
Analysis of Horizontal Gene Transfers of Potential Relevance to Microbial Virulence Fiona Brinkman...
47
Analysis of Horizontal Gene Transfers of Potential Relevance to Microbial Virulence Fiona Brinkman Simon Fraser University, Greater Vancouver, British Columbia, Canada
-
Upload
henry-floyd -
Category
Documents
-
view
219 -
download
0
Transcript of Analysis of Horizontal Gene Transfers of Potential Relevance to Microbial Virulence Fiona Brinkman...

Analysis of Horizontal Gene Transfers of Potential Relevance to Microbial Virulence
Fiona Brinkman Simon Fraser University,
Greater Vancouver, British Columbia, Canada

Overview
1. High pathogen-host protein similarities: detecting horizontal gene transfer
2. Characteristics of proteins/genes putatively horizontally acquired by bacterial pathogens
3. Implications
4. Proposal: How we should be combating bacterial pathogens

Yersinia Type III secretion system

Approach
Idea: Could we identify novel virulence factors by identifying bacterial pathogen proteins more similar to host proteins than you would expect?
1. Primary sequence similarity approach – identifies possible horizontal gene transfer
(2. Structural similarity approach)

• For each complete bacterial and eukaryote genome: BLASTP (and MSP Crunch) analysis of all deduced proteins, searched against non-redundant SWALL database
• Overlay NCBI taxonomy information
• Query database for bacterial proteins who’s top BLASTP scoring “hit” is eukaryotic (and eukaryotic proteins who’s top hit is bacterial)
• Initial Assumption: Three Domains of life (Bacteria, Eukarya, and Archaea) are so divergent that top hits to another Domain are rare
Unusual similarities between Bacteria & Eukaryote genes: Sequence similarity-based approach

• Problem: If a gene transfer occurs from a eukaryote to an ancestor of closely related bacteria top hit will be to other bacteria
• Therefore, perform similar query, but filtering different taxonomic groups from the analysis…
Unusual similarities between Bacteria & Eukaryote genes: Sequence similarity-based approach
Bacteria 1 closely related Bacteria 2 bacteria (same species,
family, etc)
Eukaryote

BAE-watch Database: Bacterial proteins with unusual similarity with Eukaryotic proteins

Problem: Proteins highly conserved in the three domains of life
Top hit to a protein from another domain may occur by chance.
“StepRatio” score helps detect these.
Example:Glucose-6-Phosphate Reductase

Example of a case with a high StepRatio:
Enoyl ACP reductase

PhyloBLAST – a tool for analysisBrinkman et al. (2001) Bioinformatics 17:385-387.
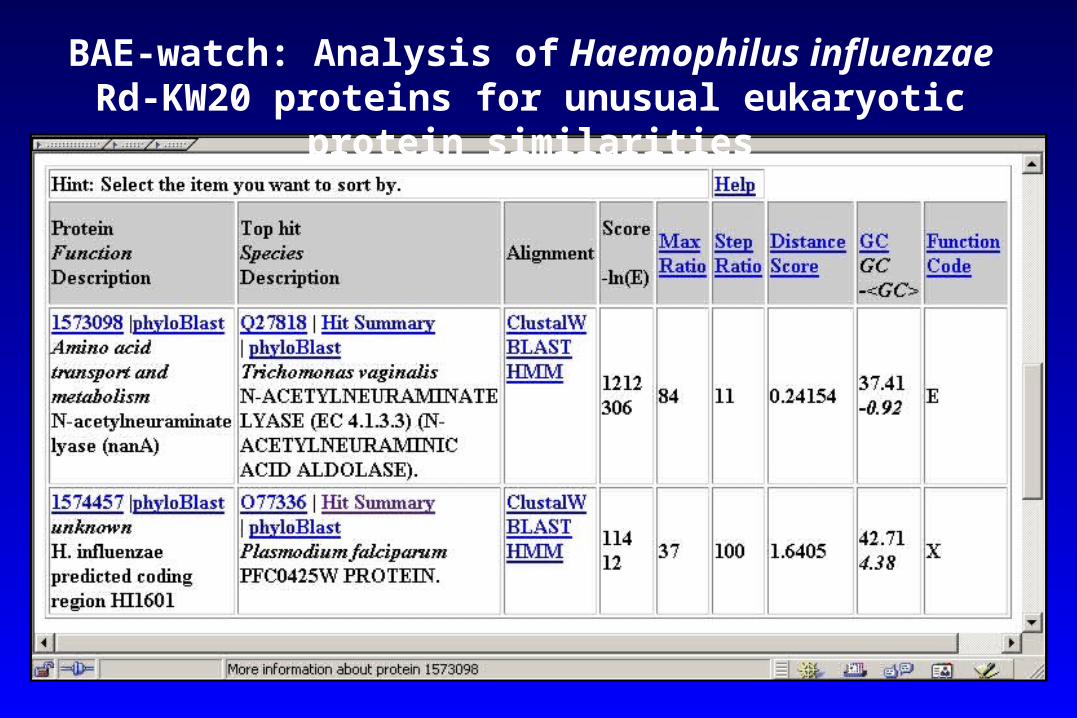
BAE-watch: Analysis of Haemophilus influenzae Rd-KW20 proteins for unusual eukaryotic protein similarities

Genome data for…
Anthrax Necrotizing fasciitis Cat scratch disease Paratyphoid/enteric feverChancroid Peptic ulcers and gastritisChlamydia Periodontal diseaseCholera PlagueDental caries PneumoniaDiarrhea (E. coli etc.) SalmonellosisDiphtheria Scarlet feverEpidemic typhus ShigellosisMediterranean fever Strep throatGastroenteritis SyphilisGonorrhea Toxic shock syndromeLegionnaires' disease Tuberculosis Leprosy TularemiaLeptospirosis Typhoid feverListeriosis UrethritisLyme disease Urinary Tract InfectionsMeliodosis Whooping cough Meningitis +Hospital-acquired infections

Bacterial Pathogens
Chlamydophila psittaci Respiratory disease, primarily in birdsMycoplasma mycoides Contagious bovine pleuropneumoniaMycoplasma hyopneumoniae Pneumonia in pigsPasteurella haemolytica Cattle shipping feverPasteurella multicoda Cattle septicemia, pig rhinitisRalstonia solanacearum Plant bacterial wiltXanthomonas citri Citrus cankerXylella fastidiosa Pierce’s Disease - grapevines
Bacterial wilt

Trends in this Sequence-based Analysis
• Identifies the strongest cases of lateral gene transfer between bacteria and eukaryotes
• Most common:
Bacteria Unicellular Eukaryote
Makes sense:
Bacteria to Multicellular eukaryote must involve germline
Eukaryote to Bacteria must not involve introns

Trends in this Sequence-based Analysis
• Identifies nuclear genes with potential organelle origins
• A control: Method identifies all previously reported Chlamydia trachomatis “plant-like” genes.

First case: Bacterium Eukaryote Lateral Transfer
0.1
Bacillus subtilis
Escherichia coli
Salmonella typhimurium
Staphylococcus aureus
Clostridium perfringens
Clostridium difficile
Trichomonas vaginalis
Haemophilus influenzae
Acinetobacillus actinomycetemcomitans
Pasteurella multocida
N-acetylneuraminate lyase (NanA) of the protozoan Trichomonas vaginalis is 92-95% similar to NanA of Pasteurellaceae bacteria.
de Koning et al. (2000) Mol Biol Evol 17:1769-1773
Pasteurellaceae

N-acetylneuraminate lyase – role in pathogenicity?
Pasteurellaceae
•Mucosal pathogens of the respiratory tract
T. vaginalis
•Mucosal pathogen, causative agent of the STD Trichomonas

N-acetylneuraminate lyase (sialic acid lyase, NanA)
Involved in sialic acid metabolism
Role in Bacteria: Proposed to parasitize the mucous membranes of animals for nutritional purposes
Role in Trichomonas: ?
Hydrolysis of glycosidic linkages of terminal sialic residues in glycoproteins, glycolipids SialidaseFree sialic acid
Transporter
Free sialic acid NanA
N-acetyl-D-mannosamine + pyruvate

Another case: A Sensor Histidine Kinase for a Two-component Regulation System
Signal Transduction “In General”
Histidine kinases more common in bacteria
Ser/Thr/Tyr kinases more common in eukaryotes
However, a histidine kinase was recently identified in fungi, including pathogens Fusarium solani and Candida albicans
How did it get there?
Candida

Neurospora crassa NIK-1
Fusarium solani FIK2 Streptomyces coelicolor SC4G10.06c
Candida albicans CaNIK1
Escherichia coli RcsC
Erwinia carotovora RpfA / ExpSEscherichia coli BarASalmonella typhimurium BarA
Pseudomonas aeruginosa GacS
Pseudomonas fluorescens GacS / ApdAPseudomonas tolaasii RtpA / PheN
Pseudomonas syringae GacS / LemA
Pseudomonas viridiflava RepAAzotobacter vinelandii GacS
0.1
Streptomyces coelicolor SC7C7.03
Xanthomonas campestris RpfCVibrio cholerae TorS
Escherichia coli TorS
Fusarium solani FIK1Fungi
Pseudomonas aeruginosa PhoQ
100
100
51100
100
100
100
100100
100
100
100
100
86
54
39
100
100
Streptomyces Histidine Kinase. The Missing Link?
Virulence Factor ( )in every organism examined to date
Brinkman et al. (2001) Infection and Immunity 69:5207-5211

“Plant-like” genes in Chlamydia• Proteins: Unusually high number most similar to plant
proteins
• Previous proposal: Obtained genes from a plant-like amoebal host?
(A relative of Chlamydiaceae infects Acanthamoeba. Chlamydiaceae: Obligate intracellular pathogens)
• However Acanthamoeba relationship to plants very controversial

“Plant-like” genes in ChlamydiaNCBI GI Protein description Subcellular localization in plants
4377270 Glycyl tRNA Synthetase Chloroplast
4376626 cADP/ATP Translocase Chloroplast
4376667 cGlycogen Hydrolase Chloroplast
4377189 GTP Cyclohydratase & DHBP Synthase Chloroplast
4377237 cBeta-Ketoacyl-ACP Synthase Chloroplast
4376686 cEnoy-Acyl-Carrier Reductase Chloroplast
4376591 cThioredoxin Reductase Chloroplast
4377185 Metal Transport P-type ATPase Chloroplast
4377346 Similar to NA+/H+ Antiporter Chloroplast
4376650 cPhosphate Permease Chloroplast
4376637 GcpE protein Chloroplast
4376637 Tyrosyl tRNA Synthetase Chloroplast
4377360 cMalate Dehydrogenase Chloroplast
4376763 GTP Binding protein Chloroplast
4376911 cADP/ATP Translocase Chloroplast
3329179 Phosphoglycerate Mutase Chloroplast
4377281 cGlycerol-3-Phosphate Acyltransferase Chloroplast
4376993 ABC Transporter ATPase Chloroplast
4376509 dDeoxyoctulonosic Acid Synthetase Chloroplast
4376872 eSugar Nucleotide Phosphorylase Chloroplast

“Plant-like” genes in Chlamydia
6578112 rRNA Methytransferase Chloroplast
3329217 HSP60 Chloroplast
3328745 cPhosphoribosylanthranilate Isomerase Chloroplast
6578104 cAspartate Aminotransferase Chloroplastf
4377328 cPolyribonucleotide Nucleotidyltransferase Chloroplastf
4377362 Putative D-Amino Acid Dehydrogenase Chloroplastg
4377331 Cytosine Deaminase Chloroplast?h
4376915 Lipoate-Protein Ligase A Mitochondrial
4377272 Glycogen Synthase N/Ai
4377065 cDihydropteroate Synthase N/Ai
4377239 cInorganic Pyrophosphatase N/Ai
4376904 Uridine 5’-Monophosphate Synthase N/Ai
4377173 cUDP-Glucose Pyrophosphorylase N/Ai
4376815 GutQ/Kpsf Family Sugar-Phosphate Isomerase Mitochondrial?j

“Plant-like” genes in Chlamydia
• Endosymbiotic theory
• Rickettsia: Many eukaryotic-like genes
• Synechocystis: Many plant-like genes
• Does Chlamydia share an ancient ancestral relationship with the ancestor of the Chloroplast?

Chlamydiaceae share an ancestral
relationship with Cyanobacteria and
Chloroplast0.1
Pyrococcus furiosus (Archaea)
Thermotoga maritima
Aquifex pyrophilus
Bacillus subtilis
Chlamydophila pneumoniae
Chlamydophila psittaci
Chlamydia muridarum
Chlamydia trachomatis1000
7041000
Chlamydomonas reinhardtii
Klebsormidium flaccidum
Zea mays
Nicotiana tabacum1000
988
998
Synechococcus PCC6301
Synechocystis PCC6803
Microcystis viridis1000
1000
1000
530
Escherichia coli
Zea mays mitochondrion
Rickettsia prowazekii
Caulobacter crescentus
868986
764
349
1000
538
Chloroplasts
Cyanobacteria
Chlamydiaceae
16S rRNA

Chlamydiaceae share an ancestral relationship with Cyanobacteria and Chloroplast
L3
L4
L23
L2
S19
L22 S3
L16 L29
S17
L14
L24
L5
S14 S8
L6
L18 S5
L30
L15S10
EscherichiaBacillusThermatogaSynechocystisChlamydia
Unique shared-derived characters unite Chlamydiaceae and Synechocystis

Chlamydiaceae “plant-like” genes reflect an ancestral relationship with Cyanobacteria and the Chloroplast
• Chlamydiaceae do not appear to be exchanging DNA with their hosts
• Existing knowledge of Cyanobacteria may stimulate ideas about the function and control of pathogenic Chlamydia?
Brinkman et al. (2002) Genome Research 12:1159-1167.

Overview
1. High pathogen-host protein similarities: detecting horizontal gene transfer
2. Characteristics of proteins/genes putatively horizontally acquired by bacterial pathogens
3. Implications
4. Proposal: How we should be combating bacterial pathogens

Horizontal Gene Transfer and Bacterial Pathogenicity
Transposons: ST enterotoxin genes in E. coli
Prophages:Shiga-like toxins in EHECDiptheria toxin gene, Cholera toxinBotulinum toxins
Plasmids:Shigella, Salmonella, Yersinia
Pathogenicity Islands:
Uro/Entero-pathogenic E. coliSalmonella typhimuriumYersinia spp.Helicobacter pyloriVibrio cholerae

Pathogenicity Islands
Associated with
– Atypical %G+C– tRNA sequences– Transposases, Integrases and other mobility genes– Flanking repeats

IslandPath: Aiding identification of Pathogenicity Islands and other Genomic Islands
Yellow circle = high %G+C
Pink circle = low %G+C
Region of unusual dinucleotide bias
tRNA gene lies between the two dots
rRNA gene lies between the two dots
Both tRNA and rRNA lie between the two dots
Dot is named a transposase
Dot is named an integrase
_
Hsiao et al. (2003) Bioinformatics 19: 418-420

Dinucleotide bias analysis
Genome divided into “ORF-clusters” of 6 consecutive ORFs
For each ORF cluster,the average absolute dinucleotide relative abundance difference is
where f (fragment) is derived from sequences in an ORF-cluster g (genome) is derived from all predicted ORFs in the genome
Dinucleotide relative abundance is *XY = f*XY/f*Xf*Y
where f*X denotes the frequency of the mononucleotide X f*XY the frequency of the dinucleotide XY
|)(*)(*|16
1),(* gfgf xyxy
See Hsiao et al. (2003) Bioinformatics 19: 418-420
and Karlin, S. and Burge, C (1995). Trends in Genetics 1995 11:283-90 for review

Dinucleotide bias analysis
ORF-clusters sampled in an overlapping manner (shift by one ORF at a time)
The mean is calculated by averaging the results from all ORF-clusters in the genome
Regions with greater than 1 standard deviation away from the mean are marked on the IslandPath graphical display with strikethrough lines
Why did we use 6 ORFs per cluster?- Not enough bp in a single ORF to get a good estimate - 4.5kb (corresponding to approximately 6-8 ORFs) is required for “reliable estimation
of nucleotide composition” (Lawrence and Ochman, J Mol Evolution 1997 44:383-97)
),(* gf

1
7
11
20
22
33
34 3536
II
I
V
IV
III
VI
VII
VIII
IX
X
32
Boxes: Known islands in the Salmonella typhi genome

What features best predict Islands?
Examined prevalence of features in over 200 known islands
• 94% of islands contain >25% dinucleotide bias (majority have >75% dinucleotide bias coverage)
• Mobility genes identified in >75% (but ID recently improved)
• Atypical %G+C (above cutoff used in Brinkman et al., 2002) not over 50% coverage on average, and tRNA genes not observed with >50% of known islands

1
37
11
18
20
22
33
34 3536
II
I
V
IV
III
VI
VII
VIII
IX
X
32
1
569
1012
13
1415
17
2122
24
323334
3536
Boxes: “Insertions” in the Salmonella typhi genome verses Salmonella typhimurium

Properties of genes in these islands?
Defined a “putative island” as
– 8 or more genes in a row with dinucleotide bias
Functional category analysis Any difference for genes in islands verses genome?

Bac
illus
sub
tilis
168
Bor
relia
bur
gdor
feri
B31
Buc
hner
a sp
. AP
S
Chl
amyd
ia tr
acho
mat
is D
Clo
strid
ium
ace
tobu
tylic
um A
TCC
824
Esc
heric
hia
coli
K12
Esc
heric
hia
coli
O1
57_E
DL9
33
Hae
mop
hilu
s in
fluen
zae
Rd-
KW
20
Hel
icob
acte
r pyl
ori 2
6695
List
eria
inno
cua
Clip
1126
2
Myc
obac
teriu
m le
prae
Myc
obac
teriu
m tu
berc
ulos
is C
DC
1551
Myc
opla
sma
pneu
mon
iae
M12
9 N
eiss
eria
men
ingi
tidis
ser
ogro
up B
stra
in M
C58
P
seud
omon
as a
erug
inos
a P
AO
1
Ric
ketts
ia p
row
azek
ii M
adrid
E
Sal
mon
ella
typh
imur
ium
LT2
S
taph
yloc
occu
s au
reus
sub
sp. A
ureu
sN
315
Stre
ptoc
occu
s pn
eum
onia
e TI
GR
4
Sul
folo
bus
solfa
taric
us
Vib
rio c
hole
rae
chro
mos
ome
I
Vib
rio c
hole
rae
chro
mos
ome
II
Yer
sini
a pe
stis
CO
92
0
10
20
30
40
50
60
70
80
Organisms
% H
ypo
thet
ical
Pro
tein
s
Hypothetical genes are more common in putative islands vs the genome
(Paired T test P= 6.8E-19)
Genome
Put. Islands
Analysis 1: COG functional category analysis

Analysis 2: SUPERFAMILY HMM search results
SUPERFAMILY: a set of HMMs built from SCOP superfamilies
Fewer ORFs in the putative islands were assigned to a SUPERFAMILY class
Genome
Put Islands
Paired T test P= 3.3E-14
B.
ha
lod
ura
ns
B.
bu
rgd
orf
eri
C.
tra
ch
om
atis
C.
ac
eto
bu
tylic
um
E.
co
li K
12
H.
influ
en
za
e
L.
mo
no
cy
tog
en
es
M.
lep
rae
M.
tub
erc
ulo
sis
M.
ge
nita
lium
N.
me
nin
giti
dis
B
P.
ae
rug
ino
sa
R.
pro
wa
ze
kii
S.
typ
him
uri
um
S.
au
reu
s
V.
ch
ole
rae
ch
I
V.
ch
ole
rae
ch
II
Y.
pe
stis
0
10
20
30
40
50
60
70
80%
OR
Fs
as
sig
ne
d t
o s
up
erf
am
ily

Analysis 3: Gene size in Putative Islands vs. “Non-Islands”
B.s
ub
tilis
B.
bu
rgd
orf
eri
B.
me
lite
ns
is c
h I
B.
me
lite
ns
is c
h I
I
Bu
ch
ne
ra
C.
tra
ch
om
atis
D
D.
rad
iod
ura
ns
ch
1
D.
rad
iod
ura
ns
ch
2
E.
co
li K
12
E.
co
li O
15
7
H.in
flue
nz
ae
L.
mo
no
cy
tog
en
es
M.
lep
rae
M.
tub
erc
ulo
sis
CD
C1
55
1
M.
tub
erc
ulo
sis
H3
7R
v
N.
me
nin
giti
dis
A
N.
me
nin
giti
dis
B
P.
ae
rug
ino
sa
R.
pro
wa
ze
kii
S.
typ
him
uri
um
S.
flex
ne
ri
S.
au
reu
s
S.
pn
eu
mo
nia
e
V.
ch
ole
rae
ch
I
V.
ch
ole
rae
ch
II
Y.
pe
stis
0
50
100
150
200
250
300
350
400
Me
an
ge
ne
le
ng
th (
bp
)ORFans (genes with no homologs among 60 microbial genomes) tend to be shorter genesAre genes in putative islands shorter as well on average?
In most cases, average ORF length in putative islands is shorter
Non Island
Put. Islands
Paired T test P= 7.1E-34

Analysis 4: COG analysis after removing ORFs <300 bp
Genes may be less well predicted in such island/atypical dinucleotide bias regions
Some genomes still show marked increase % hypothetical genes in islands verses genome
Hypothetical genes more common in islands?
Paired T test P= 0.0016
0
10
20
30
40
50
60
70
80
% h
yp
oth
eti
ca
l p
rote
ins
Genome
Islands

Summary: Bacteria gene transfer analysis
• No cases identified in our database to date of clear, recent horizontal gene transfer between bacteria and a multicellular eukaryote (involving >80% sequence similarity)
The pathogens studied are not commonly acquiring genes from their hosts, or vice versa
• Bacterial and eukaryotic pathogens may have exchanged genes
• Overall increased prevalence of hypothetical genes in putative bacterial genomic islands? Cautionary note about gene prediction accuracy

Overview
1. High pathogen-host protein similarities: detecting horizontal gene transfer
2. Characteristics of proteins/genes putatively horizontally acquired by bacterial pathogens
3. Implications
4. Proposal: How we should be combating bacterial pathogens

Implications: Evolution of Pathogenicity
Pathogen mimicry of their host: Convergent evolution or genes selectively maintained
Gene exchange between pathogens: “Arms Deals”

Pathogens and “The Art of War”
“What is of supreme importance in war is to attack the enemy's strategy. Next best is to disrupt his alliances by diplomacy. The next best is to attack his army. And the worst policy is to attack cities.”

FPMIIN
DU
STR
Y
Ani
geni
cs C
anad
a
Inim
ex P
harm
a In
c AC
AD
EM
IA
VID
O, U
Sask
UB
C, SFU
, BC
GSC
GOVERNMENTGenome CanadaGenome Prairie
Genome BCGovt of Saskatchewan
Functional Pathogenomics of Mucosal Immunitywww.pathogenomics.ca

• BC Pathogenomics group Ann M. Rose, Yossef Av-Gay, David L. Baillie, Fiona S. L. Brinkman, Robert
Brunham, Artem Cherkasov, Rachel C. Fernandez, B. Brett Finlay, Hans Greberg, Robert E.W. Hancock, Steven J. Jones, Patrick Keeling, Audrey de Koning, Don G.
Moerman, Sarah P. Otto, B. Francis Ouellette, Nancy Price, William Hsiao.
• Jeff Blanchard (NCGR, New Mexico) and Olof Emanuelsson (Stockholm Bioinformatics Center)
• Peter Wall Institute for Advanced Studies, Genome Canada


















