
Arrhythmia/Electrophysiology - Circulationcirc.ahajournals.org/content/126/17/2051.full.pdf ·...
44
Arrhythmia/Electrophysiology Transient Receptor Potential Canonical-3 Channel–Dependent Fibroblast Regulation in Atrial Fibrillation Masahide Harada, MD, PhD; Xiaobin Luo, MSc; Xiao Yan Qi, PhD; Artavazd Tadevosyan, MSc; Ange Maguy, PhD; Balazs Ordog, PhD; Jonathan Ledoux, PhD; Takeshi Kato, MD, PhD; Patrice Naud, PhD; Niels Voigt, MD; Yanfen Shi, PhD; Kaichiro Kamiya, MD, PhD; Toyoaki Murohara, MD, PhD; Itsuo Kodama, MD, PhD; Jean-Claude Tardif, MD; Ulrich Schotten, MD, PhD; David R. Van Wagoner, PhD; Dobromir Dobrev, MD; Stanley Nattel, MD Background—Fibroblast proliferation and differentiation are central in atrial fibrillation (AF)–promoting remodeling. Here, we investigated fibroblast regulation by Ca 2 -permeable transient receptor potential canonical-3 (TRPC3) channels. Methods and Results—Freshly isolated rat cardiac fibroblasts abundantly expressed TRPC3 and had appreciable nonselective cation currents (I NSC ) sensitive to a selective TPRC3 channel blocker, pyrazole-3 (3 mol/L). Pyrazole-3 suppressed angiotensin II–induced Ca 2 influx, proliferation, and -smooth muscle actin protein expression in fibroblasts. Ca 2 removal and TRPC3 blockade suppressed extracellular signal-regulated kinase phosphorylation, and extracellular signal-regulated kinase phosphorylation inhibition reduced fibroblast proliferation. TRPC3 expression was upregulated in atria from AF patients, goats with electrically maintained AF, and dogs with tachypacing-induced heart failure. TRPC3 knockdown (based on short hairpin RNA [shRNA]) decreased canine atrial fibroblast proliferation. In left atrial fibroblasts freshly isolated from dogs kept in AF for 1 week by atrial tachypacing, TRPC3 protein expression, currents, extracellular signal-regulated kinase phosphorylation, and extracellular matrix gene expression were all significantly increased. In cultured left atrial fibroblasts from AF dogs, proliferation rates, -smooth muscle actin expression, and extracellular signal-regulated kinase phosphorylation were increased and were suppressed by pyrazole-3. MicroRNA-26 was downregulated in canine AF atria; experimental microRNA-26 knockdown reproduced AF-induced TRPC3 upregulation and fibroblast activation. MicroRNA-26 has NFAT (nuclear factor of activated T cells) binding sites in the 5 promoter region. NFAT activation increased in AF fibroblasts, and NFAT negatively regulated microRNA-26 transcription. In vivo pyrazole-3 administration suppressed AF while decreasing fibroblast proliferation and extracellular matrix gene expression. Conclusions—TRPC3 channels regulate cardiac fibroblast proliferation and differentiation, likely by controlling the Ca 2 influx that activates extracellular signal-regulated kinase signaling. AF increases TRPC3 channel expression by causing NFAT-mediated downregulation of microRNA-26 and causes TRPC3-dependent enhancement of fibroblast prolifera- tion and differentiation. In vivo, TRPC3 blockade prevents AF substrate development in a dog model of electrically maintained AF. TRPC3 likely plays an important role in AF by promoting fibroblast pathophysiology and is a novel potential therapeutic target. (Circulation. 2012;126:2051-2064.) Key Words: arrhythmia calcium ion channels fibrillation remodeling A trial fibrillation (AF) is the most common arrhythmia in clinical practice, and it confers significant morbidity and mortality. 1,2 Because the utility of conventional antiarrhyth- mic agents that target cardiac ion channels is limited by inefficacy and side effects, new treatment strategies are required. 1–3 An improved understanding of underlying mech- anisms may help in the design of novel therapies. 2 Under pathological conditions, cardiac fibroblasts first proliferate and then differentiate into myofibroblasts that secrete profi- brotic extracellular matrix (ECM). 3,4 Atrial fibrotic remodel- ing causes conduction abnormalities and may promote fibroblast-cardiomyocyte electric interactions that favor AF Received May 31, 2012; accepted August 10, 2012. From the Department of Medicine and Research Center, Montreal Heart Institute and Universite ´ de Montre ´al, Montreal, Quebec, Canada (M.H., X.L., X.Y.Q., A.T., A.M., B.O., J.L., T.K., P.N., Y.S., J.-C.T., S.N.); Division of Experimental Cardiology, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany (N.V., D.D.); Department of Cardiovascular Research, Research Institute of Environmental Medicine, Nagoya University, Nagoya, Japan (K.K., I.K.); Department of Cardiology, Nagoya University Graduate School of Medicine, Nagoya, Japan (T.M.); Department of Physiology, Maastricht University, Maastricht, Netherlands (U.S.); and Department of Molecular Cardiology, Cleveland Clinic, Cleveland, OH (D.R.V.W.). The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA. 112.121830/-/DC1. Correspondence to Stanley Nattel, 5000 Belanger St E, Montreal, Quebec, H1T 1C8, Canada. E-mail [email protected] © 2012 American Heart Association, Inc. Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.112.121830 2051 by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from by guest on June 1, 2018 http://circ.ahajournals.org/ Downloaded from
Transcript of Arrhythmia/Electrophysiology - Circulationcirc.ahajournals.org/content/126/17/2051.full.pdf ·...

Arrhythmia/Electrophysiology
Transient Receptor Potential Canonical-3 Channel–DependentFibroblast Regulation in Atrial Fibrillation
Masahide Harada, MD, PhD; Xiaobin Luo, MSc; Xiao Yan Qi, PhD; Artavazd Tadevosyan, MSc;Ange Maguy, PhD; Balazs Ordog, PhD; Jonathan Ledoux, PhD; Takeshi Kato, MD, PhD;
Patrice Naud, PhD; Niels Voigt, MD; Yanfen Shi, PhD; Kaichiro Kamiya, MD, PhD;Toyoaki Murohara, MD, PhD; Itsuo Kodama, MD, PhD; Jean-Claude Tardif, MD;
Ulrich Schotten, MD, PhD; David R. Van Wagoner, PhD; Dobromir Dobrev, MD; Stanley Nattel, MD
Background—Fibroblast proliferation and differentiation are central in atrial fibrillation (AF)–promoting remodeling. Here, weinvestigated fibroblast regulation by Ca2�-permeable transient receptor potential canonical-3 (TRPC3) channels.
Methods and Results—Freshly isolated rat cardiac fibroblasts abundantly expressed TRPC3 and had appreciablenonselective cation currents (INSC) sensitive to a selective TPRC3 channel blocker, pyrazole-3 (3 �mol/L). Pyrazole-3suppressed angiotensin II–induced Ca2� influx, proliferation, and �-smooth muscle actin protein expression infibroblasts. Ca2� removal and TRPC3 blockade suppressed extracellular signal-regulated kinase phosphorylation, andextracellular signal-regulated kinase phosphorylation inhibition reduced fibroblast proliferation. TRPC3 expression wasupregulated in atria from AF patients, goats with electrically maintained AF, and dogs with tachypacing-induced heartfailure. TRPC3 knockdown (based on short hairpin RNA [shRNA]) decreased canine atrial fibroblast proliferation. Inleft atrial fibroblasts freshly isolated from dogs kept in AF for 1 week by atrial tachypacing, TRPC3 protein expression,currents, extracellular signal-regulated kinase phosphorylation, and extracellular matrix gene expression were allsignificantly increased. In cultured left atrial fibroblasts from AF dogs, proliferation rates, �-smooth muscle actinexpression, and extracellular signal-regulated kinase phosphorylation were increased and were suppressed bypyrazole-3. MicroRNA-26 was downregulated in canine AF atria; experimental microRNA-26 knockdown reproducedAF-induced TRPC3 upregulation and fibroblast activation. MicroRNA-26 has NFAT (nuclear factor of activated Tcells) binding sites in the 5� promoter region. NFAT activation increased in AF fibroblasts, and NFAT negativelyregulated microRNA-26 transcription. In vivo pyrazole-3 administration suppressed AF while decreasing fibroblastproliferation and extracellular matrix gene expression.
Conclusions—TRPC3 channels regulate cardiac fibroblast proliferation and differentiation, likely by controlling the Ca2�
influx that activates extracellular signal-regulated kinase signaling. AF increases TRPC3 channel expression by causingNFAT-mediated downregulation of microRNA-26 and causes TRPC3-dependent enhancement of fibroblast prolifera-tion and differentiation. In vivo, TRPC3 blockade prevents AF substrate development in a dog model of electricallymaintained AF. TRPC3 likely plays an important role in AF by promoting fibroblast pathophysiology and is a novelpotential therapeutic target. (Circulation. 2012;126:2051-2064.)
Key Words: arrhythmia � calcium � ion channels � fibrillation � remodeling
Atrial fibrillation (AF) is the most common arrhythmia inclinical practice, and it confers significant morbidity and
mortality.1,2 Because the utility of conventional antiarrhyth-mic agents that target cardiac ion channels is limited byinefficacy and side effects, new treatment strategies arerequired.1–3 An improved understanding of underlying mech-
anisms may help in the design of novel therapies.2 Underpathological conditions, cardiac fibroblasts first proliferateand then differentiate into myofibroblasts that secrete profi-brotic extracellular matrix (ECM).3,4 Atrial fibrotic remodel-ing causes conduction abnormalities and may promotefibroblast-cardiomyocyte electric interactions that favor AF
Received May 31, 2012; accepted August 10, 2012.From the Department of Medicine and Research Center, Montreal Heart Institute and Universite de Montreal, Montreal, Quebec, Canada (M.H., X.L.,
X.Y.Q., A.T., A.M., B.O., J.L., T.K., P.N., Y.S., J.-C.T., S.N.); Division of Experimental Cardiology, Medical Faculty Mannheim, University ofHeidelberg, Mannheim, Germany (N.V., D.D.); Department of Cardiovascular Research, Research Institute of Environmental Medicine, NagoyaUniversity, Nagoya, Japan (K.K., I.K.); Department of Cardiology, Nagoya University Graduate School of Medicine, Nagoya, Japan (T.M.); Departmentof Physiology, Maastricht University, Maastricht, Netherlands (U.S.); and Department of Molecular Cardiology, Cleveland Clinic, Cleveland, OH(D.R.V.W.).
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.112.121830/-/DC1.
Correspondence to Stanley Nattel, 5000 Belanger St E, Montreal, Quebec, H1T 1C8, Canada. E-mail [email protected]© 2012 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.112.121830
2051
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

occurrence and maintenance.5,6 Therefore, fibroblast controlmechanisms could constitute novel antiarrhythmic targets.Although it is known that cellular Ca2� homeostasis regulatesfibroblast function,6 the precise regulatory mechanisms re-main unclear.
Editorial see p 2039Clinical Perspective on p 2064
Transient receptor potential (TRP) channels, activated bycell stretch and other pathological stimuli, regulate cellularCa2� entry.6 TRP channels modulate fundamental cell func-tions such as cell proliferation and death.6–8 Mechanicalstretch and related stimuli contribute to arrhythmic sub-strates3; TRP channels are candidates to link arrhythmicremodeling stimuli to arrhythmia-promoting cardiac fibro-blast responses. In preliminary studies (online-only DataSupplement Figure I), we noted cell-selective expression ofTRP canonical-3 (TRPC3) subunits in freshly isolated fibro-blasts, along with TRPC3 downregulation on fibroblast dif-ferentiation to myofibroblasts, which identified TRPC3 as apotential modulator of fibroblast function. We undertook thepresent study to test the following hypotheses: (1) TRPC3subunits play a role in fibroblast function in normal heartsand in an AF model; (2) fibroblast TRPC3 expression isupregulated in AF under the control of discrete microRNA-related molecular mechanisms; and (3) in vivo TRPC3inhibition can suppress AF-associated remodeling.
MethodsA detailed description of methods used in this study is available inthe online-only Data Supplement.
Cardiac Fibroblast Isolation and CultureFibroblasts were isolated from adult rat or dog hearts as describedpreviously.9 Tissues were digested with collagenase II and cardio-myocytes removed by low-speed centrifugation and passage througha 20-�m nylon filter. Fibroblasts were concentrated by high-speedcentrifugation (2000 rpm, 10 minutes). Freshly isolated fibroblasts orcells in primary culture within 3 days of isolation (online-only DataSupplement Figure II-A) were used in most experiments. Long-termcultured first-passage cells (2–3 weeks), which express �-smoothmuscle actin (�-SMA) and show actin fiber organization (online-only Data Supplement Figure II-B), were used in rat myofibroblaststudies.
Atrial Tissue Samples From Humans, Goats,and DogsRight atrial (RA) appendage biopsy samples were obtained frompatients in sinus rhythm and with chronic AF during coronary arterybypass graft surgery after informed consent as approved by the ethicsreview committee of Dresden University of Technology. AF wasinduced in chronically instrumented goats by use of repetitive burstpacing for 10 days.10 Dogs with congestive heart failure (CHF) withan AF substrate were created by ventricular tachypacing (240 bpm,2 weeks).11 Normal goats and dogs were used as controls. RA tissuesamples were collected and fast-frozen in liquid N2.
AF DogsA total of 48 mongrel dogs (weight, 20–36 kg) were divided intocontrol and atrial tachypacing groups. Animal care proceduresfollowed National Institutes of Health guidelines and were approvedby the Animal Research Ethics Committee of the Montreal HeartInstitute. Dogs were anesthetized with ketamine (5.3 mg/kg IV)/diazepam (0.25 mg/kg IV)/isoflurane (1.5%), intubated, and venti-
lated. A unipolar pacing lead was inserted into the RA appendageunder fluoroscopic guidance and connected to a pacemaker in theneck. Bipolar electrodes were inserted into the right ventricular apexand RA appendage for electrogram recording. Atrioventricularblock/ventricular pacing (used in many studies of atrial tachycardiaremodeling12) was not performed, to more closely mimic spontane-ous clinical AF episodes. The atrial pacemaker was programmed tostimulate the RA at 600 bpm for 1 week, with fibrillatory atrialactivity maintained during pacing as assessed by daily ECG andintracardiac recordings. Echocardiography was performed on day 0(baseline) and day 7 to assess left atrial (LA) dimension, LA systolicfunction, and left ventricular diastolic volume.
For in vivo pyrazole-3 (Pyr3) treatment of AF dogs, ALZETosmotic pumps were implanted subcutaneously in the back tocontinuously administer intravenous Pyr3 (0.1 mg � kg�1 � d�1 inDMSO and polyethylene glycol) or vehicle during atrial tachypacing(online-only Data Supplement Figure III).
Electrophysiological StudiesA terminal open chest electrophysiological study was performed onday 7. Dogs were anesthetized with morphine (2 mg/kg SC) and�-chloralose (120 mg/kg IV, followed by 29.25 mg � kg�1 � h�1) andventilated. After performance of a median sternotomy, the atrialeffective refractory period and mean AF duration induced by burstpacing were measured as described previously.12
TaqMan Low-Density ArraysRNA was extracted with TRIzol. RNA integrity was assessed via anAgilent bioanalyzer (Agilent Technologies; RNA integrity number�7.5 required). cDNA was synthesized with a high-capacity cDNAreverse transcription kit and random hexamer primers. TaqManlow-density arrays were used in 2-step reverse transcription–poly-merase chain reaction (PCR).13 Real-time PCR was performed on the7900HT Fast Real-Time PCR System (Applied Biosystems). TRPsubunit genes were investigated by use of inventoried TaqManassays. The mean expression of genes with a cycle threshold value(Ct) �30 was used as the reference.14
Whole-Cell Patch ClampingWhole-cell voltage clamping was performed to record the nonselec-tive cation current (INSC) at 37°C. The pipette solution contained(in mmol/L) 135 CsCl, 0.1 CaCl2, 10 EGTA, 4.0 MgATP, 1.0MgCl2, 10 HEPES, 6.6 sodium phosphocreatine, 0.3 Na-GTP (pH7.4/CsOH). The bath solution contained (in mmol/L) 140 NaCl, 5.4TEA-Cl, 2.0 CaCl2, 1.0 MgCl2, 10 HEPES, 10.0 glucose (pH7.4/CsOH). Nifedipine (5 �mol/L) was used to block any L-typeCa2� current. Na� current was inactivated with a holding potentialof 0 mV. Currents were elicited with 3-second voltage-ramp proto-cols (0.07 mV/ms, from �110 mV to 100 mV, 0.1 Hz) at a holdingpotential of 0 mV.15
Cell Proliferation and Cell-Cycle AnalysisCardiac fibroblasts were cultured in T25 culture flasks (2.0�105
cells/flask, 25 cm2 growth area) for each treatment group. Afterincubation, cells were harvested after trypsinization and then fixed withice-cold 75% ethanol. Samples were stored at �20°C until analysis.Pelleted cells were resuspended and incubated (4°C, 30 minutes) instaining solution that contained propidium iodide. Cell numbers andDNA content frequency histograms were obtained via FACScan (con-stant flow rate, 60 �L/min; 5-minute acquisition time).
Western BlotsProtein was extracted, quantified, and processed.12,16 Cytoplasmicand nuclear protein fractions were extracted from fresh fibroblastswith a ProteoExtract subcellular proteome extraction kit (Calbio-chem). Protein samples were separated by gel electrophoresis andtransferred to polyvinylidene difluoride membranes. Membraneswere blocked and incubated with mouse anti-�-SMA, anti-phospho-p44/42-MAP kinase, anti-TRPM7, anti-NFATc3, anti-HSP70, anti-lamin A/C and anti-vimentin, rabbit anti-TRPC3, anti-TRPC1, anti-
2052 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

calsequestrin, and anti-NFATc4 antibodies. Secondary antibodiesconjugated to horseradish peroxidase were used for detection viachemiluminescence.
Confocal ImagingCultured, freshly isolated fibroblasts or LA tissue cryosections werefixed with 2% paraformaldehyde, washed with PBS, and incubatedwith mouse anti-�-SMA, goat anti-vimentin, mouse anti-NFATc3,and rabbit anti-NFATc4, followed by donkey anti-mouse IgG AlexaFluor 555, donkey anti-rabbit IgG Alexa Fluor 488, donkey anti-goatIgG Alexa Fluor 488, and/or TOPRO 3 iodide. Apoptosis wasassessed by TUNEL (terminal dUTP nick end-labeling) assay withApopTag. Fluorescent images were obtained via a Zeiss LSM-710 orOlympus FluoView FV1000 inverted confocal microscope.
Ca2� ImagingPrimary cultured rat fibroblasts (1-day culture) were loaded withFluo-4 (10 �mol/L) in phenol-free M199 medium in the presence ofpluronic acid (2.5 �g/mL) for 50 minutes. Ca2� imaging wasobtained with an Andor Revolution confocal system and a iXon(Andor Technologies) mounted on an upright Nikon FN-1 micro-scope. Fluo-4 was excited at 488 nm; emitted fluorescence wascollected at 495 nm. Images (512�512 pixels) were acquired at 15frames/second. F0 was determined by averaging fluorescence from10 consecutive baseline images.17
Quantitative PCRTo study microRNA, RNA/microRNA were extracted with a TRIzol/mirVana miRNA extraction kit (Ambion). Real-time reverse tran-scription–PCR was performed with 6-carboxy fluorescein–labeledfluorogenic TaqMan primers. Fluorescence signals were detected induplicate, normalized to �2-microglobulin RNA for total RNA andto U6 small nuclear RNA for microRNA, and quantified with MxProqPCR software (Stratagene).16
TRPC3 KnockdownTRPC3 knockdown lentivirus vector plasmid was obtained fromOpen Biosystems (Oligo ID: V2LMM_11490). A scrambled micro-RNA–adapted shRNA (shRNAmir)-overexpressing virus was usedas a negative control. For lentivirus infection, 3-day culturedfibroblasts were plated in T25 culture flasks at 4�103 cells/cm2,infected at 50 MOI, and incubated for 3 days before experimentswere performed.
MicroRNA (miR) Overexpression/KnockdownFor overexpression, sense and antisense oligoribonucleotides weresynthesized by Invitrogen, and the double-stranded RNA was createdby an annealing treatment. A scrambled RNA was the negativecontrol. For knockdown, antisense anti-miR oligonucleotides(AMO26a) with locked nucleic acid technology were synthesized byExiqon. Scrambled oligonucleotides with methylene bridges wereused as negative controls. Dog LA fibroblasts in primary culturewere transfected with miR-26a duplex (100 nmol/L), AMO26a (10nmol/L), and/or control sequences with Lipofectamine 2000(Invitrogen).18
Dual Luciferase Reporter AssayA fragment that contained the miR target sequence was synthesizedby Invitrogen and used as a template for PCR amplification. ThePCR product was subcloned in the pMIR-REPORT luciferase miRexpression reporter vector. HEK293 cells were transfected with 50ng of pMIR-REPORT, 0.5 ng of pRL-TK (for internal control), andmiR-26a duplex (10 nmol/L) and/or AMO26a (3 nmol/L) withLipofectamine 2000.
Statistical AnalysisData are presented as mean�SEM. A 2-way ANOVA with multiple-group comparisons (Bonferroni-corrected t test) was applied to datawith �2 main effect factors. One-way ANOVA was applied for
single main effect factor experiments. Repeated-measure analyseswere used when the same sets of subjects or materials were exposedto multiple interventions. Student t tests were used for comparisonsthat involved only 2 groups. For multiple comparisons with Bonfer-roni correction, adjusted probability values were calculated bymultiplying original probability values by the number of compari-sons (N) performed; values shown are adjusted values (N�P).Two-tailed P�0.05 was considered statistically significant. Foradditional details, see the online-only Data Supplement.
ResultsTRPC3-Mediated INSC and Ca2� EntryINSC recordings from freshly isolated rat fibroblasts areshown in Figure 1A and online-only Data Supplement FigureII-C. INSC was substantially suppressed by superfusion withthe TRPC3-selective blocker Pyr3 (Figure 1B)19 and by thegeneral TRP channel blocker, gadolinium (Gd3�;100 �mol/L; online-only Data Supplement Figure II-C).Passage-cultured myofibroblasts (online-only Data Supple-ment Figure II-B) showed larger membrane capacitance thanfibroblasts (16.8�1.4 versus 6.4�0.4 picofarads, P�0.01)but had much smaller Gd3�- and Pyr3-sensitive INSC, consis-tent with quantitative PCR data (online-only Data Supple-ment Figure II-D through II-F).
We then tested whether TRPC3 channels contribute toCa2� entry in cardiac fibroblasts. Because diacylglycerol hasbeen suggested to be a physiological activator of TRPC3,20
we examined the effect of TRPC3 blockade on Ca2� entryinduced by 1-oleoyl-2-acetyl-sn-glycerol (OAG) and angio-tensin II, which activate diacylglycerol receptors directly andindirectly, respectively. Both angiotensin II (Figure 1C and1D) and OAG (online-only Data Supplement Figure IV-Aand IV-B) induced fibroblast Ca2� entry, which was pre-vented by Pyr3, which suggests that TRPC3 channels areneeded for OAG- and angiotensin II–induced Ca2� entry.
Role in Fibroblast Proliferationand DifferentiationWe next examined whether TRPC3 channel block affectscardiac fibroblast proliferation. Rat cardiac fibroblasts weremaintained in the presence or absence of Gd3�, Pyr3, orcontrol vehicle for up to 1 day in primary culture. After24-hour culture in vehicle medium, the number of fibroblastsincreased substantially (Figure 1E). After 1- and 24-hourtreatment, fibroblasts were collected for proliferation analysisby flow cytometry. Representative DNA content histogramsshowing the G2/M phase cell content, an index of DNAduplication, are provided in online-only Data SupplementFigure V-A and V-B. Pyr3 significantly slowed the increasein fibroblast number and reduced G2/M phase cell content(Figure 1E and 1F), as did Gd3� (online-only Data Supple-ment Figure V-C and V-D). Pyr3 also significantly increasedthe percentage of TUNEL-positive fibroblasts versus control(online-only Data Supplement Figure VI). These data suggestthat TRPC3 channels regulate cardiac fibroblast survival andproliferation.
After fibroblasts proliferate, they differentiate into ECM-secreting myofibroblasts characterized by altered morphol-ogy and increased �-SMA expression. Online-only DataSupplement Figure VII-A shows confocal images of rat
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2053
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

Figure 1. A, Representative nonselective cation current (INSC) recordings with or without pyrazole-3 (Pyr3; 3 �mol/L). B, Mean�SEMGd3�- and Pyr3-sensitive INSC density (n�7 cells in Gd3� and 9 cells in Pyr3). C, Recordings of angiotensin II (AngII)–induced intracellu-lar Ca2� response in presence or absence of Pyr3. D, Mean�SEM AngII-induced Ca2� fluorescence (F/F0), normalized to baseline in0 mmol/L Ca2� (n�18 and 20 cells in AngII and AngII with Pyr3, respectively; *P�0.05 vs 0 mmol/L Ca2�, #P�0.05 vs 2 mmol/L Ca2�
in AngII). E, Mean�SEM fibroblast count after 1- or 24-hour culture with vehicle control (CTL) or 0.3 or 3 �mol/L Pyr3 (n�9/group;*P�0.05 vs CTL, #P�0.05 vs 1-hour treatment). F, Mean�SEM percentage of cells in G2/M phase after Pyr3 treatment (n�9; *P�0.05vs CTL, #P�0.05 vs 1-hour treatment). G, Representative immunoblots for �-smooth muscle actin (�SMA) and GAPDH in rat fibroblastscultured with or without Pyr3 (3 �mol/L). H, Mean�SEM �SMA/GAPDH expression ratio (n�8/group; *P�0.05 vs CTL).
2054 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

fibroblasts, cultured with or without Pyr3, stained withanti-�-SMA antibodies. The spindle-shaped expansion and�-SMA–enhanced expression associated with myofibroblastdifferentiation were inhibited by 3-�mol/L Pyr3. Figure 1Gshows representative �-SMA immunoblots on cultured ratfibroblasts; �-SMA protein expression was decreased signif-icantly by Pyr3 (Figure 1H). Thus, TRPC3 channels partici-pate in fibroblast differentiation into myofibroblasts. We alsoexamined the effect of TRPC3 blockade on already differen-tiated myofibroblasts. Consistent with downregulation ofTRPC3 in myofibroblasts, their cell number, G2/M phase cellcontent, and �-SMA expression were unchanged by Pyr3(online-only Data Supplement Figure VII-B through VII-D).Thus, once fibroblasts differentiate into myofibroblasts,
TRPC3 channels no longer appear to be involved in theirregulation.
TRPC3-Mediated Ca2� Entry, ERK-1/2Phosphorylation, and Fibroblast ActivationWe then examined whether TRPC3-mediated Ca2� entry actsby modulating Ca2�-dependent extracellular signal-regulatedkinase (ERK)-1/2 activation, which affects cell survival andfibroblast activation.21,22 Fibroblasts were cultured for 24hours in 3 conditioned media: (1) 0.4 mmol/L Ca2�, (2)2.4 mmol/L Ca2�, and (3) 2.4 mmol/L Ca2� with Pyr3.ERK-1/2 phosphorylation was decreased significantly inlow-[Ca2�] medium and with exposure to Pyr3 (Figure 2A).Next, we examined the effect of extracellular Ca2� concen-
Figure 2. A, Top, Representative immu-noblots of phosphorylated extracellularsignal-regulated kinase (ERK)-1/2(p-ERK), total ERK-1/2 (t-ERK), andGAPDH in rat fibroblasts cultured with0.4 mmol/L Ca2�, 2.4 mmol/L Ca2�, and2.4 mmol/L Ca2� medium containing3 �mol/L pyrazole-3 (Pyr3). CTL indi-cates control. Bottom, Mean�SEMp-ERK/GAPDH, t-ERK/GAPDH, andp-ERK/t-ERK (n�8/group; *P�0.05 vsCTL, #P�0.05 vs Pyr3). B, Mean�SEMfibroblast cell count after culture in M199medium containing 0.4 to 4.4 mmol/LCa2� (n�8/group; *P�0.05 vs 0.4-mmol/L Ca2�, #P�0.05 vs 1-hour treat-ment). C, Mean�SEM percentage ofcells in G2/M phase (n�8/group;*P�0.05 vs 0.4-mmol/L Ca2�, #P�0.05vs 1-hour treatment). D, Mean�SEMfibroblast cell count after 1- and 24-hourtreatment with 50 �mol/L PD98059(n�6/group; *P�0.05 vs CTL, #P�0.05vs 1-hour treatment). E, Mean�SEMpercentage of cells in G2/M phase after50 �mol/L PD98059 treatment (n�6/group; *P�0.05 vs CTL).
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2055
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

tration on fibroblast proliferation. The increase in cell numberof cultured fibroblasts and the G2/M phase cell content weresignificantly smaller with less [Ca2�] in the culture medium(Figure 2B and 2C). A selective ERK pathway inhibitor,PD98058 (50 �mol/L), also significantly reduced the numberof fibroblasts and the G2/M phase cell content after 24-hourculture (Figure 2D and 2E). The data in Figure 2 suggest thatTRPC3-mediated Ca2� influx contributes to the ERK-1/2activation that regulates fibroblast proliferation.
TRPC3 Knockdown Suppresses AtrialFibroblast ProliferationWe were unable to study TRPC3 knockdown in rat fibro-blasts because spontaneous TRPC3 downregulation in culturewas almost complete over the time period needed forlentivirus-mediated knockdown. However, we noted thatTRPC3 downregulation was slower in cultured dog atrialfibroblasts, which were used to study the effects of TRPCknockdown. TRPC3 subunit mRNA and protein expressiondecreased significantly in TRPC3 shRNA-infected fibro-blasts; TRPC6 mRNA remained unchanged (online-onlyData Supplement Figure VIII-A and VIII-B). The fold in-crease in cultured fibroblast cell number was significantlyattenuated in TRPC3 shRNA-infected fibroblasts (online-only Data Supplement Figure VIII-C), as was the G2/M phasecell content (online-only Data Supplement Figure VIII-D),which indicates that TRPC3 channels are involved in canineatrial fibroblast proliferation.
Atrial TRPC3 Expression in Large Animal/HumanAF SubstratesTo assess a potential role in the AF substrate, we examinedthe protein expression of TRPC1, TRPC3, and TRPM7subunits in RA tissue samples from AF patients, AF goats,and CHF dogs with an AF substrate. Atrial TRPC3 expres-sion increased significantly in all groups with AF substrates(online-only Data Supplement Figure IX-A through IX-C),whereas TRPC1 and TRPM7 remained unchanged (online-only Data Supplement Figure IX-D through IX-I).
Atrial Remodeling in Dogs With ElectricallyMaintained AFTo examine further the potential role of TRPC3 in AF, we useda canine model of electrically maintained AF. Representativesurface and intracardiac day 7 ECG recordings showing irregu-lar, rapid atrial activity and irregular ventricular responsestypical of AF are shown in Figure 3A. Spontaneous AF wasmaintained after pacing cessation. Fibrillatory electric activityand spontaneous postpacing AF were observed consistentlyduring the 1-week pacing period (online-only Data SupplementFigure X). Immediately after tachypacing onset (recordings at6.0�0.2 minutes after onset), ventricular activation rate in-creased by 55%, but ventricular rate then returned to controlvalues by day 3 (online-only Data Supplement Figure XI-A). Atthe end of the study, atrial and ventricular filling pressures wereslightly but significantly increased in AF dogs (online-only DataSupplement Table I). Online-only Data Supplement Figure XI-Billustrates atrial structural remodeling in AF dogs, with signifi-
cant changes in LA diastolic area and fractional area change(online-only Data Supplement Figure XI-C and XI-D).
AF dogs showed electric remodeling manifested by re-duced effective refractory periods (Figure 3B) and increasedduration of burst-pacing–induced AF (Figure 3C). LA fibro-sis and vimentin staining (an index of fibroblast density) wereassessed by histomorphometry (Figure 3D). Fibrous tissuecontent was unchanged (Figure 3E), but vimentin-positivearea (Figure 3F) and protein expression (immunoblots, Figure3G) increased significantly in AF dogs.
TRPC3 Regulation of Atrial Fibroblast Activationin AF DogsFigure 4A shows INSC before and after 3 �mol/L Pyr3 in freshlyisolated LA fibroblasts. Pyr3-sensitive current increased signif-icantly in LA fibroblasts of AF dogs (Figure 4B), correspondingto increased TRPC3 subunit protein expression in freshly iso-lated LA fibroblasts (Figure 4C). ERK phosphorylation (Figure4D) and ECM gene expression (Figure 4E) were also signifi-cantly increased in LA fibroblasts of AF dogs.
The cell number increase rate, G2/M phase cell content,and �-SMA protein expression all increased in LA fibroblastsof AF dogs (Figure 5A through 5C), which indicates in-creased fibroblast proliferation and differentiation. In vitrotreatment of AF fibroblasts with Pyr3 decreased these fibro-blast activation indexes significantly (Figure 5D through 5F).ERK activation was increased significantly in AF (Figure5G). ERK phosphorylation in AF dogs was reduced signifi-cantly after 24-hour treatment with Pyr3 (Figure 5H). Thesedata suggest that TRPC3 channels are an important contrib-utor to fibroblast activation in AF.
MiR-26 Regulation of TRPC3 ChannelsMicroRNAs posttranscriptionally regulate protein expressionin many pathological conditions.1 MiR-target prediction (Tar-getScan) suggested that miR-26 targets the TRPC3 gene(online-only Data Supplement Figure XII-A). Expression ofthe 2 miR-26 isoforms, miR-26a and miR-26b, which haveidentical seed sequences, was decreased significantly infreshly isolated LA fibroblasts from AF dogs (Figure 6A).Other miRs involved in cardiac remodeling1 were studied,and miR-133 was also downregulated; however, miR-26 wasparticularly strongly expressed in fibroblasts (Figure 6B),whereas miR-133 expression was cardiomyocyte selective(Figure 6C). Neither miR-1 nor miR-133 targets TRPC3.
We then examined posttranscriptional regulation ofTRPC3 by miR-26a with dual luciferase reporter assay.Luciferase vectors carrying the miR-26a target gene sequenceof TRPC3 were cotransfected along with miR-26a duplexand/or antisense anti-miR-26a oligonucleotides (AMO26a;online-only Data Supplement Figure XII-B) into HEK-293cells. MiR-26a overexpression significantly decreased lu-ciferase readout, whereas knockdown of endogenous miR-26a by AMO26a significantly increased luciferase fluores-cence (Figure 6D), which indicates that miR-26a regulatesTRPC3 translation.
Using immunoblotting, we further validated the effect ofmiR-26a on TRPC3 protein expression in cultured canine LAfibroblasts. MiR-26a overexpression decreased TRPC3 pro-
2056 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

tein expression significantly, whereas miR-26a knockdown tomimic AF-related miR-26 downregulation increased TRPC3protein (Figure 6E). We then assessed the regulation offibroblast proliferation by miR-26a. MiR-26a overexpressionsignificantly decreased fibroblast-number (Figure 6F), as wellas G2/M cell percentage (Figure 6G), whereas miR-26aknockdown increased these fibroblast-proliferation indexes.
These data indicate that miR-26a controls TRPC3 expressionby downregulating TRPC3 protein and produces parallelchanges in fibroblast proliferation indexes.
NFATc3 Regulation of miR-26aWe next looked for the potential mechanism of fibroblastmiR-26 control in AF. Nuclear translocation of NFAT (the
Figure 3. A, Representative surface ECG (left) and intracardiac electrograms (right) in an awake dog with atrial fibrillation (AF) on day7. B, Mean�SEM atrial effective refractory period (ERP) in control (CTL; n�11) and AF dogs (n�12) on day 7 (*P�0.05). BCL indicatesbasic cycle length. C, Mean�SEM duration of induced AF (*P�0.05 vs CTL). D, Top, Representative Masson’s trichrome–stained leftatrial images from CTL (left) and AF (right) dogs. Bottom, Representative immunofluorescent images of LA free-wall tissues stainedwith vimentin. E, Mean�SEM fibrous tissue content in left atrial free-wall tissues (n�5 CTL, 5 AF dogs). F, Mean�SEM vimentin-positive area (n�5 CTL, 5 AF dogs; *P�0.05 vs CTL). H, Representative immunoblots (top) and mean�SEM vimentin band intensitynormalized to GAPDH (bottom) in left atrial samples from CTL (n�5) and AF dogs (n�5; *P�0.05 vs CTL).
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2057
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

nuclear factor of activated T cells), subsequent to dephos-phorylation by Ca2�/calmodulin-dependent calcineurin acti-vation, is important in AF-related cardiomyocyte remodel-ing.1 The promoter regions of the host genes for miR-26a/b inhumans and dogs are predicted to have multiple bindingmotifs for NFAT (online-only Data Supplement Figure XII-D). We therefore evaluated cellular localization of NFAT infibroblasts from AF and control dogs. Figure 7A showsrepresentative confocal images of freshly isolated LA fibro-blasts stained with vimentin, TOPRO3 (nuclear stain), andNFATc3/c4 antibodies. Nuclear localization of NFATc3increased significantly in AF (Figure 7B). We also quantifiednuclear NFAT localization by immunoblotting on isolatedcanine fibroblast nuclei. AF significantly reduced the cyto-solic and increased the nuclear protein fraction of NFATc3(Figure 7C). To assess NFAT regulation of miR-26 geneexpression, we examined the effect of incubation with aselective membrane-permeable NFATc3/c4 blocker, INCA6(2.5 �mol/L), on miR-26 and TRPC3 expression in canineatrial fibroblasts. INCA6 significantly increased miR-26a/b
expression (Figure 7D) and decreased TRPC3 protein expres-sion (Figure 7E), consistent with an inhibitory effect ofNFAT on miR-26 transcription. These data suggest thatAF-induced NFATc3 translocation suppresses miR-26a tran-scription, thereby reducing miR-26a inhibition of TRPC3translation and resulting in TRPC3 protein upregulation.
Effects of In Vivo TRPC3 Blockade on theAF SubstrateTo more directly assess the role of TRPC3 in the AF-promotingremodeling of the AF dogs in the present study, additional AFdogs were treated with Pyr3 (0.1 mg � kg�1 � d�1 by continuousintravenous infusion) or vehicle for the entire period of atrialtachypacing (online-only Data Supplement Figure III). At theterminal electrophysiological study, Pyr3-treated dogsshowed significantly reduced AF duration (Figure 8A). Theyalso had slightly but significantly greater effective refrac-tory period values at short cycle lengths (Figure 8B).Consistent with in vivo TRPC3 control of fibroblastfunction, Pyr3 significantly decreased LA vimentin ex-
Figure 4. A, Representative nonselectivecation current (INSC) recordings beforeand after pyrazole-3 (Pyr3; 3 �mol/L) infreshly isolated left atrial fibroblasts fromcontrol (CTL) dogs and dogs withtachypacing-induced atrial fibrillation (AF).B, Mean�SEM Pyr3-sensitive INSC den-sity in CTL dogs (n�6 dogs, 8 cells) andAF dogs (n�6 dogs, 9 cells; P�0.05 vsCTL). C, Representative immunoblots(top) and mean�SEM TRPC3 subunitband intensity relative to GAPDH (bot-tom) in freshly isolated left atrial fibro-blasts from CTL (n�6) and AF dogs (n�6;*P�0.05 vs CTL). D, Representative im-munoblots (top) and mean�SEM data(bottom) for phosphorylated (p-ERK-1/2)and total (t-ERK-1/2) extracellular signal-regulated kinase 1/2 (ERK-1/2) relative toGAPDH. E, Mean�SEM extracellularmatrix gene mRNA expression in freshlyisolated left atrial fibroblasts (n�6 CTLand 6 AF; *P�0.05 vs CTL). Col indicatescollagen; FBN, fibrinogen; FN, fibronectin.
2058 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

pression (Figure 8C), as well as fibroblast number on day3 (Figure 8D) and G2/M cell content on day 2 (Figure 8E),in LA fibroblasts cultured from AF dogs. Pyr3 alsodecreased ECM gene expression in LA fibroblasts freshlyisolated from AF dogs (Figure 8F).
DiscussionThe present study shows that TRPC3 channels control fibro-blast function via Ca2�-dependent ERK phosphorylation,which results from Ca2� entry through INSC. In addition,atrial TRPC3 expression is upregulated in AF, and thischange induces fibroblast proliferation, differentiation, andactivation. TRPC3 upregulation in AF is caused by down-regulation of its regulatory microRNA, miR-26, under thecontrol of NFATc3. Infusion of a highly selective TRPC3inhibitor, Pyr3, suppresses development of the AF substratein a canine model.
Comparison With Previous Studies on TRPChannel–Dependent Regulation ofFibroblast FunctionTRPC3 is a Ca2�-permeable ion channel that shows 75%homology with TRPC6 and 7.20 These channels show sub-stantial Ca2� permeability and mediate receptor-activatedextracellular Ca2� entry.20,23,24 TRPC3/6/7 channels are di-rectly activated by diacylglycerol liberation into the plasmamembrane, triggered by agonist binding to G protein–cou-pled receptors, such as angiotensin II and endothelin-1receptors.20,23 TRPC channels are widely expressed and areinvolved in diverse biological functions, such as neuronal cellsurvival,24 blood vessel constriction,25 immune cell matura-tion,26 and cardiac hypertrophy.27,28 However, the regulatoryrole of TRPC3 channels in cardiac fibroblast function has notbeen reported previously.
Rose et al15 demonstrated that a TRP channel–like INSC iselicited by C-type natriuretic peptide in freshly isolated rat
Figure 5. A, Mean�SEM cell count ofleft atrial fibroblasts after 2- and 3-dayculture in control dogs (CTL; n�7) anddogs with tachypacing-induced atrialfibrillation (AF; n�7; *P�0.05 vs CTL). B,Mean�SEM percentage of cells in G2/Mphase (n�7 dogs/group; *P�0.05 vsCTL). C, Representative immunoblots(top) and mean�SEM �-smooth muscleactin/GAPDH (�SMA; bottom) in cul-tured left atrial fibroblasts from CTL(n�6) and AF dogs (n�7; *P�0.05 vsCTL). D, Mean�SEM cell count of leftatrial fibroblasts after 1- or 24-hour cul-ture with vehicle/control or pyrazole-3(Pyr3; 3 �mol/L) in AF dogs (n�6/group;*P�0.05 vs DMSO). E, Mean�SEM per-centage of cells in G2/M phase (n�6/group; *P�0.05 vs DMSO). F, Represen-tative immunoblots (top) andmean�SEM �-SMA/GAPDH expressionratios (bottom) in cultured left atrialfibroblasts from AF dogs (n�7) in thepresence of vehicle (DMSO) or Pyr3(*P�0.05 vs DMSO). G, Representativeimmunoblots (top) and mean�SEMphosphorylated extracellular signal-regulated kinase-1/2 (pERK) and totalERK-1/2 (tERK) relative to GAPDH (bot-tom) in cultured left atrial fibroblasts ofCTL and AF dogs (n�6/group; *P�0.05vs CTL). H, Representative immunoblots(top) and mean�SEM ERK (bottom) inleft atrial fibroblasts from AF dogs cul-tured with vehicle (DMSO) or Pyr3(3 �mol/L, n�6/group; *P�0.05 vsDMSO).
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2059
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

ventricular fibroblasts. Transcripts encoding TRPC3 subunitswere highly expressed in the fresh fibroblasts, and INSC wasactivated by phorbol esters; however, the role of INSC infibroblast function was not examined.
Nishida et al29 reported that TRPC1, 3, 6, and 7 mRNAsare detected in neonatal rat fibroblasts and that TRPC6channels contribute to the regulation of endothelin-1–induced myofibroblast formation through JNK and NFATsignaling. However, in contrast to the present observationsregarding TRPC3 channels, ERK-1/2 phosphorylation was notaffected by endothelin-1–activated TRPC6 currents. Of note, theNishida study was performed in neonatal cells, and TRP chan-nel–dependent regulation of fibroblast function may changeduring development from neonatal to adult conditions.
Du et al30 demonstrated that current that corresponds toTRP melastatin–related 7 (TRPM7), a Ca2�/Mg2�-permeable channel, is strongly expressed in fibroblastsisolated from RA samples of AF patients and likely playsan important role in AF pathophysiology. shRNA-basedTRPM7 knockdown decreased TRPM7-mediated Ca2�
influx in cultured atrial fibroblasts and suppressed fibro-blast differentiation induced by transforming growthfactor-� stimulation. The present results showed substan-tial TPRC3 subunit expression and associated current, aswell as significant physiological function, in fibroblaststhat were freshly isolated or kept under conditions thatlimited differentiation. Furthermore, TRPC3-dependentcurrent and fibroblast regulation were enhanced in AF dogfibroblasts. The expression and function of TRPC3 chan-nels disappeared after differentiation to myofibroblastsunder culture conditions. In contrast, mRNA expression ofTRPM7 subunits remained high in myofibroblasts, similarto the findings of Du et al,30 who also showed strongmRNA expression of TRPM7 but not TRPC3 subunits incultured human atrial fibroblasts. Cardiac fibroblast phe-notype changes dynamically during proliferation and dif-ferentiation. The present findings suggest that TRPC3controls proliferation and differentiation of fibroblasts butis downregulated in the end-cell myofibroblast. Thisnegative-feedback system may prevent excessive ECM
Figure 6. A, Mean�SEM microRNAexpression in freshly isolated left atrialfibroblasts in dogs with tachypacing-induced atrial fibrillation (AF; n�7) andcontrol dogs (CTL; n�7; *P�0.05 vsCTL). B, Mean�SEM relative microRNAexpression in freshly isolated left atrialfibroblasts in CTL (n�5; *P�0.05 vsmiR-26a). A.U. indicates arbitrary units.C, Mean�SEM relative microRNAexpression in freshly isolated left atrialcardiomyocytes in CTL (n�5; *P�0.05vs miR-26a). D, Mean�SEM relativeluciferase activity in HEK293 cells trans-fected with miR-26a overexpression(miR-26a duplex) or knockdown (anti-sense anti-miR-26a oligonucleotides[AMO26a]) probes (n�7/group; P�0.05vs cells transfected without the miR-26aprobes). E, Representative immunoblots(top) and mean�SEM TRPC3/GAPDHprotein expression (bottom) in dog leftatrial fibroblasts transfected with themiR-26a duplex or AMO26a (n�7;P�0.05 vs cells without miR-26aprobes). F, Mean�SEM fibroblast countin dog left atrial fibroblasts transfectedwith miR-26a duplex or AMO26a (n�7/group; *P�0.05 vs cells transfectedwithout miR-26a probes). G, Mean�SEMpercentage of cells in G2/M phase (n�7/group; *P�0.05 vs cells transfectedwithout miR-26a probes).
2060 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

remodeling. In contrast, TRPM7 is likely the dominantTRP channel in differentiated myofibroblasts.
Du et al30 found increased TRPM7 current in fibroblastsisolated from atrial samples of AF patients but did not havesufficient tissue to perform Western blot studies. We foundincreased protein expression of TRPC3, but not TRPC1 orTRPM7, by immunoblotting of atrial samples in AF patients,AF goats, and CHF dogs. This observation suggests that theincreased TRPM7 function noted by Du et al30 in fibroblastsfrom AF patients may have been caused by increased mem-brane trafficking of TRPM7 subunits or altered regulationrather than increased channel expression per se.
Potential MechanismsMio et al31 established the 3-dimensional structure of TRPC3channels with cryo-electron microscopy. TRPC3 channelshave both a pore-forming transmembrane domain and a largeintracellular domain, representing a “nested box” structure.The latter structure may act as a molecular anchor forsignaling complexes. TRPC3-mediated Ca2� influx and an
increase in local Ca2� concentration may trigger protein-protein interactions that activate downstream signaling path-ways that regulate fibroblast function. A recent study using Blymphocytes demonstrated that TRPC3 channels act as aplatform for protein kinase C and that the sustained scaffold-ing of protein kinase C at the plasma membrane is associatedwith activation of the ERK-1/2 signaling pathway.32
ERK-1/2 is a protein kinase and intracellular signalingmolecule involved in various biological functions, includingcell growth and survival.21,22 In the present study, selectiveinhibition of ERK-1/2 signaling attenuated rat fibroblastproliferation, which suggests that ERK-1/2 signaling contrib-utes to fibroblast function. Olson et al33 demonstrated thatangiotensin-induced increases in intracellular Ca2� and pro-tein kinase C activation in adult rat cardiac fibroblastssynergistically contribute to ERK-1/2 activation and fibro-blast proliferation. TRPC3 channels may be particularlyimportant in this situation, because angiotensin II increasescellular production of diacylglycerol, which activates TRPC3channels. The present data showed that TRPC3 blockade
Figure 7. A, Representative immunofluo-rescent images of freshly isolated leftatrial fibroblasts stained with NFATc3/NFATc4, vimentin (fibroblast marker),and TOPRO-3 (nuclear marker) antibod-ies in dogs with tachypacing-inducedatrial fibrillation (AF) and control dogs(CTL). B, Mean�SEM nuclear/cytoplas-mic (Nuc/Cyto) signal intensity ratio ofNFATc3 (top) and NFATc4 (bottom;n�5; *P�0.05 vs CTL). C, Top, Repre-sentative immunoblots of cytosolic andnuclear protein fractions of HSP70 (cyto-solic marker), lamin A/C (nuclearmarker), NFATc3, and NFATc4 in freshlyisolated left atrial fibroblasts in AF andCTL dogs. Bottom, Mean�SEM cyto-plasmic NFATc3/NFATc4 relative toHSP70 and nuclear NFATc3/NFATc4relative to lamin A/C (n�4/group;*P�0.05 vs CTL). D, Mean�SEM miR-26a and miR-26b expression in controldog left atrial fibroblasts treated withINCA6 (2.5 �mol/L) for 24 hours (n�5/group; *P�0.05 vs DMSO). E, Represen-tative immunoblots (top) andmean�SEM TRPC3/GAPDH (bottom) incontrol dog left atrial fibroblasts treatedwith INCA6 (2.5 �mol/L) for 24 hours(n�5; *P�0.05 vs DMSO).
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2061
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

suppresses Ca2� entry caused by angiotensin II, a well-known profibrotic agent, in rat cardiac fibroblasts.
Novelty and Potential SignificanceThis is the first study to show a role of TRPC3 channels incontrolling fibroblast function and the first to indicate that AFactivates fibroblasts via TRPC3-related mechanisms. Further-more, we were able to identify the mechanism of TRPC3upregulation in AF (reduced miR-26 negative control ofTRPC3 translation, caused by enhanced NFATc3 nucleartranslocation in AF fibroblasts, which caused enhanced in-hibitory NFATc3 regulation of miR-26) and found that aTRPC3 inhibitor suppresses AF-promoting remodeling. Anemerging body of evidence indicates that TRP channels act asimportant mediators for a wide variety of physiologicalfunctions and are a potential target for drug discovery. Thereis a need to develop novel approaches to AF treatment, andtherapies that prevent fibroblast activation are of potentiallygreat interest.6 The present findings point to TRPC3 as acandidate target for AF prevention.
Although it is well recognized that AF promotes atrialfibrosis, little information is available about how this
happens. In the present dog model, AF upregulated TRPC3and caused fibroblast activation that depended on TRPC3-related Ca2� entry, which activated ERK phosphorylation.Similar TRPC3 upregulation was observed in atrial sam-ples from AF patients, AF goats, and AF-prone CHF dogs.TRPC3 channels acted primarily in nondifferentiated fi-broblasts, enhancing their ability to proliferate and differ-entiate into myofibroblasts. Once myofibroblasts wereformed, TRPC3 channels became downregulated. This be-havior of TRPC3 channels has not been described previously:TRPC3 channels promote fibrosis by causing fibroblasts toproliferate and activate but are then downregulated in activatedmyofibroblasts to prevent a positive-feedback process. Theeffects of in vivo Pyr3 infusion to prevent enhanced proliferationand ECM expression of fibroblasts from AF dogs, along with theassociated suppression of AF promotion, are consistent with animportant role for TRPC3 in fibroblast-mediated AF-promotingremodeling.
Although NFAT nuclear translocation has been shown tooccur in AF cardiomyocytes and to be involved in cardio-myocyte remodeling,1,34 the present study constitutes the first
Figure 8. A, Mean�SEM duration ofinduced atrial fibrillation (AF) on day 7 inAF dogs treated throughout the AF pac-ing period with intravenous pyrazole-3(Pyr3; n�6, 0.1 mg � kg�1 � d�1) or vehi-cle (n�6, *P�0.05 vs vehicle). B,Mean�SEM atrial effective refractoryperiod (ERP; open circles indicate vehi-cle; filled circles, Pyr3; *P�0.05 vs vehi-cle). BCL indicates basic cycle length. C,Representative immunoblots (top) andmean�SEM vimentin band intensity nor-malized to GAPDH (bottom) in left atrialtissue samples from Pyr3-treated (n�5)or vehicle-treated AF dogs (n�5;*P�0.05 vs vehicle). D, Mean�SEM cellcount of left atrial fibroblasts after 2- and3-day culture in Pyr3-treated (n�6) orvehicle-treated AF dogs (n�6; *P�0.05vs vehicle). E, Mean�SEM percentage ofcells in G2/M phase (n�6/group;*P�0.05 vs vehicle). F, Mean�SEMextracellular matrix gene mRNA expres-sion in freshly isolated left atrial fibro-blasts from Pyr3-treated (n�5) orvehicle-treated AF dogs (n�5; *P�0.05vs vehicle). Col indicates collagen; FBN,fibrinogen; FN, fibronectin.
2062 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

demonstration of AF-associated NFAT changes in fibroblastsand their involvement in AF-related fibroblast remodeling.
Potential LimitationsWe cannot exclude the possibility that fibroblast propertieswere affected by cell isolation and culture. Considering thatfibroblasts lose their original properties after lengthy cultureintervals, we used cells in short-term primary culture (maxi-mum of 3 days after isolation) in most experiments. However,this approach cannot completely reproduce the complex invivo milieu. Any extrapolation of these results to humandisease should be made cautiously.
Our patch-clamp recording conditions differed from thoseused by Du et al30 in that they used pipette solutions that werevirtually Mg2�-free, whereas our pipettes contained Mg2� atconcentrations typically used for cardiac cell patch-clamprecording. TRPM7 currents are strongly suppressed by intra-cellular Mg2�, 35,36 which likely explains why most of thegadolinium-sensitive current we observed was also sensitive toPyr3. Under our conditions, INSC would not be expected tocontain substantial TRPM7 current and was strongly downregu-lated in myofibroblasts. Thus, TRPM7 channels play a muchlarger role than other TRP channels, including TRPC3, in theregulation of Ca2� influx in differentiated myofibroblasts.
The AF dogs in the present study showed increased atrialfibroblast density and signs of fibroblast activation, such asenhanced �-SMA expression (Figure 8C) and ECM geneupregulation (Figure 7E); however, we did not see increasedfibrous tissue content in AF dogs. We suspect that the lack offibrosis was related to the relatively short time (7 days) thatthe dogs were kept in AF, with longer intervals necessary forfibrosis development. When AF is maintained for longerperiods (�3 months), clear fibrosis develops, even whenexcessive ventricular rates and left ventricular dysfunctionare prevented.37 Fibroblast proliferation and differentiationcan promote AF by a range of mechanisms that do not requireenhanced fibrous tissue content, particularly fibroblast-car-diomyocyte electric interactions.3 The small atrial effectiverefractory period increases we noted in Pyr3-treated AF dogsmay reflect enhanced fibroblast effects on the electrophysi-ology of coupled cardiomyocytes.3 Alternatively, a direct rolein cardiomyocytes cannot be excluded and should be assessedin follow-up work. Our findings raise many interestingadditional questions that need to be answered but are outsidethe scope of the present study.
AcknowledgmentsThe authors thank Nathalie L’Heureux, Chantal St-Cyr, ClaudiaLiebetrau, and Louis-Robert Villeneuve for technical help andFrance Theriault for secretarial assistance.
Sources of FundingThis study was supported by the Canadian Institutes of HealthResearch (MOP 44365), Quebec Heart and Stroke Foundation, theMITACS Network, Japanese Heart Rhythm Society/Medtronic Fel-lowship, Japan Heart Foundation/Japanese Society of Electrocardi-ology Scholarship, German Center for Cardiovascular Research, andFondation Leducq (ENAFRA Network, 07/CVD/03).
DisclosuresDr Van Wagoner is the recipient of a research grant from GileadPharma to evaluate the impact of an A2b antagonist on AFinducibility/fibrosis in a canine model. Dr Nattel is listed as aninventor on a patent requested by his employer (TRPC3 channels inAF). The other authors report no conflicts.
References1. Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the
molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121:2955–2968.
2. Nattel S. From guidelines to bench: implications of unresolved clinicalissues in atrial fibrillation for basic investigations of atrial fibrillationmechanisms. Can J Cardiol. 2011;27:19–26.
3. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance inatrial fibrillation. J Am Coll Cardiol. 2008;51:802–809.
4. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissancecell. Circ Res. 2009;105:1164–1176.
5. Burstein B, Comotois P, Michael G, Nishida K, Villeneuve L, Yeh YH,Nattel S. Changes in connexin expression and the atrial fibrillation sub-strate in congestive heart failure. Circ Res. 2009;105:1213–1222.
6. Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblastelectrical function and therapeutic implications for atrial fibrillation.Cardiovasc Res. 2011;89:744–753.
7. Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito H.Transient receptor potential channels in cardiovascular function anddisease. Circ Res. 2006;99:119–131.
8. Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524.9. Shiroshita-Takeshita A, Brundel BJ, Burstein B, Leung TK, Mitamura H,
Ogawa S, Nattel S. Effects of simvastatin on the development of the atrialfibrillation substrate in dogs with congestive heart failure. CardiovascRes. 2007;74:75–84.
10. Blaauw Y, Gogelein H, Tieleman RG, van Hunnik A, Schotten U,Allessie MA. “Early” class III drugs for the treatment of atrial fibrillation:efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat.Circulation. 2004;110:1717–1724.
11. Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation byheart failure in dogs: atrial remodeling of a different sort. Circulation.1999;100:87–95.
12. Sakabe M, Shiroshita-Takeshita A, Maguy A, Dumesnil C, Nigam A,Leung TK, Nattel S. Omega-3 polyunsaturated fatty acids prevent atrialfibrillation associated with heart failure but not atrial tachycardiaremodeling. Circulation. 2007;116:2101–2109.
13. Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, DemolombeS. Regional and tissue specific transcript signatures of ion channel genes inthe non-diseased human heart. J Physiol. 2007;582:675–693.
14. Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F,Speleman F, Vandesompele J. A novel and universal method formicroRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64.
15. Rose RA, Hatano N, Ohya S, Imaizumi Y, Giles WR. C-type natriureticpeptide activates a non-selective cation current in acutely isolated ratcardiac fibroblasts via natriuretic peptide C receptor-mediated signalling.J Physiol. 2007;580:255–274.
16. Burstein B, Libby E, Calderone A, Nattel S. Differential behaviors ofatrial versus ventricular fibroblasts: a potential role for platelet-derivedgrowth factor in atrial-ventricular remodeling differences. Circulation.2008;117:1630–1641.
17. Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B,Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol1,4,5-triphosphate signaling in restricted spaces of myoendothelial proj-ections. Proc Natl Acad Sci U S A. 2008;105:9627–9632.
18. Xiao J, Lin H, Luo X, Luo X, Wang Z. miR-605 joins p53 network toform a p53:miR-605:Mdm2 positive feedback loop in response to stress.EMBO J. 2011;30:524–532.
19. Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y,Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, OjidaA, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y,Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3channels underlies biological activities of a pyrazole compound. ProcNatl Acad Sci U S A. 2009;106:5400–5405.
20. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T,Schultz G. Direct activation of human TRPC6 and TRPC3 channels bydiacylglycerol. Nature. 1999;397:259–263.
Harada et al TRPC3 Channels and Cardiac Fibroblast Function 2063
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

21. Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S,Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapkare required for fibroblast proliferation. Proc Natl Acad Sci U S A. 1993;90:8319–8323.
22. Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A syntheticinhibitor of the mitogen-activated protein kinase cascade. Proc Natl AcadSci U S A. 1995;92:7686–7689.
23. Dietrich A, Mederos y Schnitzler M, Kalwa H, Storch U, Gudermann T.Functional characterization and physiological relevance of theTRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs ArchPharmacol. 2005;371:257–265.
24. Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granuleneuron survival. Nat Neurosci. 2007;10:559–567.
25. Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3mediates pyrimidine receptor-induced depolarization of cerebral arteries.Am J Physiol Heart Circ Physiol. 2005;288:H2055–H2061.
26. Philipp S, Strauss B, Hirnet D, Wissenbach U, Mery L, Flockerzi V, HothM. TRPC3 mediates T-cell receptor-dependent calcium entry in humanT-lymphocytes. J Biol Chem. 2003;278:26629–26638.
27. Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, MoriY, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensinII-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316.
28. Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessarymediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A.2010;107:7000–7005.
29. Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, Inoue R,Mori Y, Kurose H. G�12/13-mediated up-regulation of TRPC6 nega-tively regulates endothelin-1-induced cardiac myofibroblast formation
and collagen synthesis through nuclear factor of activated T cells acti-vation. J Biol Chem. 2007;282:23117–23128.
30. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, YueL. TRPM7-mediated Ca2� signals confer fibrogenesis in human atrialfibrillation. Circ Res. 2010;106:992–1003.
31. Mio K, Ogura T, Kiyonaka S, Hiroaki Y, Tanimura Y, Fujiyoshi Y, MoriY, Sato C. The TRPC3 channel has a large internal chamber surroundedby signal sensing antennas. J Mol Biol. 2007;367:373–383.
32. Numaga T, Nishida M, Kiyonaka S, Kato K, Katano M, Mori E, KurosakiT, Inoue R, Hikida M, Putney JW Jr, Mori Y. Ca2� influx and proteinscaffolding via TRPC3 sustain PKC� and ERK activation in B cells.J Cell Sci. 2010;123:927–938.
33. Olson ER, Shamhart PE, Naugle JE, Meszaros JG. AngiotensinII-induced extracellular signal-regulated kinase 1/2 activation is mediatedby protein kinase C� and intracellular calcium in adult rat cardiac fibro-blasts. Hypertension. 2008;51:704–711.
34. Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, VilleneuveLR, Brundel BJJM, Dobrev D, Nattel S. Cellular signalling underlyingatrial tachycardia remodeling of L-type calcium-current. Circ Res. 2008;103:845–854.
35. Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalentcations but not ATP. Biophys J. 2003;84:922–927.
36. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ,Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 isa Mg.ATP-regulated divalent cation channel required for cell viability.Nature. 2001;411:590–595.
37. Avitall B, Bi J, Mykytsey A, Chicos A. Atrial and ventricular fibrosisinduced by atrial fibrillation: evidence to support early rhythm control.Heart Rhythm. 2008;5:839–845.
CLINICAL PERSPECTIVEAtrial fibrillation (AF) is the most common clinical arrhythmia, and its therapy is problematic. There is a need for noveltreatment interventions, and mechanistic insights may be helpful in developing such new therapies. Here, we examined therole of a new type of ion channel, the nonselective cation channel TRPC3 (transient receptor potential canonical-3), in AF.TRPC3 can carry a number of ions but may be particularly important as an entry pathway for calcium into nonexcitablecells such as fibroblasts. Tissue fibrosis is produced by an overproduction of extracellular matrix proteins by fibroblastsand is believed to be important in AF. In addition, fibroblasts may contribute to AF via electric interactions with excitableheart cells (cardiomyocytes). We first found that cardiac fibroblasts expressed functional TRPC3 channels, andTRPC3-mediated calcium entry into fibroblasts activated a signaling molecule (extracellular signal-regulated kinase) thatinduced fibroblast proliferation and differentiation into activated myofibroblasts. We also noted that TRPC3 expressionwas upregulated in AF patients and 2 experimental AF models. We then studied the role of TRPC3 in atrial remodelingof dogs kept in AF for 7 days by atrial tachypacing. We found that AF increased TRPC3 protein expression and current,although it activated fibroblasts, and that blocking TRPC3 suppressed fibroblast activation and the AF-promotingremodeling caused by AF. We also identified the molecular pathway that leads to TRPC3 upregulation in AF:Downregulation of a TRPC3-suppressing microRNA, miR-26, caused by AF-induced nuclear translocation/regulation bynuclear factor of activated T-lymphocytes (NFAT) in fibroblasts. This work provides novel insights into molecularmechanisms underlying AF, as well as potential new anti-AF targets.
2064 Circulation October 23, 2012
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

Wagoner, Dobromir Dobrev and Stanley NattelKamiya, Toyoaki Murohara, Itsuo Kodama, Jean-Claude Tardif, Ulrich Schotten, David R. Van
Ordog, Jonathan Ledoux, Takeshi Kato, Patrice Naud, Niels Voigt, Yanfen Shi, Kaichiro Masahide Harada, Xiaobin Luo, Xiao Yan Qi, Artavazd Tadevosyan, Ange Maguy, Balazs
Atrial FibrillationDependent Fibroblast Regulation in−Transient Receptor Potential Canonical-3 Channel
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2012 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.112.121830
2012;126:2051-2064; originally published online September 19, 2012;Circulation.
http://circ.ahajournals.org/content/126/17/2051World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2012/09/19/CIRCULATIONAHA.112.121830.DC1Data Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 1, 2018http://circ.ahajournals.org/
Dow
nloaded from

1
SUPPLEMENT MATERIAL
TRPC3-dependent Fibroblast Regulation in Atrial Fibrillation
Supplemental Methods
Rat fibroblast isolation and culture
Adult male Sprague-Dawley rats weighing 200-250 g were anesthetized with ketamine (50 mg/kg, i.p.)
and xylazine (10 mg/kg, i.p.). Hearts were quickly excised via thoracotomy into ice-cold Tyrode
solution containing (mol/L) 140 NaCl, 5.4 KCL, 2.0 CaCl2, 1.0 MgCl2, 10 HEPES, and 5.5 glucose (pH
7.35 with NaOH) and Langendorff-perfused at 37°C with 1) Tyrode solution for 5 min; 2) Ca2+-free
Tyrode solution for 5 min; 3) Ca2+-free Tyrode solution containing 0.04 mg/ml collagenase-II for
30 min. They were then removed, minced, and homogenized in Ca2+-free Tyrode solution. Isolated
cells were centrifuged (550 rpm, 3 min), separating cardiomyocytes (primarily in the pellets) from
fibroblasts (in the supernatant). Cardiomyocytes were further removed by passing the supernatant
through a 20-µm nylon filter; fibroblasts were then further concentrated via centrifugation (2000 rpm,
10 min). Pellets were resuspended and washed twice in M199-medium supplemented with 10% fetal
bovine serum for primary culture.
Atrial tissue-samples from humans, goats, and dogs
Human right-atrial appendage biopsies were obtained from patients in sinus rhythm and with chronic AF
during coronary artery bypass graft surgery. The study was approved by the ethical review committee of
Dresden University of Technology. All subjects gave informed consent. AF was induced in
chronically-instrumented goats using repetitive burst-pacing for 10 days. Congestive heart failure

2
(CHF) dogs with AF substrates were created by rapid ventricular pacing (240-bpm, 2 weeks). Normal
goats and dogs were used as controls. Right-atrial tissue samples were collected and fast-frozen in
liquid-N2.
AF-Dogs
A total of 48 mongrel dogs (20-36 kg) were divided into control and atrial-tachypacing groups. Animal-
care procedures followed National Institutes of Health guidelines (NIH Publication No. 85-23, revised
1996) and were approved by the Animal Research Ethics Committee of the Montreal Heart Institute.
Dogs were anesthetized with ketamine (5.3 mg/kg i.v.), diazepam (0.25 mg/kg, i.v.), and isoflurane
(1.5%), intubated, and ventilated. A unipolar pacing lead was inserted into the right-atrial appendage
under fluoroscopic guidance. The lead was connected to a pacemaker (Star Medical, Tokyo, Japan)
implanted in the neck. Two bipolar electrodes were inserted into the right-ventricular apex and
right-atrial appendage for internal electrocardiogram (ECG) recording. Atrioventricular-block and
ventricular pacing as employed in many studies of atrial-tachycardia remodeling1 was not performed, to
more closely mimic spontaneous clinical AF-episodes. The atrial pacemaker was programmed to
stimulate the right-atria at 600-bpm for 1 week, with fibrillatory atrial activity maintained during pacing
as assessed by daily ECG and intracardiac recordings. Echocardiography was performed on Day 0
(before atrial pacing, baseline) and Day 7 to assess changes in LA dimension, LA systolic function and
LV diastolic volume.
For in vivo treatment of the AF dogs, an Alzet osmotic pump (model 2ML1) was implanted
subcutaneously in the back and a selective TRPC3 blocker, pyrazole3 (0.1 mg/kg/day, dissolved in
DMSO and polyethylene glycol), or vehicle (DMSO and polyethylene glycol) was continuously
administrated for the entire period of atrial tachypacing (Supplemental Figure 3).

3
Dog atrial fibroblast isolation and culture
After the open chest study, the left-atrial tissues were immersed in oxygen-saturated, Ca2+-containing
Tyrode solution at room temperature. The left circumflex coronary artery was cannulated and perfused
with Ca2+-containing Tyrode-solution (37°C, 10 min), then perfused with Ca2+-free Tyrode-solution
(15 min), followed by 50-minute perfusion with Tyrode-solution containing collagenase (Worthington,
type II) and 1% bovine serum albumin (Sigma). Digested left-atrial tissue was then removed, minced,
and homogenized in M199 media. Isolated cells were centrifuged (800 rpm, 3 min) to separate
cardiomyocytes (primarily in the pellets) from fibroblasts (in supernatant). Cardiomyocytes were further
removed by filtration of the supernatant through a 20-µm nylon filter; fibroblasts were then further
concentrated via centrifugation (2000 rpm, 10 min). Pellets were resuspended and washed twice in
M199-medium supplemented with 10% fetal bovine serum for primary culture.
Terminal Open-chest Electrophysiological Study
Dogs were anesthetized with morphine (2 mg/kg, s.c.) and α-chloralose (120 mg/kg, i.v., followed by
29.25 mg/kg/h), and mechanically ventilated. Body temperature was maintained at 37°C. A median
sternotomy was performed, and bipolar electrodes were hooked into the RA appendages for recording
and stimulation. Right-atrial effective refractory period (ERP) was measured at basic cycle lengths of
150, 200, 250, 300, and 360 ms with 10 basic (S1) stimuli, followed by an S2 with 5-ms decrements (all
pulses twice-threshold, 2-ms). AF (irregular atrial rhythm > 400 bpm) was induced with 1-10 s atrial
burst-pacing (10-20 Hz, 4threshold, 2-ms pulses). Mean AF-duration was determined in each dog
based on 10 AF-inductions for AF<5 min and 5 inductions for 5-30 min AF. AF>30 min was
considered sustained: cardioversion was not performed, and electrophysiological assessment was
terminated. Haemodynamic data were obtained with fluid-filled catheters and transducers.

4
qPCR
Total RNA and microRNA were extracted with TRIzol (Invitrogen) and mirVana miRNA Extraction Kit
(Ambion) from freshly isolated fibroblasts, respectively. Real-time RT-PCR was performed with
6-carboxy-fluorescein (FAM)-labeled fluorogenic Taq-Man primers and probes (Applied Biosystem).
Fluorescence-signals were detected in duplicate, normalized to β2-microglobulin RNA for total RNA
and to U6 snRNA for microRNA, and quantified with MxPro qPCR-software (Stratagene).
TaqMan low-density arrays
Total RNA was extracted using TRIzol. RNA-integrity was assessed via Agilent Bioanalyzer. (RIN>7.5
required). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied
Biosystems) with random hexamer primers. TaqMan low-density arrays (TLDA, Applied Biosystems)
were used in two-step RT-PCR as previously reported.2 Real-time RT-PCR was performed on the
7900HT Fast Real-Time PCR System. Data were collected with SDS2.3 software and grouped with RQ
Manager software. TRP-subunit genes were investigated using inventoried Taqman assays.
Ct-methodology was applied to determine mRNA expression-levels. The mean expression of the genes
with a Ct value below 30 (Ct<30) was selected as the reference.3
Cell-Proliferation and Cell-Cycle Analysis
Isolated cardiac fibroblasts were counted with hematocytometer and placed into T25 culture-flasks
(2.0×105 cells/flask, 25 cm2 growth-area) for each treatment group. Treatment was performed on Day 2
or Day 3 unless otherwise specified. After 1-hour and 24-hour incubation, cultured fibroblasts were
totally harvested from each flask following trypsinization, then washed in ice-cold PBS and fixed in

5
1.0 ml of ice-cold 75% ethanol. Samples were stored at -20°C until analysis. Once pelleted, fibroblasts
were re-suspended and incubated (4°C, 30 minutes) in 250 µl of staining solution containing propridium
iodide (PI, Sigma) with RNAase and then 750 µl of PBS was added; the final volume in each sample
was 1000 µl. In each preparation, the numbers of cells and PI fluorescence were measured with a
FACScan (constant flow-rate, 60 µl/min, 5-min acquisition time, BD Bioscience) at 617-nm emission-
wavelength to create a DNA content-frequency histogram. Samples were gated on fibroblast population
using forward scatter vs. side scatter plot (cell size vs. granularity) and doublet discrimination gating.
The percentage of cells in each phase of the cell cycle, G0/G1, S and G2/M phases, was analyzed using
Flowjo software Dean-Jett-Fox model that fits G1 and G2 with Gaussian curves automatically (Tree Star
Inc.).4
In order to confirm the accuracy of cell-counting with flowcytometry, 3-(4,5-dimethylthiazol-2-
yl)-2, 5-diphenyltetrazolium bromide (MTT) cell proliferation assay was also performed with using Cell
Proliferation Kit I (Roche). Two-day cultured fibroblasts were resuspended and transferred into a
96-well plate (5.0×103 cells/well, 0.32 cm2 growth-area). The cells were incubated with the same
treatment protocol for the flowcytometry experiment. The assay was triplicated in each treatment group.
The absorbance (550 nm-690 nm) was quantified using a scanning multiwell spectrophotometer
(Synergy2, BioTek). The cell-counting with flowcytometry described above was also performed in
parallel with the same passage fibroblast samples. Both proliferation assays showed similar results
(Figure A and B below).

6
A, Cultured rat fibroblast proliferation estimated by MTT assay after 1-hour and 24-hour treatment with
vehicle-control, or 0.3 or 3-μmol/L Pyr3. Mean±SEM spectrophotometrical absorbance (550 nm-690
nm) (n=6, P<0.05 vs. CTL). B. Mean±SEM cell-count of rat fibroblast with flowcytometry after 1-hour
and 24-hour treatment with vehicle-control, or 0.3 or 3-μmol/L Pyr3. (n=6, P<0.05 vs. CTL).
Western Blots
Total protein was extracted from freshly-isolated/cultured fibroblasts or atrial tissues, quantified, and
processed as previously described. Cytoplasmic and nuclear protein fractions were extracted from fresh
fibroblasts with ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem, Darmstadt, Germany).
Protein-samples (20 µg/lane for total protein and 30 µg/lane for fractionated protein) were separated by
8% SDS-PAGE electrophoresis and then transferred to polyvinylidene-difluoride membranes.
Membranes were blocked and incubated with mouse anti-α smooth-muscle actin (αSMA, 1/2000,
Sigma), mouse anti-phospho p44/42-MAP-kinase (1/2000, Cell signaling), mouse anti-vimentin (1/1000,
Santa Cruz), rabbit anti-TRPC3 (1/1000, Alomone), rabbit anti-TRPC1 (1/200, Alomone), mouse anti-
TRPM7 (1/1000, Neuromab), mouse anti-NFATc3 (1/1000, Santa Cruz), rabbit anti-NFATc4 (1/1000,

7
Santa Cruz), mouse anti-HSP70 (1/1000, Stressgen), mouse anti-Lamin A/C (1/1000, Abcam),
anti-GAPDH (1/10000, Fitzgerald), and rabbit anti-calsequestrin (1/2500, Dianova). Corresponding
secondary antibodies conjugated to horseradish-peroxidase were used for detection. Staining was
detected using chemiluminescence and quantified by video densitometry. All expression data are
provided relative to GAPDH or calsequestrin staining for the same samples on the same gels.
Confocal Imaging
Cultured/freshly-isolated fibroblasts or left atrial tissue cryosections were fixed with
2%-paraformaldehyde, washed with PBS, and incubated with mouse anti-alpha SMA (1/400, Sigma),
goat anti-vimentin (1/200, Santa Cruz), mouse anti-NFATc3 (1/200, Santa Cruz), rabbit anti-NFATc4
(1/200, Santa Cruz), followed by donkey anti-mouse IgG-Alexa Fluor 555 (1/600, Invitrogen), donkey
anti-rabbit IgG-Alexa Fluor 488 (1/600, Invitrogen), donkey anti-goat IgG-Alexa Fluor 488 (1/600,
Invitrogen), and TOPRO-3 iodide (1/500, Invitrogen). Apoptosis was assessed using TUNEL-assay
with ApopTag plus fluorescein in situ apoptosis detection kits (Millipore). Fluorescent images were
obtained via Zeiss LSM-710 or OLYMPUS Fluoview FV1000 inverted confocal microscope.
Fluorescent Ca2+-Imaging
Primary cultured fibroblasts (1-day culture as described above) were loaded with Fluo-4 (10 µmol/L) in
phenol-free M199-medium in the presence of pluronic acid (2.5 µgml) for 50 min at 37C.
Ca2+-imaging was recorded with an Andor Revolution confocal system and a Xion camera (Andor
Technologies) mounted on an upright Nikon FN-1 microscope with a 60 water-immersion objective
(1.0 NA). Fluo-4 was excited with a 488-nm solid-state laser; emitted fluorescence was collected at
495 nm. Images (512512 pixels) were acquired at 15 framessec for 5 min using iQ software (Andor

8
Technologies). Ca2+-associated fluorescence was analyzed with custom-designed software (A. Bonev,
University of Vermont). Regions of interest (ROIs) were determined by cell-outlines. FO was
determined by averaging fluorescence of ROIs from 10 consecutive baseline images.5 At the end of
experiments, ATP (100-µmol/L) was applied to verify cell-viability. One-day cultured fibroblasts were
incubated in 2-mmol/L Ca2+-containing Tyrode solution followed by Ca2+-free solution to induce
Ca2+-store depletion and store-dependent Ca2+-entry. Under OAG (25-µmol/L)- or angiotensin-II
(100-nmol/L)-stimulation, cell-Ca2+ was recorded with confocal microscopy 5 minutes before and after
re-administration of 2-mmol/L Ca2+ into the extracellular solution.
TRPC3-knockdown
Plasmid constructs
The TRPC3-specific shRNAmir over-expressing pGIPZ-based lentivirus vector plasmid was obtained
from Open Biosystems (Oligo ID: V2LMM_11490).
The scrambled shRNAmir over-expressing plasmid was generated as follows. The empty pGIPZ
lentivirus vector plasmid carrying the mRNA-context sequence but no shRNA was obtained from Open
Biosystems. The EcoRI site of pGIPZ located at position 5394 was removed by partial EcoRI digestion
and Klenow fill-in, resulting in pGIPZΔEcoRI. This modification allowed the direct cloning of the
scrambled shRNAmir construct in pGIPZ between XhoI, 2654 and EcoRI, 2678 sites. In the design of
the scrambled shRNAmir and during the rest of the cloning procedure, we followed the methods by
Paddison et al.6 Briefly, a 97bp long synthetic oligonucleotide
( 5`TGCTGTTGACAGTGAGCGCCGATATCAGCAGATAATGAAATAGTGAAGCCACAGATGTA
TTTCATTATCTGCTGATATCGTTGCCTACTGCCTCGGA 3`, passanger strand, loop, guide strand)
was PCR amplified by 5`CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG sense and

9
5` CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA antisense primers, carrying XhoI and
EcoRI restriction sites, respectively. PCR product was cloned in pGIPZΔEcoRI at XhoI, EcoRI sites.
Sequence identity of the resulting clone was verified by sequencing.
The psPAX2 and pMD2.G lentivirus packaging plasmids were obtained from Didier Trono`s
laboratory (http://tronolab.epfl.ch/, Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland)
through Addgene (Addgene plasmid 12260 and 12259).
All plasmids were amplified in E. coli DH5α and purified by Nucleobond anion exchange
columns (Macherey-Nagel) following the manufacturer’s instructions.
Lentivirus production
The Hek293T/17 cell line we used for producing lentivirus production was obtained from ATCC
(Manassas, VA, USA) and were grown in DMEM (Invitrogen) supplemented with 10% FCS (Gibco).
Lentiviruses were produced by following the protocols available from Didier Trono`s laboratory
(http://tronolab.epfl.ch) with minor modifications. A subconfluent monolayer of Hek293T/17 cells
(ATCC) kept in DMEM (Invitrogen) supplemented with 10% FBS (Gibco) were transfected with a
mixture of plasmids containing one of the vector plasmids and the psPAX2 and pMD2.G packaging
plasmids in 2:2:1 weight ratio by the calcium-phosphate precipitation method. Eight hours after
transfection, the culture medium was replaced by fresh culture medium. The supernatant containing the
lentivector particles was harvested two times 32 and 56 hours post-transfection. The harvested medium
was clarified from cell debris by low-speed centrifugation and by filtration through a 0.45 μm pore size
syringe-attached filter. Virus particles were concentrated by ultracentrifugation at 47000 g (RCF
average) for 2 hours in a swinging bucket rotor, re-dissolved in sterile PBS containing 1% BSA and
stored at -80°C in aliquots. Virus preparations were titrated on Hek293T/17 cells.

10
miR-26a-Overexpression/Knockdown
For the miR-26a overexpression, the sense (5` UUCAAGUAAUCCAGGAUAGGCU 3`) and antisense
(5` CCUAUUCUUGGUUACUUGCACG 3`) oligoribonucleotides were synthesized by Invitrogen. The
both strands were annealed by mixing the same volume of each oligoribonucleotides dissolved in
annealing buffer (IDT, Coralville, IA) at a concentration of 200 µmol/L and then incubated at 95°C for
10 min, 70°C for 10 min and then 50°C for 10 min. A scrambled RNA was used as negative control
(sense: 5` UCAUAAAGCUGAUAACCUCUAGAU 3`, antisense: 5` CUAGAGGUUAU
CAGCUUUAUGAAU 3`).
For the miR-26a knockdown, the antisense anti-miRNA oligonucleotides (AMO26a: 5`
AGCCTATCCTGGATTACTTGAA 3`) was synthesized by Exiqon (Exiqon, Woburn, MA). Five
oligonucleotides on 5`-end of the antisense molecules and four oligonucleotides on 3`-end were locked
by a methylene bridge connecting the 2’-O atom and 4’ -C atom (Locked Nucleic Acid, LNA). A
scrambled oligonucleotides with the same methylene bridge were used as negative control
(5` ACTCAGAAGGACAAGTAGAGTCT 3`).
Dog left-atrial fibroblasts in primary culture were transfected with miR-26a (final concentration
of 100 nmol/L) and/or AMO26a (final concentration of 10 nmol/L), and negative control miRNAs or
AMOs with lipofectamine 2000 (Invitrogen).7 After 48-hour transfection, cells were collected for total
RNA/protein purification or proliferation analysis with a FACScan. The efficacy of miR-26a
overexpression/knockdown by the miR-26a probes was confirmed by qPCR before experiments
(Supplemental Figure 12C).

11
Dual Luciferase Reporter Assay
A fragment (5` TGACTATAGCACAAATGTGGGCAATAATATTTCTAAGTATAAAATACTT
GAAATGGTGTAAATTTTTAGTATTAACTACCTTTATCATGTGAATCTTTAA 3` miR-26a target
site) containing miR-26a target gene of TRPC3 was synthesized by Invitrogen and was used as a
template for PCR amplification with 5` GACTAGTTGACTATAGCA
CAAATGT 3` and 5` CCCAAGCTTTTAAAGATTCACATGATA 3`as forward and reverse primers,
respectively. The PCR product was ligated into HindIII and SpeI sites (3` UTR region of luciferase
gene) in the pMIR-REPORT luciferase miRNA expression reporter vector (CMV promoter-driven
Firefly luciferase expression vector, Applied Biosystems) and was subcloned.
For luciferase assay, HEK293 cells were simultaneously transfected with 50 ng of
pMIR-REPORT, 0.5 ng pRL-TK (TK promoter-driven Renilla luciferase expression vector, Promega,
Madison, WI) and miR-26a (final concentration of 10 nmol/L) and/or AMO26a (final concentration of
3 nmol/L) with lipofectamine 2000 (Invitrogen). After 48-hour transfection, luciferase activities were
measured with a dual luciferase reporter assay kit (Promega) on a luminometer (Lumat LB9507,
Berthold, Bad Wildbad, Germany).7 The pMIR-REPORT firefly luciferase signals were normalized to
the pRL-TK renilla luciferase signals as an internal control.
Statistical Analyses
Data are presented as mean±SEM. Two-way ANOVA with multiple-group comparisons (Bonferroni-
corrected t-test) was data with two or more main-effect factors like flow-cytometry with varying culture-
times (treatment and time as main-effect factors) and ERP (basic cycle length and dog-group main
factors). One-way ANOVA was applied for single main-effect factor experiments like Western blots,
qPCR, and luciferase assay. Repeated-measure analyses were used when the same set of

12
subjects/materials were exposed to multiple interventions. Students’ t-tests were used for comparisons
involving only two groups. For multiple comparisons with Bonferroni correction, adjusted P-values
were calculated as multiplying original P values for each pairwise comparison by the number of
comparisons (N) performed; values shown are adjusted values (N×P). Two-tailed P<0.05 was
considered to be statistically significant. All analyses were performed with SPSS 16.0.
Specific Analyses for Individual Figures and Tables:
Two-way ANOVA with multiple comparison (Bonferroni-adjusted t-tests) was used for data with two or
more factors as main effects and repeated measures (Figures 1D, 1E and 1F, Figures 2B-2E, Figure 3B,
Figure 4B, Figure 5A, 5B, 5D, 5E, Figure 8B, 8D and 8E, Supplemental Figure 4B, Supplemental
Figure 5C and 5D, Supplemental Figure 7B and 7C, Supplemental Fig 8C and 8D).
One-way ANOVA was applied for data with a single main-effects factor as obtained in studies of
heart rate and echocardiographic data over time, with one main factor and 3 levels (groups) or more
(Supplemental Fig 11A, 11C-11E). Repeated-measure analyses were used when the same set of
subjects/materials were exposed to multiple interventions (Figure 2A, Figure 6B-6G, Supplemental
Figure 12C).
Paired t-tests were used for Figures 1H, Figure 5F, Figures 7D and 7E, and Supplemental Figures
6B, 7D, 8A and 8B.
Non-paired t-tests were used for Figures 3C, 3E-G, Figures 4C-E, Figure 5C, 5G, Figure 7B, 7C,
Figures 8A, 8C, and 8F, and Figures 9A-I as well as Supplemental Table 1.

13
REFERENCES
1. Sakabe M, Shiroshita-Takeshita A, Maguy A, Dumesnil C, Nigam A, Leung TK, Nattel S.
Omega-3 polyunsaturated fatty acids prevent atrial fibrillation associated with heart failure but not
atrial tachycardia remodeling. Circulation. 2007;116:2101-2109.
2. Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and
tissue specific transcript signatures of ion channel genes in the non-diseased human heart.
J Physiol. 2007;582:675-693.
3. Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J.
A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol.
2009;10:R64.
4. Fox A. A model for the computer analysis of synchronous DNA distributions obtained by flow
cytometry. Cytometry. 1980;1:80-81.
5. Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI,
Nelson MT. Functional architecture of inositol 1,4,5-triphosphate signaling in restricted spaces of
myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627-9632.
6. Paddison PJ, Cleary M, Silva JM, Chang K, Sheth N, Sachidanandam R, Hannon GJ. Cloning of
Short hairpin RNAs for gene knockdown in mammalian cells. Nature methods. 2004;1:163-167.
7. Xiao J, Lin H, Luo X, Luo X, Wang Z. miR-605 joins p53 network to form a p53:
miR-605:Mdm2 positive feedback loop in response to stress. EMBO J. 2011;30:524-532.

14
Supplemental Table 1. Hemodynamic Data
Control
(n=11)
AF
(n=12)
Body weight, kg 26.8±0.3 28.2±0.2
Systolic BP, mmHg 127±2 120±2
Diastolic BP, mmHg 73±2 76±2
RAP, mmHg 1.3±0.1 2.5±0.1*
LAP, mmHg 2.6±0.1 6.5±0.3*
RVP, mmHg 22±0.4 23±0.5
RVEDP ,mmHg 1.5±0.1 4.2±0.3*
LVP, mmHg 116±1.4 101±1.9
LVEDP, mmHg 3.9±0.2 8.0±0.5*
*P<0.05 vs control.
BP indicates blood pressure; RAP and LAP, RA and LA mean pressure; RVP and LVP, RV and LV mean
pressure; RVEDP and LVEDP, RV and LV end diastolic pressure.

15
Supplemental Figure Legends
Supplemental Figure 1. A, mRNA expression of TRP channels measured by Taqman Low-density
array. B, mRNA expression of TRPC3 channel on enlarged scale.
Supplemental Figure 2. A, Representative immunofluorescent images of freshly-isolated rat cardiac
fibroblasts. Red indicates α-smooth muscle actin (αSMA); green, vimentin; blue, TOPRO-3
(nuclear staining). B, Representative immunofluorescent images of passage-cultured
myofibroblasts. C, Representative INSC-recordings with or without gadolinium (Gd3+, 100-µmol/L)
in freshly-isolated rat cardiac fibroblasts. Voltage-clamp protocol is shown in the inset. D and E,
Representative Gd3+- and Pyr3-sensitive INSC in passage-cultured myofibroblasts. F, Mean±SEM
Gd3+- and Pyr3-sensitive INSC density (n=10 cells in Gd3+ and 8 cells in Pyr3).
Supplemental Figure 3. Protocol for in vivo TRPC3-blocker (pyrazole-3) treatment study in AF dogs.
Supplemental Figure 4. A, Recordings of OAG-induced intracellular Ca2+-response in presence or
absence of Pyr3. B, Mean±SEM OAG-induced Ca2+ fluorescence (F/F0), normalized to baseline
intensity under 0-mmol/L and 2-mmol/L Ca2+ (n=14 cells in each condition, *P<0.05 vs. 0-mmol/L
Ca2+, #P<0.05 vs. 2-mmol/L Ca2+ in OAG-only).
Supplemental Figure 5. A and B, Representative DNA-content histograms and Dean-Jett-Fox model
fitting of rat cultured fibroblasts with or without 24-hour treatment of Gd3+ (100-µmol/L) or Pyr3
(3-µmol/L). C, Mean±SEM cell-count of rat fibroblasts after 1-hour and 24-hour culture with
vehicle-control, or 10 or 100-μmol/L Gd3+ (n=8/group, *P<0.05 vs. CTL, #P<0.05 vs. 1-h
treatment). D, Mean±SEM percentage of cells in G2/M-phase after Gd3+ treatment (n=8/group,
*P<0.05 vs. CTL, #P<0.05 vs. 1-h treatment).

16
Supplemental Figure 6. A, Representative images of TUNEL staining in cardiac rat fibroblasts with or
without Pyr3 treatment (3-µmol/L). Red indicates TOPRO-3 (nuclear staining); yellow,
TUNEL-positive cells. B, Mean±SEM percentage of TUNEL-positive cells. (n=5/group, *P<0.05
vs. CTL).
Supplemental Figure 7. A, Representative immunofluorescent images of rat cardiac fibroblasts
cultured with or without Pyr3 (3-µmol/L). Red indicates α-smooth muscle actin (αSMA); blue,
TOPRO-3 (nuclear staining). B, Mean±SEM cell-count of culture-passaged fibroblasts after
1-hour and 24-hour treatment with 3-µmol/L Pyr3 or vehicle DMSO (n=6/group). C, Mean±SEM
percentage of culture-passaged fibroblast cells in G2/M-phase after Pyr3 treatment or vehicle
DMSO (n=6/group). D, Representative immunoblots (left) and mean±SEM αSMA-expression in
culture-passaged fibroblasts (right, normalized to GAPDH) (n=6).
Supplemental Figure 8. A, Mean±SEM TRPC3 and TRPC6 mRNA-expression in scrambled
(Scr)-shRNA- and TRPC3-shRNA-infected dog atrial fibroblasts (n=6/group, *P<0.05 vs.
scrambled shRNA). B, Top: Immunoblots for TRPC3 subunits and GAPDH. Bottom: Mean±SEM
TRPC3/GAPDH protein-expression (n=6, *P<0.05 vs. scramble shRNA). C, Mean±SEM
fold-change over time in the number of fibroblasts, following infection with shRNA or scrambled-
probe virus (n=6/group, *P<0.05 vs. scrambled shRNA). D, Mean±SEM percentage of virus-
infected fibroblast cells in G2/M-phase (n=6/group, *P<0.05 vs. scrambled-shRNA).
Supplemental Figure 9. A, through C, Representative immunoblots (top) and mean±SEM (bottom)
TRPC3 subunit protein-expression (normalized to calsequestrin, CSQ) in right-atrial samples from
patients with AF (n=8) and in sinus rhythm (n=10), AF model goats (n=8) and normal goats (n=5)
and congestive heart failure dogs with AF substrates (n=8) and normal dogs (n=8) (*P<0.05 vs.
control). D through F, TRPC1 subunit expression. G through I, TRPM7 subunit expression.

17
Supplemental Figure 10. A-E, Representative surface ECGs (left) and intracardiac electrograms (right)
in an AF dog on Day 0 through Day 7.
Supplemental Figure 11. A, Mean±SEM heart rate over time during atrial tachypacing, ATP (n=7 CTL
and 12 AF, *P<0.05 vs. CTL). B, Representative apical 4 chamber views of echocardiography on
Day 0 (baseline, before pacing, left) and Day 7 (right) in an AF dog. C, Mean±SEM atrial diastolic
area (n=10 CTL and 11 AF, P<0.05 vs. CTL). D, atrial fractional area change. E, ventricular
diastolic volume.
Supplemental Figure 12. A, miR-26 and its complementary sequence on TRPC3 3’-UTR region.
Sequences highlighted in yellow represent the seed region of miR-26a. B, Antisense anti-miR-26a
oligonucleotide (AMO26a) sequence and its complementarity to miR-26. Sequences highlighted
in yellow represent the seed region of miR-26. Bold characters in the AMO26a sequence indicate
locked nucleic acids (LNAs). C, Results of miR-26a overexpression/knockdown by miR-26a
duplex/AMO26a in dog left-atrial fibroblasts. Lipo; lipofectamine transfection without the
miR-26a probes. (n=4, P<0.05 vs. Lipo). D, Multiple putative NFAT binding motifs in the
promoter regions of the host gene for miR-26a and miR-26b in human and canine genomes.
Ctdsp: Carboxy-Terminal Domain RNA polymerase II polypeptide A Small Phosphatase. The
numbers before the sequences represent the relative positions of predicted NFAT binding motifs to
the transcription start site (TSS) of the host gene. The numbers in the brackets stand for the scores
given by Genomatix (http://www.genomatix.de) for the similarity between the putative NFAT
cis-acting elements in the promoter regions of the host genes for miR-26a/b and the perfect NFAT
binding sequence; Two scores are given as core sequence similarity and overall sequence
similarity. Only those with overall matrix similarity >0.95 are shown. 5000 bp upstream to the
predicted TSS for miR-26a/b host genes in human and canine was analyzed.

18
TRPC1
TRPC2
TRPC4
TRPC3
TRPC5
TRPC6
TRPC7
TRPM7
TRPV6
TRPC3
40
30
20
10
0
140
120
100
80
0
40
20
60
160
Freshly isolated rat cardiomyocytes (F‐CM, n=6)
Freshly isolated rat fibroblasts (F‐FB, n=6)
Passage cultured rat myofibroblasts (P‐MFB, n=6)
F‐CM F‐
FB
P‐MFB
ND
A
B
Relative m
RNA expression
Relative m
RNA expression
Supplemental Figure 1

19
Supplemental Figure 2
FGd3+‐sensitive
currentPyr3‐sensitive
current
‐120 ‐80 ‐40 80 120
‐2.0
‐1.0
0.5
1.0
1.5
2.0
(mV)
(pA/pF)
AαSMA
TO‐PRO3 Merged
Vimentin
100 µm 100 µm
100 µm100 µm
αSMA
TO‐PRO3 Merged
Vimentin
100 µm 100 µm
100 µm100µm
B
Cm=17 pF
D
E
ControlGd3+
Current Amplitzude (pA)
0 500 1000 1500 2000 2500 3000 3500‐40
‐30
‐20
‐100
10
20
30
40
50
Cm=16 pF
Control
Pyr3
Current Amplitude (pA)
0 500 1000 1500 2000 2500 3000 3500
‐20
‐10
0
10
20
30
Time (ms)
Control
Gd3+
Current Amplitude (pA)
Time (ms)0 500 1000 1500 2000 2500 3000 3500
‐40
‐30
‐20
‐100
10
20
30
40
50
Cm=13 pF
C 0 mV0 mV
‐110 mV
+100 mV
3 s
Time (ms)

20
Pyrazole 3 (0.1mg/kg/day) n=6
Vehicle (Polyethylene glycol+DMSO) n=6
Day 0Start pacing
Surgery
Day 7
Atrial Tachypacing (600 bpm)
Drugs were continuously administrated with Alzet Osmotic Pump.
*Terminal ExperimentsOpen chest EP studyVimentin Western Blotting in LA tissue sampleqPCR for collagen gene in fresh fibroblastsProliferation assay in cultured fibroblasts
*
Supplemental Figure 3
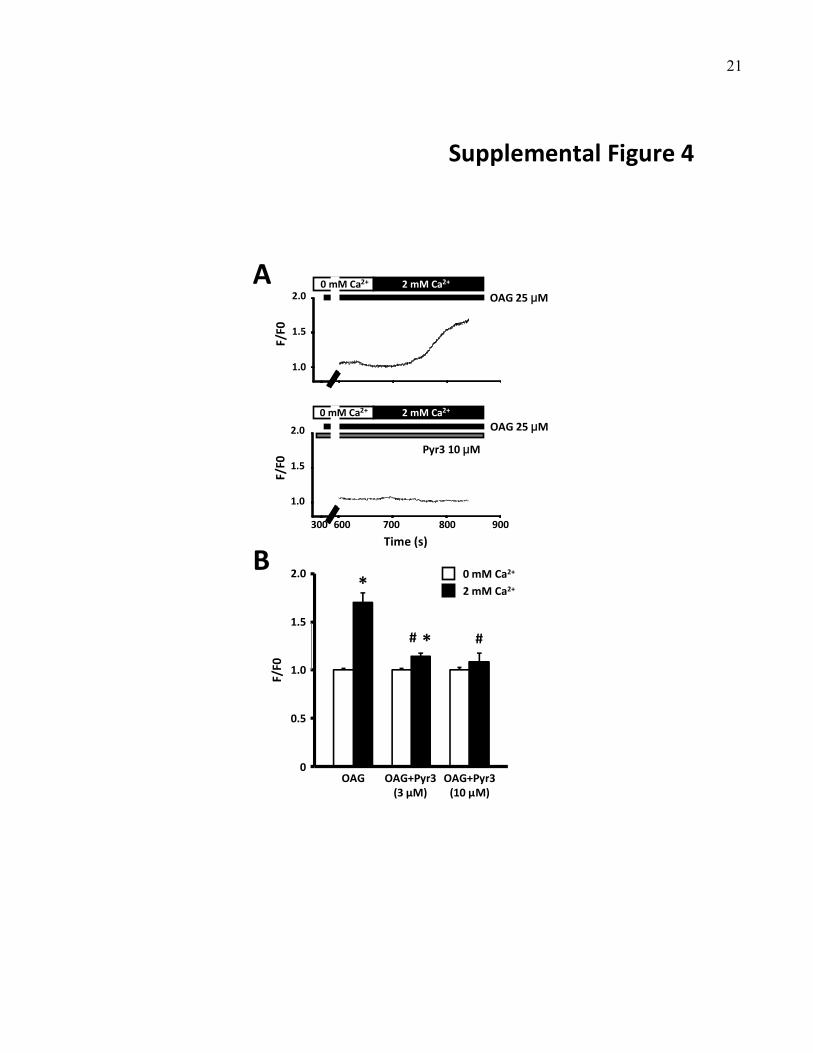
21
Supplemental Figure 4
0 mM Ca2+
OAG OAG+Pyr3 (3 µM)
F/F0
2.0
1.0
1.5
0
0.5
* 2 mM Ca2+
A
B
OAG+Pyr3(10 µM)
*# #
OAG 25 μM
Time (s)
300 600 700 800 900
2 mM Ca2+
OAG 25 μM
Pyr3 10 μM
2 mM Ca2+
F/F0
2.0
1.0
1.5
F/F0
2.0
1.0
1.5
0 mM Ca2+
0 mM Ca2+

22
A
0 200 400 600 800 1K
FL3 (PI intensity)0 200 400 600 800 1K
FL3 (PI intensity)
0
50
100
150
200
Cell count
0
50
100
150
200
Cell count
Dean‐Jett‐Fox modelFreq. G2/M = 20.86%
0 200 400 600 800 1K
FL3 (PI intensity)
0
100
200
300
400
Cell count
500
0
100
200
300
400
Cell count
500
0 200 400 600 800 1K
FL3 (PI intensity)
CTL Pyr3
CTL Gd3+
G0/G1
S
G2/M
G0/G1
S
G2/M
Supplemental Figure 5
Dean‐Jett‐Fox modelFreq. G2/M = 15.8%
Dean‐Jett‐Fox modelFreq. G2/M = 21.31%
Dean‐Jett‐Fox modelFreq. G2/M = 16.88%
B
CTL Gd3+
10 μM Gd3+
100 μM
*
G2/M cell percentage (%)
0
5
20
15
10
*
CTL Gd3+
10 μM Gd3+
100 μM
C 1‐h culture
24‐h culture1.8
1.5
1.2
0.9
0.3
Number of fibroblasts (x10
4)
0.6
0
D 1‐h culture
24‐h culture
##
#
##
#

23
A
B
50µm50µm
Control Pyr3 (3 μM)
CTL Pyr33 μM
10
12
8
6
4
2
0
TUNEL positive
cells (%
) *
Supplemental Figure 6

24
Supplemental Figure 7
DMSO Pyr3 DMSO Pyr3
αSMA
GAPDH
42 kDa
36 kDa
1.0
1.2
0.8
0.6
0.4
0.2
0
Relative expression
DMSO Pyr33 μM
Exp 1 Exp 2D
1‐h treatment
24‐h treatment
DMSO Pyr33 μM
G2/M cell percentage
(%)0.6
0.5
0.4
0.3
0.1
0.2
0
20
15
5
10
0DMSO Pyr3
3 μM
B C 1‐h treatment
24‐h treatment
Cell count (x104)
100µm100µm
25µm25µm
Control Pyr3 (3 μM)
Control Pyr3 (3 μM)
A

25
Cell n
umber increase rate
(fold change)
10
8
6
4
2
0
*
*
Day 6(Baseline)
Day 7 Day 8
TRPC3
GAPDH
96 kDa
36 kDa
Relative m
RNA expression
1.2
0
1.0
0.8
TRPC3 TRPC6
*
Scr‐shRNA
TRPC3‐shRNA
A B Scr‐
shRNA
TRPC3‐
shRNA
Scr‐
shRNA
TRPC3‐
shRNA
CScr‐shRNA
TRPC3‐shRNA
D
G2/M cell percentage
(%)
5
0
20
15
10
*
*
Day 6(Baseline)
Day 7 Day 8
Scr‐shRNA
TRPC3‐shRNA
TRPC3‐shRNAScr‐shRNA
1.0
0.8
0.6
0.4
0.2
0
1.2
0.4
0.6
0.2
*
*
Experiment 1 Experiment 2
Relative protein expression
Supplemental Figure 8

26
A C
E
B
D F
Supplemental Figure 9
HG I
TRPC3
TRPC1
TRPM7
SR SR cAF cAF
CSQ 53 kDa
100 kDaTRPC1
1.6
1.2
0.8
0.4
0
TRPC1/CSQ
(AU)
SR cAF
1.4
1.0
0.6
0.2
AF patients
CSQ
SR SR cAF cAF
53 kDa
150 kDaTRPM7
SR cAF
TRPM7/CSQ
(AU)
1.6
1.2
0.8
0.4
0
2.0
AF patients
SR SR cAF cAF
CSQ 53 kDa
93 kDaTRPC3
0.16
0.12
0.08
0.04
0
TRPC3/CSQ
(AU)
SR cAF
*0.14
0.10
0.06
0.02
AF patients
CTL CTL AF AF
CSQ 53 kDa
100 kDaTRPC1
2.5
1.5
0.5
0
2.0
1.0
TRPC1/CSQ
(AU)
CTL AF
AF goats
CSQ
TRPM7
CTL CTL AF AF
53 kDa
150 kDa
CTL AF
TRPM7/CSQ
(AU)
0.16
0.12
0.08
0.04
0
0.14
0.10
0.06
0.02
AF goats
CTL CTL AF AF
CSQ 53 kDa
93 kDaTRPC3
0.16
0.12
0.08
0.04
0
0.14
0.10
0.06
0.18
TRPC3/CSQ
(AU)
CTL AF
*
0.02
AF goats
CTL CTL CHF CHF
CSQ 53 kDa
100 kDaTRPC1
CTL
1.6
1.2
0.8
0.4
0
1.4
1.0
0.6
0.2TRPC1/CSQ
(AU)
CHF
CHF dogs
CSQ
TRPM7
CTL CTL CHF CHF
53 kDa
150 kDa
CTL CHF
TRPM7/CSQ
(AU)
1.6
1.2
0.8
0.4
0
1.4
1.0
0.6
0.2
CHF dogs
CTL CTL CHF CHF
CSQ 53 kDa
93 kDaTRPC3
0.16
0.12
0.08
0.04
0
0.14
0.10
0.06
0.02TRPC3/CSQ
(AU)
CTL CHF
*
CHF dogs

27
Supplemental Figure 10A
B
C
E
D
AEg
Intracardiac ECG
Surface ECGAF
Pacing AF
(Day 0)
Intracardiac ECG
Surface ECGAF
Pacing AF
(Day 3)
Intracardiac ECG
Surface ECGAF
Pacing AF
(Day 4)
Surface ECG
Intracardiac ECG
Pacing AF
AF
(Day 5)
Intracardiac ECG
Pacing AF
Surface ECGAF
(Day 6)
1 second
1 second
1 second
1 second
1 second
AEg
AEg
AEg
AEg
VEg
VEg
VEg
VEg
VEg

28
4
6
8
0
2
10
12
14
Diastolic area (cm2)
*
CTL AF Day 0
AF Day 7
Supplemental Figure 11
AF Day 0 AF Day 7
LA
LV
LA
LV
30
40
0
20
10
Fractional area chan
ge (%)
*
CTL AF Day 0
AF Day 7
20
30
40
0
10
50
60
70
Diastolic volume (ml)
CTL AF Day 0
AF Day 7
B
C D E
100
150
200
0
50Hea
rt r
ate
(b
pm
)
Day 0(pre-ATP)
Day 0(ATP)
CTL Day 3 Day 7
CTL
AF
A*

29
Lipo miR26amimic
AMO26a
103
101
100
10‐1
10‐2
104*
*
Relative miR26a
expression
A
B
D
C
Supplemental Figure 12


















