in humans and is caused when theadaptive immune system (T and B cells) targets self-anti-gens expressed by parietal cells and chief cells in the gastricmucosa. AIG may persist in an asymptomatic form formany years. A subset of individuals will eventually developpernicious anemia (PA). Pernicious anemia is the majorcause of vitamin B12 deficiency. AIG and pernicious anemiahave respective prevalence of 2% and 0.15% to 1% in thegeneral population (1, 2), which is increased 3- to 5-fold inindividuals with other, concomitant autoimmune diseases,such as type 1 diabetes (3, 4) and autoimmune thyroiddisease (5, 6). Gastric carcinoid tumors, evolving fromenterochromaffine-like (ECL) cell hyper/dysplasia inducedby hypergastrinemia, develop in 4% to 9% of patients withAIG/PA (7–9). Gastric carcinoid tumors are relativelybenign lesions, metastasizing in less than 10% of cases(10). Several studies have examined whether individualswith AIG/PA also have a higher risk of developing gastricadenocarcinomas, which is the second leading cause ofcancer related deaths in the world. Two recent studies,
one with 4.5 million retired male veterans in the UnitedStates and the other included 9 million individuals fromSweden, reported that individuals with pernicious anemiahad an increased risk of developing not only gastrointes-tinal carcinoids but also stomach adenocarcinomas, smallintestinal adenocarcinomas, squamous cell carcinomas(SCC), and esophageal SCCs (11, 12).
Gastric cancer is the fourth most common cancer and thesecondmost deadlymalignant neoplasia in theworld. Amodel,referred to as the Correa pathway, describes the developmentof gastric adenocarcinomas in humans from a histologicperspective (13). This model details the progression of gastriccancer through a series of pathologic steps the epitheliumundergoes starting with chronic inflammation (gastritis), fol-lowed by atrophy (especially loss of parietal cells), metaplasia,dysplasia, and eventually neoplasia. A better understanding ofhow inflammation induces gastric epithelial cell changes couldprovide potential therapeutic strategies for diagnosing andpreventing gastric cancer (14). To gain a better understandingof the progression of gastric cancer from a cellular andmolecular perspective, numerous groups have developed ani-mal models, mouse models in particular, to study gastriccarcinogenesis. Such strategies have included chronic infec-tion with Helicobacter (15), chemical depletion of parietal cells(16, 17), and several different lines of geneticallymodifiedmice.While these models have increased our understanding of theroles of infection, parietal cell loss, and genes involved inregulating epithelial cell biology, none have directly examinedthe role of chronic inflammation as the primary inducer ofepithelial cell change, whichwould be useful for understandingthe roles of cytokines and immune cells in promoting gastriccancer and for addressing the potential link between AIG andgastric cancer.
Authors' Affiliations: Departments of 1MolecularMicrobiology and Immu-nology and 2Comparative Medicine, Saint Louis University School ofMedicine; and 3Division of Gastroenterology, Washington UniversitySchool of Medicine, St. Louis, Missouri
Corresponding Author: Richard J. DiPaolo, Department of MolecularMicrobiology and Immunology, Saint Louis University School of Medicine,Saint Louis, MO 63104. Phone: 314-977-8860; Fax: 314-977-8717; E-mail:[email protected]
doi: 10.1158/0008-5472.CAN-12-3957
�2013 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 2117
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

We investigated the potential link between AIG and gastriccancer using a T-cell receptor (TCR) transgenic mouse modelof AIG (18). These transgenic CD4þT cells recognizes a peptidefrom the parietal cell specific antigen Hþ/Kþ ATPase, which isalso the major autoantigen targeted by the immune system inhumans with AIG/PA (19). All mice developed chronic gastritisthat resulted from large numbers of CD4þ T cells that infil-trated the gastric mucosa and produced large amounts of IFN-g and smaller amounts of interleukin (IL)-17. Mice developedsevere oxyntic atrophy andmetaplasia by 2 to 4 months of age.At this stage of disease, mice also developed several molecularfeatures associated with the progression of gastric cancer inhumans, including spasmolytic polypeptide expressing meta-plasia (SPEM), increased levels of mRNA for gastric cancerbiomarkers (HE4, OLFM4, TFF2), and increased levels of phos-phorylated STAT3 compared with nontransgenic controlmice.Finally, by 12 months of age, all mice with AIG developed high-grade dysplasia consistent with gastric intraepithelial neopla-sia (GIN). In summary, we report a new mouse model showingthat inflammation associated with AIG induces many of thepathologic and molecular features of gastric carcinogenesis,including the development of severe dysplasia/GIN. Thesestudies support a link between AIG and gastric cancer andhighlight the importance of localized inflammation in thedevelopment of stomach cancer. This new, immune system-induced model of gastric cancer will be useful for studyingimportant host factors that influence inflammation-inducedadenocarcinomas.
Materials and MethodsMice
TxA23 TCR transgenic mice have been previously describedand have been bredmore than 15 generations onto the BALB/cbackground (18). The BALB/c control mice described in theseexperiments are TCR transgene–negative littermates thatwereco-housed with the TxA23 TCR transgenic mice. All mice weremaintained under specific pathogen-free conditions and caredfor in our animal facility in accordance with institutionalguidelines. Our colony tested negative by PCR for the following:Helicobacter bilis, Helicobacter hepaticus, Helicobacter roden-tium,Helicobacter sp.,Helicobacter trogontum, andHelicobactertyphlonius.
HistopathologyStomachs were removed frommice, rinsed in saline, immer-
sion fixed in 10% neutral-buffered formalin (Thermo Scientif-ic), paraffin-embedded, sectioned, and stained with hematox-ylin and eosin. Pathology scores were assigned using methodsmodified from Rogers and colleagues (20). Slides were blindedand sections from individual mice were assigned scoresbetween 0 (absent) and 4 (severe) to indicate the severity ofinflammation, oxyntic atrophy, mucinous hyperplasia/meta-plasia, and dysplasia. Scores were validated by an independentsecond pathologist blinded to experimental conditions.
ImmunofluorescenceStomachs were fixed for 20 minutes with methacarn (60%
methanol, 30% chloroform and 10% glacial acetic acid; all from
Fisher), washed with 70% ethanol, embedded in paraffin, andsectioned into 0.5-mm thick sections. Slides were deparaffi-nized, rehydrated, stained, and imaged using methods mod-ified from Ramsey and colleagues (21). The primary antibodiesused for immunostaining were rabbit anti-human gastricintrinsic factor (gifts of Dr. David Alpers, Washington Univer-sity, St. Louis, MO), rabbit anti-Ki67 (Abcam), and mouse anti-Ecadherin (BD Biosciences). Secondary antibodies and GSIIlectin (Molecular Probes) labeling were as described (21).
A gastric unit is defined as an invagination of the gastricmucosa that is lined by a single layer of columnar epithelium.Each gastric unit is lined by foveolar cells at the luminal endand zymogenic cells at the base. Ki67 staining was quantifiedby counting each Ki67þ nucleus per gastric unit for more than50 units per mouse and classified into <10, 10–20, and >20positive nuclei per unit. Percentages were calculated by divid-ing the number of gastric units in each category by totalnumber of gastric units analyzed in that mouse stomachsample.
ImmunohistochemistryTissue was deparaffinized and rehydrated. Endogenous
peroxidase was blocked using a 0.3% H2O2 in methanol for15 minutes. Antigen retrieval was done in a pressure cookerwith Diva (Biocare: DV2004MX). Avidin/biotin kit (Biocare)was used to block endogenous biotin. The antibody pStat3(D3A7) fromCell Signalingwas diluted inDavinci (Biocare) andincubated over night at 4�C. The secondary antibody, biotiny-lated goat anti-rabbit, and streptavidin-HRP from Jackson Labswere each applied for 1 hour at room temperature. Visualiza-tion was done with Biocare's Betaziod DAB and slides werecounterstained in hematoxylin.
ImmunoblotA section from the stomach was homogenized with an
electric pestle tissue homogenizer. Cells were then lysed in0.5 mL of lysis buffer [20 mmol/L Tris-HCl pH 7.5, 150 mmol/LNaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% Triton, 2.5mmol/L sodium pyrophosphate, 1 mmol/L b-glyceropho-sphate, 1 mmol/L Na3VO4, 1 mg/mL leupeptin (Cell Signaling)and a protease inhibitor cocktail (Sigma)]. Lysates were vor-texed for 1 minute and sonicated for 15 seconds followed bycentrifugation for 10 minutes at 4�C. Lysates were ran on aNuPAGE 4% to 12% Bis-Tris gradient gel (Novex) and trans-ferred to a nitrocellulose membrane. Membrane was blockedfor 1 hour with 5% nonfat dairy milk. Primary antibodies (allfrom Cell Signaling) were stained for 1 hour (rabbit mAB-b-actin- and rabbit mAB-STAT3) or overnight (rabbit mABphospho-STAT3) in 5% bovine serum albumin (BSA) in 4�C.Horseradish peroxidase-linked secondary antibody (anti-rab-bit IgG) was stained for 1 hour at room temperature in 5% non-fat dairy milk. Protein was detected by chemiluminescenceusing LumiGLO (Cell Signaling) on CL-XPosure X-Ray film(Fisher).
Flow cytometryCell surface staining was conducted according to stan-
dard procedures using monoclonal antibodies against CD4,
Nguyen et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2118
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

CD19, CD11b, and Ly6G. Intracellular cytokine staining wasconducted using monoclonal antibodies against IFN-g andIL-17A. All antibodies were purchased from BD Pharmin-gen. All flow cytometry was conducted on a BD LSRII or BDFACSCalibur and analyzed using FlowJo (TreeStar). Forintracellular cytokine staining, cells were stimulated withPMA (Calbiochem) and Ionomycin (Calbiochem) for 4hours at 37�C. Golgi-stop (BD Biosciences) was added after1 hour. Cells were then washed, fixed in 4% formyl saline,washed, and permeabilized (0.5% BSA, 0.1% Triton, and 2mmol/L EDTA in PBS) for 1 hour at room temperature.After washing, cells were incubated overnight with theanti-cytokine antibodies, washed, and analyzed by flowcytometry.
Isolation of cells from the gastric lymph nodes andgastric mucosaThe method for isolating cells from the stomach tissue has
been described previously (22, 23). Briefly, the gastric lymphnodes (gLN) were removed from the stomachs, homoge-nized, and passed through a 40-mm pore nylon filter. Sto-machs were opened with an incision from the antrum to thefundus and rinsed in PBS to remove food. Cells were flushedfrom the gastric mucosa using a syringe with a 25-gaugeneedle. PBS containing 5% fetal calf serum and penicillin/streptomycin (Sigma) was repeatedly injected within themucosa causing the tissue to swell and rupture. Single-cell
suspensions were collected, gently vortexed, and passedthrough a 40-mm nylon filter. Cells were counted, stainedwith antibodies, and analyzed by flow cytometry. To detectsecreted cytokines, 1 � 106 cells were culture in vitro in24-well plates containing 2 mL of supplemented RPMI.Supernatants from cell cultures were collected after 48hours, and cytokines and chemokines were measured usingMilliplex (Millipore).
Quantitative real-time PCRTotal RNA was prepared using the RNeasy Mini Kit System
(Qiagen). The quantity and quality of RNA were determinedusing a NanoDrop 2000 spectrophotometer (Thermo Scientif-ic), and 0.5 mg of the RNA was used to generate a first-strandcDNA copy according to the manufacturer's instruction (HighCapacity cDNA Reverse Transcription Kit, Applied Biosys-tems). Quantitative PCR was carried out using TaqMan GeneExpression Assays systems (Applied Biosystems). Glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) served as an inter-nal reference standard. PCR was run on the 7500 Real-TimePCR System (Applied Biosystems).
Statistical analysisData are expressed asmeans of individual determinations�
SE. Statistical analysis was conducted using the Mann–Whit-ney test (�, P < 0.05; ��, P < 0.01; ���, P < 0.001) using GraphPadPrism 5.
Figure 1. Inflammation in TxA23mice. Representative flow cytometric plots of cell types and cytokines from a TxA23mice (n¼ 21). A, flow cytometry was usedto identify T cells (CD4þ) and B cells (CD19þ). B, the majority of CD4þ-gated T cells express the transgenic TCR (TCRVa2/TCRVb2) that recognizes apeptide from Hþ/Kþ ATPase. C, macrophages (CD11bþLy6G�) and neutrophils (CD11bþLy6Gþ) make up a large proportion of the remaining cells. D,intracellular cytokine staining showing IL-17, IFN-y, IL-2, and IL-4productionbyCD4þTcells combined from thegastricmucosa and lymphnode. E, cytokinessecreted by cells isolated from the gastric lymph nodes of TxA23 mice were measured by bead-based ELISA. Data are the mean � SE of 7 mice from3 independent experiments.
Autoimmune Gastritis Induces Gastric Carcinogenesis
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2119
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957
ResultsInflammation in TxA23 mice is characterized by CD4þ Tcells secreting IFN-g and IL-17
Our first goal was to characterize the cell types andcytokines in TxA23 mice. Cells were isolated from the gastricmucosa and gastric lymph nodes of 2-month-old mice andanalyzed by flow cytometry. The majority (>85%) ofthe hematopoietic-derived cells isolated were either CD4þ
T cells or CD19þ B cells (Fig. 1A). As expected, themajority of the CD4þ T cells that infiltrated the stomachexpressed the transgenic TCR (TCRVa2/TCRVb2) specificfor the Hþ/Kþ ATPase peptide (Fig. 1B). Macrophages(CD11bþLy6G�) and neutrophils (CD11bþLy6Gþ) and asubset of dendritic cells (CD11bþCD11cþ, data not shown)comprised the rest of the cells found in the gastric mucosa(Fig. 1C). Next, cells were restimulated and cytokine pro-
duction by CD4þ T cells was determined by intracellularcytokine staining. The majority of cytokine-producing CD4þ
T cells isolated from the stomachs and gastric lymph nodesproduced IFN-g and fewer produced IL-2 and IL-17. IL-4secretion by CD4þ T cells was not detected (Fig. 1D). Finally,total cells isolated from the gastric lymph node were cul-tured immediately after isolation. The amounts of severalcytokines secreted into the supernatants were determinedafter 48 hours. The most abundant cytokines secreted bycells were IFN-g and IL-17 (Fig. 1E). Lower levels of IL-6, IL-2,IL-10, and IL-4 were also detected. Thus, the inflammatorycell infiltrate within the gastric mucosa consists primarily ofa mixture of T-helper cells (TH)1 (IFN-gþ) and TH17 (IL-17
þ)CD4þ T and B cells. This type of inflammation is consistentwith the type of inflammation described in humans infectedwith H. Pylori and with autoimmune gastritis (24, 25).
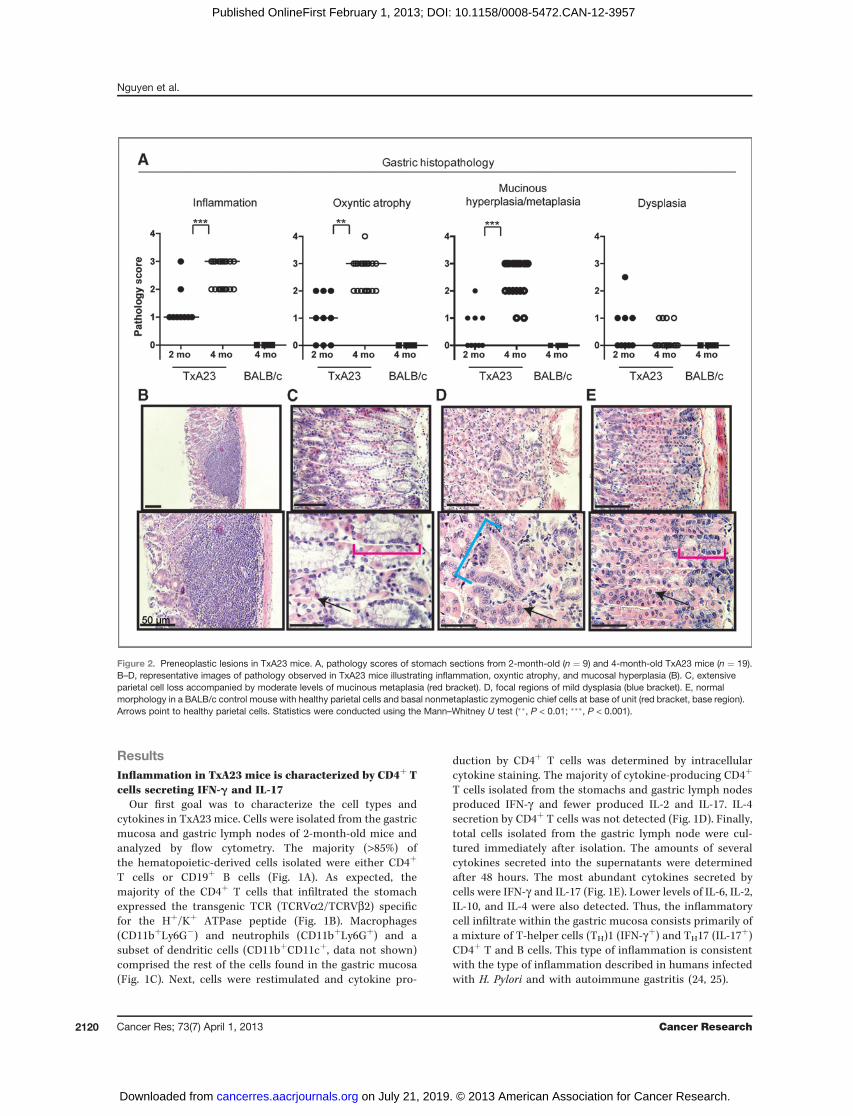
Figure 2. Preneoplastic lesions in TxA23 mice. A, pathology scores of stomach sections from 2-month-old (n ¼ 9) and 4-month-old TxA23 mice (n ¼ 19).B–D, representative images of pathology observed in TxA23 mice illustrating inflammation, oxyntic atrophy, and mucosal hyperplasia (B). C, extensiveparietal cell loss accompanied by moderate levels of mucinous metaplasia (red bracket). D, focal regions of mild dysplasia (blue bracket). E, normalmorphology in a BALB/c control mouse with healthy parietal cells and basal nonmetaplastic zymogenic chief cells at base of unit (red bracket, base region).Arrows point to healthy parietal cells. Statistics were conducted using the Mann–Whitney U test (��, P < 0.01; ���, P < 0.001).
Nguyen et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2120
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

TxA23 progress through a series of pathological changesassociated with the development of gastric cancerIn humans, the progression of intestinal-type gastric
cancer is thought to evolve through a series of discrete
steps known as the Correa pathway (13). The first step inthis pathway is inflammation (gastritis) and then loss ofparietal cells (oxyntic atrophy) and the development ofmucinous metaplasia, followed by dysplasia and finally
Figure 3. Increased epithelial cell proliferation in the gastric mucosa of TxA23 mice. A–C, images are representative of 2-month-old (A) and 4-month old (B)TxA23 mice and 4-month-old (C) BALB/c stomach sections. E-Cadherin (green) stains epithelial cells, Ki67 (red) stains proliferating cells, and Hoechst(blue)was used to stain nuclei. D, a quantitative analysis of the percentage of gastric units containing proliferating epithelial cells (E-cadherinþKi67þ) inBALB/cand 2- and 4-month-old TxA23 mice.
Autoimmune Gastritis Induces Gastric Carcinogenesis
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2121
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

cancer. We examined the pathologic features of gastricdisease in TxA23 mice. At 2 months of age, TxA23 mice hadmoderate degrees of inflammation, oxyntic atrophy, andmucosal hyperplasia/metaplasia but little or no evidence ofdysplasia (Fig. 2A). By 4 months of age, inflammation,oxyntic atrophy, and mucosal hyperplasia/metaplasia weresignificantly more severe than 2-month-old mice (Fig. 2A).Lesions in the stomachs of 4-month-old TxA23 mice com-prised large areas in which parietal cells were either reducedin number or absent from the gastric units, and the remain-ing mucosa was dominated by large, hyperplastic mucus-containing cells that expanded to the bases of gastric units(Fig. 2B and C). Four of the 19 mice had developed mild focaldysplasia (Fig. 2D). For comparison, Fig. 2E is representativeof the normal pathology observed in 11 individual controlmouse, which are transgene-negative BALB/c mice that werecohoused with TxA23 littermates. Disease severity was sim-ilar in male and female mice at all ages. These data show thatchronic inflammation resulting from autoimmune gastritisinduced the development of preneoplastic lesions in thegastric mucosa of TxA23 mice with many pathologic featuresin common with the Correa pathway.
Increased epithelial cell proliferation, phosphorylatedSTAT3, IL-6, and expression of gastric cancer–associatedbiomarkers in TxA23 mice
Next, we used immunofluorescence to compare the extent ofgastric epithelial cell proliferation in 2- and 4-month-old TxA23mice compared with BALB/c control mice (Fig. 3A–C). In wild-type BALB/c mice, the number of proliferating (marked byKi67þ immunoreactivity) epithelial cells (marked by E-cadherinþ) per individual gastric unit was always less than10. However, in TxA23mice, almost 70% of 2-month-old gastricunits had 10 or more proliferating cells, and by 4months, morethan 75% had more than 10 with about a third of those having20 or more (Fig. 3D).
Increased levels of the active (phosphorylated) pSTAT3 isinvolved in cellular transformation in numerous cancers ofepithelial origin, including gastric cancer (26). A recentstudy suggested that pSTAT3 is a significant prognosticfactor in gastric cancer in humans (27). To determinewhether the level of pSTAT3 was increased in the stomachsof TxA23 mice, we conducted Western blots on gastrictissue lysates from age-matched TxA23 and healthyBALB/c control mice. Compared with BALB/c mice, TxA23mice expressed slightly higher levels of total STAT3 and amuch higher level of pSTAT3 (Fig. 4A). Immunohistochem-ical analysis revealed a large number of pSTAT3-positiveepithelial cells present in the gastric mucosa of TxA23 miceand nearly undetectable levels in gastric tissue from BALB/ccontrols (Fig. 4B), in agreement with the results observed byWestern blotting.
Several members of the IL-6 cytokine family, including IL-6 and IL-11, activate STAT3 (28). IL-6 and IL-11 haveimportant roles in maintaining gastric homeostasis by reg-ulating mucosal proliferation, inflammation, angiogenesis,and apoptosis (29, 30). We conducted quantitative real-timePCR analysis using mRNA isolated from gastric tissue from2-month-old TxA23 and BALB/c mice to measure the rela-tive levels of IL-6 and IL-11. The levels of IL-11 mRNA wereequivalent between the 2 genotypes; however, the levels ofIL-6mRNA were approximately 40-fold higher in TxA23 micethan in BALB/c mice (Fig. 4C).
A number of genes have been described as biomarkers forprecursor lesions such as SPEM that are predisposing forgastric cancer. Some of these genes include human epidid-ymis 4 (HE4; ref. 16), Trefoil factor 2 (TFF2), and Olfactomedin4 (OLFM4; ref. 31). HE4 is absent in normal stomach andexpressed in humans and mice with SPEM (16). Increasedlevels of OLFM4, also known as GW112, have been observedin gastric cancers, including 58% of stage III/IV gastriccancers (31). TFF2 is also known as spasmolytic polypeptide,
Figure 4. Increased levels ofcancer associated markers inTxA23 mice. A, representativeWestern blotting of STAT3,pSTAT3 and b-actin on wholestomach lysates of 2-month-oldTxA23 (n¼ 9) and BALB/c mice. B,immunohistochemistry stainingfor pSTAT3 in gastric pits ofBALB/c and TxA23 stomachs(magnification,�20). C, the relativeexpression of IL-6 and IL-11 inmRNA extracted from thestomachs of TxA23 and BALB/cmice. D, the relative expression ofgenes (HE4, TFF2, and OLFM4)that serves as biomarkers for theSPEM and preneoplastic progresswas compared between mRNAisolated from the stomachs ofTxA23 mice and BALB/c controls.
Nguyen et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2122
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

and, by definition, increases when SPEM is present. Wecarried out quantitative real-time PCR analysis usingmRNA isolated from sections taken from the body of thestomachs of TxA23 mice. All of the TxA23 mice expressedhigher levels of HE4 and OLFM4, and a majority, 5 of 7 mice,expressed higher levels of TFF2 than age-matched BALB/ccontrol mice (Fig. 4D). Together, these data show thatdisease in TxA23 mice shares many of the molecular featuresof gastric cancer that have been reported in humans, includ-ing increased epithelial cell proliferation, increased levels ofpSTAT3 protein, and higher levels of IL-6, HE4, OLFM4, andTFF2 mRNA.
SPEM is present in the gastric mucosa of TxA23 miceIntestinal-type gastric cancer predominantly develops in the
setting of oxyntic atrophy and mucous cell metaplasia (13).Spasmolytic polypeptide–expressing metaplasia (SPEM) is ametaplasia in the gastric fundus resembling deep antral glandcells, and recent studies have indicated that SPEM may bedirectly linked to gastric neoplasia (25, 32). We used immu-nohistochemistry to determine whether 4-month-old TxA23mice developed SPEM. A representative section from a TxA23mouse is shown in Fig. 5A. In gastric units in which parietalcells have not yet been destroyed, chief cells are found at thebase of the unit and are identified by staining with antibodiesto gastric intrinsic factor (GIF). Of note, the antrum/pyloris ofTxA23mice were indistinguishable from BALB/c control mice.In the corpus region, neck cells are found above and identifiedby lectin GSII staining (Fig. 5B). However, we also observedmultiple gastric units inwhich themajority or all of the parietalcells had been lost (Fig. 5B and C). In these parietal cell–depleted units, there was an expansion of GSII-positive cells(mucous neck cell hyperplasia) and an emergence ofcells expressing both neck cell–specific and chief cell-specificmarkers (GIF) in the base of the units, whereas in regions withparietal cell preservation, the normal basal marker expressionpattern was maintained (Fig. 5D). Thus, GSII-positiveGIF-positive cells in the base of gastric units that lack parietalcells also stained positive for TFF2 (data not shown), showingthat TxA23 mice developed regions of SPEM by 4 months ofage.
TxA23 mice develop GINIn the next set of experiments, we allowed a cohort of
TxA23 mice to age and conducted histopathologic evalua-tions to determine whether disease in TxA23 mice pro-gressed beyond SPEM to dysplasia. Sections from stomachsof 4- and 12-month-old mice were examined by a pathologistusing a murine gastric histopathology scoring paradigmdescribed previously (ref. 20; Fig. 6A). The analysis of miceat 4 months of age revealed that 15 of 19 had dysplasia scoresof 0 and 4 of 19 mice had dysplasia scores of 1, indicatingfocal irregularly shaped gastric glands, including elongated,slit, trident, and back-to-back forms (Fig. 6B). By 12 monthsof age, disease progressed to the point at which 7 of 8 micedeveloped severe dysplasia, indicated by scores of 3.5. In thisscoring system a score of 3 is used to indicate severe loss ofgland organization and columnar orientation, marked cell
atypia, visible mitoses, GIN, and 0.5 is added for carcinomain situ or invasion without frank malignancy. We observedboth focal and widespread dysplasia and most casesinvolved pseudoinvasion into the submucosa and/or serosa(Fig. 6C–E). We also observed the formation of irregular
Figure 5. TxA23 mice have distinct regions of parietal cell loss coupledwith the emergence SPEM. Representative immunostains of the corpusregion of the stomach of TxA23 transgenic mice. A, the lamina propria isseparated from the glandular mucosa by the white dotted lines whereinparietal cells are stainedwith VEGFB (teal) and nuclei with Hoechst (blue).Note the distinct regions of parietal cell loss as highlighted by solid whitelines. B, the area highlighted by the yellow box in A was stained with GSII(green, neck cells), GIF (red, ZC), and Hoechst (blue, nuclei). The yellowbox on the left indicates a region of SPEMwhere cells coexpressGSII andGIF (white arrowheads). This region of the stomach has shownconsiderable parietal cell destruction. Higher magnification of this regionis shown in C. White arrows indicate areas of relatively normal gastricepithelial cell differentiation that correlate with regions where parietal cellnumbers are normal. Further magnification of this region is shown in D.
Autoimmune Gastritis Induces Gastric Carcinogenesis
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2123
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

glandular forms on the adventitial surface of the stomach,some of which contained papillary projections of atypicalepithelium (Fig. 6F). These data show that precancerouslesions observed in 4-month-old TxA23 mice ultimatelyprogressed to neoplastic disease.
DiscussionAlthough chronic atrophic gastritis is believed to be impor-
tant in initiating gastric carcinogenesis, the precise role(s) ofinflammation in the complex changes in gastric epithelial cellsduring the progression of gastric cancer are not understood.Furthermore, the relationship between AIG/PA and gastriccancer has been controversial and requires further investiga-tion. In this study, we describe a new mouse model showingthat autoimmune gastritis induces precancerous lesions sim-ilar to those that precede gastric cancer in humans. Mice withchronic inflammation caused byHþ/KþATPase-specific CD4þ
T cells developed severe oxyntic atrophy coupled with meta-plasia, including SPEM by 4 months of age. Similar to H. pyloriinfection and AIG in humans, inflammation in TxA23 micecontained CD4þ T cells of the TH1 (IFN-gþ) and TH17 (IL-17
þ)phenotype (33, 34). Consistent with the Correa pathway thatdescribes the progression of gastric cancer in humans, TxA23mice progressed through a series of stages that includedinflammation, atrophic gastritis, mucous neck cell hyperplasia,SPEM, which over time progressed to dysplasia, and neoplasia.These data indicate that the TxA23 model system is unique in
that it allows for the study of the development and regulation ofgastric carcinogenesis in a setting where chronic inflamma-tion, in the absence of infection, toxins, and drugs, is theprimary upstream instigator. Our findings of carcinogenesisin our mouse model are consistent with reports that humanswith AIG/PA are 3 to 6 times more likely to develop gastricadenocarcinoma and other cancers (35, 36). It has beenreported that a subset of individuals contain T cells andantibodies specific for Hþ/Kþ ATPase after they are infectedwith H. pylori (37–40). It is possible that individuals thatdevelop autoimmune responses during H. pylori infection mayremain at risk for gastric cancer even if they are treated for H.pylori infection. Studies have shown that the eradication of H.pylori reduces risk for subsequent gastric cancer by about 25%(40–42). Strategies to reduce inflammation in addition toeradicating H. pylori may further reduce the risk of gastriccancer.
With the two recent studies reporting that individuals withpernicious anemia developed gastrointestinal cancers at ahigher-than-expected rate, animal models that mimic AIG arelikely to be useful for understanding the link between AIG andGI cancers. There is no doubt that infection with H. pylori is animportant, prerequisite risk factor for gastric cancer; however,the vast majority of infected patients do not develop cancer.Therefore, it may be the types of chronic inflammation in thegastric mucosa that is triggered by H. pylori that are down-stream adjuvants or causes of actual cancer. The TxA23mouse
Figure 6. TxA23 mice develop masses with dysplastic foci as they age. A, dysplasia scores (where 1 ¼ irregular glad forms, 3 ¼ severe loss of glands andof columnar orientation of epithelium with regions of cellular atypia, increased mitotic figures, and 0.5 added when invasion of muscle or carcinomain situwas identified) for sections at given ages. Each point represents an individual mouse. B, section of a mouse at 10 months of age (magnification, �10).Note to formation of mass with abundant, dense chronic inflammatory infiltrates. C–F, images of stomach sections that represent pathology observed in12-month-old mice with various degrees of pseudoinvasion of irregular glands into the submucosa. C, section showing submucosal focus of irregulargland formations (magnification, �10). D, section showing focus of irregular glands in submucosal tissue with surrounding chronic inflammation(magnification, �20). E, section showing deep submucosal pseudoinvasion by an irregular gland (magnification, �10). F, irregular glandular form on theadventitial surface of the stomach (magnification, �20).
Nguyen et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2124
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

model described here mimics the human disease and showsthe progression of AIG to the development of SPEM andeventually severe to dysplasia. Other genetically engineeredmouse models have been useful for studying factors thatinfluence the development of gastric cancer independently ofHelicobacter infection. For example, mice expressing gastrinunder the insulin promoter (43), mice deficient in: TFF1 (44),Smad4 (45), and Hip1r (46), and mice expressing a mutatedform of the IL-6 family coreceptor gp130 (47) all develop formsof gastric metaplasia and some cases dysplasia. Our modelspecifically focuses on the immune response to Hþ/KþATPaseand its role in promoting SPEM with progression to severedysplasia. By inducing severe dysplasia in the absence ofinfection, this model will allow for a direct examination of themechanisms whereby inflammation influences gastric epithe-lial cell biology. For example, when examining disease incytokine knockout mice, using our model, we do not have tobe concerned with the potential indirect effects of the impor-tance of the cytokine in modulating Helicobacter infectionitself. Our model will be also useful for evaluating the impor-tance of immune cells, such as regulatory T cells, and how theyinfluence changes in gastric epithelial cells that are associatedwith the progression of gastric cancer. Finally, future studiesusing this model will address how various host factors, espe-cially immune-related genes, influence the risk of developinggastric cancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: T.-L.M. Nguyen, S.S. Khurana, C.J. Bellone, R.J. DiPaoloDevelopment of methodology: T.-L.M. Nguyen, S.S. Khurana, C.J. Bellone, J.E.Sagartz, J.C. Mills, R.J. DiPaoloAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): T.-L.M. Nguyen, S.S. Khurana, C.J. Bellone, B.J.Capoccia, J.E. Sagartz, R.A. Kesman Jr, R.J. DiPaoloAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): T.-L.M. Nguyen, S.S. Khurana, C.J. Bellone, J.E.Sagartz, R.A. Kesman Jr, J.C. Mills, R.J. DiPaoloWriting, review, and/or revision of the manuscript: T.-L.M. Nguyen, S.S.Khurana, J.E. Sagartz, J.C. Mills, R.J. DiPaoloAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): T.-L.M. Nguyen, R.J. DiPaoloStudy supervision: J.C. Mills, R.J. DiPaolo
AcknowledgmentsThe authors thank Joy Eslick and Sherri Koehm for assistance with flow
cytometry and Jeremy Herzog for technical assistance; Anna Cline, Lauren Kintz,Erin Touchette, Kelly Neal, Frank Speck, and Dr. Cheri West for their assistancemaintaining our mouse colonies; Dr. Deborah Rubin and Kymberli Carter fromthe Digestive Disease Research Core Center for assistance with immunohis-tochemistry; and the Washington University Digestive Disease Research CoreCenter for providing a Pilot & Feasibility grant.
Grant SupportR.J. DiPaolo was supported by ACS:RSG-12-171-01-LIB, Digestive Diseases
Research Core Center: NIH2P30 DK052574-12; J.C. Mills by ACSDDC-115769,NIHDK094989-01; AGA Funderburg Research Scholar Award; Digestive DiseasesResearch Core Center: NIH2P30 DK052574-12; and S.S. Khurana by SitemanCancer Pathways Award.
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received October 18, 2012; revised December 28, 2012; accepted January 11,2013; published OnlineFirst February 1, 2013.
References1. Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and
estimated population burden of selected autoimmune diseases in theUnited States. Clin Immunol Immunopathol 1997;84:223–43.
2. Carmel R. Prevalence of undiagnosed pernicious anemia in the elderly.Arch Intern Med 1996;156:1097–100.
3. Riley WJ, Toskes PP, Maclaren NK, Silverstein JH. Predictive value ofgastric parietal cell autoantibodies as a marker for gastric and hema-tologic abnormalities associated with insulin-dependent diabetes.Diabetes 1982;31:1051–5.
4. De Block CE, De Leeuw IH, Van Gaal LF. High prevalence of mani-festations of gastric autoimmunity in parietal cell antibody-positivetype 1 (insulin-dependent) diabetic patients. The Belgian DiabetesRegistry. J Clin Endocrinol Metab 1999;84:4062–7.
5. Centanni M, Marignani M, Gargano L, Corleto VD, Casini A, Delle FaveG, et al. Atrophic body gastritis in patients with autoimmune thyroiddisease: an underdiagnosed association. Arch Intern Med 1999;159:1726–30.
6. Irvine WJ, Clarke BF, Scarth L, Cullen DR, Duncan LJ. Thyroid andgastric autoimmunity in patients with diabetes mellitus. Lancet 1970;2:163–8.
7. Kokkola A, Sj€oblom SM, Haapiainen R, Sipponen P, Puolakkainen P,J€arvinenH, et al. The risk of gastric carcinomaandcarcinoid tumours inpatients with pernicious anaemia. A prospective follow-up study.Scand J Gastroenterol 1998;33:88–92.
8. Armbrecht U, Stockbr€ugger RW, Rode J, Menon GG, Cotton PB.Development of gastric dysplasia in pernicious anaemia: a clinicaland endoscopic follow up study of 80 patients. Gut 1990;31:1105–9.
9. BorchK, Renvall H, Liedberg G.Gastric endocrine cell hyperplasia andcarcinoid tumors in pernicious anemia. Gastroenterology 1985;88:638–48.
10. Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Currentstatus of gastrointestinal carcinoids. Gastroenterology 2005;128:1717–51.
11. Landgren AM, Landgren O, Gridley G, Dores GM, Linet MS, MortonLM, et al. Autoimmune disease and subsequent risk of developingalimentary tract cancers among 4.5 million US male veterans. Cancer2011;117:1163–71.
12. Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Effect of autoim-mune diseases on mortality and survival in subsequent digestive tractcancers. Ann Oncol 2012;23:2179–84.
13. Correa P. A human model of gastric carcinogenesis. Cancer Res1988;48:3554–60.
14. Goldenring JR, Nam KT, Wang TC, Mills JC, Wright NA. Spasmolyticpolypeptide-expressing metaplasia and intestinal metaplasia: time forreevaluation of metaplasias and the origins of gastric cancer. Gastro-enterology 2010;138:2207–10, 2210.e1.
15. Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R, et al. Hyper-trophic gastropathy in Helicobacter felis-infected wild-type C57BL/6mice and p53 hemizygous transgenic mice. Gastroenterology1996;110:155–66.
16. Nozaki K, Ogawa M, Williams JA, Lafleur BJ, Ng V, Drapkin RI, et al. Amolecular signature of gastric metaplasia arising in response to acuteparietal cell loss. Gastroenterology 2008;134:511–22.
17. Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, Mills JC, et al.Tamoxifen induces rapid, reversible atrophy, andmetaplasia in mousestomach. Gastroenterology 2012;142:21–4 e7.
18. McHughRS,ShevachEM,MarguliesDH,NatarajanK. ATcell receptortransgenic model of severe, spontaneous organ-specific autoimmu-nity. Eur J Immunol 2001;31:2094–103.
19. Callaghan JM, Khan MA, Alderuccio F, van Driel IR, Gleeson PA, TohBH, et al. Alpha and beta subunits of the gastric Hþ/K(þ)-ATPase are
Autoimmune Gastritis Induces Gastric Carcinogenesis
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2125
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

concordantly targeted by parietal cell autoantibodies associated withautoimmune gastritis. Autoimmunity 1993;16:289–95.
20. Rogers AB, Taylor NS, Whary MT, Stefanich ED, Wang TC, Fox JG,et al.Helicobacter pylori but not high salt induces gastric intraepithelialneoplasia in B6129 mice. Cancer Res 2005;65:10709–15.
21. Ramsey VG, Doherty JM, Chen CC, Stappenbeck TS, Konieczny SF,Mills JC, et al. The maturation of mucus-secreting gastric epithelialprogenitors into digestive-enzyme secreting zymogenic cells requiresMist1. Development 2007;134:211–22.
22. Alderuccio F, Toh BH, Gleeson PA, van Driel IR. A novel method forisolating mononuclear cells from the stomachs of mice with experi-mental autoimmune gastritis. Autoimmunity 1995;21:215–21.
23. DiPaoloRJ,GlassDD,BijwaardKE, Shevach EM.CD4þCD25þT cellsprevent the development of organ-specific autoimmune disease byinhibiting the differentiation of autoreactive effector T cells. J Immunol2005;175:7135–42.
24. Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. Helicobacterpylori-induced Th17 responses modulate Th1 cell responses, benefitbacterial growth, and contribute to pathology in mice. J Immunol2010;184:5121–9.
25. Lopez-Diaz L, Hinkle KL, Jain RN, Zavros Y, Brunkan CS, Keeley T,et al. Parietal cell hyperstimulation and autoimmune gastritis in choleratoxin transgenic mice. Am J Physiol Gastrointest Liver Physiol2006;290:G970–9.
26. GongW,WangL, Yao JC, Ajani JA,Wei D, AldapeKD, et al. Expressionof activated signal transducer and activator of transcription 3 predictsexpression of vascular endothelial growth factor in and angiogenicphenotype of human gastric cancer. Clin Cancer Res 2005;11:1386–93.
27. XiongH,DuW,Wang JL,WangYC, Tang JT,Hong J, et al. Constitutiveactivation of STAT3 is predictive of poor prognosis in human gastriccancer. J Mol Med (Berl) 2012;90:1037–46.
28. Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmu-nity, and cancer. J Leukoc Biol 2010;88:1145–56.
29. Jackson CB, Judd LM, Menheniott TR, Kronborg I, Dow C, Yeo-mans ND, et al. Augmented gp130-mediated cytokine signallingaccompanies human gastric cancer progression. J Pathol 2007;213:140–51.
30. Giraud AS, Jackson C, Menheniott TR, Judd LM. Differentiation of theGastric Mucosa IV. Role of trefoil peptides and IL-6 cytokine familysignaling in gastric homeostasis. Am J Physiol Gastrointest LiverPhysiol 2007;292:G1–5.
31. Oue N, Sentani K, Noguchi T, Ohara S, Sakamoto N, Hayashi T, et al.Serum olfactomedin 4 (GW112, hGC-1) in combinationwith Reg IV is ahighly sensitive biomarker for gastric cancer patients. Int J Cancer2009;125:2383–92.
32. Lennerz JK, Kim SH, Oates EL, HuhWJ, Doherty JM, Tian X, et al. Thetranscription factor MIST1 is a novel human gastric chief cell markerwhose expression is lost in metaplasia, dysplasia, and carcinoma.Am J Pathol 2010;177:1514–33.
33. Mohammadi M, Czinn S, Redline R, Nedrud J. Helicobacter-specificcell-mediated immune responses display a predominant Th1 pheno-
type and promote a delayed-type hypersensitivity response in thestomachs of mice. J Immunol 1996;156:4729–38.
34. Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD,et al. Helicobacter pylori -induced mucosal inflammation is Th1 medi-ated and exacerbated in IL-4, but not IFN-gamma, gene-deficientmice. J Immunol 2000;165:1022–9.
35. Hsing AW, Hansson LE, McLaughlin JK, Nyren O, Blot WJ, Ekbom A,et al. Pernicious anemia and subsequent cancer. A population-basedcohort study. Cancer 1993;71:745–50.
36. Brinton LA, Gridley G, Hrubec Z, Hoover R, Fraumeni JF. Cancer riskfollowing pernicious anaemia. Br J Cancer 1989;59:810–3.
37. ItoM,HarumaK, KayaS, Kamada T, KimS, Sasaki A, et al. Role of anti-parietal cell antibody in Helicobacter pylori -associated atrophic gas-tritis: evaluation in a country of high prevalence of atrophic gastritis.Scand J Gastroenterol 2002;37:287–93.
38. Claeys D, Faller G, Appelmelk BJ, Negrini R, Kirchner T. The gastricHþ,Kþ-ATPase is a major autoantigen in chronic Helicobacter pylorigastritis with body mucosa atrophy. Gastroenterology 1998;115:340–7.
39. Amedei A, Bergman MP, Appelmelk BJ, Azzurri A, Benagiano M,Tamburini C, et al. Molecular mimicry between Helicobacter pyloriantigens and Hþ, Kþ –adenosine triphosphatase in human gastricautoimmunity. J Exp Med 2003;198:1147–56.
40. D'Elios MM, Amedei A, Azzurri A, Benagiano M, Del Prete G, BergmanMP, et al. Molecular specificity and functional properties of autoreac-tive T-cell response in human gastric autoimmunity. Int Rev Immunol2005;24:111–22.
41. deVriesAC,KuipersEJ,RauwsEA.Helicobacter pylori eradication andgastric cancer: when is the horse out of the barn? Am J Gastroenterol2009;104:1342–5.
42. Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, et al.Helicobacter pylori eradication to prevent gastric cancer in a high-riskregion of China: a randomized controlled trial. JAMA 2004;291:187–94.
43. Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, RaychowdhuryR, et al. Synergistic interaction between hypergastrinemia and Heli-cobacter infection in a mouse model of gastric cancer. Gastroenter-ology 2000;118:36–47.
44. Lefebvre O, Chenard MP, Masson R, Linares J, Dierich A, LeMeur M,et al. Gastric mucosa abnormalities and tumorigenesis in mice lackingthe pS2 trefoil protein. Science 1996;274:259–62.
45. XuX,BrodieSG,YangX, ImYH,ParksWT,ChenL, et al.Haploid lossofthe tumor suppressor Smad4/Dpc4 initiates gastric polyposis andcancer in mice. Oncogene 2000;19:1868–74.
46. Jain RN, Al-Menhali AA, Keeley TM, Ren J, El-Zaatari M, Chen X, et al.Hip1r is expressed in gastric parietal cells and is required for tubulo-vesicle formation and cell survival in mice. J Clin Invest 2008;118:2459–70.
47. Judd LM, Alderman BM, Howlett M, Shulkes A, Dow C, Moverley J,et al. Gastric cancer development inmice lacking theSHP2binding siteon the IL-6 family co-receptor gp130. Gastroenterology 2004;126:196–207.
Nguyen et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2126
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957

2013;73:2117-2126. Published OnlineFirst February 1, 2013.Cancer Res Thanh-Long M. Nguyen, Shradha S. Khurana, Clifford J. Bellone, et al. Development of Gastric CancerAutoimmune Gastritis Mediated by CD4+ T Cells Promotes the
Updated version
10.1158/0008-5472.CAN-12-3957doi:
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/73/7/2117.full#ref-list-1
This article cites 47 articles, 12 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/7/2117.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/7/2117To request permission to re-use all or part of this article, use this link
on July 21, 2019. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2013; DOI: 10.1158/0008-5472.CAN-12-3957


















