
& Leachables Extractables - Dalton
10
Extractables & Leachables "To make the impossible possible. Dalton Pharma Services uses its scientific and pharmaceutical expertise to bring customer ideas to life. We develop their new drug products, optimize the synthesis of therapeutic candidates, and manufacture them at the highest level of quality." Contract Research Custom Synthesis Medicinal and Flow Chemistry API Process Development Formulation Development cGMP API Manufacturing cGMP Sterile Filling Analytical and Microbiology Services FDA, HC, & EMA approved
Transcript of & Leachables Extractables - Dalton

Extractables& Leachables
W I T H D A LT O N
[ C O M P A N Y V I S I O N ]
"To make the impossible possible. Dalton Pharma Services uses its scientificand pharmaceutical expertise to bring customer ideas to life. We develop their
new drug products, optimize the synthesis of therapeutic candidates, andmanufacture them at the highest level of quality."
Contract ResearchCustom SynthesisMedicinal and Flow ChemistryAPI Process DevelopmentFormulation DevelopmentcGMP API ManufacturingcGMP Sterile FillingAnalytical and Microbiology Services
[ S E R V I C E S ]
FDA, HC, & EMA approved

What is an impurity?
ImpuritiesA B O U T
Where do impurities come from?
Classification of impurities
Result from themanufacturing process.They are normally knownand identified.
Reagents, ligands, andcatalystsHeavy metals(elemental impuritiesi.e., Hg, Pb, Cd, As) [2]or other residualmetals Inorganic saltsOther materials (e.g.,filter aids, charcoal)
Result during the manufacturingprocess and/or storage of thenew drug substance. They can beidentified or unidentified, volatileor non-volatile.
Starting ingredient reagentsReagents and catalystsProcess by productProcess intermediatesDegradation productsi.e., PAHs, Nitrosamines, AzoDyes, Vulcanizing Agents,Phthalates, antioxidants,Elastomers, Stabilizers
Inorganic or organicliquids used for thepreparation of solutionsor suspensions in thesynthesis of a new drugsubstance. They are ofknown toxicity.
Any chemical used inthe synthesisprocess (i.e., benzene,methanol)
Organic Impurities [1] Inorganic Impurities [1] Residual Solvent Impurities [3]
"Any component of the new drug substance that is not the chemical entity defined as thenew drug substance" as defined by ICH guidelines
API synthesisAPI purificationPackaging & Storage (Extractables and Leachables)
refer to ICH Q3A Impurities in New Drug Substances/ Q3B Impurities in New Drug Products refer to ICH Q3D Guideline for Elemental Impuritiesrefer to ICH Q3C Maintenance of the Guideline forResidual Solvents
Other impurities include: (1) extraneous contaminants, (2) polymorphic forms, and (3)enantiomeric impurities.
1 - 2 - 3 -

Extractables & LeachablesA B O U T
Extractables - chemicals that extract from components of a manufacturing orpackaging system into a solvent under forced conditions.
Leachables - compounds that can migrate via contact with manufacturingsystems, container-closure systems, and drug delivery device components intothe drug product.
What is it?
The first step in an E&L study design is to collect information regarding thecomposition of the product, the delivery system, the packaging materials, andthe intended use conditions. This is followed by a safety risk assessment.Different extraction procedures and techniques are then performed to evaluatetoxicity. These toxicological assessments define the safety concern thresholdlevels for the extracted compounds.
The identification of extractables involves extensive data interpretation, librarysearches, data comparisons, confirmation by several analytical techniques, andanalytical expertise. The quantitation of any potential leachables is undertakenbased on the substances of concern that have been identified throughextractables study. Toxicity quantification and assessment of any detectedleachables that are above the analytical evaluation threshold will determine theoverall product safety.
E&L Study Design
Safety concern threshold – total daily intake threshold below which a leachablepresents negligible safety concerns from carcinogenic and noncarcinogenictoxic effectsAnalytical evaluation threshold – concentration above which E&L need to beidentified and or quantified
Limits for E&L

Impurities & ControlA B O U T
Results in lowershelf life
Changes thephysical and
chemical propertiesof the substance
Decreasestherapeutic effects
May be harmful
To meet regulatorystandards
E&L control
A controlled extraction study is a laboratory investigation into the qualitative andquantitative nature of extractable profiles from critical components of acontainer/closure system.
A critical material attribute (CMA) is a property or characteristic of an input materialthat should be within an appropriate range to ensure the desired quality of that drugsubstance, excipient, or in-process material. Below are the CMA's for E&L:
Why control impurities?
Drug Product Formulation
SolventspHEmulsions
Container closure system
LabelsList of additivesExtractable profileVendor control
Stability and Storage
TemperatureTime
Sterilization
Pre or post fillGamma irradiationHeat
CMA

As per U.S. FDA 21 CFR 211.94(a) and the European Commission Directive(2001/83/EC), regulatory standards necessitate pharmaceutical industries todevelop sensitive and accurate analytical methods to detect, identify, andquantitate extractables and leacheables in a drug product.
Dalton's ServicesA B O U T
Determination of the AnalyticalEvaluation Threshold (AET) based onSafety Concern Threshold (SCT) anddose per container.
AET
Development of tailored studydesigns for extractables andleachables, including profiling(inorganic and organic) andsimulated leachable studies.
Tailored Study Designs
Validation of analytical methodsdeveloped for determination ofextractables and leachables oncontainer closure systems.
Validation of Analytical Methods
Routine analysis of extractables andleachables in drug product bychromatographic and spectroscopictechniques.
Chromatography & Spectroscopy
Development of analytical methodsto evaluate Chemical and structuralchanges (e.g. by DSC) in polymericcontainer closure systems byspectroscopic or thermal analysis.
Spectroscopic & Thermal Analysis
For our extractables and leachables testing services, visit https://www.dalton.com/extractables-and-leachables
For a list of our full services, visit https://www.dalton.com/pharmaceutical-development-manufacturing-services
With Dalton's new E&L consultation services and our customized testingcapabilities, we can offer you a solution to your extractable and leachable needs.Our analytical team will be happy to help you overcome any concerns andchallenges. Recently Dalton has overcome the complexity of E&L testing on drugproducts that contain phosphate ions. Our solution: stable isotope labelledstandards (SILs) to quantitate leachables in the sample matrix. Dalton can also helpyou determine extractables and leachables on your next project:

The characterization of extractables provides medical-grade qualification and chemicalcharacterization information which is critical to avoid drawbacks during development.These drawbacks may include the introduction of non-intentionally added substances(NIAS) and contaminants, or even compounds migrating from printing and adhesives intothe drug products.
No single analytical technique in the study of extractables and leacheables can be usedfor all categories of compounds. As a result, testing for extractables and leachables isachieved by complementary use of a variety of analytical techniques:
E&L Studies and TestingA B O U T
GC with both flame ionization (FID) and thermal conductivity (TCD)
High-performance liquid chromatography (HPLC)-UV, HPLC-evaporative light scattering detection (ELSD), HPLC- chargedaerosol detector (CAD), ion chromatography
General compound class are typically tested by using fouriertransform infrared (FT-IR)
Thermal analysis by differential scanning calorimetry (DSC)
Volatile organic compound impurities (monomer residues, solventresidues, breakdown products) are typically tested by usingheadspace gas chromatography-mass spectroscopy (GC-MS)
Semi-volatile organic compound impurities (lubricants,plasticizers, antioxidants, polymer degradation products, solvents)are typically tested by using GC-MS or high resolution accuratemass (HRAM) GC-MS
Elemental impurities (metals) are typically tested byusing inductively coupled plasma (ICP)-MS
ID by HPLC - mass spectrometry

Guidance documents & pharmacopeia
E&L RegulationsA B O U T
Some international authorities require very detailed information on specificimpurities (e.g., Germany and France require viral clearance in biological products).Refer to the guidance documents and pharmacopeias around impurities andreporting levels.
US FDATitle 21 of the eCFR Part 211.65, states that “Equipment shall be constructed sothat surfaces that contact components, in-process materials, or drug productsshall not be reactive, additive, or absorptive so as to alter the safety, identity,strength, quality, or purity of the drug product beyond the official or otherestablished requirements” requiring the assessment of extractables andleachables.Guidance for industry: container closure systems for packaging human drugsand biologics (1999)
EMAGuideline on plastic immediate packaging materials (2005)
HCGuidance for industry: pharmaceutical quality of inhalation and nasal products(2006)
US pharmacopeiaPlastic packaging systems for pharmaceutical useAssessment of E&L associated with pharmaceutical packaging/delivery systemsAssessment of drug product leachable associated with pharmaceuticalpackaging/delivery systemsOrally inhaled nasal drug products
ISO guidelines for medical devices
Currently, there are no internationally harmonized guidelines on the assessmentand control of E&L. This current gap generates uncertainty for industry andregulators, creating potential delays in the approval of regulatory applications.
To solve this, the ICH is expected to release a guideline in November 2024 on thecontrol of E&L, Q3E EWG Impurity: Assessment and Control of Extractables andLeachables for Pharmaceuticals and Biologics.
The FDA, EMEA, and other regulatory authorities are taking an increased interest inthe interactions of various drug delivery devices, pharmaceutical productcontainers, and medical devices with a drug product and/or patient.
International standard

CTD Impurity ReportingA B O U T
CTD Amendments and Notifications
Impurity Reporting Requirements
The structure (or other identifier, if not structurallycharacterized), as well as the origin, should beprovided in the drug substance impurity table
Will include all identified impurities except for theones negotiated to be droppedInformation is provided within section 3.2.S.3.2(Impurities)
Must clearly demonstrate the purity of your drugproduct within an established rangeMust clearly demonstrate all established controls tomonitor manufacturing
The impurity name (or identifier), structure (ifcharacterized), and origin should be provided in thetable for all specified impuritiesImpurity levels for previously manufacturednonclinical and clinical batches may also besummarizedDemonstrate removal of any reagents used inmanufacture
Clearly establish a validation report to demonstrateviral clearance if applicableRaw material (animal sourced) certifications toclaim your drug product is free from TSE material(Transmittable Spongiform Encephalopathy)Removal of any reagents from the manufacturingprocess
Will include all identified impurities except for theones negotiated to be droppedInformation is provided within the developmentalsection of Module 3.2.S.3.2 (Impurities) and Module3.2.A.2; Adventitious Agents Safety Evaluation (TSEcertificates – Viral Clearance Validation)
Required where a new impurity has been identified.
Reporting
Requirements
PHASE I PHARMACEUTICAL
Reporting
Requirements
PHASE II/III PHARMACEUTICAL
Reporting
Requirements
PHARMACEUTICAL LAUNCH
Reporting
Requirements
PHASE I BIOLOGICS
Reporting
Requirements
PHASE II/III BIOLOGICS
Reporting
Requirements
BIOLOGIC LAUNCH
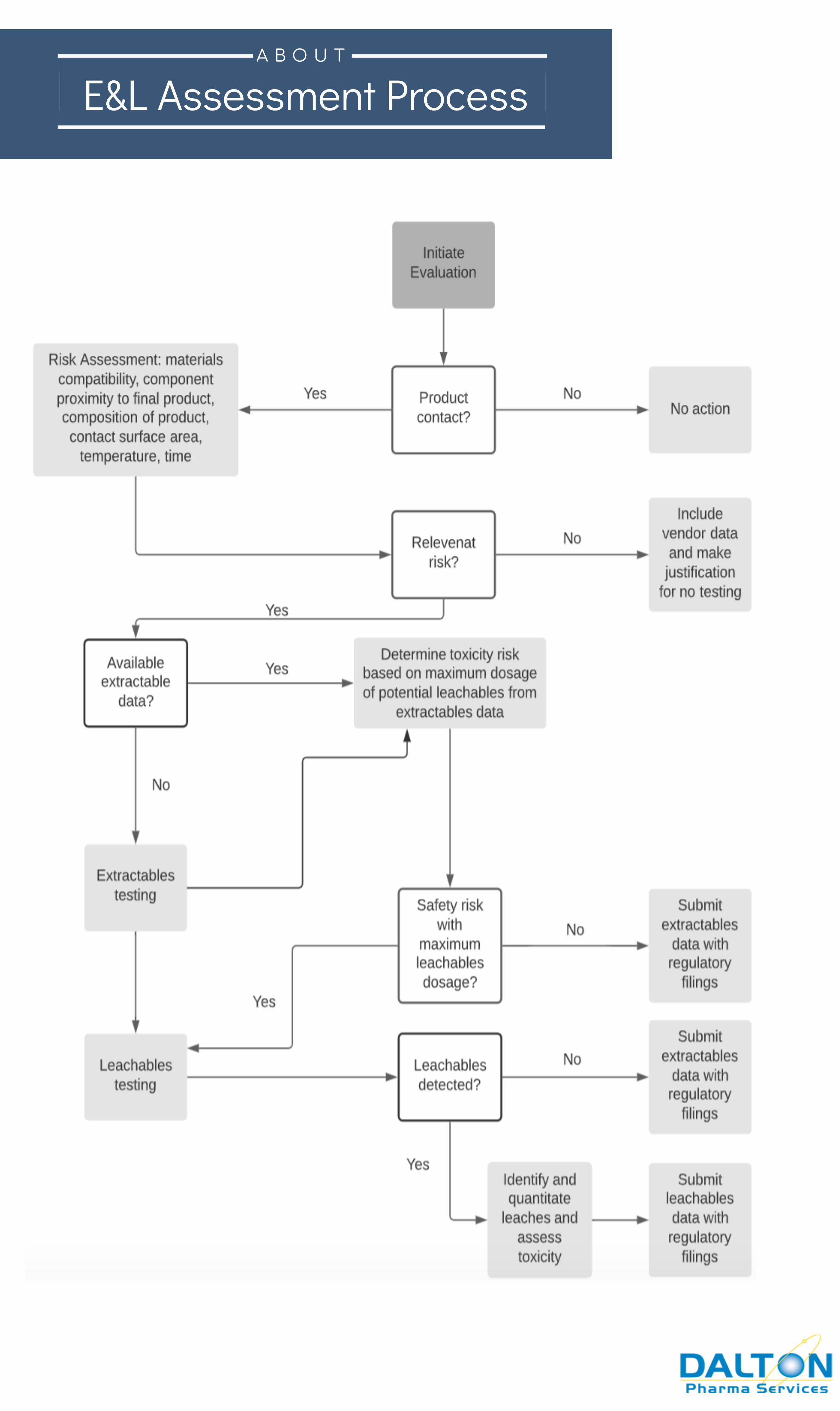
E&L Assessment ProcessA B O U T

Connect with Us
#DaltonPharmaServices
https://www.dalton.com
Special Acknowledgment to Gisel Murillo
REFERENCES
1. Hotha, K. (2017). Extractables and leachables regulatory perspectives [7]https://bit.ly/35ClhZs
2. International Conference on Harmonisation, Guideline for Impurities in New DrugSubstances Q3A(R2). Retrieved fromhttps://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf
3. Li, K., Rogers, G., Nashed-Samuel, Y., Lee, H., Mire-Sluis, A., Cherney, B., ... & Markovic, I.(2015). Creating a holistic extractables and leachables (E&L) program for biotechnologyproducts. PDA Journal of Pharmaceutical Science and Technology, 69(5), 590-619.
4. Yu LX, Amidon G, Khan MA, et al. Understanding pharmaceutical quality by design.AAPS J. 2014;16(4):771-783. doi:10.1208/s12248-014-9598-3
Call Us(416)-661-2102(800)-567-5060
Write UsDalton Pharma Services349 Wildcat Rd.Toronto, ON M�J �S�
Email [email protected]


















