
AMethod for the Improvement of Threading-Based...
19
A Method for the Improvement of Threading-Based Protein Models Andrzej Kolinski, 1,2 * Piotr Rotkiewicz, 1,2 Bartosz Ilkowski, 1,2 and Jeffrey Skolnick 1 * 1 Laboratory of Computational Genomics and Bioinformatics, Danforth Plant Science Center, CET, St. Louis, Missouri 2 Department of Chemistry, University of Warsaw, Warsaw, Poland ABSTRACT A new method for the homology- based modeling of protein three-dimensional struc- tures is proposed and evaluated. The alignment of a query sequence to a structural template produced by threading algorithms usually produces low- resolution molecular models. The proposed method attempts to improve these models. In the first stage, a high-coordination lattice approximation of the query protein fold is built by suitable tracking of the incomplete alignment of the structural template and connection of the alignment gaps. These initial lattice folds are very similar to the structures result- ing from standard molecular modeling protocols. Then, a Monte Carlo simulated annealing procedure is used to refine the initial structure. The process is controlled by the model’s internal force field and a set of loosely defined restraints that keep the lattice chain in the vicinity of the template conformation. The internal force field consists of several knowl- edge-based statistical potentials that are enhanced by a proper analysis of multiple sequence align- ments. The template restraints are implemented such that the model chain can slide along the tem- plate structure or even ignore a substantial fraction of the initial alignment. The resulting lattice models are, in most cases, closer (sometimes much closer) to the target structure than the initial threading- based models. All atom models could easily be built from the lattice chains. The method is illustrated on 12 examples of target/template pairs whose initial threading alignments are of varying quality. Pos- sible applications of the proposed method for use in protein function annotation are briefly discussed. Proteins 1999;37:592–610. r 1999 Wiley-Liss, Inc. Key words: protein modeling; homology modeling; Monte Carlo simulations; threading; lat- tice protein models; protein structure prediction INTRODUCTION In the limit of no apparent sequence similarity to any solved protein structure, threading methods are perhaps the most powerful tool for the approximate fold determina- tion of new protein sequences. 1–6 Given a library of known protein structures, threading methods attempt to find the fold that is the most compatible with a probe sequence. Various sequence-to-structure alignment methods and a variety of scoring functions (simplified potentials) have been developed to achieve this goal. 1–4,7,8 When the probe sequence is aligned onto the structure of another protein, an approximate three-dimensional model results. Unfortu- nately, the resulting models obtained from threading approaches are usually of very low quality, with gaps and insertions in threading alignments that somehow have to be connected or closed. Some fragments are significantly misaligned with respect to the true structure of the test protein. This may happen for several reasons. First, despite some general similarity in their folds, the struc- ture of the query sequence and the protein detected by a threading method may be quite different. Thus, even after the best possible superposition, the two structures may differ significantly. Second, various threading methods and their associated scoring functions only focus on as- pects of protein structure and a subset of their possible interactions. Consequently, the resulting sequence-to- structure alignments are, in general, quite different from those which result from the optimal structural superposi- tion. 6 Since standard threading procedures employ a rigid ‘‘template’’ protein scaffold, the design of a scoring function that would give rise to the best possible structural superpo- sition is, in practice, extremely difficult. Nevertheless, even crude and incomplete threading- based three-dimensional models are sometimes sufficient for the functional annotation of newly sequenced pro- teins. 9,10 However, as the quality of the model decreases, active site identification becomes more and more problem- atic. With regard to the immense number of new protein sequences of unknown functions that come from sequenc- ing various genomes, any method that improves protein structure predictions would be of great scientific as well as practical importance. When the threading alignments are of good quality, then standard homology modeling tools can be used to build useful molecular models. 11,12 In contrast, when the align- ments are poor, it is rather unlikely that classical homol- ogy modeling can significantly correct alignment errors. Grant sponsor: National Institutes of Health; Grant Number: GM-48835; Grant sponsor: KBN (Poland); Grant number: grant GR-919; Grant sponsor: Howard Hughes Medical Institute; Grant number: 75195-543402. *Correspondence to: Jeffrey Skolnick, Laboratory of Computational Genomics and Bioinformatics, Danforth Plant Science Center, CET, 4041 Forest Park Avenue, St. Louis, MO 63108 or Andrzej Kolinski, Department of Chemistry, University of Warsaw, Pasteura 1, 02–093, Warsaw, Poland. Received 8 February 1999; Accepted 20 August 1999 PROTEINS: Structure, Function, and Genetics 37:592–610 (1999) r 1999 WILEY-LISS, INC.
Transcript of AMethod for the Improvement of Threading-Based...

A Method for the Improvement of Threading-BasedProtein ModelsAndrzej Kolinski,1,2* Piotr Rotkiewicz,1,2 Bartosz Ilkowski,1,2 and Jeffrey Skolnick1*1Laboratory of Computational Genomics and Bioinformatics, Danforth Plant Science Center, CET, St. Louis, Missouri2Department of Chemistry, University of Warsaw, Warsaw, Poland
ABSTRACT A new method for the homology-based modeling of protein three-dimensional struc-tures is proposed and evaluated. The alignment of aquery sequence to a structural template producedby threading algorithms usually produces low-resolution molecular models. The proposed methodattempts to improve these models. In the first stage,a high-coordination lattice approximation of thequery protein fold is built by suitable tracking of theincomplete alignment of the structural templateand connection of the alignment gaps. These initiallattice folds are very similar to the structures result-ing from standard molecular modeling protocols.Then, a Monte Carlo simulated annealing procedureis used to refine the initial structure. The process iscontrolled by the model’s internal force field and aset of loosely defined restraints that keep the latticechain in the vicinity of the template conformation.The internal force field consists of several knowl-edge-based statistical potentials that are enhancedby a proper analysis of multiple sequence align-ments. The template restraints are implementedsuch that the model chain can slide along the tem-plate structure or even ignore a substantial fractionof the initial alignment. The resulting lattice modelsare, in most cases, closer (sometimes much closer) tothe target structure than the initial threading-based models. All atom models could easily be builtfrom the lattice chains. The method is illustrated on12 examples of target/template pairs whose initialthreading alignments are of varying quality. Pos-sible applications of the proposed method for use inprotein function annotation are briefly discussed.Proteins 1999;37:592–610. r 1999 Wiley-Liss, Inc.
Key words: protein modeling; homology modeling;Monte Carlo simulations; threading; lat-tice protein models; protein structureprediction
INTRODUCTION
In the limit of no apparent sequence similarity to anysolved protein structure, threading methods are perhapsthe most powerful tool for the approximate fold determina-tion of new protein sequences.1–6 Given a library of knownprotein structures, threading methods attempt to find thefold that is the most compatible with a probe sequence.Various sequence-to-structure alignment methods and a
variety of scoring functions (simplified potentials) havebeen developed to achieve this goal.1–4,7,8 When the probesequence is aligned onto the structure of another protein,an approximate three-dimensional model results. Unfortu-nately, the resulting models obtained from threadingapproaches are usually of very low quality, with gaps andinsertions in threading alignments that somehow have tobe connected or closed. Some fragments are significantlymisaligned with respect to the true structure of the testprotein. This may happen for several reasons. First,despite some general similarity in their folds, the struc-ture of the query sequence and the protein detected by athreading method may be quite different. Thus, even afterthe best possible superposition, the two structures maydiffer significantly. Second, various threading methodsand their associated scoring functions only focus on as-pects of protein structure and a subset of their possibleinteractions. Consequently, the resulting sequence-to-structure alignments are, in general, quite different fromthose which result from the optimal structural superposi-tion.6 Since standard threading procedures employ a rigid‘‘template’’ protein scaffold, the design of a scoring functionthat would give rise to the best possible structural superpo-sition is, in practice, extremely difficult.
Nevertheless, even crude and incomplete threading-based three-dimensional models are sometimes sufficientfor the functional annotation of newly sequenced pro-teins.9,10 However, as the quality of the model decreases,active site identification becomes more and more problem-atic. With regard to the immense number of new proteinsequences of unknown functions that come from sequenc-ing various genomes, any method that improves proteinstructure predictions would be of great scientific as well aspractical importance.
When the threading alignments are of good quality, thenstandard homology modeling tools can be used to builduseful molecular models.11,12 In contrast, when the align-ments are poor, it is rather unlikely that classical homol-ogy modeling can significantly correct alignment errors.
Grant sponsor: National Institutes of Health; Grant Number:GM-48835; Grant sponsor: KBN (Poland); Grant number: grantGR-919; Grant sponsor: Howard Hughes Medical Institute; Grantnumber: 75195-543402.
*Correspondence to: Jeffrey Skolnick, Laboratory of ComputationalGenomics and Bioinformatics, Danforth Plant Science Center, CET,4041 Forest Park Avenue, St. Louis, MO 63108 or Andrzej Kolinski,Department of Chemistry, University of Warsaw, Pasteura 1, 02–093,Warsaw, Poland.
Received 8 February 1999; Accepted 20 August 1999
PROTEINS: Structure, Function, and Genetics 37:592–610 (1999)
r 1999 WILEY-LISS, INC.

One possible way to deal with this problem was recentlyproposed by Jaroszewski et al.13 They have shown thatdetailed molecular models built from various threadingalignments (obtained by changing the parameters of thethreading algorithm) can be evaluated using knowledge-based potentials and the best alignments could be detectedin the majority of cases. This method detects and rejectsthese cases where the target and template structuresdiffer significantly. However, it does not deal with theproblem of improving the alignment subsequent to theidentification of the best probe-template pair.
In this work, we attempt to build and refine proteinmolecular models. Threading-based target-template align-ments have been obtained from one standard threadingmethod;14 but in principle any could be used.15 The model-ing technique employs a lattice model recently developedby us and tested in a different context. This SICHO (SIdeChain Only) model employs a very simple, computation-ally very efficient, yet quite accurate, representation ofprotein structure and dynamics.16,17 For the purpose of thepresent application, the model has been refined by incorpo-rating evolutionary information into the interactionscheme. Starting from an initial conformation of the modellattice chain that approximately follows the threadingtemplate, a Monte Carlo annealing procedure attempts tofind a conformation that maintains some (but not all)features of the original template and at the same time,optimizes packing and intra-protein interactions, as de-fined by the reduced model of the probe protein. This couldalso be visualized as a folding simulation in a soft tubebuilt around the threading template.
The new method has been applied to 12 target/templateprotein pairs that produce various quality models. Theparameters of the lattice model force field (more precisely,the balance between the intrinsic force field and thetemplate-related biases) have been adjusted by a trial anderror method for three of the 12 target/template proteinpairs. The obtained parameters were subsequently used inthe other nine simulations. As will become apparent afteranalysis of the simulation results, the obtained models forthe three proteins used for tuning the potential are amongthe best. This may suggest that the method was stronglytuned to these three examples. This is not the case. First,these three proteins belong to completely different struc-tural classes, so the tuning should be rather general, i.e.,applicable to the majority of single domain proteins.Second, when the tuning procedure is performed on just asingle case (the plastocyanin/azurin pair) almost the sameresults are obtained; this suggests that the optimal bal-ance between the template-related soft restraints and theintrinsic force field of the model is similar for variousproteins. Finally, the poorer results obtained for most ofthe remaining nine test proteins are simply due to the verypoor quality of the initial threading models.
The remainder of this paper is outlined as follows. InMethods, we present the reduced lattice protein modelused in the Monte Carlo sampling procedure. We describethe protein representation, the model of stochastic dynam-ics, the interaction scheme and the template-related bi-
ases and restraints. Then, in the Results section, themolecular models obtained from Monte Carlo simulatedannealing and subsequent refinement procedures are com-pared with the initial crude, threading-based models. Inthe Discussion, we analyze the improved models andattempt to identify typical underlying structural rearrange-ments. Possible developments and applications to large-scale sequence-to-structure, and structure-to-function com-putational projects are also briefly discussed. Finally, theConclusions section summarizes the main findings of thiswork. Some details that are important on a technical level,but not necessary for following the main features of thiswork, are found in the Appendix.
METHODSLattice Model
The reduced modeling of protein structure and dynamicsusually employs an alpha carbon main chain representa-tion.18,19 Side chains are either completely neglected ortreated at various levels of simplification. The choice of thealpha carbon representation is mostly motivated by thehigh level of geometric regularity of the main chains infolded proteins.19 On the other hand, the packing andinteractions between the side chains are perhaps muchmore sequence specific than are those of the main chain.
Motivated by the above reasons, we recently proposed avery simple lattice model in which we only explicitly treatthe protein side chains. Elsewhere, it has been shown thatit is possible to incorporate many protein-like features intosuch a representation.16,17 These include local conforma-tional propensities and the characteristic packing regulari-ties of protein side chains. The advantage of this model isthat the entire conformational space of quite large proteinscan be efficiently sampled. For example, assuming looseknowledge of the secondary structure and a few long-rangeside chain contacts (about N/7, where N is the number ofresidues), which may come from sparse NMR data or otherexperimental techniques, low-resolution protein structurescould be reproducibly and rapidly assembled for proteinscontaining up to 250 amino acids.17 This compares ratherfavorably with other attempts to build low-resolution modelsfrom a small number of long-distance restraints.19
The model employed here is very similar to that previ-ously described. There are small updates to the proteinrepresentation that slightly increase the geometric fidelityof the model. For the reader’s convenience, the design ofthe model is outlined below.
Reduced representation of polypeptide chains
The model chain consists of a string of virtual bondsconnecting the interaction centers that correspond to thecenter of mass of the side chains and the backbone alphacarbons. All heavy atoms have the same weight in thisaveraging. Thus, the center of glycine coincides with its Ca,the center of alanine is located in the middle of the Ca2Cb
bond, the center of valine roughly coincides with the Cb
atom, etc. These interaction centers (beads) are projectedonto an underlying cubic lattice with a lattice spacing of1.45 A. Obviously, the virtual bonds resulting from such a
593IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

projection are of various lengths that depend on theidentity of the two corresponding residues, the main chainconformation and the rotameric state of the side chain (seeFig. 1). A change in any of these variables may change thecorresponding virtual bonds (the chain vectors v). Inproteins, these distances have a quite broad distribution,ranging from 3.8 A for a pair of glycines to about 10 A forsome pairs of large side chains in their anti-parallel orienta-tion and expanded conformations. The corresponding set oflattice vectors covers this distribution with good fidelity. Theshortest vectors are of the form of (62, 62, 61) or (63, 0, 0)vectors, including all possible permutations. The length ofthese vectors corresponds to a distance of 4.35 A. The longestlattice vectors are of the (65, 62, 61) type and their lengthcorresponds to 7.94 A. Thus, the wings of the distribution arecut off. This should not have any noticeable effect on themodel’s fidelity because the small-distance cut-off error is wellbelow the resolution of the model, and the long-distance cut-offerror is not important due to very rare occurrences of distancesabove 8 A. As a result, the set of allowed lattice bondsconsists of 646 vectors. For technical reasons, sequentiallyadjacent vectors must not be identical.
A cluster of excluded volume points is associated witheach bead of the model chain. Each cluster consists of 19lattice points: the central one, six points at positions (61,0, 0), (0, 61, 0) and (0, 0, 61) with respect to the centralone, and 12 points at positions (61, 61, 0), including allpermutations. Thus, the closest approach positions ofanother cluster with respect to a given cluster are of theform (62, 62, 61) and (63, 0, 0), as measured between thecluster centers. Consequently, there are 30 closest ap-proach positions. The distance of the closest approachnicely corresponds to the smallest values of the inter-residue distances in real proteins. Since the average‘‘contact distances’’ (see the following sections) of the modelresidues are somewhat larger than the distance of the
closest approach, there are much more than 30 spatialorientations of two residues being in contact. Conse-quently, such a representation of protein structure entirelyavoids various anisotropy effects typically seen in thelower resolution lattice protein models. Figure 2 shows asmall fragment of the model chain confined to the underly-ing cubic lattice with a lattice spacing equal to 1.45 A. Theexcluded volume points are denoted by the solid and opencircles. The solid circles indicate the three lattice pointsalong the direction orthogonal to the plane of the figure:one in the plane below and one in front of the plane. Theopen circles denote points in the plane. With the abovegeometric restrictions, all PDB structures20 could be repre-sented with an average root mean square deviation (rmsd)of about 0.8 A. Again, the accuracy of the fit does not showany systematic dependence on protein length or on theorientation of the crystallographic structure with respectto the lattice coordinate system. Some features of themodel chain are illustrated in Figure 1.
Conformational updating
The simplicity of the model protein representation facili-tates the very rapid sampling of conformational space. TheMonte Carlo algorithm employs three types of conforma-tional transitions. The first type is a single bead, two-chainvector move. A random displacement of a randomly se-lected bead is generated and approved provided that thevector lengths and the excluded volume are not violated.The range of a random displacement is from 1 to 51/2 latticeunits. When accepted by the Metropolis criterion21 (see thenext section), such a move is equivalent to a collective
Fig. 1. Schematic illustration of the protein representation employedin this work. The fragment of a detailed protein structure (main chain back-bone is shown in gray and the side chains in thinner sticks). The blue stickscorrespond to the virtual bonds of the model chains, connecting the centersof mass of groups of atoms consisting of side chains and alpha carbons.
Fig. 2. Lattice representation of the model chain and its excludedvolume. The sticks correspond to the model chain virtual bonds. Excludedvolume of each model amino acid is represented by 19 points on theunderlying cubic lattice with the mesh size equal to 1.45 A. The black dotscorrespond to three lattice points along the axis orthogonal to the pictureplane (one in the plane, one below, and one above the plane). The opencircles correspond to single lattice points in the picture plane.
594 A. KOLINSKI ET AL.

rearrangement of the main chain and/or the side chaininternal coordinates in a real polypeptide chain. The forcefield of the model, especially its generic components,prevents the acceptance of nonsensical, non protein-likeconformations.16 The second type of motion involves thepermutation of three chain vectors. This is a larger scalemove that is relatively rarely accepted due to possiblesteric interactions. The last type of move involves arandomly selected fragment consisting of several chainunits. This fragment moves as a rigid body due to appropri-ate small changes in the two flanking chain vectors. Forinstance, such a move may translate a helical segment by asmall distance, thereby slightly changing the conforma-tion of the corresponding turn or loop regions.
Interaction scheme
The model force field consists of several types of poten-tials. The first are generic, sequence-independent, biasesthat penalize against non protein-like conformations. Se-quence specific contributions to the force field consist ofknowledge-based, two-body and multibody potentials ex-tracted from a statistical analysis of known protein struc-tures. Finally, there are two kinds of potentials thatcontain evolutionary information extracted from multiplesequence alignments. In all cases, all PDB structureswhose sequences are similar to the query sequence havebeen removed from the structural database used in thederivation of the potential (greater than 25% sequenceidentity). All potentials were derived from PDB structuresand then translated into proper lattice discretized form.
The generic protein stiffness potential and second-ary structure bias. As defined above, the model chain isintrinsically very flexible. A substantial fraction of itsconformations that are allowed due to the assumed simpli-fied hard core interactions do not correspond to any realpolypeptide chain conformation. In reality, proteins arerelatively stiff polymers exhibiting very characteristic dis-tributions of certain short-range distances. For example, thebimodal distribution of the distances between the i-th andi14th residues reflects the tendency to adopt either of twotypes of conformations. These correspond to expanded(b-type or expanded coil) or very compact conformations(as within helices or turns). Such generic features need tobe included in the model. We proceed in a similar fashion,as described elsewhere.16 The details are different due tothe particular protein representation that we employ.
First, for all possible two-vector sequences of the modelchain, let us define a direction w that is almost perpendicu-lar to the plane formed by the fragment. A small system-atic deviation from the exactly orthogonal direction isintroduced into w to obtain vectors that are on averageparallel to the helix axis and that also account for theaverage supertwist of b-strands.
ui 5 (vi21 ^ vi 2 vi21 2 vi) (1)
wi 5 ui / 0ui 0 (2)
where vi is the i-th vector (or virtual bond) of the modelchain, the symbol ‘‘^’’ denotes the vector cross product and
0ui0 is the length of vector ui. These ‘‘directions of secondarystructure’’ (the vectors w point along a helix or across ab-sheet) are normalized so that their length equals unity.The idea is explained in Figure 3, where the model chainvirtual bonds are shown in solid lines and the vectors wiare shown in open arrows.
The stiffness/secondary structure bias term has thefollowing form:
Estiff 5 2egen [S min 50.5, max (0, wi ● wi12)6]
2 egen [S min 50.5, max (0, wi ● wi14)6]. (3)
Where egen is a constant energy parameter, common for allgeneric potentials, and S means the summation along thechain. The above formulation means that the system isenergetically stabilized when pairs of the ‘‘direction ofsecondary structure’’ vectors are parallel (positive dotproduct). As can be read from the above equation, thestabilization energy increases in the range between 90°and 30° (the angle between appropriate vectors w) andthen maintains its extreme value. Thus, small fluctuationsof secondary structure have no influence on the value ofthis potential.
Additionally, a weak bias has been introduced towardshelix-like and b-type expanded states. All conformationsare, of course, allowed; the purpose of this bias is to mimica protein-like (average) distribution of local conforma-tions. Symbolically, this could be written as follows:
Estruct 5 S 5dH1(i) 1 dH2(i) 1 dE1(i) 1 dE2(i)6 (4)
with:
dH1(i) 5 2egen, for ri,i142 ,36 and
(vi ● vi13) . 0 and (vi ● vi12) , 25
0, otherwise (4a)
dH2(i) 5 2egen, for ri,i142 , 36 and
(vi ● vi13) . 0 and (vi11 ● vi13) , 25
0, otherwise (4b)
Fig. 3. A fragment of the model chain and a set of vectors w employedin the definition of the short-range polypeptide chain stiffness (see the textfor details).
595IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

dE1(i) 5 2egen, for 56 , ri,i142 , 135 and
(vi ● vi12) . 5
0, otherwise (4c)
dE2(i) 5 2egen, for 56 , ri,i142 , 135 and
(vi11 ● vi13) . 5
0, otherwise (4d)
The numerical values are in lattice units and are selectedto define a broad range of helical/turn conformations (forthe dH1 and dH2 contributions) or expanded conforma-tions (for the dE1 and dE2 contributions). Due to theexclusive character of the two subsets of geometricalconditions for specific chain conformations, the minimumcontribution from a residue is equal to 22egen (either thefirst two conditions or the two last conditions can besimultaneously satisfied). Let us express the last conditiona bit differently. Equation (4d) says that the system gainsan energy equal to 2egen for being in an expanded b-typeconformation. For a four-vector fragment of the chain, thisrequires that the distance between the i-th and i14th beads(the centers of mass of the side chain plus Ca units) has tolie between 10.7 and 16.8 A, and the chain vectors vi11 andvi13 have to be oriented in a parallel-like fashion (the dotproduct . 5). Additional stabilization is gained when, forthe same fragment, another pair of vectors is parallel (seeEq. (4c)). The broad ranges allow for substantial fluctua-tions around an ideal expanded state and accommodatethe variations of the model chain geometry caused bydifferences in side chain size.
We have performed computational experiments whereall interactions except the ones defined above, were turnedoff. At low temperature, the model chain forms rapidlyfluctuating local clusters of expanded and helix-like states.The persistence length and the distributions of the short-range distances along the chains mimic protein-like geom-etry.
Generic packing cooperativity. We introduce twoterms that enforce some of the most general regularities ofthe dense packing of protein structures.22 In all the moreregular elements of secondary structure (within helicesand b-sheets, but not between helices) and, to a lesserextent, in some coil-type fragments and turns, given acontact between a pair of reference residues, there is a verystrong preference to have contacts between the precedingand the following residues. Indeed, the contact maps ofglobular proteins contain very characteristic strips.23 Thosenear the diagonal correspond to the intra-helical contacts,those farther from the diagonal (parallel or antiparallel tothe diagonal) correspond to contacts between b-strandswithin b-sheets. Thus, we introduce the following ener-getic bias towards such a mode of packing:
Emap 5 2egen 5SS (di, j ● di11, j11 ● di21, j21) dpar
1 SS (di, j ● di21, j11 ● di11, j21) dapar6 (5)
where the summations are over all pairs of residues i, j,and di,j is equal to 1 (0) when residues i and j are (are not) incontact. dpar is equal to 1 only when the correspondingchain fragments are oriented in a parallel manner, i.e.,when the chain vectors satisfy the following condition(vi21 1 vi)●(vj21 1 vj) . 0, otherwise dpar 5 0. Similarly,dapar is equal to 1 when the chain fragments are anti-parallel, and it is equal to zero otherwise. For a givencontact of a pair of residues, the maximal energeticstabilization due to regular side chain packing is thereforeequal to 2egen, which has the same value as in thepreviously defined potentials.
The packing cooperativity of the model protein is furtherenhanced by a term that mimics main-chain hydrogenbonds. The geometry of protein hydrogen bonds is trans-lated into a specific range of the model chain geometry.First, let us define a vector that is likely to connect themodel beads that are within motifs that represent regularsecondary structure elements. Such a vector should con-nect beads i and i13 in a helix and the appropriate beadsin a b-sheet. An optimization procedure leads to thefollowing definition of this vector:
hi 5 3.3(vi21 ^ vi )/ 0 (vi21 ^ vi ) 02 vi21/ 0vi21 0 . (6)
The value of the 3.3 pre-factor has been found to beoptimal for reproducing the internal main chain hydrogenbonding in the lattice projected PDB structures. However,due to the wide distribution of the model chain bondlengths, there are always some hydrogen bonds that aremissed in the model. The coordinates of the vectors hi arerounded-off to the nearest integer value. Thus, in a helixthe hi vectors have a component whose length is about 3lattice units in the direction perpendicular to the three-residue plane (the first term in the above sum). They arealso tilted back by a lattice unit (the last term of Eq. (6)).The projection along the helix axis is also about 3 latticeunits; this nicely coincides with the 1.5 A longitudinalincrement per residue in a real helix. Residue i is consid-ered to be hydrogen bonded with residue j when the vectorhi points to any of the 19 points of the excluded volumecluster of residue j. Correspondingly, the vector 2hi maypoint to another cluster. Such a situation is illustrated inFigure 4, where residue i is hydrogen bonded with residuesj and k because the hydrogen bond vectors coincide withthe excluded volume of both residues. The excluded vol-ume clusters are symbolically represented by open spheres.Since the excluded volume clusters never overlap, themaximum number of these ‘‘hydrogen bonds’’ originatingfrom residue i is equal to 2. The total energy of the‘‘hydrogen bond network’’ can be written as:
EH-bond 5 2eH-bond S (d1 1 d2 1 d1,2) (7)
where d1 (d2) equals 1 when the vector hi (2hi) connectswith an excluded volume cluster, and d1,2 5 1 when bothvectors connect to some clusters, respectively. Otherwise,the corresponding terms are equal to zero. The cooperativecontribution, d1,2, corresponds to the local saturation ofthe hydrogen bond network.
596 A. KOLINSKI ET AL.

Again, a computational experiment has been done tocheck the effect of these generic potentials on the behaviorof the model system. When only the interactions outlinedup to this point are included (all the above short- andlong-range generic potentials), the model lacks sequencespecific information. At sufficiently low temperatures, thechain adopts either of the following two types of struc-tures: a long (sometimes broken) helical structure, or ab-sheet with a right-handed supertwist. These motifsfluctuate and are not structurally unique. In a long chain,these two classes of secondary structure elements some-times form separate domains.
Sequence specific short-range interactions. For thesequence of interest from the structural database, one mayextract the statistics of distances between a pair of aminoacids (with their interaction centers as defined in themodel) Ai and Bi1k, where A and B denote the identities ofthe amino acids and i is the position in the chain. Here, weconsider k 5 1, 2, 3, 4, 6 and 8. The terms for k 5 3 and k 56 are treated as chiral variables. This means that thedistance between Ai and Bi13 is stored as a positive ornegative number, depending on the handedness of thecorresponding three-bond segment. For the k 5 6 case, thechirality is defined for three subsequent supervectors (thedoublets of vectors between beads i and i12, i12 and i14,and from i14 to i16). As was done here, the sequence ofinterest can be divided into overlapping short fragments.These could be aligned to the sequences of known struc-tures. The highest scoring fragments provide a set ofstructural templates. The obtained statistics could berelated to a random distribution and the statistical poten-tial of mean force could be appropriately derived. The k 51, 2, 3, and 4 terms were weighted equally, while the termsfor k 5 6 and k 5 8 had weights reduced by a factor of two,
with respect to the lower order terms. Homologous pro-teins were always excised from the structural database forthe purpose of these test calculations. As previously shown,this type of potential very nicely reproduces the localconformational propensities of globular proteins.16
The short-range potentials could be made even moresequence specific when one employs evolutionary informa-tion encoded in homologous sequences. In such a case, thealigned fragments of highly homologous sequences (fromthe sequence database) are treated as the original testsequence, thereby increasing the strength of the statistics.The details of the derivation procedure are given inAppendix 1. Encoding such evolutionary information im-proves performance of the proposed method; however, it isnot crucial. Simulations without homology-enhanced poten-tials lead to slightly worse results. Most of the testsequences employed in this work belong to relatively largefamilies of proteins; however, the criterion of the numberof similar sequences was not taken into consideration inthe selection process. Also, in this respect, the selected setis rather representative of all small single domain pro-teins.
Sequence specific pairwise interactions. The pair-wise interactions between model residues are defined bycontact potentials in the form of a square well function.
`, for rij , 3
Eij 5 Erep, for 3 # rij , Ri, jrep
eij, for Ri, jrep # rij , Ri, j
0, for Ri, j , ri, j (8)
where eij are the pairwise interaction parameters, rij is thedistance between chain beads i and j, Erep 5 3kT is aconstant repulsive term operating at very short distances,and Ri,j
rep and Ri,j are the cut-off values that depend onamino acid type. The values of these cut-off parameters areprovided in Table I.
Because the derivation of the potentials uses evolution-ary information, the interaction parameters depend notonly on amino acid identity, but also on their positions inthe polypeptide chain. A more detailed description of the
Fig. 4. Schematic illustration of the main chain’s ‘‘hydrogen bonds.’’Residue i is hydrogen bonded to residue j and k because the vectors h i
and 2h i (see the definition in the text) connect with any of the pointsforming of the excluded volume clusters (the clusters are symbolicallyshown as large spheres) of these residues.
TABLE I. Compilation of Pairwise Cut-OffDistances for Pairwise Interactions
Ai Aj Ri,jrep (Å) Ri,j (Å)
Smalla Small 4.35b 5.97Largec Large 4.83 6.80Other Combinationsd 4.57 6.32aSmall amino acids are: Gly, Ala, Ser, Cys.bThis value corresponds to the excluded volume radius ofthree lattice units; therefore, for pairs of small amino acids,the soft-core envelope does not exist.cLarge amino acids are Phe, Tyr, Trp.dSmall-large, other (than small or large)-large, other-small.
597IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

derivation of these potentials may be found elsewhere.17
The total energy contribution from the pairwise interac-tions is therefore calculated as follows:
Epair 5 S S Eij (9)
where the summations are over all j . i pairs of residues.Multibody potentials. The hydrophobic interactions
in our model are partially accounted for by pairwiseinteractions between residues; however, this is not suffi-cient to generate well-packed proteins. Thus, a surfaceexposure based statistical potential has been developed.The scheme is as follows: Each model residue has beenassigned 24 surface contact points. A specific subset ofthese contact points becomes occupied upon contact withother residues. The main-chain Ca atoms contribute sepa-rately to the coverage of a given residue. The positions ofthe Ca atoms could be quite well approximated given thepositions of three consecutive side chain beads.16 Somecontact points could be multiply occupied. The fraction ofnon-occupied surface points defines the exposed fraction ofa given side chain. Potentials could be derived from astatistical analysis of the protein structures for which thesolvent exposure has been determined on the atomic level.The total surface energy is computed as follows:
Esurface 5 S Eb(Ai, ai) (10)
where ai is the covered fraction of the residue Ai and Eb(Ai,ai) is the statistical potential when amino acid type A has ai
of its surface points occupied, i.e., the covered fraction of itssurface is equal to ai/24.
Studying the distribution of inter-residue contacts inglobular proteins, we have found that various amino acidshave different tendencies to pack in a parallel or antiparal-lel fashion. A contact between residues i and j is consideredto be ‘‘parallel’’ when (vi21 2 vi)●(vj21 2 vj) . 0, and‘‘antiparallel’’ otherwise. Moreover, for a given residuethere are strong correlations between the number ofparallel and antiparallel contacts given the total numberof contacts. Due to the reduced character of our model, theother contributions to the force field do not properlyaccount for such effects. Therefore, the model force fieldhas been supplemented by the following multibody poten-tial:
Emulti 5 S Em(A,np,na) (11)
where Em(A,np,na) is the value of the statistical potentialfor residue type A having np parallel and na antiparallelcontacts. The reference state is a random distribution ofcontacts. The values along particular diagonals (np 1 na 5
nc) have been re-normalized such that the lowest energyfor a diagonal was exactly equal to the value of statisticalpotentials derived from the distribution of the total num-ber of contacts nc for a given type of residue.
Total intrinsic conformational energy. The totalinternal conformational energy of the model chain wasequal to:
Etotal 5 Estiff 1 Emap 1 0.875EH-bond 1 0.75Eshort
1 1.25Epair 1 0.5Esurface 1 0.5Emulti (12)
with the value of generic parameter egen 5 1 kT.The relative scaling of various potentials has been
adjusted by a trial and error method in ab initio foldingexperiments performed for the following selected smallproteins: 1fna, the B domain of protein A and the B1domain of protein G. The objective was to maintain lowsecondary structure content in the random coiled state anddense packing with a proper level of secondary structure inthe collapsed globular state. For instance, the small 56-residue a/b protein G domain folded ab initio in about 30%of simulated annealing Monte Carlo simulations to anative-like structure with an rmsd from native in therange of 4 A. The majority of the remaining misfoldedconformations had native-like secondary structures, butthey had topological errors, usually involving the wrongorder of b-strands in the four-member b-sheet. The modelis not sensitive to small variations in these scaling param-eters.
Building the Starting Lattice Model
A separate algorithm was used to build an initial latticemodel from a given target sequence alignment to a tem-plate structure. Such alignments contain gaps and inser-tions. First, interaction centers are computed from thetemplate. Then, starting from the first aligned position,the lattice chain is sequentially built. At each step in thealigned region, the new vectors are selected so as tominimize the distance of the lattice chain from the equiva-lent template points. In the gap regions, the distance fromthe last residue of the preceding aligned fragment to thefirst residue of the next is divided to generate a set ofcheckpoints. The number of these checkpoints is equal tothe number of target sequence residues that have to bemounted to span the gap. The checkpoints outside theentire alignment are randomly generated. The set of allcheckpoints provides the target for the starting latticemodel. The model chain maintains the excluded volumeand satisfies the other geometric restrictions discussedbefore.
Implementation of the Template Restraints
The template (more precisely the structural fragmentsof the template protein that correspond to the alignedresidues of the probe sequence) is projected onto theunderlying cubic lattice. The corresponding three-dimen-sional array, initially filled with zeros, is then updated tostore a loose trace of the template. All elements of thearray that are closer than 61/2 lattice units from templateresidues are assigned the corresponding residue numbers.When a lattice point is within a distance of 61/2 from two ormore residues, the number of the closest residue is as-
598 A. KOLINSKI ET AL.

signed to the corresponding element of the occupancyarray. In the direction towards the center of mass of thetemplate, the cut-off distance for creating the template‘‘tube’’ is equal to 141/2 instead of the 61/2 value in the otherdirection. This fills in most of the volume occupied by thetemplate structure. Figure 5 schematically shows suchtubes surrounding the aligned fragments of the templatechain (in solid lines). To illustrate the above-mentioneddifferent width of the tube in the directions towards thecenter (versus the outside) of the template structure, theblobs forming the tube are shifted towards the center ofmass of the template. This facilitates the close packing ofthe query (target) chain that wanders within the tube.
As described in the previous section, the starting modelis placed into the template tube. The initial alignmentprovides an equivalence list between the template andtarget residue indices. This is called ‘‘the old assignment’’in contrast to the ‘‘new assignment’’ which will be gener-ated by the program. Both the old and the new assign-ments are then evaluated and updated in the followingway:
a) At the very beginning of the simulation process, the oldassignment (the original alignment) is copied into thenew assignment list. The entries of these lists identifythe tube compartments and the equivalent residues ofthe template chain. Then, all residues for which thetotal number of long distance (i 2 j . 4) contacts for athree-residue fragment (with the residue of interest asa central one) is smaller than two become non-assignedboth in the old and new assignment lists. This erasesthose template fragments that do not interact with the
rest of the model protein. Thus, ‘‘non compact’’ frag-ments of the template are ignored.
b) The new assignment is then updated when, for a stericreason (or due to local stiffness), the initial query chainresidue simultaneously satisfies the following two crite-ria: (i) the bead of the query chain is farther away thanfive lattice units from the corresponding template resi-due of the original equivalence assignment (‘‘old assign-ment’’), and, (ii) the position of the query chain residue(the central point of the excluded volume cluster)coincides with a lattice point that is assigned to anyother template residue. The number read from theappropriate element (occupied by the lattice chain) ofthe occupancy array that corresponds to the beadcoordinates becomes the updated entry of the newequivalence list.
c) For all residues of the starting query chain that arefarther away than nine lattice units from the equiva-lent (according to the old assignment) template resi-dues, both old and new assignments are erased. Theseresidues also become non-assigned. All allowed updatesof the old assignments can only remove some entriesfrom the equivalence list, which means that some partof the threading alignment is erased. The new assign-ments are dynamic (due to the updates described in b),and they have the character of a structural superposi-tion, which is not sequential in many places.
This updated pair of assignments of the query chainresidues to the template defines a flexible tube around thetemplate chain. To keep the moving query chain in theneighborhood of the template, a set of biases is introduced.First, the model chain is kept in the broad vicinity of theoriginal template (according to the updated old assign-ment list) by
Etemp,o 5 S do(i)fr max 50, (0ri 2 roi 0 2 9)6 (13)
where fr is a constant (equal to 1kT in all simulations), ri isthe position of the query chain, roi is the position of thetemplate and do(i) is equal to 1 (0) when residue i isassigned (non assigned) according the old alignment.
Then, the residues of the query chain are similarlybonded to the template residues in the new assignment by
Etemp,n 5 S dn(i)fr max 50, (0ri 2 rni 0 2 Rt)6 (14)
where rni is the position of the initial template according tothe new assignment and dn(i) is equal to 1 (0) when residuei is assigned (non assigned) according the new assignment.The constant Rt is equal to 7 (4) when residue i occupiesany point of the template tube (the residue is outside thetube, i.e., the occupancy array at position ri has value 0).
Additional restraints are the following:
Etube 5 2Erep S 5do(i)d3(i) 1 dn(i)dt(i) 1 dn(i)dc(i)6 (15)
where d3(i) is equal to 1 when residue i of the query chain isat a distance smaller than 3 lattice units from the template
Fig. 5. Fragment of the model template chain (shown in the blacksticks) and the template tube formed by the chain of spheres. The targetchain (not shown in the drawing) is allowed to move in the tube with apenalty associated with all excursion from the tube.
599IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

according to the old assignment, otherwise d3(i) 5 0. Thesecond component, dt(i), is equal to 1 (0) when the residueis anywhere in the template tube (is outside). dc(i) is equalto 1 for a ‘‘quasi-continuous’’ alignment on the tube, i.e.,when 5al(i21) 1 al(i11)6/2 2 al(i) , 2, where al(i) is thevalue of occupancy array in the tube for residue i of thequery chain, otherwise dc(i) 5 0.
A small energy reward is also provided when the second-ary structure of the query chain is consistent with thetemplate structure. For all residues that are in extended orhelical states (as defined in the loose conformationaldefinition used for the generic short-range potentials) andthat are in agreement with the secondary structure readfrom the corresponding fragments of the template protein,the system is stabilized by an energy equal to 2egen.
With the above restraints, the system only pays a smallenergetic penalty for moving along the template tube(shifts in the alignment with possible lateral adjustment);however, the penalty is large for escaping from the looselydefined volume occupied by the template. For instance, itis possible (and this happened in a couple of cases studiedhere) that continuous fragments of the original alignmentspermute (this cannot be called an alignment in the conven-tional sense) by swapping their original tube compart-ments. This only occurs when the potential strongly favorssuch a rearrangement of the topology. The two assign-ments, carried out by the algorithm, play a different role.The ‘‘old’’ one bonds the model chain to the broad vicinity ofthe threading-based template. The ‘‘new’’ dynamic assign-ment is a compromise between the template restraints andpacking requirements of the model chain.
Summary of the Threading Model RefinementProtocol
The entire model building procedure is illustrated in aflow-chart (see Figure 6) and can be outlined as follows:
a) Generate the threading alignment between the querysequence and the template structure.
b) Derive the sequence similarity-based short and long-range pairwise potentials. (Structures of proteins ho-mologous to the query sequence are excised from thestructural database; however, multiple alignments withhomologous sequences of unknown structures wereused in the potential derivation procedures.)
c) Build the starting continuous model chain onto thelattice-projected template structure.
d) Build the tube around the aligned fragments of thetemplate structure. Then, perform the first stage ofMonte Carlo refinement, where simulated annealing isdone over a temperature range of 2–1. Since the MonteCarlo algorithm corrects unlike fragments of the align-ment, the simulated annealing run is repeated twotimes. Subsequent runs have no systematic effect onthe obtained models.
e) Refinement of the structure. The model obtained fromthe above simulations is assumed to be the new tem-plate, with a full length, complete self-alignment. Thedistance restraints from the new template are nar-
rowed to 4 lattice units, and simulated annealing isperformed over a narrower temperature range (1.5 to1.0).
f ) Selection of the lowest energy structures, by shortisothermal simulations at T 5 1, followed by buildingall-atom models using MODELLER.24
RESULTSTest Proteins, Templates and Starting Alignments
Twelve pairs of target/template proteins of very lowsequence similarity were selected for the present study.These proteins belong to various classes of small globularproteins, with the selected set being rather representative.As described in the Methods section, the relative scaling ofthe various potentials of the model force field has beenadjusted in a series of ab initio folding simulations on
Fig. 6. Flow chart illustrating the molecular modeling proceduredescribed in the text.
600 A. KOLINSKI ET AL.
several (different from described here) small proteins. Forthe tuning of the template restraint contribution, weselected three proteins: 2pcy, 256b, and 1hom. Theseproteins belong to rather different structural classes: 2pcyis a quite irregular b-type protein with a very poor initialthreading-based model, when the 2azaA template is used.256b is a compact, four-helix bundle, where the originalalignment appears to be quite good; however, the templateand target structures have a different packing of helicesthat needs to be significantly readjusted to obtain areasonable model. A very different example is 1hom. Here,the target fold is not very compact, and it is important tosee if the proposed procedure can handle such small openstructures. All proteins were subject to the previouslydescribed model building/refinement procedure. The list ofthese proteins is given in Table II. The threading align-ments have been generated by a standard threadingalgorithm.14 These alignments are compiled in Table III.
Compilation of the Modeling Results
Due to its stochastic character, the entire simulationprocedure has been repeated several times for each case ofthe target template chains. The resulting structures werethen subject to a refinement run. Namely, the algorithmemployed in the first stage of the Monte Carlo modeling(starting from the initial, ‘‘old’’ threading-based alignmentand performing all the updates of the alignment describedin the ‘‘implementation of the template restraints’’ section)has been used in short isothermal runs at low (T 5 1)temperature, with the final structure obtained at the endof the first stage of Monte Carlo used as input. At thistemperature, the model does not change any of its globalfeatures, rather only local fluctuations are seen. Theaverage conformational energy, which includes the intrin-sic force field of the model and the effect of templaterestraints, was then used to select the ‘‘best’’ structure.The model has quite a strong, root-mean-square deviation,rmsd, versus energy correlation far from the native state.Closer to native state, the two quantities become uncor-rected or the correlation is weak, depending on the particu-lar case. It should be pointed that out that this refers to theentire force field (intrinsic terms and the template biases).
A quite different situation is observed for the intrinsicforce field alone; this has the strongest correlation of rmsdversus energy near the native structure (unpublishedresults). Since all our models are, at best, of moderateresolution, this criterion is no better than that based onthe total energy. The lowest average (total) energy confor-mation from these short isothermal runs was selected forfurther consideration. For example, in the case of 1tlk, astructure that has a rmsd of 4.4 A from native wasselected, while several simulations resulted in structuresabout 3 A from native.
Tables IV and V contain a compilation of the simulationresults. In Table IV, the Ca rmsd from the native arecompared for two kinds of molecular models. The first wasgenerated using the initial threading template followed byautomated modeling using MODELLER. We realize thatthe use of this homology-modeling tool in such a naive wayis not the best practice; however, we wanted some means ofcomparison for two automated methods of model buildingfrom poor initial data. The second set of rmsd values is forthe present lattice models, which for convenient compari-son were converted into the full-atom models via anautomatic application of MODELLER (with lattice modelsof the Ca backbones used as templates). As one may see,the most significant improvement of the model qualityoccurs when the threading alignment produces a ratherpoor but not nonsensical initial model (compare Table IVand Table V). As shown in Table V, for small globularproteins, such initial threading-based models have a rmsdin the range of 6–8 A from native (over the alignedfragments). When the threading models are really bad,e.g., for 1cewI or 2azaA, the improvement is rather small.At the other extreme are those cases when the alignmentis good and the resulting rmsd relatively small. Here also,the changes are small because the models are alreadygood. Importantly, the procedure essentially does no harmto these models; thus, it can be applied to all situationswith impunity. Moreover, as it is illustrated in Figure 7,the proposed modeling method systematically improvesthe entire threading-based model. The number of residuesin structurally very accurate fragments (say less than 2 Afrom native) is the same as for threading based models.
TABLE II. List of Target/Template Pairs Studied in This Work
Target protein Template proteinPDB code Name Length PDB code Name Length
1aba_ Glutaredoxin 87 1ego_ Glutaredoxin 851bbhA Cytochrome C 131 2ccy_ Cytochrome C 1271cewI Cystatin 108 1molA Monellin 941hom_ Antennapedia protein 68 1lfb_ Transcription factor 771stfI Papain 98 1molA Monellin 941tlk_ Telokin 103 2rhe_ Immunoglobulin 114256bA Cytochrome c 106 1bbh_ Cytochrome C 1312azaA Azurin 129 1paz_ Pseudoazurin 1202pcy_ Plastocyanin 99 2azaA Azurin 1292sarA Ribonuclease 96 9rnt_ Ribonuclease 1043cd4_ T-cell surface glycoprotein 178 2rhe_ Immunoglobulin 1145fd1_ Ferrodoxin 106 2fxd_ Ferrodoxin 81
601IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

TABLE III. StartingAlignments Employed in Model Building
602 A. KOLINSKI ET AL.

For the remaining residues, a large average decrease ofthe distance from target coordinates could be observed inall cases (see Fig. 7). We have also run a number of testsimulations employing the structure of the query proteinas a template (with full and randomly selected ‘‘align-
ments’’ fragments). In all cases, the resulting structureswere 2.5–4.0 A rmsd (depending on protein size) from thenative structures. Thus, given a good initial alignment, themodel stays in the vicinity of that structure, therebydemonstrating that this approach ‘‘does no harm’’.
In summary, in 6 of 9 test cases (in 9 of 12, including thethree proteins employed in the model tuning procedure),the models generated by the method proposed here givelower values of rmsd over the set of aligned residues thanthat found in the initial structure. In the three remainingcases, the changes in rmsd are insignificant (essentially inthe range of the statistical fluctuations). In five cases,qualitative improvements were observed (for the alignedresidues as well as for entire models; compare data givenin Table IV): from 5.6 A to 3.5 A for 1hom, from 7.1 A to 4.7A for 1stfI, from 7.9 A to 3.9 A for 1tlk, from 6.9 A to 4.4 Afor 256b or from 6.6 A to 4.4 A for 2pcy. These numbers arefor the initial threading and final lattice (refined withMODELLER) models, respectively. It should be noted thatthe MODELLER refinement of the final lattice modelschanges their rmsd very little (in the range of 0.2 A), whilethe improvement of the initial threading models by theapplication of MODELLER is more noticeable.
TABLE IV.Alpha Carbon Rmsd FromNative for Models Built From the InitialThreadingAlignments and Refined by
Lattice Simulations†
Targetprotein
Threading1MODELLER
SICHO1MODELLER
1aba_ 4.43 4.861bbhA 6.77 6.821cewI 14.96 14.381hom_ 7.82 3.701stfI 6.40 5.951tlk_ 7.23 4.17256bA 6.09 4.362azaA 21.95 10.772pcy_ 6.56 4.412sarA 10.28 7.833cd4_ 6.74 6.395fd1_ 25.67 12.40†Note: The threading 1MODELLER models use thethreading alignments (for the aligned residues) asthe target for all-atom reconstruction. SICHO mod-els are the reduced lattice models obtained by themethod described in this work. The final all-atommodel is also built by MODELLER using as a targetthe lattice model alpha carbon positions estimatedfrom the SICHO lattice model. The values of thermsd for alpha-carbon traces (in Å) are given for thestructured parts of the target molecules (1hom_:residues 7–59, 1tlk_: residues 9–103, 3cd4_: residues1–97 i.e., the first domain).
TABLE V.Alpha Carbon Rmsd (in Å) From Native forThreading-Based Models and Lattice SICHO Models Built
by MODELLER. Comparison for Threading-AlignedFragments Only†
Targetprotein
StartingRMSD
ThreadingRMSD
SICHORMSD LENGTH
1aba_ 4.37 3.89 4.40 691bbhA 7.03 6.35 6.69 1161cewI 12.88 12.37 10.74 691hom_ 5.59 5.34 3.45 401stfI 7.05 6.04 4.73 831tlk_ 7.88 7.15 3.94 86256bA 6.92 6.06 4.37 1042azaA 11.04 13.53 9.94 802pcy_ 7.64 6.65 4.36 942sarA 8.28 8.07 7.60 733cd4_ 5.72 5.56 5.22 825fd1_ 12.38 12.18 11.94 69†Note: The starting rmsd is for the set of threading-aligned residues ofthe template from the equivalent native target coordinates. TheMODELLER models use the threading alignments and an all-atomtarget. SICHO models are the all-atom models built by MODELLERusing the lattice models (only Ca) as a target. The length of thealignments is given in the last column, and the same sets of residuesare compared for both methods.
Fig. 7. Comparison of the accuracy of the threading/MODELLERstructure with SICHO/MODELLER structures. The number of alphacarbon atoms whose distance from the native structure is less than agiven cut-off is plotted as a function of the cut-off value for four exampleproteins.
603IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

It is very interesting to see how the proposed proceduredeals with the non-aligned part of the model. Comparisonof the rmsd values for the aligned parts (Table V) and forthe entire structured parts (Table IV) of the model showsthat the algorithm builds rather reasonable models of theentire structure, provided there is a well-defined fragmentof good geometrical fidelity in the original alignment.Again, in all but two cases, the present method leads tomore accurate models. For both the aligned part of themolecules and for entire chains (Table IV), good models aregenerated in about half of the studied cases (including allthree proteins used in the model-tuning procedure). In theremaining cases, one may see models that are marginallyimproved as for 3cd4 or that remain rather poor finalmodels as for 2azaA or 5fd1; this is true despite an rmsddecrease of more than 10 A, as compared to modelsgenerated automatically by MODELLER from the initialthreading results. However, as demonstrated in the secondpanel of Figure 7, it should be pointed out that in thesecases, large fragments of the structure qualitatively im-prove. Indeed, about 40 residues of 2azaA in our model arecloser than 6 A from the native structure, while thethreading/MODELLER model structure has only five resi-dues in this range. A possible way to improve the perfor-mance of the method in the case of very bad templates is toloosen the template restraints and perform the annealingfrom higher temperature. This, however is closer to abinitio folding with weak homology based restraints andwill be addressed elsewhere.
DISCUSSIONMeans of the Model Improvement
There are several ways in which the described algorithmchanges the protein model from the original fragmentarythreading model. The first is rather trivial in that thenon-aligned parts (mostly loops) are added and readjustedaccording to packing requirements and the preferencesencoded in the force field. Then, the entire chain has somefreedom of movement within the template tube withoutany changes in its template-target sequence assignment.Furthermore, parts of the chain can slide along the tube,thereby allowing for a quite substantial modification of theinitial alignment and, consequently, the resulting struc-ture. Finally, the aligned fragments can leave the tube in alateral direction. These segments can enter a different partof the template tube or remain outside of it. Such motionsof the model chain could result in a large change of thestructure, or even change the fold topology. The last,rather radical mode of the model rearrangements, hap-pened in several cases. In other words, the most effectiveway of model improvement was by neglecting a part of thethreading alignment, even at the expense of varioustemplate-related energetical penalties. Interestingly, thosesections of the threading-based model consistent with thetarget structure undergo only very minor changes in allcases, and the alignment remains unchanged. As dis-cussed below, this observation may help identify thosemodels that should be of good quality from those for which
improvement of the starting threading model is not satis-factory.
Below, for three selected cases, we analyze in more detailspecific rearrangements of the initial threading modelsthat take place during the Monte Carlo simulations.
2pcy case
The threading alignment of the 2pcy sequence on 2azaAcovers a substantial part of the sequence. There are gaps ofsubstantial length. As a result, the threading model hasthe wrong topology, and two-edge strands of the eight-member b-barrel (one in each of the two b-sheets) arelocated in the wrong sheets. This is the reason for theresulting 7.6 A rmsd from native for the models built solelyfrom the threading alignment. During the simulations, thethree C-terminal strands remain almost unchanged. Simi-larly, the three N-terminal strands undergo only smalladjustments; however, in several models, one or twostrands slide along the tube by a distance that sometimeschanges the original alignment by one or two positions.The central fragment of the model chain (two putativeirregular strands, with a couple of short helices breakingthese strands) is responsible for the large rmsd in theinitial model. The algorithm erases most of the template-target assignments in this part of the molecule. Partly thisoccurs because of the compactness criterion; several resi-dues do not have any long-range contacts in the threadingmodel. During the simulated annealing process, residues30–37 (small differences in the extension of this fragmentcan be seen between the particular runs) switch theirsheet assignment, and join the tube fragment associatedwith one of the C-terminal b-strands, the third one fromthe C-terminus. This is seen in the final ‘‘new assignment,’’or pseudo alignment. At the same time, the second strand(completely helical in the threading model) moves to thesecond sheet, and the long helix breaks and becomesdistorted, as actually occurs in 2pcy’s native structure.Most of the displaced residues join the tube fragmentsgenerated by various secondary structural elements of thetemplate, but only a few maintain their original assign-ments to the template tube. This way the internal forcefield of the lattice model overrides the target interactions,significantly correcting the threading model. The initialmodel and the final model are compared with the nativestructure in Figure 8, where stereo alpha-carbon tracesare displayed in their best mutual superposition, using theMOLMOL25 drawing program.
256bA case
This molecule is a four-helix bundle and the threadingalignment has a few gaps. The template structure is verysimilar to the target, but the threading model is not verygood. During the simulations, most of the C-terminalhelical hairpin remains almost unchanged, except for theloop region that is very mobile. The third (first helix of theC-terminal hairpin) helix of the model is the most stable.The N-terminal hairpin undergoes a large-scale rearrange-ment. The second helix undergoes a rotation that changesits packing angle with respect to the remainder of the
604 A. KOLINSKI ET AL.

molecule. As a result, the end of this helix moves by about 7A in a lateral direction, while the beginning of this helixstays close to its original position. The largest changes areobserved for the first N-terminal helix. It moves along thetube, changing assignment indices by several (up to 8)residues; a lateral adjustment takes place as well. Theinitial model and the final model (superimposed onto thenative structure) are compared in Figure 9. The helicalregions of the final model are very close to the nativestructure; the largest errors that account for most of thestructure errors are in the central turn/loop region.
1tlk case
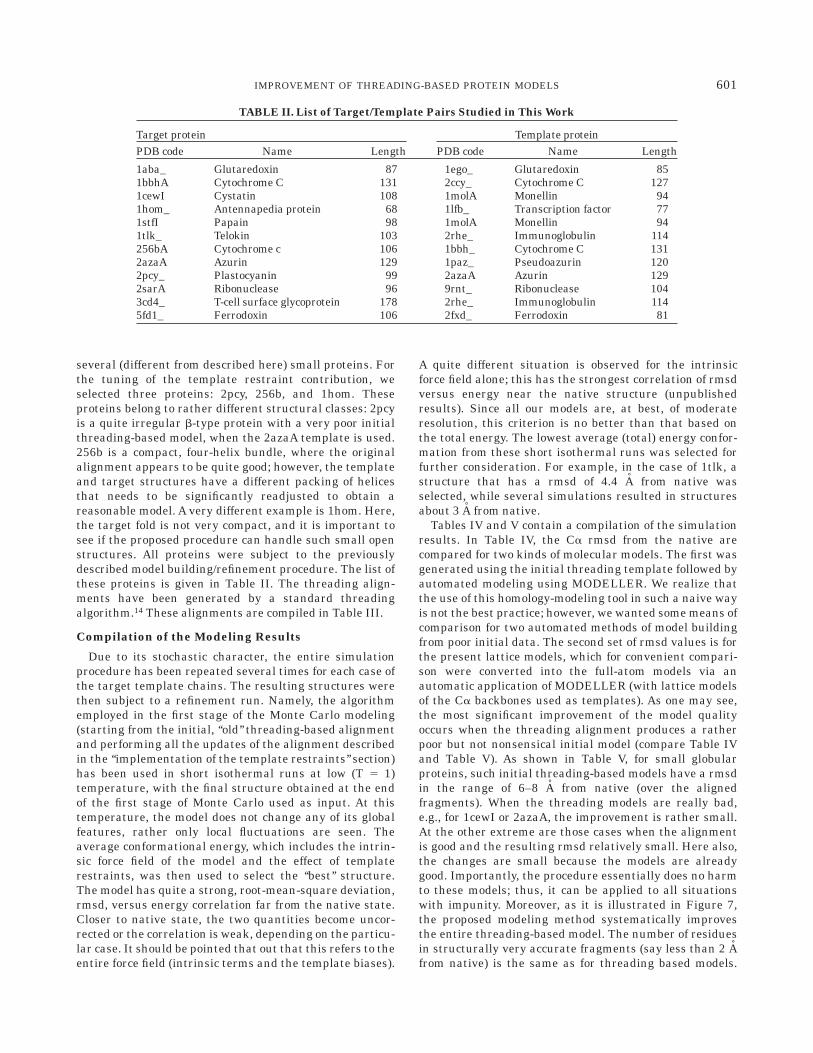
Telokin is a quite regular b-protein. Again, due to gapsand insertions, the threading model has produced a modelwhose topology is wrong. During the simulations, one ofthe b-strands from the original model leaves the initialassignment and sticks to the tube of a strand from the
opposite sheet. Two b-strands that are not in the threadingmodel (lack of the alignment assignments) are built in thesimulated annealing procedure, and they join tubes associ-ated with existing strands. The entire structure, except forthe last C-terminal b-strand that remains essentiallyunchanged, rearranges substantially. Mostly lateral (or-thogonal to the local direction of the template tube)displacements occur in the range of 6 A for about half of allthe residues. As a result, the model improves its rmsd byalmost 4 A. The initial model and the final model (super-imposed onto the native structure) are compared in Fig-ure 10.
How to Identify Good Models
As mentioned before, the proposed method generateslow to moderate resolution models of correct topology inthose cases when the initial threading-based alignmentleads to at least a partially correct structure, i.e., when a
Fig. 8. Stereo drawings of the two models of plastocyanin (in red)superimposed onto crystallographic structure 2pcy (in green). The upperpanel shows the model obtained by MODELLER from the threading
alignment, the lower panel shows the model obtained by the proceduredescribed in this work. For the sake of readability, only the alpha carbontraces are displayed.
605IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

part of the identified template is close to the targetstructure. How to (a priori) distinguish between a good(threading-based) alignment from a poor one is a non-trivial question. Unfortunately we do not have a generalsolution to this problem.
For example, it might appear that the cases where theinitial alignment is more continuous should lead to betterfinal model. In reality, no such correlation was observed.For instance similar qualitative (by more than 2 A)improvements of the model were observed for two helicalproteins, 1hom and 256bA. This is true in spite of the factthat in the first case the fraction of aligned residues is
much less than in the second case where threading aligns104 of the 106 residues in the target sequence.
The intrinsic force field of the reduced model correctlyidentifies the native structure (the lattice projection) asthe lowest energy conformation when compared with themodels generated by MODELLER from the initial thread-ing alignments. The models obtained by lattice homologymodeling are described in this work. In all cases, exceptone (1bbhA, where MODELLER gave a slightly betterresult than the present method) the energy of the modelsbuilt by the present method is significantly lower thanother worse models (including these built by automatic use
Fig. 9. Stereo drawings of the two models of the cytochrome 256b (inred) superimposed onto crystallographic structure (in green). The upperpanel shows the model obtained by MODELLER from the threading
alignment, the lower panel shows the model obtained by the proceduredescribed in this work. For the sake of readability, only the alpha carbontraces are displayed.
606 A. KOLINSKI ET AL.
of MODELLER). These are interesting results; however,our goal is to identify those target /template pairs wherethe final model is of reasonable quality from those caseswhere, despite a sometimes large improvement of theinitial models, the resulting structures are still far fromthe native target conformation. Unfortunately, simpleenergetic criteria (conformational energy per residue inthe final model, decrease of energy from the starting modelto the final model, etc.) do not enable identification of thesepoor quality structures.
In the previous section, we discussed how the proposedmodeling procedure improves the initial, threading-basedmodel. This could actually be used for a qualitative identi-fication of better models. Consider the displacement ofparticular residues (as a function of their position alongthe chain) during the entire simulation procedure. In those
cases where the final model is of good quality, the plotsindicate relatively well separated regions where the chainmodifications were small and also indicated regions oflarge modifications. This is consistent with the previouslymentioned characteristic behavior of ‘‘good’’ models, forwhich some fragments of alignments are recognized by theprocedure as being very good and behave as a scaffold forreadjustment of the remainder of the protein. In contrast,poor models are characterized by random fluctuations ofthe spatial amino acid displacements along the sequence.In such cases there is no pattern. Perhaps there is a hugeenergy barrier between the starting model and the better,near native models that cannot be surmounted by partialreadjustment of the initial alignment. Examples of bothsituations are given in Figures 11 and 12. The lowest (andlocally similar) displacement (during the modeling proce-
Fig. 10. Stereo drawings of the two models of telokin (in red)superimposed onto crystallographic structure 1tlk (in green). The upperpanel shows the model obtained by MODELLER from the threading
alignment, the lower panel shows the model obtained by the proceduredescribed in this work. For the sake of readability, only the alpha carbontraces are displayed.
607IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

dure) regions identify the regions of an optimal (or veryclose to optimal) alignment. While the above is not easy fora simple quantification, it still can be used as a heuristiccriterion for the identification of cases where the methodproposed in this work is likely to provide relatively good,low resolution models. Figure 13 shows the plot of modelaccuracy (measured as the alpha carbon rmsd from native)as a function of the variability in the model chain mobilityduring the simulations. Unfortunately, the correlation isnot very strong. Consequently, the mobility criterion has tobe used with caution. Rather, plots as given in Figures 11and in 12 can be used to identify the best fragments of thethreading models. Indeed, there are very strong correla-tions between the lowest mobility and the best structuralfidelity (to the target structure) of the model chain frag-ments. This may have some other applications, where
assessment of the reliability of various parts of a modelstructure is needed.
SUMMARY AND CONCLUSION
In this work, we proposed a novel approach to predictionof low-resolution protein structures that is based on homol-ogy or sequence-structure compatibility. The method em-ploys templates obtained from threading procedures. Itshould be noted that the alignments used in this workbelong to the best alignments available from threadingprocedures.5 Of course, the alignments can also be ob-tained from sequence alignments. Such templates are usedto guide Monte Carlo simulations that employ a reducedprotein chain representation. In about a third of thestudied cases, the procedure is capable of making largestructural rearrangements that lead to qualitative improve-ments in the initial poor models. In some other cases,despite a huge decrease in the rmsd between the modeland the target native structure, the final model was stillunsatisfactory. Analysis of the simulation trajectories al-lows for a plausible identification of those cases where thefinal models qualitatively improved with respect to theinitial, threading-based model.
While the described method needs further improvement(better resolution, better procedure for the model valida-tion), even now it may be useful for large-scale proteinstructure and function prediction. A complete series ofsimulations for a single target /template pair could be nowperformed in 24 hours on a single state-of-the-art com-puter. The process itself could easily be automated. Thus,predictions on a genomic scale are quite feasible and willbe attempted in the near future. In this regard, it ispossible to identify the biochemical function of a proteinfunction having a model with a 5–6 A backbone rmsd.9,10
Certainly, that would be much more difficult, if not impos-sible, for a model with an 8 A Ca rmsd from native. Forexample, the model of plastocyanin (2pcy) generated bythe proposed method has its four copper-binding residues
Fig. 12. Displacement of the model chain units during the Monte Carlosimulation as a function of the position along the chain for the alignedportion of the 5fd1 molecule. In contrast to the case of 256b (see Fig. 10)the displacements of the chain elements are essentially random. This kindof pattern suggests a rather poor quality final model.
Fig. 11. Displacement of the model chain units during the Monte Carlosimulation as a function of the position along the chain for the alignedportion of the 256b molecule. The very stable (most of the second helixand C-terminal hairpin) regions and very mobile regions (the first helix andthe central loop region) are clearly separated. This is the pattern typical forsuccessful modeling (relatively low final rmsd from the native structure).
Fig. 13. Accuracy of the final models, measured as the Ca rmsd fromthe native structure, as a function of displacement variation. The variationis defined as a ratio of the number of passages of the residue displace-ment plot (as given in Figs. 10 and 11) through the line of averagedisplacement to the total number of protein residues.
608 A. KOLINSKI ET AL.

much closer to their native position than the threading-based model does. Thus, having a structural template ofthis active site, the model structure can be identified withhigh fidelity as a copper-binding protein. In a substantialfraction of cases, function annotation based on structuresprovided by the proposed method would certainly fail, dueto the above-discussed problems in the identification ofgood quality models. Nevertheless, it appears from thepresent studies that for many new proteins that cannot beannotated by other simpler methods, their function couldbe identified. Thus, the proposed method is complemen-tary to sequence-based and threading methods and pro-vides a means for improvement of initially poor andincomplete models. On the other hand, it is also complemen-tary to standard homology modeling tools, enabling homol-ogy modeling in those cases where the template is structur-ally very far from the target structure.
ACKNOWLEDGMENTS
This work was partially supported by NIH Grant No.GM-48835 and KBN (Poland) grant GR-919. AK is anInternational Scholar of the Howard Hughes MedicalInstitute (HHMI grant #75195-543402). We would like toexpress our thanks to an anonymous referee for his/hercomments that helped us to greatly improve the presenta-tion of this work.
REFERENCES1. Bowie JU, Luethy R, Eisenberg D. A method to identify protein
sequences that fold into a known three dimensional structure.Science 1991;253:164–170.
2. Jones DT, Taylor WR, Thornton JM. A new approach to proteinfold recognition. Nature 1992;358:86–89.
3. Godzik A, Skolnick J, Kolinski A. A topology fingerprint approachto the inverse folding problem. J Mol Biol 1992;227:227–238.
4. Miller RT, Jones DT, Thornton JM. Protein fold recognition bysequence threading tools and assessment techniques. FASEB1996;10:171–178.
5. Zhang B, Jaroszewski L, Rychlewski L, Godzik A. Similarities anddifferences between nonhomologous proteins with similar folds:evaluation of threading strategies. Fold Des 1997;2:307–317.
6. Hu W-P, Godzik A, Skolnick J. On the origin of sequence-structurespecificity. How does an inverse folding approach work? ProteinEng 1997;10:317–331.
7. Bryant SH, Lawrence CE. An empirical energy function forthreading protein sequence through folding motif. Proteins 1993;16:92–112.
8. Madej T, Gibrat JF, Bryant SH. Threading a database of proteincores. Proteins 1995;23:356–369.
9. Fetrow JS, Skolnick J. Method for prediction of protein functionfrom sequence using the sequence to structure to function para-digm with application to glutaredoxins/thioredoxins andT1ribonucleases. J Mol Biol 1998;281:949–968.
10. Fetrow J, Godzik A, Skolnick J. Functional analysis of theEscherichia coli genome using the sequence-structure-functionparadigm: identification of proteins exhibiting the glutaredoxin/thioredoxin disulfide oxireductase activity. J Mol Biol 1998;282:703–711.
11. Sali A, Overington JP, Johnson MS, Blundell TL. From compari-son of protein sequences and structures to protein modeling anddesign. TIBS 1990;15:235–250.
12. Aszodi A, Tylor WR. Homology modeling by distance geometry.Fold Des 1996;1:325–334.
13. Jaroszewski L, Pawlowski K, Godzik A. Multiple model approach:exploring the limits of comparative modeling. J Mol Modelling1998;4:294–309.
14. Jaroszewski L, Rychlewski L, Zhang B, Godzik A. Fold prediction
by a hierarchy of sequence, threading, and modeling methods.Protein Sci 1998;7:1431–1440.
15. Wodak SJ, Rooman MJ. Generating and testing protein folds.Curr Opin Struct Biol 1993;3:247–259.
16. Kolinski A, Jaroszewski L, Rotkiewicz P, Skolnick J. An efficientMonte Carlo model of protein chains. Modeling the short-rangecorrelations between side groups centers of mass. J Phys Chem1998;102:4628–4637.
17. Kolinski A, Skolnick J. Assembly of protein structure from sparseexperimental data. An efficient Monte Carlo model. Proteins1998;32:475–494.
18. Kolinski A, Skolnick J. Lattice models of protein folding, dynamicsand thermodynamics. Austin, TX: RG Landes; 1996. 200 p.
19. Skolnick J, Kolinski A, Ortiz AR. MONSSTER: A method forfolding globular proteins with a small number of distance re-straints. J Mol Biol 1997;265:217–241.
20. Bernstein FC, Koetzle TF, Williams GJB, et al. The protein databank: a computer-based archival file for macromolecular struc-tures. J Mol Biol 1977;112:535–542.
21. Binder K. The Monte Carlo method in condensed matter physics:Institut Fur Physik, Johannes Gutenberg-Universitat, Mainz,1991.
22. Godzik A, Skolnick J, Kolinski A. Regularities in interactionpatterns of globular proteins. Protein Eng 1993;6:801–810.
23. Milik M, Kolinski A, Skolnick J. Neural network system for theevaluation of side chain packing in protein structures. ProteinEng 1995;8:225–236.
24. Sali A, Blundell TL. Comparative protein modelling by satisfac-tion of spatial restraints. J Mol Biol 1993;234:779–815.
25. Koradi R. MOLMOL: a program for display and analysis ofmacromolecular structures. J Mol Graph 1996;14:51–55.
26. Altschul SF, Madden TL, Schaefer AA, et al. Gapped BLAST andPSI-BLAST: a new generation of protein database search pro-grams. Nucleic Acid Res 1997;25:3389–3402.
27. Hobohom U, Scharf M, Schneider R, Sander C. Selection of arepresentative set of structures from the Brookhaven ProteinData Bank. Protein Sci 1992;1:409–417.
28. Henikoff S, Henikoff JG. Amino acid substitution matrices fromprotein blocks. Proc Natl Acad Sci USA 1992;89:10915–10919.
APPENDIX ADerivation of the Homology-Enhanced Short-RangeStatistical Potentials
The input sequence is compared to the non-redundantsequence database (containing over 300,000 sequences)using the PSI-BLAST program.26 This process is iterateduntil a stable multiple sequence alignment is obtained.Next, the sequence profile is built using this set of homolo-gous sequences. The profile is a two-dimensional array ofsize 203N where N is the input sequence length and eachof 20 positions corresponds to the frequency of a givenamino acid in the multiple sequence alignment. Let oa(i)will be a number of occurrences of amino acid type a atmultiple sequence alignment position i. Then profile P(a, i)can be defined in the following way:
P(a, i) 5 oa(i)/M i 5 1,2, . . N (A1)
where M is a number of aligned sequences.In the next step, a representative set of PDB files27 of
proteins of known structure (PDBSELECT’97) is scannedto find the fragments of sequences similar to the profile.The comparison is done using a 21-amino acid window ofsequences and the BLOSUM80 mutation matrix28 as thescoring function.
si, j 5 Sk5210,10P(D( j 1 k), i 1 k) (A2)
609IMPROVEMENT OF THREADING-BASED PROTEIN MODELS

where si,j is the score of a given fragment (at position i ofthe profile and position j of database sequence D), D is agiven database sequence, k denotes the scanning windowposition, D( j 1 k) is the amino acid type at position j 1 k ofsequence D.
The 200 best scoring sequence fragments (and corre-sponding pieces of structures) are stored. These presum-ably exhibit significant structural similarity to the corre-sponding fragments of the query sequence. Then, the sidegroup atom positions of these fragments are used in thecalculation of the potentials. For each of these fragments,various distances (based on side chain 1 Ca centers ofmass) are calculated. For computational convenience, thesedistances are discretized into a number of bins. Five binshave been assumed for the distances between the i-th andi11st residue; six bins for the distances between i-th andi12nd residue; eight bins for the distances between thei-th and i13rd residues; seven bins have been assumed forthe i, i14 distances; nine bins for the i, i16 distances andseven bins for the i, i18 distances. The distances i, i13 andi, i16 have been assumed to be ‘‘chiral,’’ i.e., negativevalues have been assigned to the left handed fragmentsand positive to right-handed fragments, respectively. For
the two shortest distances, conservation of the identity ofthe flanking residues was enforced in the fragment align-ment procedure. Then, weighted histograms are built bysummation of the number of occurrences of a particularbin at a given sequence position (using the mutationmatrix score as a weight).
H(rlx,b) 5 1/200 Sm51,200sm(rlx,b) (A3)
where H(r1x,b) is the histogram value, r1x,b is the b-th bin ofdistance r1x, (1x is the short-hand notation for the dis-tances between residues i and i1x), sm (r1x,b) is the score offragment that belongs to the bin b and m is the number offragments.
Finally, the potentials for particular distances along thechain are calculated using the obtained histograms and arandom statistical distribution as the reference state.
E 5 2ln (H(r1x,b)/Ho(r1x,b)) (A4)
where the denominator corresponds to the histogramsaveraged over the database (ignoring sequence similarityand amino acid identity).
610 A. KOLINSKI ET AL.


















