
Amyloidosis of Lower Genitourinary Tract: A...
7
Amyloidosis of Lower Genitourinary Tract: A Review Sudhanshu Chitale a, *, Mo Morsey a , Danielle Peat b , Ralph Webb a a Department of Urology, Norfolk & Norwich University Hospital NHS Trust, Colney Lane, Norwich, NR4 7UY, United Kingdom b Department of Histopathology, Norfolk & Norwich University Hospital NHS Trust, Colney Lane, Norwich, NR4 7UY, United Kingdom 1. Introduction Virchow first used the term amyloid [1] meaning ‘‘starchlike’’ to describe tissue deposits that stained with iodine solutions. Although a carbohydrate moiety was rightly identified in the amyloid tissue, it is well established that the principal constituent of the amyloid fibril is protein. Many different proteins (20) may form amyloid under diverse clinical conditions, all characterised by extracellular accu- mulation of fibrillar protein deposits. Their tissue distribution and consequent clinical manifestations vary widely. A single amyloid protein deposit occurring in different clinical conditions indicates a common pathologic mechanism (e.g. persistent inflammation) but each amyloid protein is usually associated with a unique clinicopathological condi- tion [2]. Thus, amyloidogenesis depends upon whether a single common pathologic mechanism is involved or diverse mechanisms are operational in a given disease process. How different proteins organise themselves into a common structure such as ‘‘amyloid’’ is not clearly understood. There is strong evidence that some derangement in the immune apparatus underlies this process and being a systemic disorder, it cannot be assigned to a single organ [3]. 2. Morphology Amyloid deposition occurs insidiously and its clinical recognition depends upon morphologic identification of this protein in the extracellular space in appropriate biopsy specimens. With pro- gressive accumulation it produces pressure atrophy of adjacent cells [3]. Light microscopy with standard tissue stains shows amorphous eosinophilic hyaline eau-ebu update series 5 (2007) 70–76 available at www.sciencedirect.com journal homepage: www.europeanurology.com Article info Keywords: Amyloidosis Genitourinary Urethral Vesical Abstract Amyloidosis is a pathological process characterized by extracellular deposition of proteinaceous substance. This is an endpoint for a variety of disease processes and can be generalised or localised. Amyloidosis of the lower urinary tract is unusual and has varied presentation. It is noted more commonly than expected and may be seen in patients with a background of chronic inflammatory disorders. In those with no pre- existing systemic disorder, an incidental finding of an amyloid deposit during investigations for urinary tract symptoms should alert the urol- ogist to arrange appropriate referral/investigations. # 2006 European Association of Urology and European Board of Urology. Published by Elsevier B.V. All rights reserved. * Corresponding author. Department of Urology, Norfolk & Norwich University Hospital, Colney Lane, Norwich, NR4 7UY, UK. Tel. +44 1603 286774; Fax: +44 1603 287884. E-mail address: [email protected] (S. Chitale). 1871-2592/$ – see front matter # 2006 European Association of Urology and European Board of Urology. Published by Elsevier B.V. All rights reserved. doi:10.1016/j.eeus.2006.11.002
Transcript of Amyloidosis of Lower Genitourinary Tract: A...

e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 6
Amyloidosis of Lower Genitourinary Tract: A Review
Sudhanshu Chitale a,*, Mo Morsey a, Danielle Peat b, Ralph Webb a
aDepartment of Urology, Norfolk & Norwich University Hospital NHS Trust, Colney Lane, Norwich, NR4 7UY, United KingdombDepartment of Histopathology, Norfolk & Norwich University Hospital NHS Trust, Colney Lane, Norwich, NR4 7UY, United Kingdom
avai lable at www.sc iencedi rect .com
journal homepage: www.europeanurology.com
Article info
Keywords:AmyloidosisGenitourinaryUrethralVesical
Abstract
Amyloidosis is a pathological process characterized by extracellulardeposition of proteinaceous substance. This is an endpoint for a varietyof disease processes and can be generalised or localised. Amyloidosis ofthe lower urinary tract is unusual and has varied presentation. It is notedmore commonly than expected and may be seen in patients with abackground of chronic inflammatory disorders. In those with no pre-existing systemic disorder, an incidental finding of an amyloid depositduring investigations for urinary tract symptoms should alert the urol-ogist to arrange appropriate referral/investigations.
# 2006 European Association of Urology and European Board of Urology.
Published by Elsevier B.V. All rights reserved.
* Corresponding author. Department of Urology, Norfolk & Norwich University Hospital,Colney Lane, Norwich, NR4 7UY, UK. Tel. +44 1603 286774; Fax: +44 1603 287884.E-mail address: [email protected] (S. Chitale).
1. Introduction
Virchow first used the term amyloid [1] meaning‘‘starchlike’’ to describe tissue deposits that stainedwith iodine solutions. Although a carbohydratemoiety was rightly identified in the amyloid tissue,it is well established that the principal constituent ofthe amyloid fibril is protein. Many different proteins(20) may form amyloid under diverse clinicalconditions, all characterised by extracellular accu-mulation of fibrillar protein deposits. Their tissuedistribution and consequent clinical manifestationsvary widely. A single amyloid protein depositoccurring in different clinical conditions indicatesa common pathologic mechanism (e.g. persistentinflammation) but each amyloid protein is usuallyassociated with a unique clinicopathological condi-tion [2]. Thus, amyloidogenesis depends uponwhether a single common pathologic mechanism
1871-2592/$ – see front matter # 2006 European Association of UrologPublished by Elsevier B.V. All rights reserved.
is involved or diverse mechanisms are operationalin a given disease process. How different proteinsorganise themselves into a common structure suchas ‘‘amyloid’’ is not clearly understood. There isstrong evidence that some derangement in theimmune apparatus underlies this process and beinga systemic disorder, it cannot be assigned to a singleorgan [3].
2. Morphology
Amyloid deposition occurs insidiously and itsclinical recognition depends upon morphologicidentification of this protein in the extracellularspace in appropriate biopsy specimens. With pro-gressive accumulation it produces pressure atrophyof adjacent cells [3]. Light microscopy with standardtissue stains shows amorphous eosinophilic hyaline
y and European Board of Urology. doi:10.1016/j.eeus.2006.11.002

Fig. 1 – TEM: Amyloid fibrils randomly arranged with
coarse spike formation (�10000).
e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 6 71
material deposited between cells of the respectivetissue. Congo red stain under ordinary light impartspink/red colour to the deposits but under polarisedlight, the specific green birefringence of the stainedamyloid is noted. Transmission electron microscopy(TEM) (Fig. 1) reveals the amyloid to be 95% [3]nonbranching fibrils of 7.5 to 10 nm in diameter.X-ray crystallography and infrared spectroscopyshow a characteristic cross beta-pleated sheetconfiguration. A minor second component calledthe P component (5%); a pentagonal doughnutshaped glycoprotein seen under EM is alwayspresent in addition to the fibrils.
3. Chemistry
Amyloid is not a chemically distinct entity despitethe uniform appearance of all amyloids. There aretwo major and around 13 minor biochemical forms.The two most common or major forms are: AL(amyloid light chain) and AA (amyloid-associated).AL is an immunoglobulin derived from plasma cellsand AA is a unique nonimmunoglobulin synthe-sized by the liver [3].
The amyloid protein of the AL type is associatedwith monoclonal B cell proliferation such as multi-ple myeloma, whereas the AA protein is depositedas secondary amyloidosis in chronic inflammatorydisorders including rheumatoid arthritis and osteo-myelitis.
Apart from AL and AA, other biochemicallydistinct forms of amyloid have been found indistinct clinical settings: amyloid transthyretin(ATTR; prealbumin) is deposited in familial amyloidpolyneuropathies, b2-microglobulin may be seen inpatients on long term haemodialysis and in cerebralamyloid angiopathy; and in Alzheimer’s disease the
main protein is Ab2. Amyloid in medullary carci-noma of the thyroid is formed from the hormonecalcitonin [3].
4. Classification
Amyloids can be classified based on their chemicalfibril constituent as AL, AA or ATTR (biochemicalclassification) or based on clinical syndromes assystemic and localised (clinical classification). Acombined biochemical-clinical or clinico-pathologi-cal classification is thought to be more practical as agiven biochemical form of amyloid may be asso-ciated with diverse clinical settings. Clinically, thesystemic/generalised and localised patterns couldbe subclassified into primary amyloidosis whenassociated with some immunocyte dyscrasias (AL)and secondary amyloidosis when it occurs as acomplication of chronic inflammation or tissuedestructive process (AA) [3]. Hereditary or familialamyloidosis constitutes a separate group of condi-tions with distinctive pattern of organ involvement[3].
5. Pathomorphology
In any of the amyloid categories, there are nospecific patterns of organ/tissue involvement. As ageneral observation, primary or immunocyte asso-ciated (AL) amyloidosis more often involves heart,gastrointestinal tract, carpal tissue, peripheralnerves, tongue and skin whereas the secondaryamyloidosis (AA) tends to involve kidneys, spleen,liver, GI tract, adrenal, thyroid and lymph nodes[2,3]. Familial ATTR amyloid predominantly involvesperipheral and autonomic nervous system andcarpal tunnel. b2M amyloid chiefly affects muscu-loskeletal system. The gross appearance of theinvolved organ depends upon the volume of amyloidand the site of deposition within the tissue. Theaffected organ is enlarged, pale and waxy; if there isvascular involvement there may be atrophy. Thestaining characteristics are shared by all forms ofamyloid and are attributable to the cross-betapleated configuration of amyloid fibrils. Stainingdyes include Congo red, potassium permanganate,methyl violet thioflavine and Alcian blue. Thespecific fibril protein components of the amyloidcan be identified by immunohistochemistry orelectron microscopy [2,3].
Amyloid is located extracellularly in vessels orparenchyma. Progressive deposition of amyloidfollows the stromal architecture of the organ.

Fig. 2 – A single glomerulus shows expansion of mesangial
matrix by acellular eosinophilic amyloid (H & E �400)
which is congophilic.
e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 672
Amyloid isolates parenchymal cells and interfereswith the intrinsic circulation of the organ causingatrophy in advanced cases. A significant amount ofamyloid deposition is needed before the function ofthe organ is impaired; the kidney is the exceptionwhere glomerular function is compromised at anearly stage of the disease.
5.1. Renal amyloidosis
The kidney may be normal, enlarged or shrunken inadvanced cases due to vascular narrowing inducedby amyloid deposits within arterial and arteriolarwalls. Deposits are primarily in the glomeruli as asubtle nodular increase in mesangial matrix andwidening of capillary basement membrane as wellas in the interstitiium, arteries and arterioles (Fig. 2).The glomerular vascular tuft may be distorted bymasses of amyloid leading to obliteration of capil-lary lumina.
5.2. Vesical amyloidosis
Deposits of amyloid are found in the arteries,arterioles, veins and suburothelial connective tis-sue. Foreign body giant cell reaction and calcifica-tion may be present.
Similar deposits have also been described in therenal pelvis, ureter and urethra.
6. Clinical features and correlation
Amyloidoses are a significant cause of morbidityand mortality in the Western world. Its presentation
may vary from an unsuspected anatomic changewith no clinical manifestations to causing death.The symptoms depend upon the particular organ/site involved and the magnitude of the deposits.Initial symptoms could be nonspecific such asweakness, weight loss or syncope. Specific symp-toms are related to renal, gastrointestinal andcardiac involvement as well as hepato-splenome-galy and altered serum protein levels. Clinicalfeatures in an individual are determined by principalsite of deposition in that individual. Different typesof amyloidosis have certain peculiarities about theirorgan involvement. Isolated deposits of amyloid canvirtually develop anywhere in the body but the mostcommon sites of localized amyloidosis are thegenitourinary system, lungs, skin and stroma ofmedullary carcinoma of the thyroid. These depositsalthough composed of light chains are rarely amanifestation of systemic AL amyloidosis [2].
6.1. Kidney
The hallmark of renal amyloidosis is heavy protei-nuria which may be associated with nephroticsyndrome; there may be progression to chronic renalfailure. Bence Jones proteinuria is a feature of ALamyloidosis [4,5]. Renal amyloidosis is one of thedominant features of systemic amyloidosis (AA), themost common being rheumatoid arthritis and otherchronic conditions including familial Mediterraneanfever (Heredofamilial amyloidosis). Ab2M amyloido-sis isan important extrarenalcomplication of chronichaemodialysis, its incidence approaching 100% inpatients on haemodialysis for over 15–20 years [6–9].
Diagnosis of renal amyloid is by renal biopsy.Although renal biopsies have a high positive yield inmost forms of systemic amyloidosis [10,11], hae-morrhage is a significant complication of biopsyingamyloidotic organ due to vascular involvement byamyloid and abnormalities of haemostasis asso-ciated with amyloidosis [12]. In patients withsuspected systemic (AA) amyloidosis, a rectal orgingival biopsy may be helpful [10]. Patients withsuspected (AL) amyloidosis should have serum andurine protein electrophoresis performed. Needleaspiration of subcutaneous abdominal fat may beused [13–16].
Treatment of renal amyloid is symptomatic or ofits associated complications [17].
Amyloidosis of the pelvicalyceal system withoutinvolvement of the renal parenchyma has also beenreported as easily confused with urothelial tumour[18]. Diagnosis of urothelial amyloidosis may bepossible if presence of amyloid is noted in the urinecytology sample [19].

e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 6 73
6.2. Ureter
Primary amyloidosis of the lower ureter has beendescribed in the literature presenting as hydrone-phrosis secondary to ureteric stricture [20–22] andbilateral involvement presenting as anuria [23].Treatment with ureteric stenting and occlusivedressing technique using dimethyl sulfoxide (DMSO)for 6 months leading to complete resolution of thelesion has been described [20].
6.3. Bladder
Amyloidosis of the urinary bladder is uncommon[24]. However amongst the various sites in the lowerurinary tract, bladder is the most commonlyinvolved organ [26]. Both sexes are equally affectedbetween the fifth and seventh decade. Painless grosshaematuria is the main presenting symptoms inmost (>75%) cases [24,25]. Both primary and sec-ondary amyloidosis may involve the bladder, iso-
Fig. 3 – (a & b): Vesical amyloidosis: (a): Deposits of
perivascular eosinophilic (H & E �100) amyloids are
present in the submucosal tissue. (b): These are
congophilic (�400).
lated primary vesical amyloidosis (Fig. 3) beingmore common than secondary involvement [32]and the lesions can be easily confused with a tumour[25–32].
Diagnosis depends upon specific staining char-acteristics of the bladder tissue and immunohisto-chemical studies identifying the amyloid type.Presence of amyloid light chain (AL) indicates thatthe protein originated outside the urinary bladder butthe protein subtyping by sequence analysis suggestsit has no role in restricting the deposited protein tothe bladder [26]. Repeated workups for systemicamyloidosis are unnecessary in AL amyloidosis of thebladder [25] but early eradication of the lesion withresection/fulguration/laser ablation is advisable withregular cystoscopic follow up. Prognosis remainsexcellent in primary amyloidosis [27–31].
6.4. Urethra
Presentation of urethral amyloid deposit (Fig. 4)mimics primary tumour of the urethra or nonspe-cific urethritis [33]. It has been described in bothsexes [34,35]. Isolated amyloid of the penile urethra[36] and corpus spongiosum have also beenreported, presenting with painful erection [37] orhaemospermia [38]. MRI may be useful in suggestinga diagnosis [39]. Symptomatic and expectant man-agement has been shown to be appropriate in thesecases [39,40].
6.5. Prostate & seminal vesicle
Clinical presentation of prostatic amyloid as a partof systemic amyloidosis resembles tumour of theprostate [41]. Localized secondary amyloidosis of theseminal vesicle presenting with chronic perinealpain mimics seminal vesiculitis [41] and although itsprevalence is higher in those who had hormonaltherapy for prostatic carcinoma, there has been nodirect association between seminal vesicle amyloi-dosis (Fig. 5) and occurrence of prostatic carcinoma[42].
6.5.1. Diagnostic techniques
A definitive diagnosis requires histological identifi-cation of amyloid deposits. Identification of thespecific subtype of amyloid is important as it hasprognostic and therapeutic implications. Biochem-ists use ancillary techniques such as serum andurine protein electrophoresis and immuno-histo-chemistry for specific subtype identification ofamyloid. Diagnostic molecular genetics has becomeimportant for the heredofamilial type of amyloidosisand the data used for genetic counselling and
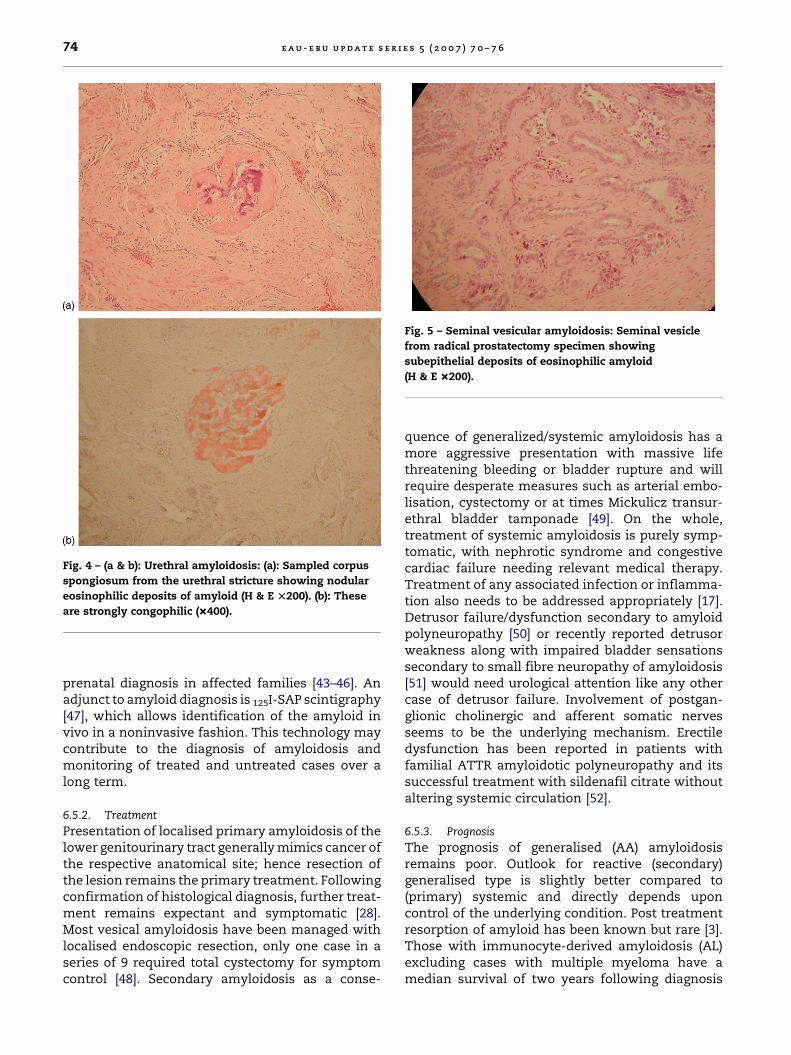
Fig. 5 – Seminal vesicular amyloidosis: Seminal vesicle
from radical prostatectomy specimen showing
subepithelial deposits of eosinophilic amyloid
(H & E �200).
Fig. 4 – (a & b): Urethral amyloidosis: (a): Sampled corpus
spongiosum from the urethral stricture showing nodular
eosinophilic deposits of amyloid (H & E T200). (b): These
are strongly congophilic (�400).
e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 674
prenatal diagnosis in affected families [43–46]. Anadjunct to amyloid diagnosis is 125I-SAP scintigraphy[47], which allows identification of the amyloid invivo in a noninvasive fashion. This technology maycontribute to the diagnosis of amyloidosis andmonitoring of treated and untreated cases over along term.
6.5.2. Treatment
Presentation of localised primary amyloidosis of thelower genitourinary tract generally mimics cancer ofthe respective anatomical site; hence resection ofthe lesion remains the primary treatment. Followingconfirmation of histological diagnosis, further treat-ment remains expectant and symptomatic [28].Most vesical amyloidosis have been managed withlocalised endoscopic resection, only one case in aseries of 9 required total cystectomy for symptomcontrol [48]. Secondary amyloidosis as a conse-
quence of generalized/systemic amyloidosis has amore aggressive presentation with massive lifethreatening bleeding or bladder rupture and willrequire desperate measures such as arterial embo-lisation, cystectomy or at times Mickulicz transur-ethral bladder tamponade [49]. On the whole,treatment of systemic amyloidosis is purely symp-tomatic, with nephrotic syndrome and congestivecardiac failure needing relevant medical therapy.Treatment of any associated infection or inflamma-tion also needs to be addressed appropriately [17].Detrusor failure/dysfunction secondary to amyloidpolyneuropathy [50] or recently reported detrusorweakness along with impaired bladder sensationssecondary to small fibre neuropathy of amyloidosis[51] would need urological attention like any othercase of detrusor failure. Involvement of postgan-glionic cholinergic and afferent somatic nervesseems to be the underlying mechanism. Erectiledysfunction has been reported in patients withfamilial ATTR amyloidotic polyneuropathy and itssuccessful treatment with sildenafil citrate withoutaltering systemic circulation [52].
6.5.3. Prognosis
The prognosis of generalised (AA) amyloidosisremains poor. Outlook for reactive (secondary)generalised type is slightly better compared to(primary) systemic and directly depends uponcontrol of the underlying condition. Post treatmentresorption of amyloid has been known but rare [3].Those with immunocyte-derived amyloidosis (AL)excluding cases with multiple myeloma have amedian survival of two years following diagnosis

e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 6 75
whereas those with associated myeloma have apoorer prognosis.
However, the prognosis of localised primaryamyloidosis involving the genitourinary tract, anunusual clinical entity, remains excellent withconservative therapy as long as a definitive histo-logical diagnosis is established and causes ofsecondary (generalized) amyloidosis are ruled out.Repeated work ups for systemic amyloidosis are notnecessary in cases of primary (AL) localised amy-loidosis of the lower genitourinary tract becauselocal recurrence is more commonly found than anycauses for secondary/systemic amyloidosis [25].
References
[1] Virchow R. Zur Cellulosefrage. Virchows Arch Pathol Anat
Physiol 1854;6:416–26.
[2] Kisilevsky R, Young ID. Amyloidosis. In: Damjanov I,
Linder J, editors. Anderson’s Pathology. 10th ed. St. Louis:
Mosby; 1996. p. 448–59.
[3] Mitchell RN, Kumar V. Diseases of Immunity. In: Kumar
V, Cotran RS, Robbins SL, editors. Robbin’s Basic Pathol-
ogy. 7th ed. Philadelphia: WB Saunders; 2003. p. 103–64.
[4] Triger DR, Joekes AM. Renal amyloidosis: a fourteen year
follow-up. Q J Med 1973;42:15–40.
[5] Stone MJ, Frendel EP. The clinical spectrum of light chain
myeloma: a study of 35 patients with special reference to
the occurrence of amyloidosis. Am J Med 1975;58:601–19.
[6] Gejyo F, Homma N, Arakawa M. Long-term complications
of dialysis: pathogenic factors with special reference to
amyloidosis. Kidney Int Suppl 1993;41:S78–82.
[7] Koch KM, Gennari FJ, Vanypersele C, et al. Dialysis –
related amyloidosis. Kidney Int 1992;41:1416–29.
[8] Floege J, Schaffer J, Koch KM, et al. Dialysis related amy-
loidosis: a disease of chronic retention and inflammation?
Kidney Int Suppl 1992;38:S78–85.
[9] Honkanen E, Gronhagen-Riska C, Teppo AM, et al. Acute –
phase protein during haemodialysis: correlation with
serum interleukin-1ß levels and different dialysis mem-
branes. Nephron 1991;57:283–7.
[10] Blum A, Sohar E. The diagnosis of amyloidosis: ancillary
procedures. Lancet 1962;1:721–4.
[11] Stauffer MH, Gross JB, Foulk WT, et al. Amyloidosis: diag-
nosis with needle biopsy of the liver in 18 patients. Gas-
troenterology 1961;41:92–6.
[12] Yood RA, Skinner M, Rubinow A, et al. Bleeding manifes-
tations in 100 patients with amyloidosis. JAMA 1983;249:
1322–4.
[13] Duston MA, Skinner M, Meena RF, et al. Sensitivity, spe-
cificity and predictive value of abdominal fat pad aspira-
tion for the diagnosis of amyloidosis. Arthritis Rheum
1989;32:82–5.
[14] Blumenfeld W, Hildebrandt RH. Fine needle aspiration of
abdominal fat for the diagnosis of amyloidosis. Acta Cytol
1993;37:170–4.
[15] Libbey CA, Skinner M, Cohen AS. Use of abdominal fat
tissue aspirate in the diagnosis of systemic amyloidosis.
Arch Intern Med 1983;143:1549–52.
[16] Duston MA, Skinner M, Shirahama T, et al. Diagnosis of
amyloidosis by abdominal fat aspiration: analysis of four
year’s experience. Am J Med 1987;82:412–4.
[17] Falk RH, Comenzo RL, Skinner M. The systemic amyloi-
dosis. New England J of Medicine 1997;337:898–909.
[18] Murphy MN, Alguacil-Garcia A, MacDonald RG. Primary
amyloidosis of renal pelvis with duplicate collecting sys-
tem. Urology 1986;27:470–3.
[19] Takahashi T, Miura H, Matsu-ura Y, Iwana S, Maruyama
R, Harada T. Urine cytology of localized primary amyloi-
dosis of the ureter: a case report. Acta Cytol 2005;49:319–
22.
[20] Kato Y, Sue Y, Fujii H, Numata A, Yachiku S. Localized
amyloidosis of the ureter and bladder treated effectively
by occlusive dressing technique therapy using dimethyl
sulfoxide: a case report. Hinyokika Kiyo 2000;46:421–4.
[21] Andreas BF, Oosting M. Primary amyloidosis of the ureter.
J Urol 1958;79:929–31.
[22] Kitsukawa S, Hosoda S, Otsuru N, Matsumoto T, Yama-
moto Y, Matsumoto T. Localized amyloidosis of the
ureter: a case report. Hinyokika Kiyo 2006;52:131–4.
[23] Paraskevas KI, Anagnostou D, Bouris C. Anurea caused by
primary amyloidosis of the lower third of the ureters, the
ureterovesical junction and the urinary bladder: a case
report and review of the literature. Int Urol Nephrol
2004;36:339–42.
[24] Johansson SL, Cohen SM. Lower urinary tract: Diseases of
the urogenital and reproductive systems. In: Damjanov I,
Linder J, editors. Anderson’s Pathology, 10th ed. St. Louis:
Mosby; 1996. p. 2154.
[25] Tirzaman O, Wahner-Roedler DL, Malek RS, Sebo TJ, Li CY,
Kyle RA. Primary localized amyloidosis of the urinary
bladder: a case series of 31 patients. Mayo Clin Proc
2000;75:1264–8.
[26] Livneh A, Shtrasburg S, Martin BM, Baniel J, Gal R, Pras M.
Light chain amyloidosis of the urinary bladder: A site
restricted deposition of an externally produced immuno-
globulin. J Clin Pathol 2001;54:920–3.
[27] Zaman W, Singh V, Kumar B, Mandhani A, Srivastava A,
Kumar A, et al. Localized primary amyloidosis of the
genitourinary tract: does conservatism help? Urol Int
2004;73:280–2.
[28] Grainger R, O’Riordan B, Cullen A, Kelly D, Heaney J.
Primary amyloidosis of lower urinary tract. Urology
1988;31:14–6.
[29] Reddy VC. Primary amyloidosis of the lower urinary tract:
a case report. Mo Med 1991;88:39–40.
[30] Duffau P, Imbert Y, De Faucal P, Fleury D, Arlet P, Camou F,
et al. Primary localized amyloidosis of the urinary tract. A
case seriesof five patients. Rev Med Interne 2005;26:288–93.
[31] Chamatan A, Peters C, Ngendahayo P, Maquet JH. Isolated
primary amyloidosis of the bladder. Prog Urol 2004;
14:218–20.
[32] Palmero Marti JL, Budia Alba J, Arlandis Guzman S, Bene-
dicto Redon A, Hernandez Marti M, Jimenez Cruz JF. Sec-
ondary vesical amyloidosis. Actas Urol Esp 2004;28:238–42.

e a u - e b u u p d a t e s e r i e s 5 ( 2 0 0 7 ) 7 0 – 7 676
[33] Williams OE, Kynaston H, Dixon G, Arya OP. Amyloid
tumour of the urethra presenting as non-specific urethri-
tis. Genitourin Med 1992;68:332–3.
[34] Kageyama S, Suzuki K, Ushiyama T, Fujita K, Kawabe K.
Primary localized amyloidosis of the urethra in a woman.
Br J Urol 1998;81:918–9.
[35] Zimmer G, Weber G, Braun HP, Bersch W. Report of 2 cases
of isolated primary amyloidosis of the male urethra and
urinary bladder of a female. Urologe A 1989;28:363–6.
[36] Sakuma S, Miyazaki T, Hirata H. A case of primary loca-
lized amyloidosis of the penile urethra. Int J Urol
1996;3:163–4.
[37] Lau PC, Norman RW, Murphy DM. Localised amyloidosis
of the urethra presenting as painful erection. Can J Urol
1995;2:148–9.
[38] Trivez Boned MA, Blas Martin M, Garcia Garcia MA, Gil
Martinez P, Garcia de Jalon Martinez A, Rioja Sanz LA.
Urethral amyloidosis. Actas Urol Esp 2002;26:46–9.
[39] Ichioka K, Utsunomiya N, Ueda N, Matsui Y, Yoshimura K,
Terai A. Primary localized amyloidosis of urethra: mag-
netic resonance imaging findings. Urology 2004;64:376–8.
[40] Crook TJ, Koslowski M, Dyer JP, Bass P, Birch BR. A case of
amyloid of the urethra and review of this rare diagnosis,
its natural history and management, with reference to the
literature. Scand J Urol Nephrol 2002;36:481–6.
[41] Carris CK, McLaughlin 3rd AP, Gittes RF. Amyloidosis of
the lower genitourinary tract. J Urol 1976;115:423–6.
[42] Harvey I, Tetu B. Amyloidosis of the seminal vesicles: a
local condition with no systemic impact. Ann Pathol
2004;24:236–40.
[43] Nordvag BY, Husby G, Ranlov I, et al. Molecular diagnosis
of the transthyretin (TTR) Met III mutation in familial
amyloid cardiomyopathy of Danish origin. Hum Genet
1992;89:459–61.
[44] Nichols WC, Padilla LM, Benson MD. Prenatal detection of
a gene for hereditary amyloidosis. Am J Med Genet
1989;34:520–4.
[45] Ii S, Minnerath S, Ii K, et al. Two-tiered DNA-based diag-
nosis of transthyretin amyloidosis reveals two novel
point mutations. Neurology 1991;41:893–8.
[46] Morris M, Nichols W, Benson M. Prenatal diagnosis of
hereditary amyloidosis in a Portuguese family. Am J
Med Genet 1991;39:123–4.
[47] Hawkins PN, Lavender JP, Pepys MB. Evaluation of
systemic myloidosis by scintigraphy with I-123-labeled
serum amyloid-P component. N Engl J Med 1990;323:
508–13.
[48] Merrimen JL, Alkhudair WK, Gupta R. Localise amyloido-
sis of the urinary tract: case series of nine patients.
Urology 2006;67:904–9.
[49] Farina Perez LA, Ortis Rey JA. Secondary bladder amyloi-
dosis with severe recurrent haematuria: transurethral
Mickulicz procedure as hemostatic option. Arch Esp Urol
2005;58:665–8.
[50] Andersson R, Bjerle P. Studies of urinary bladder dysfunc-
tion in amyloidosis with polyneuropathy. Acta Med Scand
1975;197:117–23.
[51] Ito T, Sakakibara R, Yamamoto T, et al. Urinary dusfunc-
tion and autonomic control in amyloid neuropathy. Clin
Auton Res 2006;16:66–71.
[52] Obayashi K, Ando Y, Terazaki H, et al. Effect of sildenafil
citrate (Viagra) on erectile dysfunction in a patient with
familial amyloidotic polyneuropathy ATTR Val30Met. J
Auton Nerv Syst 2000;80:89–92.
CME questions
Please visit www.eu-acme.org/europeanurologyto answer these CME questions on-line. The CMEcredits will then be attributed automatically.
1. A
myloid is stained in the laboratory usingA. Congo greenB. Congo blueC. Congo redD. Congo yellow2. U
ltrastructurally, amyloid is made up of non-branching fibrils ofA. 7.5–10 cmB. 7.5–10 mmC. 7.5–10 mmD. 7.5–10 nm3. A
A deposits are characteristically laid down inA. Chronic inflammatory disordersB. Myeloproliferative diseasesC. Epithelial malignanciesD. Alzheimer’s disease
4. D
eposition of amyloid isA. IntracellularB. ExtracellularC. IntranuclearD. In basement membranes5. T
he core protein in patients on long-termhaemodialysis developing amyloid isA. CalcitoninB. Ab amyloidC. ALD. b2-microglobulin6. T
he main presenting symptom of amyloidosis ofthe urinary bladder isA. PainB. Urinary tract infectionC. HaematuriaD. Acute retention

















