
Analysis and speciation of organic phosphorus in environmental …748881/... · 2014-09-22 ·...
58
Analysis and speciation of organic phosphorus in environmental matrices: Development of methods to improve 31 P NMR analysis Johan Vestergren Doctoral Thesis, Department of Chemistry Umeå University, 2014
Transcript of Analysis and speciation of organic phosphorus in environmental …748881/... · 2014-09-22 ·...

Analysis and speciation of organic phosphorus in
environmental matrices: Development of methods to improve 31P NMR analysis
Johan Vestergren
Doctoral Thesis, Department of Chemistry
Umeå University, 2014

Responsible publisher under swedish law: the Dean of the Faculty of Science and
Technology
This work is protected by the Swedish Copyright Legislation (Act 1960:729)
ISBN: 978-91-7601-132-4
Electronic version available at http://umu.diva-portal.org/
Tryck/Printed by: Service Center KBC
Umeå, Sweden, 2014

“I have wished to see chemistry applied to domestic objects, to malting, for
instance, brewing, making cider, to fermentation and distillation generally,
to the making of bread, butter, cheese, soap……..”
- Thomas Jefferson


i
Table of Contents
Table of Contents i Abstract iii List of Abbreviations iv List of Publications v Introduction 1
Phosphorus in nature 1 Phosphorus limitation of ecosystems’ plant productivity 4 Phosphorus: Its way to bioavailability 6 31P NMR Spectroscopy: an innovative tool for studying the speciation of organic
phosphorus and the need for advanced techniques to study P-species. 8 Liquid NMR in soil science 10 Solid state 31P MAS NMR on intact soils 13
Aim of thesis 15 Materials and Methods 16
NMR spectroscopy 16 The NMR signal 16 The chemical shift 18 Indirect spin-spin couplings 18 Line broadening contributing factors 19 The 2D 1H-31P HSQC NMR experiment 21 Inductively coupled plasma optical emission spectrometry (ICP-OES) 22 X-ray fluorescence spectroscopy (XRF) 23 Other analytical methods used in this thesis 24 Site descriptions 24 Sampling and general preparations 25 Preparations for solid state NMR 26 Sample preparations for liquid NMR by conventional extraction 26 Sulfide treatment 27 NMR experiments 28 SIMCA analysis 29
Summary of Papers 30 Paper I; High Resolution Characterization of Organic Phosphorus in Soil Extracts
using 2D 1H-31P NMR Correlation Spectroscopy 30 Paper II; Soil organic phosphorus transformations in a boreal forest
chronosequence 32 Paper III; Effect of Trees on Forms of Soil Phosphorus in an Agroforestry
Parkland in Burkina Faso (manuscript) 33 Paper IV; Changes in organic P-composition in boreal forest humus soils: the
role of iron and aluminium 34 Conclusions and future work 36

ii
Acknowledgements 38 References 42

iii
Abstract
Phosphorus (P) is an essential element for life on our planet. It is central in
numerous biochemical processes in terrestrial and aqueous ecosystems
including food production; and it is the primary growth-limiting nutrient in
some of the world’s biomes. The main source of P for use as agricultural
fertilizer is mining of non-renewable mineral phosphate. In terrestrial
ecosystems the main source is soil P, where the largest fraction is organic P,
composed of many species with widely differing properties. This fraction
controls the utilization of P by plants and microorganisms and influences
ecosystem development and productivity. However, there is only scarce
knowledge about the molecular composition of the organic P pool, about the
processes controlling its bioavailability, and about its changes as soils
develop. Therefore, the aim of this thesis was to develop robust solution- and
solid-state 31P nuclear magnetic resonance spectroscopy (NMR) methods to
provide molecular information about speciation of the organic P pool, and to
study its dynamics in boreal and tropical soils. By studying humus soils of a
groundwater recharge/discharge productivity gradient in a Fennoscandian
boreal forest by solution- and solid-state NMR, it was found that P speciation
changed with productivity. In particular, the level of orthophosphate diesters
decreased with increasing productivity while mono-esters such as inositol
phosphates increased. Because the use of solution NMR on conventional
NaOH/EDTA extracts of soils was limited due to severe line broadening
caused by the presence of paramagnetic metal ions, a new extraction method
was developed and validated. Based on the removal of these paramagnetic
impurities by sulfide precipitation, a dramatic decrease in NMR linewidths
was obtained, allowing for the first time to apply modern multi-dimensional
solution NMR techniques to soil extracts. Identification of individual soil P-
species, and tracking changes in the organic P pools during soil development
provided information for connecting P-speciation to bioavailability and
ecosystem properties. Using this NMR approach we studied the
transformation of organic P in humus soils along a chronosequence (7800
years) in Northern Sweden. While total P varied little, the composition of the
soil P pool changed particularly among young sites, where also the largest
shift in the composition of the plant community and of soil microorganisms
was observed. Very old soils, such as found Africa, are thought to strongly
adsorb P, limiting plant productivity. I used NMR to study the effect of
scattered agroforestry trees on P speciation in two semi-arid tropical
woodlands with different soil mineralogy (Burkina Faso). While the total P
concentration was low, under the tree canopies higher amounts of P and
higher diversity of P-species were found, presumably reflecting higher
microbial activity.

iv
List of Abbreviations
ATP Adenosine Triphosphate
CSA Chemical Shift Anisotropy
D2O Deuterium oxide or “heavy water”
DNA Deoxyribonucleic Acid
EA-IRMS Automated Elemental Analyzer Isotope Ratio Mass Spec.
EDTA Ethylenediaminetetraacetic Acid
FID Free Induction Decay
HSQC Heteronuclear Single Quantum Coherence Spectroscopy
HR High Resolution
I Magnetic Spin
ICP-OES Inductively Coupled Plasma-Optical Emission Spectroscopy
LRC Land Rising Coast (translation from Swedish)
MAS Magic Angle Spinning
MDPA Methylene Diphosphonic Acid
MPA Methyl Phosphonic Acid
N Nitrogen
NaOH Sodium Hydroxide
Na2S Sodium Sulfide
NMR Nuclear Magnetic Resonance Spectroscopy
P or 31P Phosphorus
PC Phosphatidylcholine
ppb Parts per billion (µg/L)
ppm Parts per million
RF Radio Frequency
RNA Ribonucleic Acid
T1 Longitudinal Relaxation or Spin-Lattice Relaxation
T2 Transversal Relaxation or Spin-Spin Relaxation
XRF X-Ray Fluorescence Spectroscopy

v
List of Publications
This thesis is based on the following papers:
I: Vestergren J., Vincent AG., Ilstedt U., Jansson M., Persson P., Gröbner G.,
Giesler R., Schleucher J. “High Resolution Characterization of Organic
Phosphorus in Soil Extracts using 2D 1H, 31P NMR Correlation”. Environ. Sci.
Technol, 46, 3950−3956, (2012)
II: Vincent AG., Vestergren J., Gröbner G., Persson P., Schleucher J., Giesler R.
“Soil Organic Phosphorus Transformations in a Boreal Forest
Chronosequence” Plant Soil 367:149–162 (2013)
III: Vestergren J., Vincent A.G., Ouattara K., Persson P., Gröbner G., Giesler R.,
Schleucher J., Ilstedt U. “Agroforest Parkland Influence on Phosphorus
Speciation – A 31P NMR Study”, Manuscript
IV: Vincent AG., Schleucher J., Gröbner G., Vestergren J., Persson P., Jansson
M., Giesler R. “Changes in Organic Phosphorus Composition in Boreal Forest
Humus Soils: the Role of Iron and Aluminium”, Biogeochemistry, 108, 1-3, 485-499 (2011)
Additional papers which are not included in the thesis:
Vestergren J., Sundman A., Persson P., Giesler R., Schleucher J., Jonsson
A., Jansson M., Gröbner G. ”Absorption Dynamics of Phosphate Species on
Ion Exchange Resin – A 31P NMR study”. Manuscript
Kruse J., Abraham M., Amelung W., Baum C., Bol R., Kühn O., Lewandowski
H., Niederberger J., Oelmann Y., Rüger C., Santner J., Siebers M., Siebers N.,
Spohn M., Vestergren J., Vogts A., Leinweber P. “: Advanced Methods in
Soil Phosphorus Research: A Review”. submitted to Journal of Plant
Nutrition and Soil Science
Vincent A., Ilstedt U., Vestergren J., Giesler R., Persson P., Gröbner G,
Schleucher J. “Phosphorus in Forest Soils - Disentangling the Chemistry of an
Essential Nutrient” Fakta Skog (Forest Facts) 4 (2013)

1
Introduction
Phosphorus in nature
Life on this planet is in all its aspects in need of phosphorus (P). Life would
not persist without this unique chemical element. P is well known for its
essential role in biochemical processes, its use as fertilizer in agricultural
production, and its notorious role as pollutant in aqueous environments [1,
2]. In general, organic P is the dominant form of P found in soils, but also in
stream, lake and sea water. Organic P is present as numerous P-species and
controls primary production in many terrestrial and aquatic ecosystems, as
found in tropical areas as well as in boreal locations [3]. In these systems
restricted availability of P occurs not only through low P-levels but in many
cases through P-fixation; a process which includes physical adsorption of P
to organic and mineral material and precipitation into chemically bound
complexes of P [4-8].

2
Figure 1: Change of phosphorus use over time. Demand for rock phosphate
has increased dramatically while importance of manure has declined.
Adapted from [9]; based on concept from [10])
Therefore, in many parts of the world, food and forestry production is
severely restricted by the limited availability of P for the plants. Due to this
fact large-scale mining and manufacturing of P-fertilizer from rock
phosphate has increased dramatically in the last century (s. Figure 1) to
improve productivity, especially for food crops. Since human demand for P
will grow due to an increase in population and climate warming [11] a
dramatic increase in demand for P-fertilizer is expected in the future.
However, due to the current non-sustainable P-management and the
expected decline in P-mining, the reduced availability of P-fertilizer is
predicted to become an imminent problem in the next 50 years (Figure
2)[9].

3
Figure 2: The request for Phosphorus in the future (picture by Magnus
Bard; published in “Dagens Nyheter” 2011-12-31). Translation of text in
picture: “Yes, there is one more thing. Phosphorus - we are starting to run
out of that, too.”
P-availability will therefore impose fundamental constraints on production
capacity affecting resource availability, and even social and economic
development. Politics and science therefore have to provide solutions for
avoiding P-scarcity but also P-pollution, to achieve a sustainable P-economy
for ensuring food security as well as a stable society and economy in the
future. Establishing a sustainable P-management and sustainable
agriculture, demands profound knowledge of the ability of plants and
microorganisms to utilize P; a process which is an important part of the
global P-dynamics and with wide ranging implications for ecosystems [7].
However, at present, there is a severe lack in knowledge about the molecular
processes controlling the speciation and reactivity of the organic P-pool and
its resulting P-bioavailability in ecosystems [12]. Therefore, identification of
the different organic P-species is crucial in tracking their individual origin
and understanding the species-dependent dynamics and bioavailability of P
in soils, and ultimately, to ascertain their role in ecosystems and their effects
on soil fertility. Related questions like these need to be answered, in
particular because little is known about the response of P biogeochemistry to
global environmental changes, and the possibility that P fertilizer for

4
agriculture may soon become scarce [13, 14]. Therefore, the general aim of
this thesis was to carry out a multidisciplinary study to provide not only new
basic knowledge about the speciation and dynamics of organic P-species in
boreal and tropical ecosystems but also to generate the necessary
methodology and knowledge for the sustainable management.
Figure 3: Sustainability and phosphorus dynamics: Main factors are
environment, society and economy (adapted from [9]).
Phosphorus limitation of ecosystems’ plant productivity
In tropical parts of the world with their many millions year old soils, P is
often the limiting nutrient since primary P-containing minerals are depleted
with soil age [15, 16]. In these old tropical soils, as typical found in Africa (s.
Figure 4), the remaining P is often bound strongly to secondary iron and
aluminum minerals and is considered not to be bioavailable. Furthermore
atmospheric input of P is quite low [17]. As a consequence, P limitation
limits agricultural and forest productivity and is - besides social, political and
economic imbalances - a major cause of famine in this part of the world [18].
However, P limitation is not only associated with highly weathered soils in
the tropics but can also occur in boreal systems (s. Figure 4), as recent

5
studies have demonstrated [19-21]. As the second largest biome, the boreal
forest accounts for a third of the global woodland cover and is crucial for
biodiversity, economy and as an important carbon sink [22]. A large body of
research has studied the factors behind growth of this boreal ecosystem and
ca. 25 elements of soil nutrients were identified with N and P often the most
important ones. Fertilization with these elements is expected to increase tree
growth.
Figure 4: Example of tropical environment (left: Burkina Faso) and boreal
environment (right: forest in northern Sweden); (photos: Burkina Faso:
Andrea Vincent and boreal forest: Johan Vestergren)
For a long time, N was seen as being the main limiting nutrient in boreal
forests. Therefore most research focused on understanding the role and
mechanism of N for forest growth [23]. For P much less research was carried
out, partially due to the appearence of soil P in many different molecular
species. Only recently it was found out that P can also act as an important
limiting factor for plant growth in boreal ecosystems [15, 20, 24] . The likely
explanation for this locally limiting role of P is that a large fraction of soil P is
chemically bound in stable complexes, in particular on soil surfaces. This
holds also for the organic P-pool, which mostly consists of mono- and
diesters of phosphoric acid. This is a direct consequence of the unusually
large reactivity of phosphate groups towards environmental particles in
comparison with other nutrients [8, 25, 26]. These mineral and organic soil
particles are, together with amorphous aluminium and iron compounds, the
main factors determining soil sorption capacity for organic P [27, 28]. Soil
adsorption stabilizes the organic P-compounds against microbial
degradation; a process which prevents biological utilization of this P-pool by
plants [12]. However, there is only sparse knowledge about the mechanisms
of these surface reactions and how they affect the organic P-speciation,
enzymatic degradation and bioavailability. Adsorption of P-species probably

6
also affects losses of P from terrestrial P into aquatic systems, because soils
with surface reactive properties causing P-limitation are also found at the
transition zone between terrestrial and aquatic ecosystems. In freshwater
ecosystems the organic P-availability also affect primary production and the
heterotrophic energy mobilization rate [29]. A likely climate impact in the
northernmost ecosystems will result in an increased leaching loss of
dissolved organic P into these aquatic systems.
Phosphorus: Its way to bioavailability
All the mineral P located in the surface layer of our planet is found in its
oxidized phosphate form in primary minerals such as the minable apatite
family [16]. In contrast N compounds can be biologically created from
atmospheric N2 by different N-fixing organisms. Weathering and microbial
breakdown of P-containing minerals releases the P to the soil. Here, plants
and organisms in the soil microbial community consume P. By converting
inorganic P such as phosphate, pyro- and polyphosphate (s. Figure 5) e.g.
into organic P, they generate the P-containing molecular building blocks
needed for their growth. Most input of organic P to the soils originates from
P-diesters [12]. Even so, P monoesters are the prevailing form of organic P in
soil. Organic P is continuously degraded into plant accessible phosphate by
enzymes and acids, produced by roots and microbes, so that over time P
cycles between being fixed in organisms and being released into the soil as P-
monoesters. (s. Figure 5).
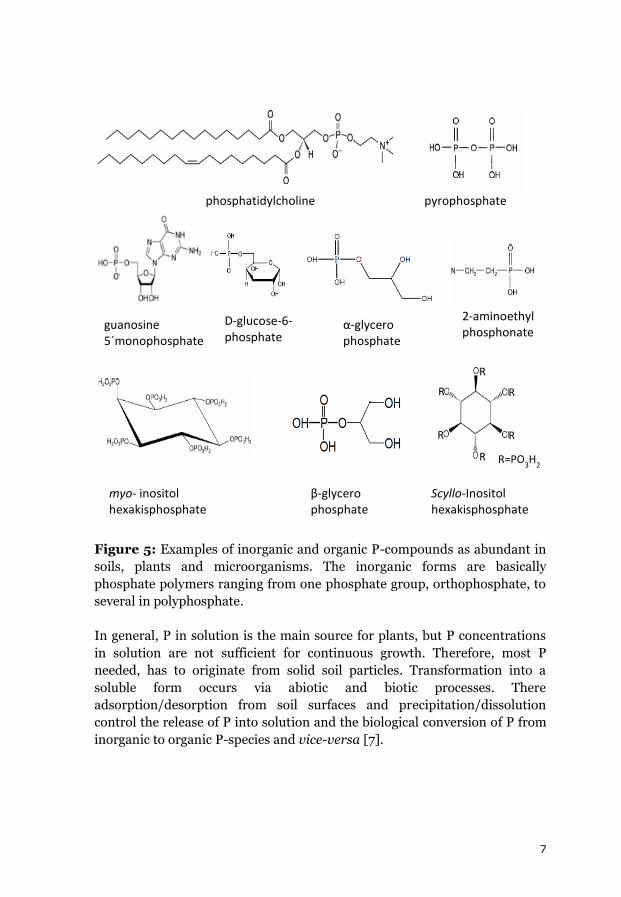
7
Figure 5: Examples of inorganic and organic P-compounds as abundant in
soils, plants and microorganisms. The inorganic forms are basically
phosphate polymers ranging from one phosphate group, orthophosphate, to
several in polyphosphate.
In general, P in solution is the main source for plants, but P concentrations
in solution are not sufficient for continuous growth. Therefore, most P
needed, has to originate from solid soil particles. Transformation into a
soluble form occurs via abiotic and biotic processes. There
adsorption/desorption from soil surfaces and precipitation/dissolution
control the release of P into solution and the biological conversion of P from
inorganic to organic P-species and vice-versa [7].
D-glucose-6-phosphate
guanosine 5´monophosphate
myo- inositol hexakisphosphate
pyrophosphate
2-aminoethyl phosphonate
Scyllo-Inositol hexakisphosphate
phosphatidylcholine
α-glycero phosphate
β-glycero phosphate
R
R R
R R
R R=PO3H
2

8
Boreal forest soils contain a range of organic P compounds, often as a major
fraction of total soil P [30]. Much of this organic P occurs in small organic
molecules with high energy content and high nutrient values. These
molecules include numerous molecular species, often monoesters such as
inositol-phosphate or nucleotides, diesters such as DNA or RNA,
phospholipids from cell membranes of organisms and phosphonates. All of
them, as well as inorganic polyphosphate and pyrophosphate (s. Figure 5)
require hydrolysis before biological utilization. Organic P is continuously
degraded into plant accessible phosphate by enzymes and acids, which are
produced by roots and microbes. After some time of repeating death and
degradation cycles of plants and microbes, the P is released once more into
the soil as P-monoesters.
However, the release of phosphate and thereby its bioavailability for plants is
controlled by the degradation rates of the organic P-compounds and their
release by desorption from mineral surfaces in the soil matrix and presence
of multivalent cations (Fe, Al, Mn…) in the surfaces. However, the strength
of adsorption, which can prevent release completely in very mineral rich soil
as found in the tropics [31], depends not only on the soil matrix but also
crucially on the type of organic P- species.
The tendency for adsorption of a P-species increases with the number of
phosphate groups and therefore its negative charge at a given pH. Therefore
phosphate diesters have a lower propensity for adsorption than monoesters
and are easier accessible for microbes, making them more bioavailable than
e.g. monoesters [32]. Monoesters like inositol phosphates but also inorganic
phosphates can due to their high charge bind much stronger to mineral
surfaces and even form very stable complexes with multivalent metal ions
[28].
31P NMR Spectroscopy: an innovative tool for studying the speciation of organic phosphorus and the need for advanced techniques to study P-species.
While the role of N in boreal forests and other ecosystems has been studied
in large detail [23], much less is known about the role of P as a nutrient and
a limiting factor in these systems [20, 21, 24, 33]. The main reason for this
limited knowledge about soil P is the sheer complexity and heterogeneity of
soils. As a Science editorial stated “ The processes occurring in the top few
centimeters of the earth’s surface are the basis of all life on dry land, but the
opacity of soil has severely limited our understanding of how it functions”
[34]. The problem with P stems from the fact that P is abundant in many
different molecular species, which can in addition also be bound physically

9
to soil surfaces and form complexes with the metal ions present in these soils
[35-37]. Therefore, identification of these numerous organic P-species,
elucidating their origin and understanding their chemical and biological
dynamics in soils (and in aquatic environments which is not the main scope
here) is crucial in ascertaining their role in ecosystems and their effects on
soil fertility. In addition, comprehensive information about soil P is essential
in understanding the response of P-biochemistry to global environmental
changes and in sustainable P-fertilizer management in agriculture [11, 13,
14]. Therefore, it is quite obvious that advanced experimental techniques
with the capabilities of analyzing the speciation and dynamics of organic P in
these complex systems at the molecular level are greatly needed.
NMR has emerged in recent years as the most powerful and versatile
technique to obtain molecular information on the nature of soil components
and their interactions [38]. NMR can now also be used to elucidate soil
processes at various scales. While liquid-state NMR on soil extracts can be
applied to extract structural information of individual components, diffusion
NMR and related techniques can study the formation of colloidal soil
aggregates from organic matter. Solid-state NMR techniques such as magic
angle spinning (MAS) NMR and high resolution (HR) MAS NMR add
another layer of information by being able to explore a soil matrix directly
and to extract details about larger complexes and their internal arrangement,
in addition, the adsorption behavior of smaller molecules (organic
molecules, contaminants) to soil interfaces and aggregates can be studied.
Finally, also magnetic resonance imaging as known from its medical
applications, is now applied in exploring distributions of specific
components across soil layers, as is localized spectroscopy of specific spots in
soil profiles [38].
Phosphorus consists of just one isotope, 31P, which is NMR-visible with a
high sensitivity. Already basic liquid 31P NMR on soil extracts can be used to
provide identification of various organic P-species of unknown origin [39].
As a result, liquid NMR studies on extracts have been widely employed not
only to identify P-containing molecules in complex mixtures (metabolomics
approach [38]) but also to determine their structure (molecular
identification approach [38]). More recently, also solid state NMR
techniques (31P MAS NMR) gained an increased interest in soil science, since
these approaches can directly study unperturbed solid soil matrices, as well
as complexes of small organic P-species adsorbed to mineral- or biological
surfaces, or colloidal particles [40-44].

10
Liquid NMR in soil science
NMR on liquid samples containing the molecules of interest in solution, is
the most commonly used type of NMR experiments in chemistry, structural
biology and even soil science. In solution, molecules tumble fast which leads
to well resolved and easy-to-analyze isotropic NMR signals. Thus, liquid
NMR provides molecular details that allow structure determination of
molecules, or their identification, because the positions values of these
signals are then indicative for specific P-compounds [45].
NMR as an approach for studying soil organic P has been employed since the
80’s to identify different species of organic P in soil matrices [46]. Because
the sample has to be liquid, soil specimen have to be treated in a way which
releases their P-species into the solution to be used in the NMR experiments.
For this purpose various extraction procedures have been developed, based
on strong acids or bases to extract organic P from soils in a quantitative way
[47, 48]; or step-wise using different extraction conditions to extract
different portions of P in each step [49]. A single-step extraction using
sodium hydroxide/ethylenediaminetetraacetic acid (NaOH/EDTA)
treatment was developed by Cade-Menun, and colleagues [47, 48, 50, 51],
and has become the most widely used extraction protocol, suitable for most
soils and even other matrices such as marine particles and sediments [52,
53]. Extracts produced this way presumably contain nearly all bio-available
P, and are routinely studied by liquid NMR to identify different organic P-
species and to compare them quantitatively [47, 48, 50]. However, the
conditions used in most extraction protocols alter the chemical structures of
various P-species [54-56]. A typical 1D 31P NMR spectrum of a boreal soil
extract is shown in figure 6.
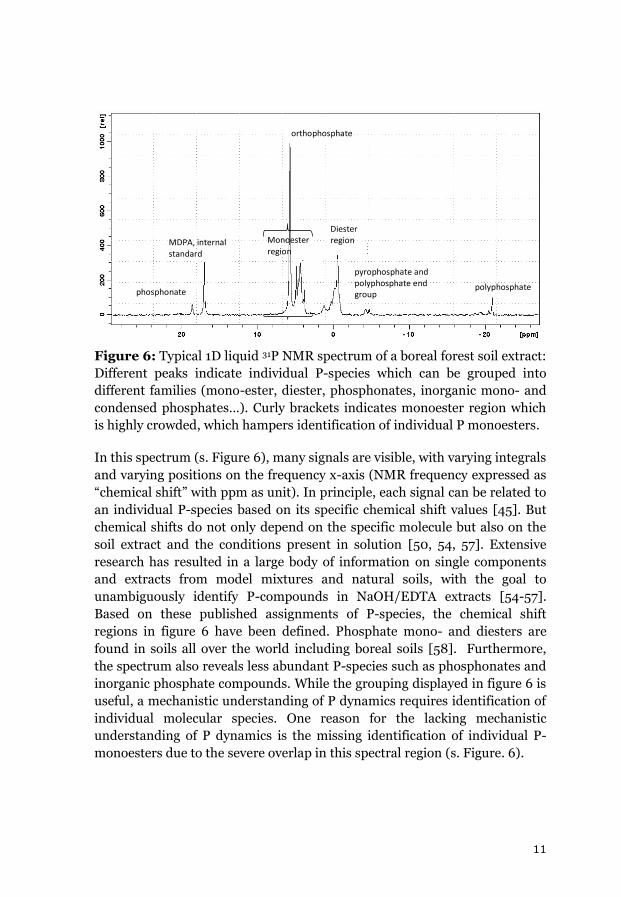
11
Figure 6: Typical 1D liquid 31P NMR spectrum of a boreal forest soil extract:
Different peaks indicate individual P-species which can be grouped into
different families (mono-ester, diester, phosphonates, inorganic mono- and
condensed phosphates…). Curly brackets indicates monoester region which
is highly crowded, which hampers identification of individual P monoesters.
In this spectrum (s. Figure 6), many signals are visible, with varying integrals
and varying positions on the frequency x-axis (NMR frequency expressed as
“chemical shift” with ppm as unit). In principle, each signal can be related to
an individual P-species based on its specific chemical shift values [45]. But
chemical shifts do not only depend on the specific molecule but also on the
soil extract and the conditions present in solution [50, 54, 57]. Extensive
research has resulted in a large body of information on single components
and extracts from model mixtures and natural soils, with the goal to
unambiguously identify P-compounds in NaOH/EDTA extracts [54-57].
Based on these published assignments of P-species, the chemical shift
regions in figure 6 have been defined. Phosphate mono- and diesters are
found in soils all over the world including boreal soils [58]. Furthermore,
the spectrum also reveals less abundant P-species such as phosphonates and
inorganic phosphate compounds. While the grouping displayed in figure 6 is
useful, a mechanistic understanding of P dynamics requires identification of
individual molecular species. One reason for the lacking mechanistic
understanding of P dynamics is the missing identification of individual P-
monoesters due to the severe overlap in this spectral region (s. Figure. 6).
MDPA, internal standard
Monoester region
orthophosphate
Diester region
pyrophosphate and polyphosphate end group
polyphosphate phosphonate

12
With this type of 1D liquid 31P NMR approach on soil extracts, knowledge
have dramatically increased in the last decades about P-speciation, and our
understanding of the biogeochemistry of soil P, and its role in the relevant
ecosystems [47, 48]. This methodology was even applied to other P-
containing matrices ranging from lake sediments to manure, and even to
colloidal particles in aquatic systems [59-61].
Despite its huge success in soil science in the last decades, 1D 31P NMR using
soil extracts has major shortcomings which restrict the exploitation of the
full power of liquid NMR in this field. One main reason is the appearance of
spectral overlap in crowded regions, due to signals with broad line widths
and the small chemical shift dispersion of the 31P nucleus in these
compounds. Because of that analysis of these 1D 31P NMR spectra is often
complicated; a problem which severely hampers e.g. a complete
identification and quantification of all individual components in the
interesting mono- and diester regions. Furthermore, the sample matrix
influences the chemical shifts of P-species to a varying degree; which can
make unambiguous identification difficult. To relieve this problem partially,
spiking experiments have been carried out which enabled at least the
identification of the main components [54, 62-64]. However, this procedure
is not possible for less abundant P-species whose signals would be hidden
under these larger peaks. Furthermore, different species of the same P-
family, such as monoesters can have the same chemical shift, so that the full
spectral integral would be attributed to the P-species used for spiking, while
the other component would be overlooked. Therefore, many NMR studies
report not individual P-species but only the monoester region in total, thus
abandoning all biologically most interesting information on specific,
components of this P- species family.
In general, the occurrence of spectral overlap in these NMR spectra is not
primarily due to the sheer number of individual P-species e.g. in the
monoester region, but due a side effect of the NaOH/EDTA extraction
procedure used routinely for extracting P-species from soil matrices [47, 48].
While this method extracts presumably most of the bioavailable inorganic
and organic P, it also transfers high amounts of paramagnetic metal ions
(e.g. Fe3+, Mn2+) into solution due to the strong complexing properties of
EDTA. Despite being complexed by EDTA, these paramagnetic impurities
still broaden P signals much beyond their intrinsic line width of under 3 Hz,
thus causing overlap and degrading sensitivity, making identification of most
individual P impossible [39].
Finally, the routine extraction protocol itself has biological drawbacks which
have to be carefully considered in any quantitative analysis of organic P by

13
1D liquid 31P NMR. The harsh NaOH/EDTA extraction method with its high
pH and duration causes hydrolysis and degradation of organic P-species
[47]. Depending on the type of P-compound and the time used for
extraction, the degree of degradation varies between P-species [55], making
not only quantitative comparison difficult but also masking the original P-
species present in the soil matrix prior extraction.
Solid state 31P MAS NMR on intact soils
While solution NMR relies on extracts from soil samples, solid state MAS
(magic angle spinning) NMR approaches can probe soil material directly.
This method, still much less used than liquid NMR in soil science, can access
large complexes and molecules adsorbed to immobile matrices, where the
reduced tumbling rates exclude the used of solution NMR. These solid state
NMR methods are able to characterize interactions between molecules and
solid material, and that not only from the point of the adsorbed molecule [8,
65] but (if suitable NMR isotopes and conditions exist) also from the point of
the adsorption matrix [66]. However, due to the missing tumbling and the
presence of heterogeneous material and paramagnetic impurities, the
obtained NMR spectra suffer from poor resolution and often only 2 or 3
major P-species families can be identified (s. Figure 7). Due to the
differential line-broadening effect on various P-groups, only a semi-
quantitative analysis is possible if at all. However, the main advantage of this
method is the probing of the complete P-pool in the soil matrix and not only
the extractable fraction, which might often be less than half of the total P,
with debate ongoing if the extractable fraction can be considered to
represent the bio-available P-pool [37, 48, 49].
Nevertheless, 31P MAS NMR has been employed in studying a wide range of
samples and ecosystems and has generated useful information despite the
limitations of the obtained NMR spectra. Using model minerals the surface
speciation of specific P components could be studied under controlled
conditions, as recently shown with phosphate on Boehmite mineral [8]. This
information could then be transferred to real soil matrices to understand the
dynamics and sorption pattern of individual P-species there; information
essential for understanding the mobility and biological availability of these
P-species to plants [47]. But also dissolved organic matter in aquatic system
has been studied by 31P MAS NMR upon enrichment by ultrafiltration [67].
By investigating dried material from high-molecular weight organic material
originating from various sea depths (0-4000 m) they found a constant ratio
between P esters and phosphonates. They concluded that dissolved organic P
is preferentially remineralized from dissolved organic matter, and its
selective removal form this matter indicates the demand of marine

14
microorganisms for this nutrient. To avoid lyophilization of huge amounts
of water for this type of research, enrichment procedures using small
amounts of water where suspended colloidal particles based on polymeric
ion exchange material adsorb P-species, are investigated after simple
centrifugation as pellets by solid state 31P MAS NMR ([68], ongoing
development in the lab). Nevertheless 31P MAS NMR is not only employed
on natural P-species but is also ideal in tracing the behavior of P-based
xenobiotics (warfare components, pesticides etc) in soil matrices, especially
their mobility there and any transformation processes which can cause
alteration of their toxicity and bioavailability [65].
Figure 7: A comparison of spectral resolution between solid state 31P MAS
NMR (top trace, 4 kHz spinning speed) and liquid (bottom trace) 31P NMR.
In 31P MAS NMR the large peak in the middle consists of several P
components e.g. monoesters, diesters and inorganic P, concealed by the poor
resolution associated with solid state 31P MAS NMR on intact soil samples.
To the left and to the right of this main peak are two emerging peaks that are
the MAS spinning side bands and arise due to the spinning of the sample
during the NMR experiment. The bottom trace reveals several P peaks
although they are not completely resolved. The same chemical shift range is
present in both spectral traces and differences in chemical shift are due to
differences in pH between soil and soil extract.

15
Aim of thesis
While much research has been carried out to elucidate the role of nitrogen as
nutrient in ecosystems, much less knowledge exists about the role of P. The
main reason for this is not only the complexity of soils as the main P source
in terrestrial ecosystems, but also the appearance of P in many different
molecular species. And these P-species can even be adsorbed to soil surfaces
and interact with multivalent metal ions (e.g. Fe3, Al3+, Mn2+ …) present in
these soils. Therefore, advanced experimental molecular techniques are
required for identification of the individual P constituents composing the
organic P-pool in these soils, and for tracking their behavior during soil and
ecosystem development. And one of the most promising method for this
purpose is liquid 31P NMR on soil extracts. This approach provides a wealth
of information on P-speciation and dynamics in soils, but its wider
application in soil sciences is severely hampered by the fact that the presence
of paramagnetic soil impurities causes poor spectral resolution and prevents
application of multi-dimensional NMR methods for identification of
unknown P-components and separation and quantification of all P-species
present in the soil´s organic P-pool.
The general objective of this thesis was therefore to carry out a
multidisciplinary study to develop and implement the necessary 31P NMR
methodology for providing new basic knowledge on a molecular level about
the speciation and dynamics of organic P-species in various ecosystems. The
specific objectives were:
Developing biologically meaningful new methods in organic P specification
in agricultural and forest soils based on solid state/liquid 31P NMR
techniques. In particular, to implement 2D liquid NMR experiments for
unambiguous P-species assignment and quantification; an objective which
requires development of soil extraction methods which remove NMR
interfering paramagnetic impurities.
Analyzing the speciation and dynamics of organic P in soils in different
ecosystems: i) boreal forest productivity gradient, ii) humus soil
chronosequence (7800 years) in Northern Sweden, and iii) Semi-arid
tropical agricultural woodlands with different soil mineralogy.
Understanding the chemical and biological dynamics of organic P-species in
these ecosystems and how these contribute to soil fertility and to plant and
microbial growth.

16
By addressing these objectives we will not only develop new NMR based
molecular techniques for soil science, but we will generate molecular
information important to understand the environmental behavior of organic
P released from the terrestrial environment and its microbial availability; key
issues in future climate related effects on our high-latitude ecosystems and
in combating P-restriction in food/forestry production, especially in tropical
third world countries.
Materials and Methods
NMR spectroscopy
The NMR signal
Most, but not all nuclei have a magnetic spin (I) which allows them to be
susceptible to an external magnetic field. The most commonly used nuclei in
chemistry are 1H, 13C, 15N and 31P which all have I=1/2. Putting the sample in
an NMR spectrometer with a large internal static magnetic field with a
magnetic field strength B0, all magnetically susceptible nuclei align parallel
or antiparallel to this external field much like a compass needle aligning to
the earth’s magnetic field (s. Figure 8). These orientations reflect a lower (α)
and a higher (β) energy state, respectively. The discrete difference between
these two energy states is proportional to the gyromagnetic constant, γ. The
lower energy state is marginally more populated resulting in a bulk
magnetization called Mz or the longitudinal magnetization. However the
energy difference is quite small and this causes NMR to be a rather
insensitive technique that requires a sample with high concentration. In
NMR experiments, radiofrequency (RF) signals induce transitions between
the two states, when the radiofrequency fulfills the resonance condition.

17
Figure 8: In its relaxed state, the macroscopic sample magnetization is
found aligned to B0 parallel to the z- axis in a three dimensional coordinate
system.
When a RF pulse is applied along the x-axis it creates a small magnetic field
perturbing the position of Mz angling it towards the x- y- plane (s. Figure 9).
Here the Mx,y components rotate around the z-axis with the frequency ν,
which is called the Larmor frequency. The rotating magnetization generates
a radiofrequency field which is picked up by a detection coil. The signal
detected is at this point in a time domain and is known as Free Induction
Decay (FID). This can be transformed using a Fourier transformation
converting the time-domain signal into a frequency-domain signal, which is
the NMR spectrum. After the Mz component has been perturbed in an NMR
experiment, is relaxes back to equilibrium with a characteristic time constant
T1, also called the spin-lattice relaxation time, which is experimentally
important to allow quantification of NMR spectra.
Figure 9: Here the magnetization vector, M0, to the left is before the 90˚ RF
pulse and to the right after the RF pulse. From that position it will relax back
to the static magnetic field, B0, at the z axis.

18
The RF pulse is short, generally in the µs time regime and adjusting pulse
length together with pulse power can give maximum signal when Mz is
completely rotated into the x, y plane, by a so-called 90º pulse.
The chemical shift
The chemical shift is the position of one signal in an NMR spectrum, relative
to the position of a specific standard in a ppm scale. The chemical shift is
created by the fact that each nucleus experiences a slightly modified B0
leading to slight variation in , reflecting its chemical surrounding i.e.
electron density and how it is affected by other atoms in the molecule. A
change in the structure of the molecule means a change in the electronic
environment and will move its position along the frequency axis [45].
Because the variation in is small, it is usually given in parts per million
(ppm) relative to a reference compound, according to equation δ = (νmolecule -
νstandard)/ νstandard. This equation can be used to calculate the chemical shift. δ
is the chemical shift, ν is the resonance frequency and ν standard is the
frequency of the reference compound. For 31P NMR, 85 % phosphoric acid is
used as an external standard with chemical shift δ =0.0 ppm.
Experimentally, it is essential that the B0 field is as homogenous across the
sample volume as possible, so that identical molecules in any volume
element will contribute signals with the same frequency, so that (i) the signal
of each P-species has maximal intensity, and (ii) the signals of all P-species
are as sharp as possible for optimal resolution in the spectrum. Homogeneity
of B0 is optimized using a set of coils (“shim coils”) situated around the
sample in the NMR magnet. Currents in these shim coils are manipulated in
such a way that the magnetic field becomes as homogeneous as possible.
Indirect spin-spin couplings
Indirect spin-spin couplings - also known as J-couplings or scalar couplings -
are crucial for structure determination and identification of unknown
compounds. Interaction of one spin with the spin states of other NMR-active
nuclei through up to four connecting bonds causes splitting of the peak of the
observed nucleus. When n is the number of equivalent neighboring nuclei,
this effect causes a splitting of the signal in a pattern of n+1 multiplet lines.
The lines follow a specific pattern intensity ratio that for a spin system of
I=1/2 is given by Pascal´s triangle, [69], which is valuable for structure
determination. Within a group of chemically equivalent nuclei, splitting due
to spin-spin couplings does not occur. The distance between lines within a
multiplet is called the J-coupling constant. This constant is independent of

19
magnetic field strength and it is measured in Hertz (Hz). The size of J
couplings depends on a molecule’s conformation and therefore helps to
identify a molecule and determine its structure. The splitting effect between
different nuclei can be cancelled out using a broadband decoupling sequence
that irradiates the nucleus not being observed continuously. Examples of
such sequences are WALTZ-16 and GARP [70]. Spin-spin couplings are an
important factor in many experiments because they can be used to create
magnetization transfer between nuclei, which is the basis for a large group of
multidimensional correlation experiments.
Line broadening contributing factors
In order to analyze NMR spectra properly all individual resonances have to
be separated, i.e. the spectra have to be fully resolved. Resolution is strongly
affected by the line widths of the signals, which are specified through the
peaks’ width at half height, Δν1/2, measured in Hz. The line width of an NMR
signal is reverse proportional to the spin-spin relaxation time T2, so any
mechanism that accelerates T2 relaxation increases line width. Important
factors affecting relaxation and therefore line width are the size of the
chemical shift anisotropy (CSA), dipolar interactions and paramagnetic
impurities [45]. Dipolar interactions and CSA are molecular properties; their
contribution to relaxation depends on B0, temperature, and the viscosity of
the NMR sample, all parameters that can only be modified in a limited
range. But for small molecules, the contribution of these relaxation
mechanisms to 31P relaxation is small, so that line widths of approximately 1
Hz can be obtained.
Paramagnetic impurities such as iron (Fe3+) and manganese (Mn2+) cause
severe line broadening if present. The unpaired electrons in the outer shell of
these ions have a magnetic momentum that can affect the spin of the
observed nucleus, causing faster relaxation and thus broader lines. This
problem is exacerbated by the high affinity of highly charged metal ions for
phosphate groups.

20
Table 1: Table adapted from Vincent et al. 2013 [58] where the chemical
shifts of both 31P and 1H can be seen. This is a direct result of removing the
paramagnetic ions and thus being able to perform 2D 1H-31P HSQC NMR on
soil extracts. In [39] fine structures are also included for additional
information. Unknowns and internal standards (MPA and MDPA) have been
removed from the table.
Label Compound 31P-shift (ppm)
1H-shift (ppm)
P family
A β glycerophosphate 4.95 4.07 Monoester
B α glycerophosphate 5.27 3.68, 3.75 Monoester
C Myo-Inositol hexakisphosphate 4.65 (C4,6) 4.52 Monoester
Cʹ “ 4.55 (C5) 4.27 “
Cʹʹ “ 5.03 (C1,3) 4.48 “
Cʹʹʹ “ 5.97 (C2) 4.48 “
D Guanosine 2´ monophosphate 4.33 4.89 Monoester
E Guanosine 3´ monophosphate 4.90 4.58 Monoester
F 2-aminoethyl phosphonate 18.98 1.59, 3.28 Phosphonate
G Uridine 2’ monophosphate 4.62 4.54 Monoester
H Uridine 3’ monophosphate 4.85 4.38 Monoester
I Choline phosphate 4.25 4.08 Monoester
J Ethanolamine phosphate 4.91 3.69 Monoester
K α-D-glucose-1-phosphate 3.39 5.34 Monoester
L Guanosine 5´ monophosphate 4.78 3.84, 3.89 Monoester
M DNA –0.5 to 0.0 3.80–4.20
4.62–4.98
Diester
N Adenosine 5´ monophosphate 4.77 3.85, 3.90 Monoester
O Phosphatidyl choline 0.69 4.23, 3.79 Diester
P Phosphatidyl ethanolamine 1.79 3.81 Diester

21
Q Adenosine 2´, 5´ diphosphate 4.54 (2´) 4.93 Monoester
R “ 4.81 (5´) 3.88, 3.94 “
T Scyllo-Inositol hexakisphosphate 4.14 4.47 Monoester
A1 Orthophosphate 6.29 Orthophosphate
A4 Polyphosphate (end-chain) –3.70 to–4.27 Polyphosphate
A5 Pyrophosphate -4.14 Pyrophosphate
A6 Polyphosphate (mid-chain) –18.5 to–21.0 Polyphosphate
The 2D 1H-31P HSQC NMR experiment
In 1D 31P NMR, acquisition of the FID starts directly after the RF pulse, and
the FID is Fourier transformed and the spectrum analyzed after suitable
post-processing. For 2D NMR, an evolution time and magnetization mixing
are inserted between the initial excitation pulse and data acquisition. It is of
significant importance that all pulses are accurately calibrated. The HSQC
was developed in 1980 by Bodenhausen and Ruben [71] and has quickly
become the mainstay of analytical tools in protein chemistry. The HSQC also
has potential as a valuable tool in other disciplines such as environmental
research [39]. In a 1H- 31P HSQC experiment, 1H magnetization is excited
and transferred via J couplings to 31P. Then 31P chemical shifts are recorded
in the evolution time, and finally 31P magnetization is transferred back to 1H
for detection. The evolution time, t1, is systematically varied starting with
t1=0 and incrementing for each of the n experiments that create the raw data
of the 2D experiment. Thus, NMR signals are obtained that depend on two
time axes, the “indirect” time domain created by incrementing t1, and the
“real” time domain t2 corresponding to recording of the FIDs. Modulations of
the signals as function of t1 and t2 encode 31P and 1H chemical shifts,
respectively. Two Fourier transformations are performed and the result is a
spectrum with two dimensions [72, 73]. Fourier transformation of t2 creates
the frequency dimension for the more sensitive nucleus, usually 1H, and
transformation along of t1 creates a second frequency dimension of the less
sensitive nucleus e.g. 31P. We now have two chemical shifts, and because the
HSQC transfers magnetization between J-coupled nuclei, the 2D spectrum
correlates the chemical shifts of J-coupled 31P and 1H nuclei. The spectra are
commonly presented as contour plots like topographical maps (s. Figure 10).

22
The combination of two chemical shifts vastly increases resolution compared
to 1D 31P NMR, because P-species with identical 31P chemical shifts can be
resolved based on different 1H chemical shifts. Table 1 lists 1H and 31P
chemical shifts of some typical P compounds found in soils.
Figure 10: A typical 2D 1H-31P HSQC spectrum showing the monoester
region, with a 1D 31P NMR spectrum as overlay. This overlay shows how the
increased resolution of the 2D spectrum allows identification of individual P-
species which are not resolved in the 1D spectrum. The circled peak X is an
unidentified P-species present in both boreal and tropical samples.
Inductively coupled plasma optical emission spectrometry (ICP-OES)
ICP-OES is a well-established and frequently used technique. Today over 70
elements of a total 92 can simultaneously be analyzed in a matter of minutes.
The main feature of this method is the plasma ionization and the optical
emission detector. Samples are introduced generally as a liquid and are
promptly nebulized into an aerosol before entering the plasma torch. The
sample is transported through the center of the plasma torch by argon gas.
This argon gas is also part of the plasma discharge. The argon gas passes a
RF copper coil that applies an oscillating RF current creating an electric and
magnetic field. As argon gas passes through the RF field electrons are
removed from the argon atoms and caused to crash into other argon atoms

23
by the magnetic field creating a continuous ionization process of the argon
gas. This adds energy to the process and is known as inductive coupling. This
converts the otherwise non-reactive gas argon to turn into argon plasma with
temperatures between 6 000 and 10 000 K.
The sample when passing through the center of the plasma torch spends
approximately 2 milliseconds in the center creating optimal conditions for
low matrix interference. As the sample subsequently passes through the
plasma it is first removed of all solvent and then the remaining salts are
vaporized and molecules broken into atoms. These atoms are then excited by
the high consistent temperature in the plasma. Excitation is the process in
which one to several electrons is moved from their ground state into an
excited state. When they go back from the excited state they release energy in
the form of a photon. This energy is specific for each element and each
elements different excited states and this creates a highly accurate method
for analyzing elements. In the optical emission detector the light created by
the released photon is detected and analysis is complete. The detection limit
for ICP-OES is in the parts per billion (ppb) range and this is applicable to
the majority of the more than 70 elements possible to analyze with this
method. ICP-OES, Optima 200 DV (PerkinElmer, Überlingen, Germany);
equipped with Cross-flow nebulizer and double spray chamber was used for
elemental analysis. A portion of 50 mg of lyophilized extract was dissolved in
20 ml of 2% HNO3 (aq.). If needed steps were taken to insure there was no
particulate matter left. Standards with concentrations of 2 ppm and 10 ppm
were made for Al, Fe, Mn and 31-P and used in all ICP measurements
throughout my PhD project.
X-ray fluorescence spectroscopy (XRF)
XRF is a non-invasive analytical technique widely used in archaeological,
industrial, geological, and environmental studies [74]. A problem for XRF is
the matrix effects. That is most commonly caused by the composition and
grain size of the observed sample. By grinding the samples into a fine powder
these effects can be minimized. XRF on powder, pellets, or fused beads
needs quite a large sample. The sample sometimes need to be in the range of
several grams for accurate elemental quantification [75]. The latest of
wavelength-dispersive XRF (WD-XRF) machinery gives the possibility to
minimize the amount of sample by specific calibration procedures on small
sample volumes. The XRF measurements in paper 3 were made on a Bruker
S8 Tiger WWD machine.

24
Other analytical methods used in this thesis
The EA-IRMS elemental analysis for carbon and nitrogen, in paper 3, was
made using an elemental analyzer (Flash 2000 Organic Elemental Analyzer,
Thermo Fisher Scientific Inc., Bremen Germany). Multi variate analysis was
performed on Simca 13.0 32-bits (SIMCA v.13.0 Umetrics, Umeå, Sweden),
with the 1D 31P NMR integration data set combined with elemental analysis
data from XRF and EA-IRMS.
Site descriptions
In this thesis several sites in different ecosystems were investigated. At each
site, there was a hypothesis that P-speciation might change as a function of
variation in site conditions. Therefore the sites allowed us to investigate the
influence of abiotic and biological parameters on P biogeochemistry. The
first site was Betsele, where a study on six boreal humus layer soils was
performed. Betsele is situated in the Umeå river valley northwest of the town
of Betsele in northern Sweden. It is a 90 m productivity gradient with a
groundwater recharge area and a groundwater discharge area. The gradient
area has a slope of 2% and has been thoroughly described in literature [24,
76, 77]. The transition between groundwater recharge and discharge is
associated with a pronounced change in soil properties and in the vegetation.
Therefore the goal of paper IV was to investigate relations between the
parameters changing along the gradient and P biogeochemistry [78].
The second sample area consisted of eight sites on the coast outside Umeå in
northern Sweden. These sites were chosen to perform a chronological
gradient due to the landrising occurring there (9 mm year-1). The coast
outside Umeå is rising as a result of the last ice age. The age of the sites could
therefore be calculated from the elevation above sea level for each site. The
youngest site was approximately 90 years and the oldest 7800 years. Here a
total of eight sites were included and from all sites we collected five boreal
humus layer soil samples. From these chronosequence soil samples were also
used for paper I [39] as well as for paper II [58].
Finally, the last two sites are representative of sub-sahelian tropical soils and
are taken in Burkina Faso, representative for old soils where adsorption of P-
species to surfaces of secondary minerals is thought to strongly limit P-
bioavailability. The two Burkina sites represent one oxisol soil and one
Ferralsol soil. Outside the village of Saponé the soils are sandy and very poor
in P. The other soil comes from Finlande, another village that is situated in
an area where the soils are richer in P and organic material than Saponé. The
samples were taken under tree canopies and outside tree canopies in a

25
previously determined pattern, with the aim to test the hypotheses that soil
type and presence of trees might influence P-speciation. This studies are
described in paper III.
Figure 11: On the left is a typical soil profile from a boreal soil in northern
Sweden. On the right is a soil profile from Saponé in Burkina Faso
containing depleted soil with only small amounts of nutrients. (Pictures
taken by Reiner Giesler, boreal soil; Ulrik Ilstedt Saponé soil).
Sampling and general preparations
At each individual site four to six replicate samples were taken and pooled.
Samples were taken with a soil auger at a depth of 5 to 15 cm except for the
Burkina Faso samples which were taken in 10 cm segments at 0 to 50 cm and
100-120 cm. The Burkina Faso study in paper III focused on the 0 cm to 10
cm layer. The boreal soil samples from Betsele and the land rising coast
chronosequence (LRC) were stored under cold conditions directly after
sampling. These samples were then sieved using a 4 or 5 mm steel mesh and
subsequently dried. After drying, a sufficient portion was ball-milled into a
fine powder ready for extraction or direct analysis. Betsele samples were
milled by hand in a mortar. The Burkina Faso samples were dry already on
sampling and were transported and kept dry prior to analysis. The tropical
samples were sieved and milled in the same fashion as boreal soils.

26
Preparations for solid state NMR
The dried and milled soil sample was carefully packed in a 7.5 mm zirconia
MAS sample rotor without any further treatment. Each rotor was weighted
prior and upon sample loading. Solid state 31P NMR spectra were acquired
on a Chemagnetics 400 MHz spectrometer (Chemagnetics/Varian, Fort
Collins, CO, USA) equipped with a 7.5 mm double resonance probe. A 31P
frequency of 162 MHz together with a proton frequency of 400 MHz was
used for all experiments and a MAS spinning speed of 4 kHz at 293 K. A
single pulse excitation sequence together with proton decoupling was used
[79] All spectral processing was done using the Spinsight software
(Chemagnetics).
Sample preparations for liquid NMR by conventional extraction
For liquid NMR further preparations are necessary. Specifically for liquid 31P
NMR a common extraction scheme using NaOH-EDTA was developed by
Cade-Menun and Preston[50]. The suggested mechanism for solubilizing
and extracting soil organic P is derived from the proposed adsorption of
anions such as organic P on soil mineral surfaces by Russel [80] (s.
Figure.12). The hydroxide ions are drawn into the surface layer by the
metallic cations, rendering the net surface charge more negative with
increasing pH, and thus causing an electrostatic repulsion between the
surface and phosphate groups. At the same time the sodium ions replace the
monovalent cations and also neutralize the net surface charge. In practice, a
portion of 1.5 g of the dried and milled soil is extracted using a 1:20 mass
ratio between soil and extraction solution (250 mM sodium hydroxide and
50 mM ethylenediaminetetraacetic acid disodium salt (EDTA). Soil and
extraction solution were shaken together for either 4 or 16 hours. After
centrifugation, a smaller portion was set aside for other wet chemical
analysis. The majority of the extract was frozen at -80˚C or flash frozen
using liquid nitrogen and then lyophilized. In paper IV the lyophilized
material was directly dissolved in heavy water (D2O) for 31P NMR analysis in
a 10 mm NMR glass tube.

27
Figure 12: Cartoon representing the extraction mechanism of organic P as
proposed in [80]
Sulfide treatment
The conventional NaOH-EDTA extraction co-extracts a significant amount of
paramagnetic metal ion such as Fe and Mn, which are known to cause line
broadening in NMR spectra. Therefore we hypothesized that physical
removal of the paramagnetic ions would strongly improve resolution in NMR
spectra. The method that we developed adds an extra step and by that also
extra time to the NMR sample preparation but is crucial for obtaining
sharper lines in the 31P NMR spectra. The sample preparation towards liquid 31P NMR starts by dissolving the lyophilized material in D2O as stated in the
previous paragraph. In the protocol developed and reported in paper I [39],
we used a total of three D2O solutions for NMR sample preparation: i) The
bulk solvent is pure D2O, ii) an approximated 5 molar equivalents solution of
sodium sulfide (Na2S) in D2O, iii) methyl phosphonic acid (MPA) in D2O
with a concentration giving 50 µg in the NMR sample with a 10 µl addition.
To prepare a sample, 100 mg of the lyophilized sample is weighed into an
Eppendorf cap, and 10 µl MPA solution and 50 µl sulfide solution are added.
The choice of 50 µl is calculated to have sulfide ions in excess, compared to
the expected or measured amount of paramagnetic metals in the sample.
Thirdly, D2O is added to give a total volume of 500 µl. The sample mixture is
homogenized by vortexing the Eppendorf, then sulfide precipitation is
allowed to occur at room temperature overnight, approximately 18 hours.
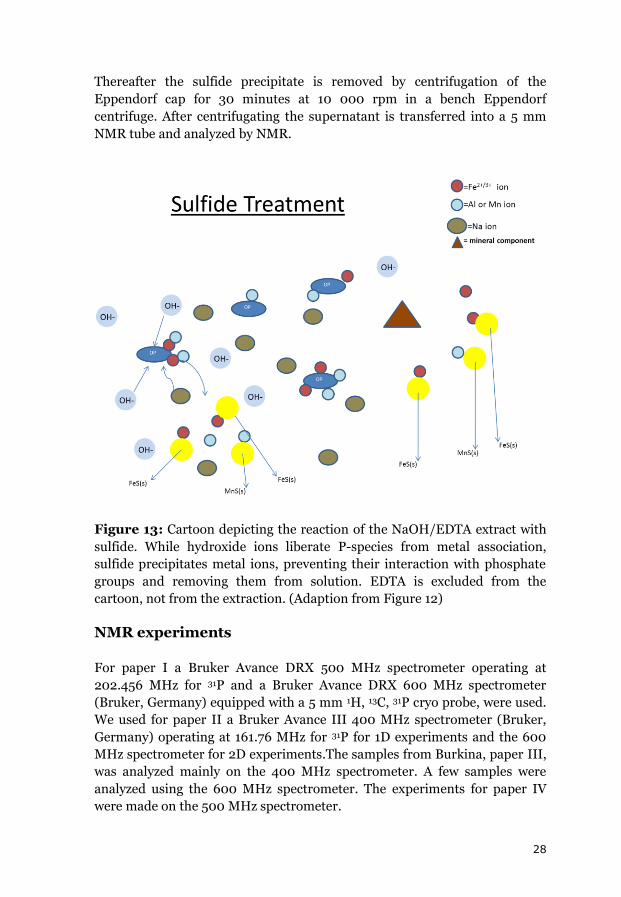
28
Thereafter the sulfide precipitate is removed by centrifugation of the
Eppendorf cap for 30 minutes at 10 000 rpm in a bench Eppendorf
centrifuge. After centrifugating the supernatant is transferred into a 5 mm
NMR tube and analyzed by NMR.
Figure 13: Cartoon depicting the reaction of the NaOH/EDTA extract with
sulfide. While hydroxide ions liberate P-species from metal association,
sulfide precipitates metal ions, preventing their interaction with phosphate
groups and removing them from solution. EDTA is excluded from the
cartoon, not from the extraction. (Adaption from Figure 12)
NMR experiments
For paper I a Bruker Avance DRX 500 MHz spectrometer operating at
202.456 MHz for 31P and a Bruker Avance DRX 600 MHz spectrometer
(Bruker, Germany) equipped with a 5 mm 1H, 13C, 31P cryo probe, were used.
We used for paper II a Bruker Avance III 400 MHz spectrometer (Bruker,
Germany) operating at 161.76 MHz for 31P for 1D experiments and the 600
MHz spectrometer for 2D experiments.The samples from Burkina, paper III,
was analyzed mainly on the 400 MHz spectrometer. A few samples were
analyzed using the 600 MHz spectrometer. The experiments for paper IV
were made on the 500 MHz spectrometer.

29
SIMCA analysis
A set of difficulties appeared proving to be unreasonably time-consuming to
solve. The inherent factors in the sample affecting pH and thus giving a small
but noticeable change in chemical shift of some P compound peaks in the
NMR spectra. This made alignment between individual sample spectra’s
impossible to achieve within a reasonable timeframe. The lack of alignment
meant that the data from the integration of a full 1D 31P NMR spectra could
not be imported into SIMCA since severe differences in the data appeared
caused by this lack of alignment. We therefore, in paper III, chose to
integrate peaks manually and then export the integration data to SIMCA. A
strategy that worked in the sense that individual peaks assigned to the same
P compound could be matched to each other and as such provide better data
for analysis in SIMCA.

30
Summary of Papers
Paper I; High Resolution Characterization of Organic Phosphorus in
Soil Extracts using 2D 1H-31P NMR Correlation Spectroscopy
Aim of the study
For many years 31P NMR spectra on soil extracts have been suffering from
poor resolution which impedes identification and quantification of soil
organic P-species. In this paper we aimed to resolve and improve the key
factors behind this problem. We intended to: First, to develop a method to
remove the paramagnetic ions that degrade the spectral resolution by
inducing line broadening. Second, to apply 2D 1H-31P correlation
spectroscopy to soil extracts, to improve identification of P-species, in
particular when very similar 31P chemical shifts preclude unambiguous
identification; third, to produce a worldwide accessible table of both 31P
chemical shifts and 1H chemical shifts of common soil P-species.
Methods and Results
Alkaline NaOH/EDTA extraction solution, the most commonly used
extraction for soil organic P, usually extracts between 30 and 85 % of soil P.
However this standard extraction creates several problems for the intended
analysis. Hydrolysis of the phosphate ester bond causes different P-species,
in particular of phosphodiesters, to degrade into smaller components and
other P-species. One example is RNA that degrades into eight
mononucleotides, because isomeric 2’ and 3’ monophosphates are formed
from each nucleotide. This hydrolysis is caused by the high pH in the
extraction solution and has so far not successfully been addressed. The
second big problem is line broadening in the NMR experiments. This is
mainly caused by the co-extracted paramagnetic metal ions. We
hypothesized that the paramagnetic ions had to be removed completely from
solution because they still affect NMR spectra even when complexed by
EDTA. By adding sulfide ions this goal could be achieved. The sulfide first
reduces Fe3+ to Fe2+ which reduces its affinity for forming complexes with
phosphate groups. Secondly, the sulfide forms insoluble metal sulfides
allowing for a convenient removal. The decrease in line width is significant,
from typically above 10 Hz down to 2-3 Hz. The longer T2 relaxation times
associated with sharper line width make it possible for the first time to
obtain good 2D 1H-31P spectra of soil extracts, significantly improving
resolution. The increased resolution is not only from sharper lines but also
from the added 1H dimension. This extra dimension adds an extra set of

31
chemical shifts for identification of each P-species, and also provides fine
structure information extractable from the J-coupling information in the 1H
lines. Now a comprehensive list with 31P and 1H chemical shifts and 1H fine
structures of soil organic P-species is available.
Conclusion
By treating the soil extracts with sulfide and subsequently removing the
paramagnetic metal ions we were able to dramatically reduce the line widths
in 1D liquid 31P NMR spectra. This proves that physically removing the ions
is essential for optimizing NMR resolution. When paramagnetic ions were
removed, the line width for all P-species decreased to 2-3 Hz, which is
comparable to “clean” samples. The decrease in line width also allowed for
the use of two-dimensional 1H-31P NMR. This is a significant breakthrough in
the analysis of the speciation of soil P, as it allows not only for the
unambiguous identification of most P-species but also gives valuable
information on the structure of yet unidentified species. Furthermore,
quantification is more reliable since overlap caused by poor resolution is
avoided.

32
Paper II; Soil organic phosphorus transformations in a boreal forest chronosequence
Aim of the study
It is known that the composition of soil organic P changes in ecosystems, as
they develop over time. We wanted to investigate how that composition
changes over time as a boreal ecosystem develops. We hypothesized that
there would be a decrease in total P and in easily mineralizable P-species
over time. We also hypothesized that P-compounds with a higher sorption
affinity would increase with progressing site age.
Methods and Results
This is the first study of soil organic P where both one-dimensional 31P NMR
and two-dimensional 1H-31P correlation NMR were used in an environmental
study. Humus horizon samples from a chronosequence spanning over 7800
years were analyzed using the commonly used NaOH/EDTA extraction to
prepare samples for 1D 31P NMR, while for the 2D experiments the extracted
material was treated with sulfide to meet the criteria of line width for multi-
dimensional NMR experiments.
Overall no clear trends supporting our hypothesis could be seen. Although
site age is not replicated results are still consistent with other similar studies
in the region. Total P showed no clear trend but was almost constant across
the gradient. Easily mineralizable P increased with the two youngest sites
and after that declined. Although soil Fe and Al content varied with site age,
no significant correlation of P sorption with age was observed.
Conclusion
The key results are that monoesters other than inositol phosphates are
predominantly RNA and phospholipid degradation products, indicating that
approximately 40 % of the extracted soil organic P originates from microbial
biomass. Secondly 2D 1H-31P NMR contains information leading us to the
possible identification of compounds not possible to identify with 1D 31P
NMR alone. These results would not have been obtainable if only 1D 31P
NMR had been used.

33
Paper III; Effect of Trees on Forms of Soil Phosphorus in an
Agroforestry Parkland in Burkina Faso (manuscript)
Aim of the study
We hypothesized that we would see a difference in organic P composition, in
a tropical agroforest parkland in Burkina Faso, between soil samples taken
under the canopies of trees, compared toin the middle between trees, due to
influences of trees on soil properties. In this study we included two different
tree species in two locations that differ in soil iron and clay content. We also
hypothesized that close to trees there would be more P forms of microbial
origin due to changes in the activity of the microbial community as an effect
of increased amounts of organic matter, and that this difference would be
more pronounced in the area with more Al/Fe clays.
Methods and Results
The main analytical techniques in this study were 31P NMR and XRF. The
data from these techniques were analyzed with multivariate analysis.
Between the two locations a clear difference in the amount of P and also in P-
composition was visible. Looking at each location, there is a difference
between under and outside tree canopies which gives good separation in the
multivariate analysis. This separation is driven by different soil parameters
at both locations, but is at both locations linked to an increased fraction of
organic P in the total soil P pool.
Conclusion
The presence of trees gives higher amounts of P and organic matter under,
compared to outside tree canopiesand it also gives a greater diversity of P-
species, probably caused by a larger microbial community. Thus, presence of
trees seems to increase the bioavailability of P.

34
Paper IV; Changes in organic P-composition in boreal forest humus soils: the role of iron and aluminium
Aim of the study
This study aimed to improve the understanding of factors controlling soil
organic P-dynamics and speciation in boreal forest soils. More specific the
objectives were to a) see if compounds with a high adsorption affinity, due to
higher number of phosphate groups creating a larger overall negative charge
on the molecule, dominate in highly sorbing soils in groundwater discharge
areas: amd b) of weakly sorbing compounds, with low number of phosphate
groups and/or sterical hindrance, occur in higher levels in a low adsorption
soils in groundwater recharge areas?
Methods and Results
Liquid- and solid-state 31P NMR methods were used to probe the soil organic
P of the humus layer at six sampling sites covering two groundwater
recharge and groundwater discharge areas in northern Sweden. The dynamic
properties of these areas make them appear as a productivity gradient with
low productivity in the recharge area and higher productivity in the
discharge area. Along this 90-meter gradient a large change in P composition
was observed. Moving from groundwater recharge to groundwater discharge
area the levels of soil organic P compounds with high affinity for adsorption
increased. At the same time the concentration of compounds with low
adsorption affinity declined. The concentrations of myo- and scyllo-, inositol
hexakisphosphates, correlate strongly with soil Al and Fe concentrations.
The correlation between P diesters and Al/Fe concentrations on the other
hand is weak.
Conclusion
The results from this study are consistent with other studies that find
positive correlation between the concentrations of especially myo-inositol
hexakisphosphate and Al/Fe. This furthermore coincides with the fact that
the greater quantity of myo-inositol hexakisphosphate is more strongly
correlated between soil/mineral surface adsorption capacity than with
parameters such as N and organic carbon concentrations and microbial
biomass. In conclusion we can see that soil organic compounds that have a
higher propensity for adsorption are found in higher concentrations in
groundwater discharge areas that have a high adsorption capacity.
Simultaneously we find that in groundwater recharge areas with low

35
adsorption capacity we have a larger relative proportion of compounds with
low propensity for adsorption, such as P diesters. Furthermore,
polyphosphate levels decline along the productivity gradient going from
recharge to discharge which could be connected to the fact that
polyphosphate is found in higher levels in fungi than in bacteria together
with the fact that the microbial community shifts from fungi-oriented
towards bacteria-oriented along the gradient.

36
Conclusions and future work
Although the commonly used extraction with NaOH/EDTA extracts a large
fraction of soil P, it suffers from severe drawbacks. Alkaline conditions with
pH above 13 cause rapid hydrolysis of especially phospholipids and RNA.
The high pH also lyses cells of microorganisms and plants, thus blending the
pools of free soil P and of cellular P. This extraction also co-extracts a
significant amount of paramagnetic metal ions like iron and manganese.
These paramagnetic ions cause a dramatic increase in 31P line width, strongly
reducing spectral resolution. The EDTA that is used to increase
solubilization of soil P also keeps the co-extracted paramagnetic ions in
solution making them constantly close to the extracted P species to be
observed by NMR. The results presented in paper I solve the problem with
paramagnetic ions, so that optimal resolution can be achieved, together with
the possibility to apply 2D 1H-31P NMR for unambiguous assignment or
identification of P-species. The problem of hydrolysis is on the other hand
still very much present and it has been claimed that the extra time added by
the sulfide treatment is of more negative effect since it adds to the hydrolysis
time. However, we consider an increase in the fraction of hydrolysis
irrelevant, because substantial hydrolysis occurs even using the usual
NaOH/EDTA extraction conditions. Given this fact, we consider application
of 2D 1H-31P NMR as a significant advance, because it is possible to
differentiate between naturally occurring metabolites and hydrolysis
products [39]. Still, for some hydrolysis products it is not possible to deduce
the original P species. Optimizing the NaOH/EDTA extraction with the
sulfide treatment, to minimize hydrolysis, may reduce this problem. Future
work should be focused on developing extraction protocols that avoid
hydrolysis, while maintaining the efficiency in extracting soil organic P.
Application of the new methodology produced well-resolved 1D 31P NMR
spectra for all soils that were analyzed. The 2D 1H-31P spectra revealed that
overlap can occur even in seemingly well-resolved 1D 31P spectra, allowed
identification of several new organic P species. Furthermore, 2D spectra gave
hints concerning the structure of some unknown P species, which remain to
be identified to extend the chemical shift collection for soil P species, and to
increase our understanding of soil P speciation. Results on boreal soils
indicated that concentrations of P species can be related to their sorption
properties, and that a large fraction of extractable P originated from living
biomass. Similar conclusions could be inferred from a completely different
system, dry tropical soils from Burkina Faso, where P species originating
from microbes were more abundant under the canopies of scattered trees in
the agroforestry parklands in soils with high sorption properties. Thus, I
finally conclude that despite remaining challenges in the 31P NMR method, it

37
can reveal interesting insight concerning ecosystem properties and
processes.

38
Acknowledgements
Fem år ganska precis har passerat och jag, projektet och mycket annat har
utvecklats och förändrats. Om det är till det bättre låter jag andra avgör. Vi
är ju ändå i en bransch där ”granskning av ens jämlikar” utgör en av
hörnstenarna. Under denna tid har jag träffat på många människor, sett nya
platser av vår planet och även fått delta i olika aktiviteter som äggskjutning,
hundspann och konferenser.
Inkörsporten till allt det här kommer från en av mina studiekompisar som
också sökte sig till det akademiska livet som doktorand. Shahin
Noorbakhsh, som han heter, hade i maj 2009 blivit kallad till intervju hos
professor Gerhard Gröbner men hade samtidigt också blivit erbjuden en
doktorandtjänst på SLU i Uppsala. Han visste att jag också letade tjänst och
ringde därför mig och frågade om han skulle lämna mitt namn istället. Jag
svarade naturligtvis ja. Efter ett tag fick jag ett samtal eller ett mail från
Gerhard som undrade om jag kunde komma förbi universitetet på en
intervju med honom och Ulrik Ilstedt, en av mina assisterande handledare.
På den vägen är det. I början på juni erbjöds jag tjänsten, tackade ja och
kunde sedan börja planera för ett nytt kapitel i mitt liv. Tillsammans med
tjänsten var det viktigaste för mig just då att jag hann berätta nyheten för
min pappa som då var svårt sjuk och senare dog i mitten på juli. Jag är
mycket tacksam mot Shahin för att han tänkte på mig och lämnade mitt
namn till Gerhard. I samband med det vill jag också uttrycka min
tacksamhet för Centrum för Miljöforskning som slängt upp en ansenlig
summa pengar för att betala för åtminstone de första fyra åren av min tid
som doktorand. Ett tack också till andra donatorer.
Gerhard, du tog in mig i ett projekt som utvecklades på vägen till att handla
om nästan allt annat än ditt eget huvudområde, fast fas NMR. Trots det har
det alltid funnits en generös, snäll och rolig handledare nära till hands med
”immer” något att säga om vad som än avhandlades. Jag fick en chans till
något spännande av dig och jag hoppas att du inte blivit besviken utan att jag
på något sätt bidragit till dig och ditt arbete på samma sätt som du har gett
mig.
Jag har över åren kommit att jobba mycket med Jürgen Schleucher då
flera av delprojekten inneburit att han är inblandad från start. Han har i
perioder varit huvudhandledare tillsammans med Gerhard kan man säga och
för det är jag tacksam.

39
Redan från början var det bestämt att ett av mina delprojekt skulle innefatta
prover från Burkina Faso och det var Ulrik Ilstedts bord. Tyvärr har det
projektet hamnat lite i bakvattnet av de andra projekten och har aldrig riktigt
fått fart. Nu är det med ändå med ett manuskript i avhandlingen och
kommer snart vara klart. Ulrik tog med mig till Burkina en vecka och det var
mycket intressant och något jag kommer att minnas med glädje.
När jag kom till universitetet i oktober 2009 fick jag börja jobba direkt
tillsammans med en post doc från Costa Rica. Ett intensivt energiknippe vid
namn Andrea Vincent. I followed her around from place to place, from
building to building in our daily work sampling, preparing, extracting,
cleaning and drying and all the different things needed to be done in our
project. I learned a lot from you and for that I am grateful.
Solomon Tesfalidet, vill jag tacka för hjälpen med ICP delarna. En
värdefull hjälp som mottagits med tacksamhet.
I mitt kontor satt det en kille vid namn Marcus Wallgren som jämt
jobbade och som i framtiden definitivt kommer bli professor. Han är en
riktigt bra kille som har hjälpt mig med mycket under min tid här och vi har
tagit en eller två öl tillsammans vid olika tillfällen. Trots att du själv
doktorerat, Marcus och flyttat till annan avdelning finner du ändå tid att
hjälpa mig och andra med allt möjligt, t.ex. korrekturläsning av min
avhandling. Jag har också haft stort utbyte av alla diskussioner då vi pratat
om allt mellan himmel och jord. Jag hoppas att det också har varit av nytta
för dig. Det har varit ett lufthål för mig.
Med tiden flyttade Martin Lidman in i kontoret, in i Gerhards grupp och
han sitter här än. Väldigt kunnig med många tankar i huvet. Roligt att du gav
dig på att brygga öl också. Det ger oss ju möjligheten till en ”collaboration
brew” i framtiden. Tack Martin och lycka till.
Till Gerhards grupp har det hört till flera andra personer med tiden. Artur
Dingeldein som är nyaste doktoranden. En glad och trevlig kille med huvet
på skaft. Lycka till i fortsättningen, Artur.
When I started in Gerhards group, Tofeeq Ur-Rehman was with us. A
modest, pleasant and bright man from Pakistan whom with his family spent
a number of years in Umeå, Sweden for his PhD. We had some interesting
discussions him and me. I wish him all the best.
Recently came Ilona Dudka to our group. Yes, she is another person not
working with soil and stuff like me, but a nice easy going person. She also

40
jumps up fast and eager when we come in the corridor when it is fika time. A
good swedish quality. Good luck, Ilona.
Ximena Aguilar en härlig tjej som efter ett tag försvann upp till Med chem
vilket var en förlust för oss men en vinst för Med chem. Vi peppade varandra
när det gick tungt och en klapp på axeln när det gick bättre. Jag hade ändå
turen att få ta del av Ximenas kunskaper från hennes expertisområde i
pikobryggeriet Umeå Brygghus där hon gjorde ett positivt avtryck, tack
Ximena.
Alexander Christiansen som tillbringade ett par trevliga nyårsaftnar hos
oss och Moritz Niemiec hans kompanjon som fortfarande jobbar på. Tack
för den här tiden.
Lina Nilsson som kom, såg och for vidare. En till som helt plötsligt
hamnade hos Med chem. Glad och trevlig och saknad hos oss. Lycka till,
Lina.
Jörgen Åden och jag har spenderat mången lunch och fikastund
snackandes om allt från vapen till takplåt. Hjälp med både det ena och det
andra har han också kunnat erbjuda. Tack för den här tiden Jörgen och lycka
till.
Therese Mikaelsson och jag hamnade till slut i samma kontor och glad för
det är jag. Vi har också spenderat många luncher vid samma bord och det
har blivit mycket jaktprat med tiden. Lycka till, Therese och jag hoppas att
du hittar något bra jobb i Vindeln eller närområdet.
Jag har också fått spela lite innebandy under åren. Med betoning på lite. Det
var Tobias Sparrman, NMR-oumbärlig och fika-kungen som tyckte att jag
skulle vara med och spela och javisst tänkte jag. Sen bröt jag knät och sen var
det slut med den aktiviteten. Jag är skyldig Tobias ett stort tack för all hjälp
jag fått av honom under åren med oscilloskop, prober, och allt annat NMR
relaterat du hjälpt mig med. I owe you, som man säger i vissa delar av
världen. Tack för allt, Tobias.
Thank you, Ina Ehlers for the conversations here, in Germany and in
Austria. We have both been under the supervision of Jürgen and we started
almost the same time. I hope your work moves along and that your
dissertation is just around the corner.
Sen är det en hel del andra personer som passerat under åren. Xiang, Linh,
Dat, Konrad, Agnieszka, Lenka, Janek, Robert, Matteus, Ulrika, Christoph,

41
osv osv. Kan vara någon som jag just nu inte kommer ihåg som jag efter
tryck kommer att tänka på och då känns det jobbigt men vet detta;
Någonstans är det någon som kommer ihåg dig.
När jag började var det mycket folk i korridoren. Vi var ett helt gäng
forskargrupper som fikade, samtalade, hade kul och arbetade tillsammans.
Trots att det inte var någon som jobbade med jord lärde jag känna många av
er och kommer att komma ihåg många av er med värme.
Till sist vill jag också tacka min familj, min fru Karin mina söner David,
Hannes och Adam för att ni inte gjort det hela för svårt för mig utan
funnits där i vått och i torrt.
”Chansen kom och jag tog den……..”

42
References
1. Carpenter, S.R., et al., Nonpoint Pollution Of Surface Waters With Phosphorus And Nitrogen. Ecological Applications, 1998. 8(3): p. 559-568.
2. Ryther, J.H. and W.M. Dunstan, Nitrogen,Phosphorus, And
Eutrophication In Coastal Marine Environment. Science, 1971. 171(3975): p. 1008.
3. Elser, J.J., et al., Global Analysis of Nitrogen and Phosphorus Limitation of Primary Producers in Freshwater, Marine and Terrestrial Ecosystems. Ecology Letters, 2007. 10(12): p. 1135-1142.
4. Hinsinger, P., Bioavailability Of Soil Inorganic P In The Rhizosphere As Affected By Root-Induced Chemical Changes: A Review. Plant and Soil, 2001. 237(2): p. 173-195.
5. Brown, G.E., et al., Metal Oxide Surfaces And Their Interactions
With Aqueous Solutions And Microbial Organisms. Chemical Reviews, 1999. 99(1): p. 77-174.
6. Goldberg, S. and G. Sposito, On The Mechanism Of Specific Phosphate - Adsorption By Hydroxylated Mineral Surfaces - A Review. Communications in Soil Science and Plant Analysis, 1985. 16(8): p. 801-821.
7. Frossard, E., et al., Processes Governing Phosphorus Availability In Temperate Soils. Journal of Environmental Quality, 2000. 29(1): p. 15-23.
8. Li, W., et al., Surface Speciation of Phosphate on Boehmite
(gamma-AlOOH) Determined from NMR Spectroscopy. Langmuir, 2010. 26(7): p. 4753-4761.
9. Wyant, K.A., J.R. Corman, and J.J. Elser, eds. Phosphorus, Food, And Our Future. 2013, Oxford University Press: New York.
10. Ashley, K., D. Cordell, and D. Mavinic, A Brief History Of Phosphorus: From The Philosopher's Stone To Nutrient Recovery And Reuse. Chemosphere, 2011. 84(6): p. 737-746.
11. Tilman, D., et al., Forecasting Agriculturally Driven Global
Environmental Change. Science, 2001. 292(5515): p. 281-284.

43
12. Magid, J., H. Thiessen, and L.M. Condron, Dynamics Of Organic Phosphorus In Soils Under Natural And Agricultural Ecosystems., in Humic Substances In Terrestrial Ecosystems, A. Piccolo, Editor. 1996, Elsevier: Amsterdam. p. 429-466.
13. Cordell, D., J.O. Drangert, and S. White, The Story Of Phosphorus:
Global Food Security And Food For Thought. Global Environmental Change-Human and Policy Dimensions, 2009. 19(2): p. 292-305.
14. Gilbert, N., The Disappearing Nutrient. Nature, 2009. 461(7267): p. 1041-1041.
15. Wardle, D.A., L.R. Walker, and R.D. Bardgett, Ecosystem Properties And Forest Decline In Contrasting Long-Term Chronosequences. Science, 2004. 305(5683): p. 509-513.
16. Walker, T.W. and J.K. Syers, Fate Of Phosphorus During
Pedogenesis. Geoderma, 1976. 15(1): p. 1-19.
17. Chadwick, O.A., et al., Changing Sources Of Nutrients During Four Million Years Of Ecosystem Development. Nature, 1999. 397(6719): p. 491-497.
18. Sanchez, P.A., Ecology - Soil Fertility And Hunger In Africa. Science, 2002. 295(5562): p. 2019-2020.
19. Giesler, R., et al., Phosphate Sorption In Aluminum- And Iron-Rich
Humus Soils. Soil Science Society of America Journal, 2005. 69(1): p. 77-86.
20. Giesler, R., T. Petersson, and P. Hogberg, Phosphorus Limitation In Boreal Forests: Effects Of Aluminum And Iron Accumulation In The Humus Layer. Ecosystems, 2002. 5(3): p. 300-314.
21. Giesler, R., et al., Microbially Available Phosphorus In Boreal Forests: Effects Of Aluminum And Iron Accumulation In The Humus Layer. Ecosystems, 2004. 7(2): p. 208-217.
22. FAO, Production Yearbook 1999, in Statistical Series. 1999, Food
And Agricultural Organisation Of The United Nations: Rome.
23. Pettersson, E., Predictive Functions For Impact Of Nitrogen Fertilization On Growth Over Five Years. 1994, Forest Research Institute of Sweden. p. 1-56.

44
24. Giesler, R., M. Hogberg, and P. Hogberg, Soil Chemistry And Plants In Fennoscandian Boreal Forest As Exemplified By A Local Gradient. Ecology, 1998. 79(1): p. 119-137.
25. Liu, Y.-T. and D. Hesterberg, Phosphate Bonding on Noncrystalline
Al/Fe-Hydroxide Coprecipitates. Environmental Science & Technology, 2011. 45(15): p. 6283-6289.
26. Lindegren, M. and P. Persson, Competitive Adsorption Between Phosphate And Carboxylic Acids: Quantitative Effects And Molecular Mechanisms. European Journal of Soil Science, 2009. 60(6): p. 982-993.
27. Turner, B.L., et al., Soil Organic Phosphorus Transformations During Pedogenesis. Ecosystems, 2007. 10(7): p. 1166-1181.
28. Turner, B.L., et al., Inositol Phosphates In The Environment.
Philosophical Transactions of the Royal Society of London Series B-Biological Sciences, 2002. 357(1420): p. 449-469.
29. Jansson, M., et al., Terrestrial Carbon And Intraspecific Size-Variation Shape Lake Ecosystems. Trends in Ecology & Evolution, 2007. 22(6): p. 316-322.
30. Cross, A.F. and W.H. Schlesinger, A Literature-Review And Evaluation Of The Hedley Fractionation - Applications To The Biogeochemical Cycle Of Soil-Phosphorus In Natural Ecosystems. Geoderma, 1995. 64(3-4): p. 197-214.
31. Turner, B.L., Soil Organic Phosphorus In Tropical Forests: An
Assessment Of The NaOH-EDTA Extraction Procedure For Quantitative Analysis By Solution P-31 NMR Spectroscopy. European Journal of Soil Science, 2008. 59(3): p. 453-466.
32. Celi, L. and E. Berberis, Abiotic Stabilization Of Organic Phosphorus In The Environment, in Organic Phosphorus In The Environment, B.L. Turner, E. Frossard, and D.S. Baldwin, Editors. 2005, CAB International: Wallingford. p. 113-132.
33. Lagerstrom, A., et al., Soil Phosphorus And Microbial Response To A Long-Term Wildfire Chronosequence In Northern Sweden. Biogeochemistry, 2009. 95(2-3): p. 199-213.
34. Sugden, A., R. Stone, and C. Ash, Ecology In The Underworld -
Introduction. Science, 2004. 304(5677): p. 1613-1613.

45
35. Martin, C.J., Reaction Of The Coordinate-Complexes Of Inositol Hexaphosphate With First Row Transition Series Cations And Cd(Ii) With Calf Intestinal Alkaline-Phosphatase. Journal of Inorganic Biochemistry, 1995. 58(2): p. 89-107.
36. Martin, C.J. and W.J. Evans, Phytic Acid - Divalent-Cation
Interactions .3. A Calorimetric And Titrimetric Study Of The Ph-Dependence Of Copper(Ii) Binding. Journal of Inorganic Biochemistry, 1986. 28(1): p. 39-55.
37. Condron, L.M., B.L. Turner, and B.J. Cade-Menun, Chemistry And Dynamics Of Soil Organic Phosphorus, in Phosphorus: Agriculture And The Environment, J.T. Sims and A.N. Sharpley, Editors. 2005, American Society of Agronomy: Madison, WI. p. 87-121
38. Simpson, A.J., D.J. McNally, and M.J. Simpson, NMR Spectroscopy In Environmental Research: From Molecular Interactions To Global Processes. Progress in Nuclear Magnetic Resonance Spectroscopy, 2011. 58(3-4): p. 97-175.
39. Vestergren, J., et al., High-Resolution Characterization Of Organic
Phosphorus In Soil Extracts Using 2D 1H-31P NMR Correlation Spectroscopy. Environ Sci Technol, 2012. 46(7): p. 3950-3956.
40. Van Emmerik, T.J., et al., P-31 Solid-State Nuclear Magnetic Resonance Study Of The Sorption Of Phosphate Onto Gibbsite And Kaolinite. Langmuir, 2007. 23(6): p. 3205-3213.
41. Lindstrom, F., P.T.F. Williamson, and G. Grobner, Molecular Insight Into The Electrostatic Membrane Surface Potential By N-14/P-31 MAS NMR Spectroscopy: Nociceptin-Lipid Association. Journal of the American Chemical Society, 2005. 127(18): p. 6610-6616.
42. Nordgren, A., A Method For Determining Microbially Available-N
And Available-P In An Organic Soil. Biology and Fertility of Soils, 1992. 13(4): p. 195-199.
43. Ilstedt, U. and S. Singh, Nitrogen And Phosphorus Limitations Of Microbial Respiration In A Tropical Phosphorus-Fixing Acrisol (Ultisol) Compared With Organic Compost. Soil Biology & Biochemistry, 2005. 37(7): p. 1407-1410.
44. Bonev, B., et al., Electrostatic Peptide-Lipid Interactions Of Amyloid-Beta Peptide And Pentalysine With Membrane Surfaces

46
Monitored By P-31 MAS NMR. Physical Chemistry Chemical Physics, 2001. 3(14): p. 2904-2910.
45. Derome, A.E., Modern Nmr Techniques For Chemistry Research.
Tetrahedron Organic Chemistry Series. Vol. 6. 1991: Pergamon Press.
46. Newman, R.H. and K.R. Tate, Soil-Phosphorus Characterization By P-31 Nuclear Magnetic-Resonance. Communications in Soil Science and Plant Analysis, 1980. 11(9): p. 835-842.
47. Cade-Menun, B. and C.W. Liu, Solution Phosphorus-31 Nuclear Magnetic Resonance Spectroscopy of Soils from 2005 to 2013: A Review of Sample Preparation and Experimental Parameters. Soil Science Society of America Journal, 2014. 78(1): p. 19-37.
48. Turner, B.L., et al., Extraction Of Soil Organic Phosphorus. Talanta,
2005. 66(2): p. 294-306.
49. Hedley, M.J., J.W.B. Stewart, and B.S. Chauhan, Changes In Inorganic And Organic Soil-Phosphorus Fractions Induced By Cultivation Practices And By Laboratory Incubations. Soil Science Society of America Journal, 1982. 46(5): p. 970-976.
50. Cade-Menun, B.J. and C.M. Preston, A Comparison Of Soil Extraction Procedures For P-31 NMR Spectroscopy. Soil Science, 1996. 161(11): p. 770-785.
51. Cade-Menun, B.J., et al., Soil And Litter Phosphorus-31 Nuclear
Magnetic Resonance Spectroscopy: Extractants, Metals, And Phosphorus Relaxation Times. Journal Of Environmental Quality, 2002. 31(2): p. 457-465.
52. Cade-Menun, B.J., et al., Refining P-31 Nuclear Magnetic Resonance Spectroscopy For Marine Particulate Samples: Storage Conditions And Extraction Recovery. Marine Chemistry, 2005. 97(3-4): p. 293-306.
53. Dong, L., et al., Investigation Into Organic Phosphorus Species in Sediments of Baiyangdian Lake in China Measured by Fractionation and P-31 NMR. Environmental Monitoring and Assessment, 2012. 184(9): p. 5829-5839.
54. Doolette, A.L., R.J. Smernik, and W.J. Dougherty, Spiking Improved
Solution Phosphorus-31 Nuclear Magnetic Resonance Identification

47
of Soil Phosphorus Compounds. Soil Science Society of America Journal, 2009. 73(3): p. 919-927.
55. Turner, B.L., N. Mahieu, and L.M. Condron, Phosphorus-31 Nuclear
Magnetic Resonance Spectral Assignments Of Phosphorus Compounds In Soil NaOH-EDTA Extracts. Soil Science Society of America Journal, 2003. 67(2): p. 497-510.
56. Makarov, M.I., L. Haumaier, and W. Zech, Nature Of Soil Organic Phosphorus: An Assessment Of Peak Assignments In The Diester Region Of P-31 NMR Spectra. Soil Biology & Biochemistry, 2002. 34(10): p. 1467-1477.
57. McDowell, R.W. and I. Stewart, Peak Assignments For Phosphorus-31 Nuclear Magnetic Resonance Spectroscopy In Ph Range 5-13 And Their Application In Environmental Samples. Chemistry and Ecology, 2005. 21(4): p. 211-226.
58. Vincent, A.G., et al., Soil Organic Phosphorus Transformations In A
Boreal Forest Chronosequence. Plant and Soil, 2013. 367(1-2): p. 149-162.
59. Hupfer, M., B. Rube, and P. Schmieder, Origin And Diagenesis Of Polyphosphate In Lake Sediments: A P-31-NMR Study. Limnology and Oceanography, 2004. 49(1): p. 1-10.
60. Paytan, A., et al., Selective Phosphorus Regeneration Of Sinking Marine Particles: Evidence From P-31-NMR. Marine Chemistry, 2003. 82(1-2): p. 55-70.
61. He, Z.Q., et al., Phosphorus Forms In Conventional And Organic
Dairy Manure Identified By Solution And Solid State P-31 NMR Spectroscopy. Journal of Environmental Quality, 2009. 38(5): p. 1909-1918.
62. Cade-Menun, B.J., J.A. Navaratnam, and M.R. Walbridge, Characterizing Dissolved And Particulate Phosphorus In Water With P-31 Nuclear Magnetic Resonance Spectroscopy. Environmental Science & Technology, 2006. 40(24): p. 7874-7880.
63. Doolette, A.L., R.J. Smernik, and W.J. Dougherty, Overestimation Of The Importance Of Phytate In NaOH-EDTA Soil Extracts As Assessed By (31)P NMR Analyses. Organic Geochemistry, 2011. 42(8): p. 955-964.

48
64. Smernik, R.J. and W.J. Dougherty, Identification Of Phytate In Phosphorus-31 Nuclear Magnetic Resonance Spectra: The Need For Spiking. Soil Science Society of America Journal, 2007. 71(3): p. 1045-1050.
65. Chamignon, C., et al., Mobility Of Organic Pollutants In Soil
Components. What Role Can Magic Angle Spinning NMR Play? European Journal of Soil Science, 2008. 59(3): p. 572-583.
66. Pascaud, P., et al., Interaction between a Bisphosphonate, Tiludronate, and Biomimetic Nanocrystalline Apatites. Langmuir, 2013. 29(7): p. 2224-2232.
67. Clark, L.L., E.D. Ingall, and R. Benner, Marine Phosphorus Is Selectively Remineralized. Nature, 1998. 393(6684): p. 426-426.
68. Vestergren, J., et al., Adsorption Dynamics Of Phosphate Species On
Ion Exchange Resin – A 31p Nmr Study. in preparation.
69. Lash, T.D. and S.S. Lash, The Use Of Pascal-Like Triangles In Describing 1st-Order NMR Coupling Patterns. Journal of Chemical Education, 1987. 64(4): p. 315-315.
70. Smith, S.A. and N. Murali, Relaxation Effects In A System Of A Spin-1/2 Nucleus Coupled To A Quadrupolar Spin Subjected To RF Irradiation: Evaluation Of Broadband Decoupling Schemes. Journal of Magnetic Resonance, 1999. 136(1): p. 27-36.
71. Bodenhausen, G. and D.J. Ruben, Natural Abundance N-15 NMR by
Enhanced Heteronuclear Spectroscopy. Chemical Physics Letters, 1980. 69(1): p. 185-189.
72. Aue, W.P., E. Bartholdi, and R.R. Ernst, 2-Dimensional Spectroscopy - Application To Nuclear Magnetic-Resonance. Journal of Chemical Physics, 1976. 64(5): p. 2229-2246.
73. Braunschweiler, L. and R.R. Ernst, Coherence Transfer By Isotropic Mixing - Application To Proton Correlation Spectroscopy. Journal of Magnetic Resonance, 1983. 53(3): p. 521-528.
74. West, M., et al., Atomic Spectrometry Update-X-Ray Fluorescence
Spectrometry. Journal of Analytical Atomic Spectrometry, 2010. 25(10): p. 1503-1545.

49
75. Cultrone, G., et al., Iberian Ceramic Production From Basti (Baza, Spain): First Geochemical, Mineralogical And Textural Characterization. Archaeometry, 2011. 53: p. 340-363.
76. Nordin, A., P. Hogberg, and T. Nasholm, Soil Nitrogen Form And
Plant Nitrogen Uptake Along A Boreal Forest Productivity Gradient. Oecologia, 2001. 129(1): p. 125-132.
77. Högberg, P., Interactions Between Hillslope Hydrochemistry, Nitrogen Dynamics And Plants In Fennoscandian Boreal Forest, in Global Biogeochemical Cycles In The Climate System, E.D. Schulze, M. Heimann, and S. Harrison, Editors. 2001, Academic Press: San Diego, CA. p. 237-256.
78. Vincent, A.G., et al., Changes In Organic Phosphorus Composition In Boreal Forest Humus Soils: The Role Of Iron And Aluminium. Biogeochemistry, 2012. 108(1-3): p. 485-499.
79. Shand, C.A., et al., Solid-Phase P-31 NMR Spectra Of Peat And
Mineral Soils, Humic Acids And Soil Solution Components: Influence Of Iron And Manganese. Plant and Soil, 1999. 214(1-2): p. 153-163.
80. Russel, E.W., Soil Conditions and Plant Growth. 11th Ed. ed. 1988, Harlow, UK: Longman Scientific & Technical.


















