
Acute Promyelocytic Leukemia: From Genetics to Treatment
17
REVIEW ARTICLE Acute Promyelocytic Leukemia: From Genetics to Treatment By Francesco Grignani, Marta Fagioli, Myriam Alcalay, Letizia Longo, Pier Paolo Pandolfi, Emilio Donti, Andrea Biondi, Francesco Lo Coco, Fausto Grignani, and Pier Giuseppe Pelicci A CUTE PROMYELOCYTIC leukemia (APL) has aroused interest well beyond the hematologic field during the last 4 to 5 years. Two features, both of which are unique to APL, have attracted the attention of various sectors of biomedical research: (1) the remission of the dis- ease obtained with retinoic acid (RA) treatment, whose mechanism of action consists in inducing the APL blasts to differentiate term in all^"^; and (2) the presence in the APL blasts of an anomalous protein, the PML/RARa protein, a mutant of one of the retinoic acid receptor^.^-^ The high sensitivity of the promyelocytic blasts to RA makes APL a unique model for differentiation therapy. Be- cause this antineoplastic strategy is radically different from conventional cytotoxic chemotherapy, it potentially pro- vides a further tactic for controlling neoplastic growth. Sev- eral attempts were made to treat hematologic neoplastic dis- orders with differentiation therapy in the past.’ They were based on the observation that the differentiation block is a striking feature of the leukemic phenotype*and that it can be reversedin vitro by a number of substances(eg, low-dose Ara-C).’ Unfortunately, on the whole, these clinical trials led to disappointing results.” In contrast, differentiation therapy with RA induces clinical remission in the great ma- jority of APL patient^."^ The PML/RARa fusion protein is formed as the conse- quence of a chromosomal translocation, [t(15; 17)] that in- volves the PML and RARa genes.4s5 The anomalous protein is thought to play a crucial role in promyelocytic leukemo- genesis. RARa is a member of the super-family ofnuclear hormone receptors that are involved in fundamental bio- logic processes such as development and differentiation.” The discovery of a putative oncogenic RARa mutant, PML/RARa, raises questions of great importance concern- ing the normal function and oncogenic potential of all members of the nuclear receptor super-family. The association of these two unique features of APL is paradoxical. On one hand, the alteration in the RA signaling pathway by PML/RARa could contribute to the leukemic From Istituto di Clinica Medica I, University ofperugia. Policlin- ico Monteluce, Perugia; Dipartimento di Biopatologia, Divisione di Ematologia, I University of Rome; and Clinica Pediatrica, Univer- sity of Milan, Ospedale S. Gerardo. Monza. Italy. Submitted September 21, 1993; accepted October 4, 1993. Supported in part by grants from the Italian Association for Can- cer Research (AIRC), Italian Council of Research (CNR, ACRO project), and EC (Biomed and Biotech projects). Address reprint requests to Pier Giuseppe Pelicci, MD, Istituto di Clinica Medica I, University of Perugia, Policlinico Monteluce, Via Brunarnonti, 06100 Perugia, Italy. The publication costs of this article were defrayed in part by page charge payment.This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section I734 solely to indicate this fact. 0 1994 by The American Society of Hematology. 0006-4971/94/8301-0034$3.00/0 10 phenotype, particularly the differentiation block and/or the outgrowth of the APL blasts. On the other hand, activation of the same signaling pathway by RA causes the disappear- ance of the leukemic blasts by inducing terminal differen- tiation. The story is evenmore intriguing when one consid- ers that the differentiation block is the most distinctive component of the APL phenotype, as demonstrated by the modest proliferative rate ofthe promyelocyticblasts12*” and the correlation between RA-induced differentiationand dis- ease remi~sion.”~ Although the puzzle remains unresolved, should the PMLJRARa protein, as expected, turn out to be responsible for both the differentiationblock and sensitivity to RA, APL will be the first example of a neoplastic disease that can be treated by specifically targeting therapy to the transforming protein. This review willsummarize available information on the genetics of APL by trying to group what is known according to the three main questions that require an answer: ( 1)What is the role of PMLJRARa in promyelocytic leukemia? (2) What is the mechanism of action of the PML/RARa pro- tein? (3) What is the molecular basis of RA treatment? The aim is to provide the hematologist with the background in- formation required to evaluate the numerous findings that will emerge in the near future. The more directly clinical implications of such studies will bealso discussed. THE 15; 17 TRANSLOCATION APL is characterizedby a reciprocal translocation that in- volves chromosomes 1 5 and 1 7.14 As a consequence of this translocation two recombinant chromosomes are formed: 15q+ and 17q- (Fig 1). The chromosome breakpoints have been variously mapped to 15q22-q24and 17qll-q2 1 .l5 Be- cause the quality of the metaphases obtained from neoplas- tic promyelocytes is often poor, the reported breakpoint het- erogeneity may reflect technical difficulties rather than molecular heterogeneity of the translocation. By combining chromosome banding and in situ hybridization techniques, our group mapped the translocation breakpoints to 15q24 and 17q2 l .l6 The chromosome break sites were isolated by four groups using distinct experimental appro ache^.'^." The chromo- some 15 breakpoint falls within a previously unknown gene, whichwasoriginallydesignatedmyl, but then renamed PML, for promyelocytes. The chromosome 17 breakpoint is located in the locus that encodes the retinoic acid receptor (RARCY).~~.” Two fusion genes are formed as a conse- quence of the translocation: the PML/RARa gene on the recombinant 15q+ chromosome and its reciprocal RAW PML on the recombinant 17q- chromosome (Fig Expression of the t( 15; 17) is strictly limited to neoplastic promyelocytes; it has never been reported in any other neo- plasia. There have been descriptionsof chronic myeloid leu- kemia in blast crisis with a t( 15; 17) that, however, displayed the promyelocytic phenotype and rearrangements in PML and RARa.21-23 Blood, Vol83, No 1 (January l), 1994: pp 10-25 For personal use only. on January 2, 2019. by guest www.bloodjournal.org From
Transcript of Acute Promyelocytic Leukemia: From Genetics to Treatment

REVIEW ARTICLE
Acute Promyelocytic Leukemia: From Genetics to Treatment By Francesco Grignani, Marta Fagioli, Myriam Alcalay, Letizia Longo, Pier Paolo Pandolfi, Emilio Donti, Andrea Biondi,
Francesco Lo Coco, Fausto Grignani, and Pier Giuseppe Pelicci
A CUTE PROMYELOCYTIC leukemia (APL) has aroused interest well beyond the hematologic field
during the last 4 to 5 years. Two features, both of which are unique to APL, have attracted the attention of various sectors of biomedical research: (1) the remission of the dis- ease obtained with retinoic acid (RA) treatment, whose mechanism of action consists in inducing the APL blasts to differentiate term in all^"^; and (2) the presence in the APL blasts of an anomalous protein, the PML/RARa protein, a mutant of one of the retinoic acid receptor^.^-^
The high sensitivity of the promyelocytic blasts to RA makes APL a unique model for differentiation therapy. Be- cause this antineoplastic strategy is radically different from conventional cytotoxic chemotherapy, it potentially pro- vides a further tactic for controlling neoplastic growth. Sev- eral attempts were made to treat hematologic neoplastic dis- orders with differentiation therapy in the past.’ They were based on the observation that the differentiation block is a striking feature of the leukemic phenotype* and that it can be reversed in vitro by a number of substances (eg, low-dose Ara-C).’ Unfortunately, on the whole, these clinical trials led to disappointing results.” In contrast, differentiation therapy with RA induces clinical remission in the great ma- jority of APL patient^."^
The PML/RARa fusion protein is formed as the conse- quence of a chromosomal translocation, [t( 15; 17)] that in- volves the PML and RARa genes.4s5 The anomalous protein is thought to play a crucial role in promyelocytic leukemo- genesis. RARa is a member of the super-family of nuclear hormone receptors that are involved in fundamental bio- logic processes such as development and differentiation.” The discovery of a putative oncogenic RARa mutant, PML/RARa, raises questions of great importance concern- ing the normal function and oncogenic potential of all members of the nuclear receptor super-family.
The association of these two unique features of APL is paradoxical. On one hand, the alteration in the RA signaling pathway by PML/RARa could contribute to the leukemic
From Istituto di Clinica Medica I, University ofperugia. Policlin- ico Monteluce, Perugia; Dipartimento di Biopatologia, Divisione di Ematologia, I University of Rome; and Clinica Pediatrica, Univer- sity of Milan, Ospedale S. Gerardo. Monza. Italy.
Submitted September 21, 1993; accepted October 4, 1993. Supported in part by grants from the Italian Association for Can-
cer Research (AIRC), Italian Council of Research (CNR, ACRO project), and EC (Biomed and Biotech projects).
Address reprint requests to Pier Giuseppe Pelicci, MD, Istituto di Clinica Medica I, University of Perugia, Policlinico Monteluce, Via Brunarnonti, 06100 Perugia, Italy.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section I734 solely to indicate this fact. 0 1994 by The American Society of Hematology. 0006-4971/94/8301-0034$3.00/0
10
phenotype, particularly the differentiation block and/or the outgrowth of the APL blasts. On the other hand, activation of the same signaling pathway by RA causes the disappear- ance of the leukemic blasts by inducing terminal differen- tiation. The story is even more intriguing when one consid- ers that the differentiation block is the most distinctive component of the APL phenotype, as demonstrated by the modest proliferative rate ofthe promyelocytic blasts12*” and the correlation between RA-induced differentiation and dis- ease remi~sion.”~ Although the puzzle remains unresolved, should the PMLJRARa protein, as expected, turn out to be responsible for both the differentiation block and sensitivity to RA, APL will be the first example of a neoplastic disease that can be treated by specifically targeting therapy to the transforming protein.
This review will summarize available information on the genetics of APL by trying to group what is known according to the three main questions that require an answer: ( 1) What is the role of PMLJRARa in promyelocytic leukemia? (2) What is the mechanism of action of the PML/RARa pro- tein? (3) What is the molecular basis of RA treatment? The aim is to provide the hematologist with the background in- formation required to evaluate the numerous findings that will emerge in the near future. The more directly clinical implications of such studies will be also discussed.
THE 15; 17 TRANSLOCATION
APL is characterized by a reciprocal translocation that in- volves chromosomes 1 5 and 1 7.14 As a consequence of this translocation two recombinant chromosomes are formed: 15q+ and 17q- (Fig 1). The chromosome breakpoints have been variously mapped to 15q22-q24 and 17qll-q2 1 .l5 Be- cause the quality of the metaphases obtained from neoplas- tic promyelocytes is often poor, the reported breakpoint het- erogeneity may reflect technical difficulties rather than molecular heterogeneity of the translocation. By combining chromosome banding and in situ hybridization techniques, our group mapped the translocation breakpoints to 15q24 and 17q2 l .l6
The chromosome break sites were isolated by four groups using distinct experimental appro ache^.'^." The chromo- some 15 breakpoint falls within a previously unknown gene, which was originally designated myl, but then renamed PML, for promyelocytes. The chromosome 17 breakpoint is located in the locus that encodes the retinoic acid receptor
(RARCY).~~.” Two fusion genes are formed as a conse- quence of the translocation: the PML/RARa gene on the recombinant 15q+ chromosome and its reciprocal R A W PML on the recombinant 17q- chromosome (Fig
Expression of the t( 15; 17) is strictly limited to neoplastic promyelocytes; it has never been reported in any other neo- plasia. There have been descriptions of chronic myeloid leu- kemia in blast crisis with a t( 15; 17) that, however, displayed the promyelocytic phenotype and rearrangements in PML and RARa.21-23
Blood, Vol83, No 1 (January l) , 1994: pp 10-25
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT 11
The percentage of cases with the t( 15; 17) varies from 70% to 90%.15 It would be of great importance if the remaining 10% to 30% had, indeed, a normal karyotype, because it would imply heterogeneity in the genetic lesions underlying the disease. However, molecular analysis has shown re- arrangements of PML and RARa genes in all cases with an apparently normal karyotype examined thus far." We re- cently analyzed two APL cases with apparently normal karyotypes using the fluorescence in situ hybridization tech- nique (FISH) and found a submicroscopic translocation of PML on chromosome 17 in one and of RARa on 15 in the other (our unpublished results in collaboration with M. La- fage, Institut Paoli-Calmettes, Marseille, France). In sum- mary, combined cytogenetic and molecular findings strongly suggest that t( 15; 17) is present in 100% APL cases.
The t( 15; 17) is often (70% to 90%) the only chromosomal anomaly seen in the neoplastic metaphases. However, addi- tional karyotypic changes may accompany the t( 15; 17) and, as in other types of myeloid leukemia, trisomy 8 is the most commonz5 and our unpublished results, February, 1993).
Variant translocations are very rare in APL and usually involve 17 with chromosomes other than l 5.15 Breakpoints in a 1 1 ; 17 chromosome translocation have recently been cloned from a case of APL. The chromosome 17 breakpoint has been shown to lie within the RARa gene, while in chro- mosome 1 1 it is located in a previously unknown gene named PLZF (promyelocytic leukemia zinc finger), which has certain structural similarities with PML.26*27
To conclude, the t( 15; 17) is associated exclusively with a promyelocytic phenotype and there are no other known chromosome alterations able to express this phenotype [(aside from the t( 15; 17)], which suggests that the t( 15; 17) is implicated in the pathogenesis of APL. Furthermore, it would seem that of the two chromosomes, 17 is the only one indispensable for expression of the promyelocytic pheno- type. However, this does not necessarily imply that only al- terations in RARa are crucial in the pathogenesis of APL, because concomitant alterations in a second gene, almost always PML, occasionally PLZF, are invariably present. Therefore, it would seem that PML, PLZF, and other puta- tive translocation partners of RARa must share common properties necessary for the pathogenesis of the disease.
PML/RARa AND ITS EFFECTS ON DIFFERENTIATION AND GROWTH OF HEMATOPOIETIC PRECURSORS
The centromere-telomere transcriptional orientation of both the PML and RARa loci is 5'-3'.6*20 As a result, in the PML/RARa chimeric gene, the PML and RARa portions are fused in a head-to-tail configuration and are under the transcriptional control of the PML promoter (Fig 1).
The chimeric PMLIRARa gene is transcriptionally active in all cases of APL.28*29 The fusion transcript can be identi- fied by Northern blotting using either PML or RARa cDNA probes which both hybridize to transcripts of 4.4 and 3.6 kb in approximately half of APL cases, and 4.0 and 3.2 kb in the remaining cases. The variable position of the chromo- some 15 breakpoints (see below) and the alternative usage of two RARa polyadenylation sites are responsible for the different sizes of the PMLfRARa transcript^.^' The PMLf
RARa fusion transcript has the potential to encode a PML/ RARa fusion protein.3a34
Because the expression of the PML/RARa protein and the emergence of the APL phenotype coincide, the working hypothesis is that PML/RARa is responsible for the transformed phenotype in APL; however, direct proof is still lacking (eg, ectopic expression of the fusion protein into normal hematopoietic precursors). Indirect evidence has, instead, been obtained from expressing PML/RARa in he- matopoietic precursor cell lines. Results have provided evi- dence of biologic activities of PML/RARa that recapitulate critical features of the promyelocytic leukemia phenotype.35 Efects on dzferentiation. Hematopoietic precursor cell
lines can be driven to terminal differentiation by various in- ducer~. '~ For example, vitamin D3 (VD) or combined VD and transforming growth factor @ l (TGFBI) treatment in- duces terminal monocytic differentiation of the U937 pro- monocytic cell line.37 U937 cells that express the PML/ RARa protein fail to terminally differentiate, as shown by both surface marker analysis and functional tests (prolifera- tion, phagocytosis). The effect of PML/RARa protein ex- pression is dose dependent: differentiation is effectively blocked in U937 cells only when the PML/RARa expres- sion levels are higher than those of the normal RARa pro- tein, a situation similar to that found in the APL blasts.35 The same inhibitory activity of PML/RARa is observed when PMLfRARa-expressing cells are exposed to low RA concentrations, comparable with the physiologic levels.38 Whereas the control cells undergo limited differentiation, the hematopoietic precursor cells containing PML/RARa do not differentiate at all.35
The situation is completely reversed when the cells ex- pressing the fusion protein are exposed to high RA concen- trations, comparable with the peak levels achieved in the bloodstream of APL patients treated with R A . 3 8 The per- centage of cells that enter the differentiation program is higher in the PMLfRARa-expressing cell population than in control cells. Therefore, a high concentration of RA con- verts the activity exerted by the fusion protein on differen- tiation from inhibitory to st irnulat~ry.~~
Overall these results show that the PML/RARa fusion protein could alone account for two major features of the APL phenotype: the block of differentiation and the high sensitivity to RA.
At what level of the cascade of events associated with the differentiation process does PML/RARa exert its activity? In traditional models, stem cells undergo commitment pro- cesses that limit their self-renewal and direct their differen- tiation potential. Maturation begins as the cells exhaust their capacity to replicate.36 Hematopoietic precursor cell lines make good in vitro models for studying differentiation. Their potential to grow indefinitely reflects their capacity for self-renewal. When they are treated with various inducers, precursor cell lines proceed to commitment, lose their ca- pacity for self-renewal, and become mature36 (Fig 2). In this setting, PMLfRARa could interfere with the differentiation process during either the commitment or maturation pro- cesses. PMLfRARa-expressing U937 cells treated with VD not only lose their capacity to differentiate but, to a large extent, also their self-renewal properties (our unpublished
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

12 GRIGNANI ET AL
U U '
+l5 y U U L +l 7
X15 #l%+ X17 #17q-
Fig 1. Cytogenetic and molecular architecture of APL t ( 17). (A) FISH using a chromosome 17 painting probe on a metaphase from an APL patient. The larger hybridization spot identifies the re- sidual normal chromosome 17, smaller spots the chromosome markers 15q+ and 17q-. (B) The normal chromosome 15 (#l 5) and 17 (#l 7) as well asthe APLderivative chromosomes 15q+ (#l 5q+) and 17q- (#l 7q-) are shown diagramatically. The PML and RARa loci and the fusion aenes PMURARa on 15a+ and RARalPML on
t
1 5;
Fig. 6.
17q- are indicated-
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT 13
Fig 2. Schematic represen- tation of the effects of PML/ RARa on hematopoiesis. Be- cause the PMLpARatarget cell population in vivo is unknown, this scheme is imprecise and does not account for all hemato- poietic cell populations (stem cells, progenitor cells, imma- ture cells) that can be function- ally identfied. This scheme is based on the assumption that the PML/RARa target popula- tion is, nevertheless, a ”precur- sor” cell. The main in vitro effects of PML/RARa expres- sion on hematopoietic precur- sor cells are indicated within a box.
Normal hemopoiesis
H.mmopoi.tiopr#unor
PMURARa
APL pnarnor
0 U
Matured
Q . ’.. . .
results, April 1993). This suggests that PML/RARa acts by blocking maturation (Fig 2). Instead, in the presence of RA, the percentage of cells that enter the differentiation process without fully differentiating is higher than that of control cells, suggesting that PML/RARa acts by increasing the fre- quency of commitment events (our unpublished results) (Fig 2) . Therefore, PML/RARa seems to exert dual func- tions on differentiation, one inhibitory and RA-indepen- dent, the other stimulatory and RA-dependent. The two activities are not apparently antagonistic because simulta- neous addition of VD and RA induces terminal differenti- ation in PML/RARa-expressing cells (our unpublished re- sults, August 1993). Eflects on cell growth. U937 cells expressing the PML/
RARa protein do not undergo programmed cell death in conditions that induce apoptosis in control cells, like growth factor d e p r i ~ a t i o n . ~ ~ Therefore, it seems that the PML/ RARa protein promotes cell survival by inhibiting endoge- nously programmed death. Cell-cycle analysis of control and PMLIRARa-expressing cells showed no significant variations of proliferation indices. Similarly, in the granulo- cyte-macrophage colony-stimulating factor (GM-CSF)-de- pendent cell line TF-1, survival after withdrawal of the growth factor is prolonged by the expression of PMLIRARa (our unpublished results, March 1993).
Fig 6. tight microscopy APL morphology. (A) Classic APL (bone marrow). Most blasts display heavy granule content. Bilobate nu- clei can be seen despite the granules. (B) ”Mixed” or ”intermedi- ate“ APL (bone marrow). Several nongranular basophilic promyelo- cytes with bilobate nuclei are mixed with more tipically granular cells. One cell has multiple Auer rods. (C) M3v APL (peripheral blood). Three basophilic blasts without granules show the typical bilobate nuclei. These cells were strongly positive for Sudan black, myeloperoxidase, and chloroacetate esterase. (Courtesy of Dr Da- vid Swirsky. Department of Haematology, RPMS, Hammersmith Hospital. London, UK).
U U M.tu-
In vivo labeling experiments have shown that APL blasts have a lower proliferation rate than blasts from other types of leukemia^.'^^^^ By prolonging cell survival, PML/RARa could sustain the expansion of a population of leukemic cells with a low proliferative index. This effect, combined with the differentiation block, provides a cellular mecha- nism to account for the oncogenic potential of PML/RARa (Fig 2 ) .
STRUCTURE AND MECHANISMS OF ACTION OF THE PML/RARa FUSION PROTEIN
On the basis of its structure and preliminary biochemical data it appears that PML/RARa may act as multifunctional protein with the potential to interfere with the endogenous activation pathways of PML, RARa, and other members of the nuclear receptor family, such as thyroid hormone recep- tors (TR) and VD receptors (VDR).
A first step in understanding the mechanism(s) of action of PML/RARa is to define which RARa regions are re- tained in the fusion protein and to establish whether their corresponding functions are maintained or modified when they fuse with PML (Fig 3).
Wild-Type RARa Protein
RARa is a member of a retinoic acid receptor family that also includes RARP and RARy.” A separate retinoid recep- tor family, the X receptors, includes RXRa, RXRP, and R X R Y . ~ ~
Physiologic Role Retinoids include vitamin A, its natural and synthetic de-
rivatives, and its metabolites. They exert multiple crucial effects on embryogenesis, cell differentiation, and growth in vertebrates?’ However, the specific role of the various RARs or RXRs in processes controlled by retinoids in vivo is unclear.
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom
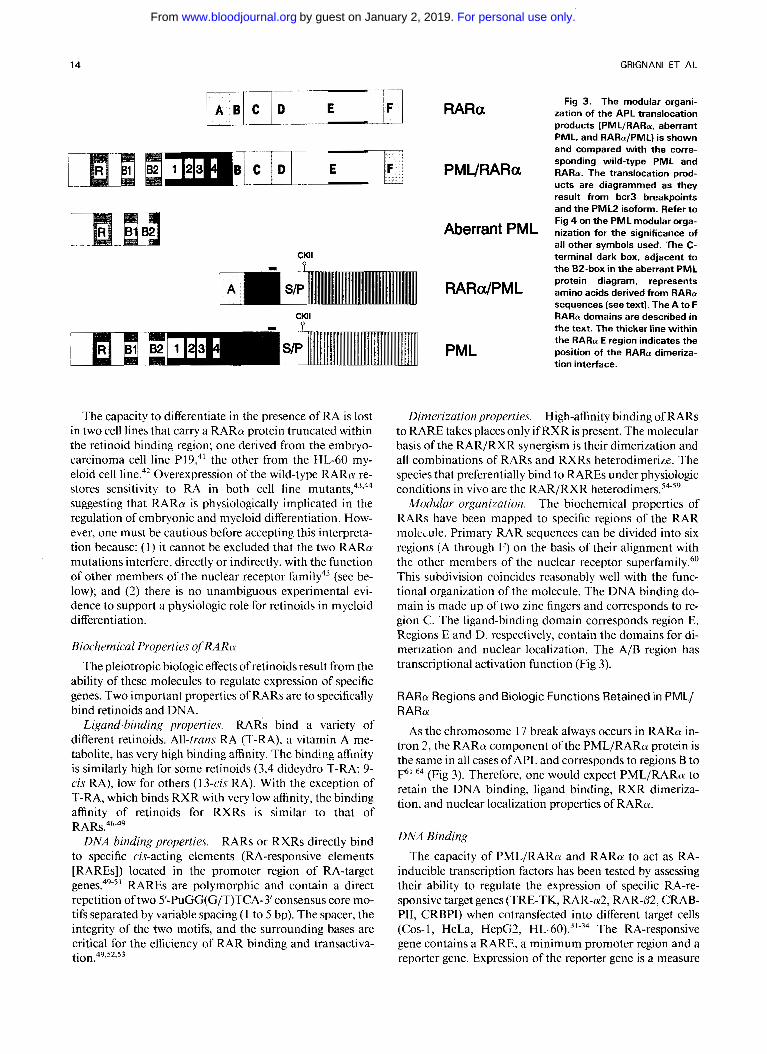
14 GRlGNANl ET AL
CKll
CKll
The capacity to differentiate in the presence of RA is lost in two cell lines that carry a RARa protein truncated within the retinoid binding region; one derived from the embryo- carcinoma cell line P19,41 the other from the HL-60 my- eloid cell line.42 Overexpression of the wild-type RARa re- stores sensitivity to RA in both cell line mutant^,^^.^^ suggesting that RARn is physiologically implicated in the regulation of embryonic and myeloid differentiation. How- ever, one must be cautious before accepting this interpreta- tion because: ( 1 ) it cannot be excluded that the two RARa mutations interfere, directly or indirectly, with the function of other members of the nuclear receptor family4' (sec be- low); and (2) there is no unambiguous experimental evi- dence to support a physiologic role for retinoids in myeloid differentiation.
Biochernicul Pro1pertie.v qfRARn
The pleiotropic biologic effects of retinoids result from the ability of these molecules to regulate expression of specific genes. Two important properties of RARs are to specifically bind retinoids and DNA.
Ligund-binding properties, RARs bind a variety of different retinoids. All-truns RA (T-RA), a vitamin A me- tabolite, has very high binding affinity. The binding affinity is similarly high for some retinoids (3,4 dideydro T-RA; 9- cis RA), low for others ( 1 3 4 s RA). With the exception of T-RA, which binds RXR with very low affinity, the binding affinity of retinoids for RXRs is similar to that of R A R s . ~ ' - ~ ~
DNA binding propertie.7. RARs or RXRs directly bind to specific cis-acting elements (RA-responsive elements [RAREs]) located in the promoter region of RA-target gene^!^.^' RAREs are polymorphic and contain a direct repetition of two S-PuGG(C/T)TCA-Y consensus core mo- tifs separated by variable spacing ( l to 5 bp). The spacer, the integrity of the two motifs, and the surrounding bases are critical for the efficiency of RAR binding and transactiva- tion.49,52,53
RARa
PMURARa
Aberrant PML
RARdPML
PML
Fig 3. The modular organi- zation of the APL translocation products (PMLIRARa, aberrant PML, and RARaIPML) is shown and compared with the corre- sponding wild-type PML and RARa. The translocation prod- ucts are diagrammed as they result from bcr3 breakpoints and the P M L 2 isoform. Refer to Fig 4 on the P M L modular orga- nization for the significance of all other symbols used. The C- terminal dark box, adjacent to the B2-box in the aberrant P M L protein diagram, represents amino acids derived from RARa sequences (see text). The A to F RARa domains are described in the text. The thicker line within the RARa E region indicates the position of the RARa dimeriza- tion interface.
Dimerization properties. High-affinity binding of RARs to RARE takes places only if RXR is present. The molecular basis of the RAR/RXR synergism is their dimerization and all combinations of RARs and RXRs heterodimerize. The species that preferentially bind to RAREs under physiologic conditions in vivo are the RAR/RXR h e t e r ~ d i m e r s . ~ ~ . ~ ~
Modular orgunizution. The biochemical properties of RARs have been mapped to specific regions of the RAR molede . Primary RAR sequences can be divided into six regions (A through F) on thc basis of their alignment with the other members of the nuclear receptor superfamily.60 This subdivision coincides reasonably well with the func- tional organization of the molecule. The DNA binding do- main is made up of two zinc fingers and corresponds to re- gion C. The ligand-binding domain corresponds region E. Regions E and D, respectively, contain the domains for di- merization and nuclear localization. The A/B region has transcriptional activation function (Fig 3 ) .
RARa Regions and Biologic Functions Retained in PML/ RARa
As the chromosome 17 break always occurs in RARa in- tron 2, the RARa component ofthe PML/RARa protein is the same in all cases of APL and corresponds to regions B to
(Fig 3 ) . Therefore, one would expect PMLIRARa to retain the DNA binding, ligand binding, RXR dimeriza- tion, and nuclear localization properties of RARa.
DNA Binding
The capacity of PML/RARa and RARa to act as RA- inducible transcription factors has been tested by assessing their ability to regulate the expression of specific RA-re- sponsive target genes (TRE-TK, RAR-a2, RAR-(32, CRAB- PII, CRBPI) when cotransfected into different target cells (Cos- l , HeLa, HepC2, HL-60).31-34 The RA-responsive gene contains a RARE, a minimum promoter region and a reporter gene. Expression of the reporter gene is a measure
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL FROM GENETICS TO TREATMENT 15
of the capacity of RARa and PML/RARa to bind the RARE and activate the promoter, assuming that they have equal affinity for retinoids. The results showed that PML/ RARa and RARa have different transactivating properties. The effect on transcription of both PML/RARa and RARa can be either stimulatory and inhibitory, depending on the promoter and cell type used, either in presence or absence of RA. However, PMLjRARa is consistently more active than RARcx.'"'~ Unfortunately, because cultured promy- elocytes and the RARa target genes involved in regulating myeloid differentiation are not available, these results do not allow a prediction of the effects of PMLjRARa on RA- target genes in the APL blasts.
The t( l 5 ; 17) causes an interruption in the A/B region of RARa, so that the A region is lost in PML/RARa. Because the A/B region contains a transcription-activation function with promoter-context- and cell-type-dependent the loss of the RARa A region may alter the transcription regulation function of the fusion protein. However, an RARa mutant that lacks the A region does not exert the same activity on transcription as PML/RARCY.~*,~~ Alterna- tively, the diverse transcription regulation of RARa and PMLIRARa could be caused by the presence of a region with transcriptional regulation function in PML, which may influence the activating or silencing functions of RARCY.~~ However, variable portions of PML fused to the GAL4 DNA binding domain did not show any transactivat- ing function of PML on GAL4 reporter gene (our unpub- lished results, January 1993).
Ligand Binding The RA binding affinity of PML/RARa and its specificity
for various retinoids are similar to that of RARa as shown by Scatchard binding experiments on Cos cells transfected with PML/RARa or RARa expression vect0rs.6~
RXR Dimerization PMLjRARa dimerizes with RXR in vitro and this asso-
ciation results in the binding of PML/RARa-RXR hetero- dimers to RAREs. The PML/RARa-RXR association can be visualized in intact cells and, in fact, overexpressed PML/ RARa and RXR colocalize in Cos cells.68
Wild-Type PML Protein The information available on PML is limited to its ex-
pression pattern, analysis of the primary protein sequence, and its homology with other proteins of known function. The hypothesized functions of PML are based on these ho- mologies.
PML expression is characterized by its ubiquity and its extreme complexity: 13 isoforms have been i ~ o l a t e d . ~ ~ . ~ ~ The PML locus comprises nine coding exons. The primary PML transcript can be alternatively assembled into 13 sep- arate transcripts that encode an equal number of PML iso- forms. The processing of the PML primary transcript in- cludes the variable assemblage of whole exons, portions of exons, or retained introns. Variable amounts of the PML transcripts have been identified in all histologically diverse human cell lines examined.69
Modular Organization of the PML Protein On the basis of its homology with other proteins, PML
can be regarded as consisting of regions with distinct puta- tive functional relevance (Fig 4).
The RINGfinger, the B1 and B2 boxes: The PML putative DNA binding domain. The N-terminal region of PML contains three clusters rich in cysteine and histidine residues that are retained in all PML and PML/RARa isoforms (see below). The first cluster corresponds to a new zinc finger motif (RING motif) that defines a recently recognized fam- ily of proteins with functions, where known, that require DNA binding.7@73 These functions include: (1) regulation of development (eg, the XNF-7 gene in Xenopus); (2) regula- tion of gene expression (eg, RPT-1) that affects the expres- sion of the interleukin-2 (IL-2) RPT-l) that affects the ex- pression of the interleukin-2 (IL-2) receptor gene; (3) repair ofUV-damaged DNA (eg, Radl8); (4) DNA recombination (eg, RAG- 1). RING genes other than PML have been impli- cated in tumorigenesis; examples are T18 and Rfp, two transforming proteins that result from the fusion between a RING protein and the B-Raf and ret proteins, respectively, and Bmi- 1, a gene that cooperates with myc in lymphoma development7' (and references therein). That RING is a zinc finger domain with DNA binding activity is supported by the fact that a RING peptide binds zinc with tetrahedral co-ordination to cysteines and DNA in a zinc-dependent fashion.73
A group of RING genes contains a cysteinejhistidine-rich region, termed the B box, which corresponds to an addi- tional putative zinc finger d ~ m a i n . ~ ' . ~ ~ PML belongs to the RING + B box family, which also includes XNF7, T 18, rfp, rptl, and the human RNA-binding autoantigen SS-A/Ro. PML contains two B boxes, which correspond to the second and third cysteinejhistidine-rich cluster (Fig 4).
Indirect support for the putative DNA-binding function of PML derives from its nuclear localization. The protein gives a typically speckled pattern within the nucleus by im- muno~taining.~~.'~ The region responsible for nuclear loca- tion has been preliminarly mapped within the a-helix (Fig 4), whereas the region responsible for the speckled distribu- tion has been mapped within the RING
The a-helix and the dimerization domain. Immediately C-terminal to the fingerlike region is a region that can form multiple a-helices. A portion ofthe a-helix has the potential to assume a coiled-coil configuration and contains four clus- ters of heptad repeats with hydrophobic amino acids at first, fourth, and eighth position^.^^,^' Similar repeats are found in the ligand-binding domain of TR, RAR, and VDR and are considered to be dimerization interface^.^^ The a-helix without the heptad repeats is variably retained in the differ- ent PML isoforms, whereas the fingerlike and coiled-coil re- gions are retained in all, suggesting that they all have the potential to bind DNA and form homodimers and/or het- e r ~ d i m e r s . ~ ~
The serine/proline region, the CKII phosphorylation site, and the variable C-termini. Carboxyterminal to the a-he- lix is a region rich in serine and proline. It contains several X-S-P-X type repeats that have been identified as the mini- mum recognition sequence ofa serinejthreonine kinase and
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

16
Bcr3 Bcr2 Bcrl (45%) (10%) (45%)
I I I
GRlGNANl ET AL
Putative Dimerization DNA-binding domain
domain NLS CKll / l I P
PML 1
Phosphorylation - SiteS \ = l
~ PML4 L"
l . I l 4 ._ S.. , t"' -. .. . "H'
Regions common to all PML isoforms .X'
Variable C-termini
Fig 4. Modular organization of the PML proteins. The diagram shows the modular organization of the various PML isoforms so far identi- fied. All PML isoforms have three regions in common, as indicated: (1) the putative zinc-finger DNA binding domain (the RING domain [R] and the two B boxes [B1 and B21); (2) the dimerization domain (the coiled-coil region with the four heptad clusters [ l , 2, 3, and 41); (3) a portion of the serine/proline rich (S/P) domain that contains the phosphorylation site for CKII. The non-coiled-coil portion of the a-helix is of variable length in the different PML isoforms. Four alternative C-termini of 41 (PMLI isoform), 259 (PML2). 63 (PML3). and 312 (PML4) amino acids have been identified. The arrows indicate the Dosition of the chromosome 15 translocation breakpoints. The frequency of each bcr is indicated between brackets. NLS, nuclear localization signal.
a casein kinase I1 (CKII) phosphorylation site" (and our un- published results, June 1993) (Fig 4). Phosphorylation by CKll is a posttranslational modification shared by many transcription factors and usually associated with modifica- tions in their biologic a~tivity.~' Four alternative C-termini have been identified. Only the CKII site is retained in all PML isoforms."
PML Regions and Functions Retained in PML/RARa
The chromosome 15 breakpoint of the t( 15; 17) is varia- bly located in three regions ofthe PML locus. In 90% to 95% of cases, it is equally distributed between intron 6 (break- point cluster region 1; hcrl) and intron 3 (hcr3). In the re- maining 5% to I O % cases, it is located within exon 6 (hcr2)" (Fig 4). Regardless of the extreme variability of the PML break sites, PMLIRARa genes that have the potential to en- code for a fusion PMLIRARa protein are consistently se- lected by the Icukemia. In hcrl or her3 cases, the 5' portion of PML intron 6 or 3, respectively, fuse with the 3' portion of RARa intron 2. During assemblage of the PML/RARa junction in the fusion transcript the chimeric intron is spliced out and the longest PML and RARa open reading frames ORFs become aligned. The operative mechanism of bcr2 is more complex: a cryptic donor site of the retained portion of PML exon 6 and the RARa intron 2 physiologic acceptor site take part in the assernblag~.~~
The Pututivt~ DNA Binding Domuin Because of the chromosome I5 breaksite heterogeneity, the
retained PML portion differs in each PML/RARa (Fig 4). If the
various PMLIRARa proteins are compared, it will be seen that the only portions of PML consistently retained in the fusion protein are the fingerlike and the coiled-coil region.30
Nuclear Localization ofthe PML/RARa Protein The localization of PMLIRARa differs from that of PML
and RARa. PML/RARa has micropunctated nuclear pat- tern and PML has a speckled nuclear pattern, whereas RARa is finely dispersed in the nucleus.34@,74,77 Anti-PML antibodies show that APL cells, which express all three pro- teins, display the PML/RARa-like micropunctated nuclear pattern, indicating that the fusion protein localization dom- inates over the two wild-type proteins.68 Strikingly, the treatment of APL blasts with RA converts the micropunc- tated nuclear pattern to the speckled, PML-like att tern.'^ Ilornodimerization and Heterodimerization Potential of the PMI,/RARa Protein
Analysis of the RA-binding proteins in PML/RARa-ex- pressing cells has shown that the fusion protein is part of multiple nuclear complexes with molecular weights of 600 and 1,200 kD (the apparent molecular weight of PML/ RARa is 1 10 kD).67 Although little is known of the proteins that in vivo belong to these complexes, it has been shown in vitro that PML/RARa dimerizes with itself, with PML and, as already discussed, with RXR.6X
Mechanisms of PML/RARa Action: PML/RARa Is a Multifunctional Protein
PML/RARa can heterodimerize with PML and the nuclear localization of the heterodimer in APL cells is
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT
different from that of PML h ~ m o d i m e r s . ~ ~ Because it is un- likely that the PMLIRARa-PML heterodimers perform the functions of wild type PML, it is conceivable that PML/ RARa interferes with PML functions.
PML/RARa also forms homodimers that bind to RAREs with a different specificity than RAR/RXR heterodimers." In addition, preliminary results indicate that PML/RARa homodimers interact with the responsive elements of other nuclear receptors, such as the TR or the VDR (J. Jansen, A. Mahfoudi, A. Dejean, personal communication, July 1993) (Fig 5).
Finally, PML/RARa forms heterodimers with RXR. The PML/RARa-RXR complexes are similar to the RAR-RXR dimers in terms of binding activity to RAREs.~' This is probably caused by the fact that, in the presence of RXR, PML/RARa binds RAREs through one RARa and one RXR DNA-binding domain, as in the RAR/RXR hetero- dimers, and not through two RARa DNA-binding do- mains, as in the PML/RARa homodimers.6' However, the formation of PMLIRARa-RXR heterodimers can indi- rectly influence the functional activity of other nuclear re- ceptors like RARs, TR, and DR, which physiologically dimerize with RXR allowing efficient formation of stable RE-bound complexes'4~'' (Fig 5). Because PMLIRARa ex- pression is high in APL,30 dimerization of PML/RARa with RXR could indirectly influence the activity of other nuclear hormone receptors. In vitro experiments have shown that an excess of PML/RARa prevents both VDR binding to VDRE in vitro, and activation of a reporter gene by VDR.68
These multiple biochemical interactions may represent the molecular basis of the biologic activities of the PML/ RARa protein (Fig 5) . In the presence of low near-physio- logic concentration of RA3* in vitro, PMLIRARa acts as a transcription repressor of certain RA-target gene^.^"^^ Fur- thermore, PMLIRARa may also function as a VD signaling antagonist by acting directly on VD-target genes or, indi- rectly, by sequestering a cofactor, like RXR. essential for
Fig 5 . PML/RARa is a multi- functional protein. The figure highlights the putative func- tions of PMLIRARa: (1) binding to PML putative DNA target se- quences; (2) heterodimerization with PML; (3) homodimeriza- tion with PMLIRARa; (4) bind- ing to RA and, possibly, VDR and TR target genes; (5) dimer- ization with RXR and indirect in- teraction with VDR and, pre- sumably, TR through the RXR cellular pool. Also, the effect of the RA-binding on the activity of the RARa DNA binding domain and, possibly, on the PML DNA- binding and coiled-coil domains are indicated.
PML putative DNA target sequences
17
D3 receptor activity. As both RA and VD are implicated in myeloid differentiati~n,~' these effects may be responsible for the inhibition of differentiation by PML/RARa.
OTHER TRANSLOCATION PRODUCTS: ABERRANT PML AND RARa/PML PROTEINS
Because PML/RARn is not the only abnormal product of the t( 15; 17), one should be cautious in assigning it a solitary role in promyelocytic leukemogenesis (Fig 3).
The chimeric PML/RARa gene encodes not only the PML/RARu protein, but also an aberrant PML protein. The aberrant PML protein is encoded by alternatively spliced PML/RARa transcripts that are assembled in such a way that the longest (ORFs) of PML and RARa are not aligned. Consequently, a stop codon is found a few base pairs 3' to the PML/RARtv junction and the transcript has the potential to encode a C-terminal truncated PML pro- tein. This protein retains the PML putative DNA binding domain and, therefore, the potential to interact with PML target sequences (Fig 3). An aberrant PML protein is present in 100% of APL cases with the t( 15; l 7).30
The RARaIPML fusion gcne codes an RARa/PML fu- sion p r~ te in .~" .~ ' As the RARa/PML is the reciprocal of PML/RARa it contains neither PML nor RARa DNA- binding domains. Instead it includes the RARa A domain, and a variable portion of the PML a-helix tract, phosphory- lation sites, and variable C-termini (Fig 3). On the basis of these structural characteristics, it is far from easy to predict whether RARaIPML has a biologic function. Staining of cells that overexpress the RARaIPML fusion protein with anti-RARa A domain polyclonal antibodies has shown that RARa/PML is a nuclear protein.34 The RARa/PML pro- tein is cxpressed in 70% to 80% of APL cases.20
The t( 15; 17) is the only known translocation with the po- tential to encode three abnormal proteins. The fact that, de- spite the considerable molecular heterogeneity of the t( 15; 17), the leukemic phenotype selects chromosome
RAR target genes VDR, TR target genes ? //
t PMURARa hornodimers and
PML heterodirners t S
RXR heterodimers
I H
RA-dependent cis-activation RA
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

18 GRlGNANl ET AL
breaks that ultimately lead to the generation of three abnor- mal proteins, is, to say the least, intriguing. Two other points are worthy of consideration: ( I ) the t( 15; I7), unlike other leukemia-associated chromosomal lesions, has never been documented in preleukemic syndromes ( I 5); and (2) the al- terations associated with the multistep transformation pro- cesses, that are common to other types of myeloblastic leu- kemia (eg, additional chromosome abnormalities, ras and p53 mutations), have never been seen in APL." Could it be that the t( 15; 17), with its three abnormal proteins, is all that is required to fully transform the APL target cells?
DIAGNOSIS OF APL
It is essential that APL be diagnosed accurately and rap- idly because it can benefit from specific treatment such as RA and because, if not recognized and treated promptly, it cames the risk of a potentially fatal coag~ lopa thy .~ ' -~~
Diagnosis of APL is on the whole easy, certainly easier than diagnosing other myeloblastic leukemias. APL has two distinguishing features: hypergranular morphology and the t( 15; 17). The granulations, which correspond to primary granules that contain lysosomal enzymes, are pathogno- monic for APL and are clearly visible under the light mi- croscopyS4 (Fig 6). However, both markers have limitations. Morphologically, there is a subset of APL, called M3 APL variant ( M ~ v ) , in which the granules can only be visualized by electron-microscopy.85 Cytogenetically, only 70% to 80% cases display the t( 15; 17) and good quality bone marrow metaphases cannot always be ~ b t a i n e d . ' ~ , ~ ~ When these two limitations co-exist, diagnosis becomes problematic and Southern blot analysis of the RARa and PML genes is in- dispensable.
A diagnosis of APL by Southern blot analysis rests on the capacity of the technique to recognize the rearrangements in the PML and RARa genes that occur during transloca-
particular value because: ( I ) It is easy to perform, because it can be limited to the intron 2 region where the RARa consistently break^.^^,^^ Only two to three RARa genomic DNA probes are required to identify rearrangements in 100% of cases.63-64.87 (2) Rearrangements are found in both classical caseswith a normal karyotype and M3v.24,87 (3) Re- arrangements in RARa are also found in the rare APL cases with the 1 I ; I7 variant t rans l~ca t ion .~~,~ '
APL diagnosis can also be performed by polymerase chain reaction (PCR) analysis of the PML/RARa tran- script. However, with respect to Southern blotting, RNA manipulation and PCR technology is probably less appro- priate as a routine approach in nonexperienced diagnostic laboratories.
RARa rearrangements or PCR identification of PML/ RARa transcripts have the further advantage of being a pathogenetic marker of APL. For example, a number of cases, diagnosed as APL according to French-American- British (FAB) morphologic criteria, have displayed no RARa rearrangements and have failed to respond to RA treatment.89 Therefore, beyond the morphologic concepts, APL should probably be defined as a leukemia character- ized by abnormalities of the RARa gene and sensitivity to RA treatment. Leukemias that exhibit no abnormalities in
tion.24,28.63-66,87.88 Southern blotting of the RARa gene is of
RARa are not APL and will, presumably, be refractory to RA therapy.
Monitoring the Leukemic Clone During Treatment Monitoring of residual disease is particularly important
in APL because the incidence of complete remissions (CR) is higher and long-term survival and cure after standard che- motherapy are more frequent in APL than in other myelo- blastic leukemia^.^@^^ When one considers that APL pa- tients achieve a CR easily, and a number will be definitively cured, the value ofa test that predicts which CR patients will relapse is obvious.
The presence of a known genetic marker of disease, like the PMLIRARa fusion transcript, permits the leukemic clone to be unambiguously distinguished from normal cells. The PCR amplification of the PML/RARa transcript iden- tifies one neoplastic cell against a background of IO5 normal
Several groups have set up tests for PCR amplifi- cation of the PML/RARa fusion transcript that are easy to perform, need few sets of primers, recognize the transcript in 100% of cases, and show the heterogeneity of the chro- mosome 15 break during the t r a n s l o ~ a t i o n . ~ ~ ~ ~ ~ " ~
Minimal residual disease has been evaluated in a recent retrospective study on 35 APL patients in apparent CR after different therapeutic regimens.Iw The APL clone was de- tected in all cases when the PCR test was performed l to 3 months after treatment. Because this "early PCR positivity" did not correlate with response to treatment, it could reflect the presence of differentiated elements likely to disappear in later checks rather than resistant residual blasts.'oo3'o' How- ever, when PCR was performed later after treatment, the prognostic significance of residual disease became clear. Pa- tients who were PCR-positive in two consecutive determi- nations relapsed within 2 to 4 months, whereas the PCR- negative cases remained free of disease for 3 to 60 months.100 The predictive value of PCR positivity as an in- dicator of relapse in APL received further support from two independent studies.'02,103 Furthermore, residual disease was not detected in nine APL patients in long-term re- mission (4 to 12 years).95 Although these findings require confirmation in prospective longitudinal studies, they indi- cate that PCR methodology is a reliable predictor of cure and relapse in APL and that PCR negativity should be con- sidered the therapeutic goal in APL patients. Therefore, PCR can also be applied to assessing the potential capacity of different antileukemic strategies to eradicate the disease. In retrospective studies, RA never induced PCR negativity, bone marrow transplantation (BMT), and the combination T-RA plus chemotherapy did, and chemotherapy alone did so in a percentage ~ f c a s e s . ~ ~ ~ ~ ~ ~ ~ ' ~ ~
While PCR monitoring studies appear to be clinically rel- evant in APLs, their value in other leukemias remains con- troversial. For example, in t(9;22) chronic myeloid leuke- mia and in t(8;21) AML patients, cells carrying the abnormal hcrlabl or AMLIIETO fusion genes are fre- quently found after several years of long-term re- m i s s i ~ n . ' ~ ~ , ' ~ ~ The persistence of residual leukemia cells in these cases has been interpreted as the consequence of the fact that bcrlabl in CMLs and AMLI/ETO in AMLs are not sufficient for the expression of a clinically aggressive disease.
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT 19
The finding that in APL PCR positivity for PML/RARa correlates with impending relapse and, conversely, long- term remission patients show no detectable transcript, may indicate that PML/RARa is by itself able to confer a fully transformed phenotype to the APL cell.
The M3 Variant APL (M3v) M3v blasts are microgranular promyelocytes with a dis-
tinct folding and lobulation of the nucleus (Fig 6).85 Ultra- structural investigations have shown the presence of dense granules with an average size significantly less than the 250 nm resolution of light microscopy. Despite these morpho- logic differences, patients with M3v bear the t( 15; 17), ex- press the PML/RARa protein, and respond to RA treat- ment94 (and our unpublished results, January 1993). However, M3v exhibits other distinctive clinical, pheno- typic, and molecular features, suggesting that it evolves ge- netically from APL.
The hematologic characteristic of M3v at onset is hyper- leukocytosis, whereas "classic" APL usually presents with a low leukocyte count. A very severe coagulopathy and death from central nervous system and lung hemorrhage are more frequent in M3v than in APL during the first 10 days after diagnosis.'"
The immunophenotype of APL is considered to be highly specific (HLA-DR-, CD34-, CD1 1 b-, CD1 5Y, CD9+, CD33+, CD13+). Although the M3v blasts have the same phenotype, they also express the T-cell-associated antigen CD2.10"109 In a recent survey of 37 APL cases, I4 of 15 M3v displayed the CD2 antigen, whereas 0 of 22 APL did not (our unpublished results, June 1993).
Molecular analysis of the t( 15; 17) translocation showed that the incidence of bcr3 type breakpoints is higher in M3v than in APLS.~',"~
TREATMENT OF APL
Results obtained with standard chemotherapy in APL differ considerably from those obtained in other myeloblas- tic leukemias. The incidence of CR is much higher.9w93 Sen- sitivity to anthracyclines (daunorubicin, idarubicin, rubida- zon) is also much higher, to the point that it is not clear whether the anthracyclines + AraC combination is superior to anthracyclines alone in APL as it is in other forms of AML.93s"' Failure to achieve CR is not normally caused by drug resistance, but rather by death from coagulation disor- ders during the first days of treatment or from sepsis during postchemotherapy aplasia. Survival is often Over the last few years, T-RA has been proposed as an addi- tional tool for treating APL.1"*"2-"8
T-RA Treatment Produces a High Rate of CR Since it was first introduced in Shangai in 1986, about
1,500 patients have been treated with T-RA. A recent review of 565 patients treated at first diagnosis or in relapse gives body to the impression, held since the earliest studies, that T-RA is very effective in inducing complete remission: 84% overall and 95% in patients in whom t( 15; 17) was docu- mented by cytogenetic or molecular analysis. ' '* The hema- topoietic reconstitution in CR patients is polyclonal."9 The
response rate is also high in patients resistant to chemo- therapy, whereas results are less favorable in second or subsequent relapse. Failure to reach CR with T-RA is gen- erally due to early death rather than to leukemic resis- tance.l-3,112-118
CR Is Reached Without a Phase of Bone Marrow Aplasia T-RA-treated patients achieve CR in 1 to 3 months (me-
dian 35 to 45 days) without suffering severe bone marrow depression. Patients generally continue treatment during this period and in most cases the dose is 45 mg/m2/d.
T-RA Is Also Effective for Treating APL-Associated Coagulopathy
APL is associated with severe coagulation disorders, char- acterized by bleeding diathesis, which is present in 80% pa- tients at onset, usually becomes more serious during chemo- therapy, and may also appear in the other 20% of patients. Bleeding diathesis in APL has been attributed to dissemi- nated intravascular coagulation (DIC) and excessive fibro- nolysis, which are the result of the release of procoagulants and tissue plasminogen activator from neoplastic promy- elocytes during senescence and chemotherapy-induced cell lysis.83 Traditional treatment for APL-associated coagulop- athy during chemotherapy is fresh-frozen plasma transfu- sion, intensive platelet support, and tranexamic acid, whereas the value of heparin is debatable.83.93*'20 T-RA rap- idly resolves the coagulopathy within the first 48 hours of treatment, before any morphologic effect is ~ e e n . " ~ , ' ~ ~ - ' ' ~ T- RA probably acts by reducing the mass of neoplastic pro- myelocytes without provoking cell lysis.
T-RA Treatment Is Generally Well Tolerated Toxicity caused by treatment is, in most cases, modest.
Dry skin and mucosae are the most common; cheilosis, na- sal stuffiness, and itching less frequent; whereas other symp toms such as headache, flaxing, clogged ears, cervical and tonsillar lymphoadenopathy, bone pain, and arthralgias are rarely seen. These side effects are mostly short-term and eas- ily controlled by appropriate symptomatic therapy. Bio- chemically, treatment is sometimes associated with hyper- triglyceridemia, and an increase in serum aminotransferase, alkaline phosphatase, and bilirubin. Intracranial hyper- tension, with cerebral pseudotumor, occasionally occurs, primarily in pediatric patients, and can be dose-limit- ing.l-3,112-l18
The Only Severe T-RA Side Effect is the So-called "Retinoic Acid Syndrome," But This Can Be Treated or Prevented
This syndrome, which occurs in about a quarter of pa- tients, usually early in the course of treatment (between the second day and the third week), is clinically very similar to the capillary leak syndrome observed in patients treated with various cytokines (fever, respiratory disease, radio- graphic pulmonary infiltrates, pleural effusions, renal im- pairment and, in some patients, cardiac f a i l ~ r e ) . ' ~ ' - ' ~ ~ It is more frequent in APL patients who initially present with high tumor burden or develop rapid increase in the leuko-
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

20 GRlGNANl ET AL
cyte count during treatment (myeloblasts, promyelocytes, cells in various stage of differentiation and neutrophils). If not promptly treated, the retinoic acid syndrome can lead to death from progressive hypoxemia and multiorgan failure. There is no agreement as to the best treatment. Some find steroid therapy efficacious, others prefer to prevent its onset by treating all patients that tend toward leukocytosis with cytoreductive therapy (high- or intermediate-dose chemo- therapy, leukapheresis). Administration ofstandard chemo- therapy (AraC + daunorubicin) when the white blood cell count of patients in treatment with T-RA rapidly increases is effective in preventing the retinoic acid ~yndrome."~ High doses of corticosteroids early in the course of the syndrome have led to prompt improvement in some patients.lz2
The etiology of the retinoic acid syndrome is unknown. It may be a consequence of PML/RARa activity on ICAM- 1, an adhesion molecule that regulates homotypic and endo- thelial cell adhesion of l e ~ k o c y t e s . ' ~ ~ We have data (unpub- lished, August 1993, in collaboration with A. Pinto, Centro Riferimento Oncologico, Aviano, Italy) to show that PML/ RARa induces a marked, RA-dependent, increase in IC- AM- I expression in maturing myeloid cells both in vivo and in vitro.
Duration of the Hematologic Remission Afer T-RA Is Generally Brief
Few patients are still in CR 1 year post T-RA therapy; mean duration reported in different studies varies from I month to 2 year^.'-^^"^-"' R andomized studies that com- pare RA plus chemotherapy with chemotherapy alone are in progress and should show whether RA plus chemother- apy is superior to chemotherapy alone in terms of remission frequency, toxicity, and treatment of complications and survival rate.'24,'26 A pilot study on 26 newly diagnosed APL patients shows that T-RA followed by intensive chemother- apy (daunorubicin plus AraC) slightly increases the rate of CR and reduces the relapse rate.124
Post T-RA Therapy Relapse Is Resistant to T-RA Despite the fact that de novo resistance to T-RA is very
rare, relapsing patients are invariably resistant to further treatment with T-RA. However, the acquisition of T-RA re- sistance does not imply the acquisition of chemotherapy re- sistance.l-3.112-118
Choice of Treatment in APLs Two treatment strategies are successful in inducing CR in
APLs: T-RA (>90%) and anthracycline-based chemother- apy (75% to 80%). As intrinsic resistance to both T-RA and anthracyclines is very low in APLs, the effect T-RA exerts on the coagulopathy is probably responsible for these differences. Although, in terms of cure, anthracyclines are superior (35% to 45%) to T-RA (practically O%), the combi- nation of the two induces PCR negativity of PML/RARa and appears to increase the CR and cure rates of those ob- tained with either treatment Moreover, the use of high-dose chemotherapy after T-RA is in keeping with principles of modern chemotherapy, ie, to administer max-
imum doses when the tumor mass is the lowest possible, and to use agents that do not induce cross-resistance.
A major unanswered question is: what strategy should be followed in APL patients who have already entered CR? There is no agreement on the optimal postinduction che- motherapy. Whereas intensive consolidation chemotherapy seems to be useful,93 the value and modality of maintenance chemotherapy is still in doubt, but studies are underway that should provide an answer (Gimema Italian cooperative group; in progress). To date no studies have compared post- induction chemotherapy with allogenic BMT in APLs. However, because 35% to 45% of APL CR patients are in fact already cured, the choice of submitting all patients to the risk of BMT is, at best, debatable.
PCR analysis of APL residual disease should be able to solve this problem. If its value in predicting cure will be con- firmed, PCR should be considered fundamental for iden- tifying those CR patients who have not obtained cure and to rapidly evaluate the eradicative potential of the various intensification regimens (BMT or high-dose chemotherapy) and select the most appropriate one.
Mechanism O f R A Sensitivity The cellular mechanism ofT-RA sensitivity in vivo seems
to be the terminal differentiation of the APL blasts. This interpretation is in agreement with the fact that APL blasts differentiate in the presence of T-RA in vitro and that the in vivo response can be predicted by in vitro differentiation tests. It is also supported by the findings of: ( 1) absence of bone marrow aplasia during treatment; ( 2 ) appearance, during treatment, of cells having the morphologic charac- teristics of maturation stages intermediate between promy- elocytes and neutrophils3; (3) presence, during treatment, of PML and RARa rearrangements in peripheral blood neu- trophils that disappear after treatment.'28
Although the molecular mechanism awaits formal proof, there is experimental evidence that PML/RARa underlies the sensitivity of the promyelocytic blasts to T-RA. Indeed, there is a strict correlation between the expression of the PML/RARa fusion transcript and response to treatment." In addition, PMLIRARa increases the sensitivity to RA in vitro,35 and clones of the APL NB4 cell line become T-RA- resistant when they lose PML/RARa expre~s ion . '~~
Any attempt to explain the biochemical mechanism through which PMLIRARa mediates differentiation in the presence of RA runs into the same perplexities encountered in trying to describe the role of PMLIRARa in the induction of the differentiation block. As previously mentioned, the PML/RARa fusion proteins retain both the RARa DNA and retinoid binding domains and, therefore, they could di- rectly influence the RARa-dependent endogenous pathway that controls terminal myeloid differentiation. Transactiva- tion experiments have shown that, in certain experimental conditions, PML/RARa overstimulates the expression of RARa target genes when RA is p r e ~ e n t . ~ ' - ~ ~ This PML/ RARa function might explain RA sensitivity of APL blasts.
Alternative explanations for sensitivity to RA are based on the claim that PML/RARa is localized, at least in part, in the c y t o p l a ~ m . ~ ~ , ~ ~ The cytoplasmic PMLIRARa would constitute a barrier for RA, that can be bypassed by high
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT 21
concentrations of RA. However, it cannot be excluded that RA may exert its effect on PML/RARa by triggering the release of the hypothetical dominant negative action over PML. It should be kept in mind that the interaction between RA and the RA binding domain of PML/RARa could cis- activate the PML portion of the fusion protein, and that RA changes the PMLIRARa nuclear localization pattern to one that is indistinguishable from that of PML.76
Sensitivity to RA could, instead, be an intrinsic property of promyelocytes and, therefore, also of leukemic promy- elocytes. The promyelocytes that occupy the bone marrow in congenital agranulocytosis are, in fact, sensitive to RA.130 The translocation products would be involved in RA sensi- tivity only to the extent that they cause a differentiation block at the promyelocytic stage.
Mechanism o f R A Resistance Resistance to RA that develops post-therapy at the time
of relapse can be partially explained on a pharmacokinetic basis. As treatment proceeds, the bioavailability of the drug is lowered by an as yet unidentified m e ~ h a n i s r n . ~ ~ . ' ~ ' How- ever, in most cases of post RA therapy relapse, the promy- elocytic blasts are resistant in vitro to the differentiation ac- tion of RA, which suggests that during treatment they have accumulated mutations in molecules involved in the re- sponse to RA (C. Chomienne, personal communication and our unpublished results, May 1993). Candidate molecules are those dedicated to the intracellular transport of RA (cell- ular-retinoic-acid-binding proteins [CRABP]) and those in- volved in the response to RA (PML/RARa, RARa, RXR). RA-resistant subclones of the NB4 cell line were shown to have lost the expression of the PML/RARa protein, suggest- ing that an alteration of the fusion protein is associated to retinoic acid
FUTURE PROSPECTS
It is apparent from this review that there is still much to be learned on the molecular mechanisms through which PML/ RARa induces a differentiation block and through which APL cells are sensitive to RA. In vivo, PML/RARa could interfere with many of the signaling pathways activated by hormone receptors such as RARs, RXR, VDR, and TR. But how they regulate terminal differentiation and interfere with other pathways known to be implicated in hematopoietic regulation (eg, the cytokine signaling pathways) cannot, as yet, be explained. Clarification of these issues should pro- vide a picture of the interdependence among multiple mechanisms that regulate differentiation. In the end, they should also be able to establish if other, and which signaling pathways, can be activated to release or bypass the APL differentiation block or synergize with RA. Within the same framework, much should be learned about the PML/ RARa-activated pathways that regulate cell survival, their relation with those that regulate differentiation, and their dependence on RA or other effectors.
PML/RARa influences differentiation at more than one level. The model derived by the various biologic activities of PML/RARa would predict two leukemic cell populations: one less differentiated than promyelocytes, with self-re-
newal potential and probably the true leukemic reservoir; and another of committed precursors with promyelocytic morphology (Fig 2). These two populations may not be equally sensitive to RA, which would explain some of the characteristics of the response to RA therapy (eg, failure to eradicate the disease).
Studies already underway should lead to new therapeutic strategies for treating APL. They are based on the fact that the PML/RARa junction creates a new tumor-specific se- quence in APL. It has been shown that the PML/RARa fu- sion protein contains an antigenic site, not present in the normal parent molecules and recognizable by human CD4' T-lymphocyte clones. One of these clones had cytotoxic ac- tivity against autologous cells expressing the PML/RARa p1-0tein.l~~ These findings provide the background for future adoptive immunotherapy investigations. Furthermore, the PML/RARa transcript is a putative target for therapy based on the use of antisense oligonucleotides. Similar therapies are being tested for other disorders (eg, chronic myeloid leu- kemia) in experimental animals.
ACKNOWLEDGMENT
Some of the studies cited and summarized in this review are the fruit of a rewarding collaboration between various laboratories in Italy. Therefore, we thank the many researchers who have contrib- uted to these studies, among them Drs A. Rambaldi, D. Zangrilli, G. Talamo, D. Rogaia, L. Tomassoni, P. F. Fermcci, M. Ruthardt, D. Diverio, V. Rossi, C. Peschle, U. Testa, C. Gambacorti, G . Av- visati, F. Mandelli, and C. Nervi. We also thank L. Luzzatto for critical reviewing of the manuscript.
REFERENCES 1. Huang M, Yu-Chen Y, Shu-Rong C, Chai J, Lin Z, Long J,
Wang Z: Use of all-trans retinoic acid in the treatment of acute pro- myelocytic leukemia. Blood 72567, 1988
2. Degos L, Chomienne C, Daniel MT, Berger R, Dombret H, Fenaux P, Castaigne S: Treatment of first relapse in acute promy- elocytic leukemia with all-trans retinoic acid. Lancet 336:1440, 1990
3. Warrell RP Jr, Frankel SR, Miller WH Jr, Scheinberg DA, Itri LM, Hittelman WN, Vyas R, Andreeff M, Tafuri A, Jakubowski A, Gabrilove J, Gordon MS, Dmitrovsky E: Differentiation therapy for acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N En@ J Med 324: 1385, 199 1
4. de Th; H, Chomienne C, Lanotte M, Degos L, Dejean A: The t( 15; 17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor (Y gene to a novel transcribed locus. Nature 347558, 1990
5. Borrow J, Goddard AD, Sheer D, Solomon E: Molecular anal- ysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 249: 1577, I990
6. Alcalay M, Zangrilli D, Pandolfi PP, Longo L, Mencarelli A, Giacomucci A, Rocchi M, Biondi A, Rambaldi A, Lo Coco F, Di- verio D, Donti E, Grignani F, Pelicci PG: Translocation breakpoint of acute promyelocytic leukemia lies within the retinoic acid recep- tor (Y locus. Proc Natl Acad Sci USA 88: 1977, 1991
7. Young CW, Warrel R P Differentiating agents, in De Vita VT, Hellman S, Rosemberg SA (eds): Cancer Principle & Practise of Oncology (ed 4). Philadelphia, PA, Lippincott, 1993, p 2636
8. Sawyers CL, Denny CT, Witte ON: Leukemia and the disrup tion of normal hematopoiesis. Cell 64:337, 199 1
9. Degos L, Castaigne S, Tilly H, Sigaux F, Daniel MT: Treat-
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

22 GRlGNANl ET AL
ment of leukemia with low dose ara-C. A study of 160 cases. Semin Oncol 12: 196, 1985 (suppl3)
IO. Cheson BD, Jasperse DM, Simon R, Friedman MA: A criti- cal appraisal of low dose cytosine arabinoside in patients with acute non-lymphocytic leukemia and myelodysplastic syndrome. J Clin Oncol 4:1857, 1986
1 1. Evans RM: The steroid and thyroid hormone receptor super- family. Science 240:889, 1988
12. Raza A, Maheshwari Y , Preisler HD: Differences in cell cycle characteristics amongst patients with acute nonlymphocytic leuke- mia. Blood 69: 1647, 1987
13. Raza A, Yousuf N, Abbas A, Umerani A, Mehdi A, Bokhari SKJ, Sheikh Y, Qadir K, Freeman J, Masterson M, Miller MA, Lampkin B, Browman G, Bennett J, Goldberg J, Grunwald H, Lar- son R, Vogler R, Preisler H D High expression of transforming growth factor-8 prolong cell cycle times and a unique clustering of S-phase cells in patients with acute promyelocytic leukemia. Blood 79: 1037, I992
14. Rowley JD, Golomb HM, Dougherty C: 15/17 Transloca- tion: A consistent chromosomal change in acute promyelocytic leu- kaemia. Lancet 1549, 1977
15. Mitelman F Catalog ofChromosome Aberrations in Cancer (ed 3). New York, NY, Liss, 1988
16. Donti E, Longo L, Pelicci PG: Chromosomal localization of the APL t( 15; 17) breakpoints by molecular cytogenetic analysis. Cancer Genet Cytogenet 54:265, 199 I
17. Lemons RS, Eilender D, Waldmann RA, Rebentisch M, Frej AK, Ledbetter DM, Willmann C, McConnell T, O’Connell P: Cloning and characterization of the t( 15; 17) translocation break- point region in acute promyelocytic leukemia. Genes Chromosom Cancer 2:79, 1990
18. Petkovich M, Brand NJ, Krust A, Chambon P A human retinoic acid receptor which belongs to the family of nuclear recep- tors. Nature 330444, 1987
19. Giguere V, Ong ES, Segui P, Evans RM: Identification of a receptor for the morphogen retinoic acid. Nature 330:624, 1987
20. Alcalay M, Zangrilli D, Fagioli M, Pandolfi PP, Mencarelli A, Lo Coco F, Biondi A, Grignani F, Pelicci PG: Expression pattern of the RARa-PML fusion gene in acute promyelocytice leukemia. Proc Natl Acad Sci USA 89:4840, 1992
2 I . Kadam PR, Merchant AA, Advani SH: Cytogenetic findings in patients with acute promyelocytic leukemia and case of CML blast crisis with promyelocytic proliferation. Cancer Genet Cyto- genet 5 0 109, I990
22. Lai JL, Fenaux P, Zandecki M, Savary JB, Estienne MH, Jouet JP, Bauters F, Deminatti M: Promyelocytic blast crisis of Philadelphia-positive thrombocythemia with translocations (9;22) and (1 5 ; 17). Cancer Genet Cytogenet 29:3 1 1, 1987
23. Takaku F, Ogawa S, Hirai H, Yamada K: Rearrangement of retinoic acid receptor a and PML in promyelocytic blast crisis of Ph chromosome positive chronic myelocytic leukemia with normal copies of chromosome 15. Blood 8 1:2469, 1993
24. Lo Coco F, Diverio D, DAdamo F, Avvisati G, Alimena G, Nanni M, Alcalay M, Pandolfi PP, Pelicci PG: PML/RAR-a rearrangement in acute promyelocytic leukaemias apparently lack- ing the t( 15; 17) translocation. Eur J Haematol48: 17, I992
25. Berger R, Le Coniat M, Der& J, Vecchione D, Jonveaux P Cytogenetic studies in acute promyelocytic leukemia. Genes Chro- rnosom Cancer 3:332, 1991
26. Chen Z, Brand NJ, Chen A, Chen SJ, Tong JH, Wang ZY, Waxman S, Zelent A: Fusion between a novel Kruppel-like finger
‘ gene and the retinoic acid receptor-a locus due to a variant t( 1 1 ; 17) translocation associated with acute promyelocytic leukemia. EMBO J 12:1161, 1993
27. Chen SJ, Zelent A, Tong JH, Yu HQ, Wang ZY, Derrk J, Berger R, Waxman S, Chen Z: Rearrangements of the retinoic acid
receptor alpha and promyelocytic leukemia zinc finger genes result- ing from t( I I; 17) (q23;q2 1) in a patient with acute promyelocytic leukemia. J Clin Invest 91:2260, 1993
28. Longo L, Pandolfi PP, Biondi A, Rambaldi A, Mencarelli A, Lo Coco F, Diverio D, Pegoraro L, Avanzi G, Tabilio A, Zangrilli D, Alcalay M, Donti E, Grignani F, Pelicci PG: Rearrangements and aberrant expression of the retinoic acid receptor a gene in acute promyelocytic leukemia. J Exp Med 172: 157 1, 1990
29. Miller WH Jr. Warrell RP Jr, Frankel SR, Jakubowski A, Gabrilove JL, Muindi J, Dmitrovsky E: Novel retinoic acid receptor a transcript in acute promyelocytic leukemia responsive to all- trans-retinoic acid. J Natl Cancer Inst 82: 1932, 1990
30. Pandolfi PP, Alcalay M, Fagioli M, Zangrilli D, Mencarelli A, Diverio D, Biondi A, Lo Coco F, Rambaldi A, Grignani F, Ro- chette-Egly C, Gaube MP, Chambon P, Pelicci PG: Genomic vari- ability and alternative splicing generate multiple PMLIRARa tran- scripts that encode aberrant PML proteins and PML/RARa isoforms in acute promyelocytic leukaemia. EMBO J 1 : 1397, 1992
3 1. Pandolfi PP, Grignani F, Alcalay M, Mencarelli A, Biondi A, Lo Coco F, Grignani F, Pelicci PG: Structure and origin of the acute promyelocytic leukemia myl/RARa cDNA and characterization of its retinoid-binding and transactivation properties. Oncogene 6: 1285, 1991
32. Kakizuka A, Miller WH Jr, Umesono K, Warrel RP Jr, Fran- kel SR, Murty VVVS, Dmitrovsky E, Evans RM: Chromosomal translocation t( 15; 17) in human acute promyelocytic leukemia fuses RARa with a novel putative transcription factor, PML. Cell 66:663, 1991
33. de Th6 H, Lavau C, Marehio A, Chomienne C, Degos L, Dejean A: The PML/RARa fusion mRNA generated by the t( 15; 17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 66:675, 1991
34. Kastner P, Perez A, Lutz Y, Rochette-Egly C, Gaub MP, Durand B, Lanotte M, Berger R, Chambon P: Structure, localiza- tion and transcriptional properties of two classes of retinoic acid receptor a fusion proteins in acute promyelocytic leukemia (APL): Structural similarities with a new family of oncoproteins. EMBO J 1 1:629, 1992
35. Grignani Fr, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, Mencarelli A, Grignani F, Peschle C, Nicoletti I, Pelicci PG: The acute promyelocytic leukaemia specific PML/RARa fu- sion protein inhibits differentiation and promotes survival of my- eloid precursor cells. Cell 74:423, 1993
36. Metcalf D The molecular control of cell division, differen- tiation commitment and maturation in haemopoetic cells. Nature 339:27, 1989
37. Testa U, Masciulli R, Tritarelli E, Pustorino R, Mariani G, Martucci R, Barberi T, Camagna A, Valtieri M, Peschle C: Transforming growth factor-@ potentiates vitamin D3 induced ter- minal monocytic differentiation of human leukemic cell lines. J Im- munol I50:24 18, I993
38. Muindi J, Frankel SR, Miller WH, Jakuboski A, Scheinberg DA, Young CW, Dmitrovsky E, Warrel R P Continuous treatment with all-trans retinoic acid causes progressive reduction in plasma drug concentrations: Implication for relapse and retinoid “resis- tance” in patients with acute promyelocytic leukemia. Blood 79: 299, 1992
39. Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM: Nuclear re- ceptors that identifies a novel retinoic acid response pathway. Na- ture 345:224, 1990
40. Tabin CJ: Retinoids, homeoboxes and growth factors: To- ward molecular models for limb development. Cell 66: 199, 199 1
41. Pratt MAC, Kralova J, McBurney MW: A dominant nega- tive mutation of the alpha retinoic acid receptor gene in a retinoic acid-nonresponsive embryonal carcinoma cell. Mol Cell Biol IO: 6445, 1990
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT
42. Robertson KA, Emami B, Collins SJ: Retinoic acid-resistant HL-60R cells harbor a point mutation in the retinoic acid receptor ligand binding domain that confers dominant negative activity. Blood 8 0 1885,1989
43. Kruyt FAE, van der Verr LJ, Mader S, van den Brink CE, Feijen A, Jonk UC, Kruijer W, van der Saag PT: Retinoic acid resistance of the variant embryonal carcinoma cell line RAC65 is caused by expression of a truncated RARa. Differentiation 49:27, 1992
44. Collins SJ, Robertson K, Mueller L Retinoic acid induced granulocytic differentiation of HL60 myeloid leukemia cells is me- diated directly through the retinoic acid receptor (RAR-a). Mol Cell Biol 102154, 1990
45. Robertson KA, Emami B, Mueller LM, Collins SJ: Multiple members of retinoic acid receptor family are capable of mediating granulocytic differentiation of HL60 cells. Mol Cell Biol 12:3743, 1992
46. Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P, Levin A: Retinoic acid receptors (RARs) and retinoid X receptors (RXRs): Interactions with endogenous retinoic acids. Proc Natl Acad Sci USA 90:30, 1993
47. Levin AA, Sturzenbecker U, Kazmer S, Bosakowski T, Hu- selton C, Allenby G, Speck K, Kratzeisen C, Rosenberger M, Lovely A, Grippo JF: 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXRa. Nature 255:359, 1992
48. Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 68:397, 1992
49. Leroy P, Nakshatri H, Chambon P Mouse retinoic acid re- ceptor a2 isoform is transcribed from a promoter that contains a retinoic acid response element. Proc Natl Acad Sci USA 88: 10 138, 1991
50. de Thi H, Vivanco-Ruiz MDM, Tiollais P, Stunnenberg H, Dejean A: Identification ofa retinoic acid responsive element in the retinoic acid receptor 0 gene. Nature 343: 177, 1990
5 1 . Smith WC, Nakshatri H, Leroy P, Rees J, Chambon P: A retinoic acid response element is present in the mouse cellular reti- nol binding protein I (mCRBPI) promoter. EMBO J 102223, 1991
52. Vivanco-Ruiz M, Bugge TH, Hirschmann P, Stunnenberg HG: Functional characterization of natural retinoic acid responsive elements. EMBO J 10:3829, 1991
53. Umesono K, Murakami KK, Thompson CC, Evans RM: Di- rect repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 65: 1255, 1991
54. Yu VC, Delsert C, Andersen B, Holloway JM, Devary OV, Naar AM, ffim SY, Boutin JM, Glass CK, Rosenfeld MG: RXRB: a coregulator that enhances binding of retinoic acid, thyroid hor- mone, and vitamin D receptors to their cognate response elements. Cell67:1251, 1991
55. Zhang XK, Hoffmann B, Tran PBV, Graupner G, Pfahl M: Retinoid X receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature 355:44 I , 1992
56. Marks MS, Hallenbeck PL, Nagata T, Segars JH, Appella E, Nikodem NM, Ozato K: H-2RIIBP (RXRB) heterodimerization provides a mechanism for combinatorial diversity in the regulation ofretinoic acid and thyroid hormone responsive genes. EMBO J 1 1 : 1419,1992
57. Kliewer SA, Umesono K, MangelsdorfDJ, Evans RM: Reti- noid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Cell 67: 125 I , 199 1
58. Bugge TH, Pohl J, Lonnoy 0, Stunnenberg HG: RXRa, a promiscuous partner of retinoic acid and thyroid hormone recep- tors. EMBO J I I : 1409, I992
59. Leld M, Kastner P, Lyons R, Nakshatri H, Saunders M, Za- charewsky T, Chen JY, Staub A, Gamier JM, Mader S, Chambon
23
P Purification, cloning and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell 68:377, 1992
60. Krust A, Green S , Argos P, Kumar V, Walter P, Bornet JM, Chambon P The chicken oestrogen receptor sequence: Homology with v-erbA and the human oestrogen and gluococorticoid recep- tors. EMBO J 5:89 I , 1986
61. Diverio D, LoCoco F, DAdamo F, Biondi A, Fagioli M, Grignani Fr, Rambaldi A, Rossi V, Avvisati G, Petti MC, Testi AM, Lis0 V, Specchia G, Fioritoni G, Recchia A, Frassoni F, Ciolli S, Pelicci PG: Identification of DNA rearrangements at the RARa lo- cus in all patients with acute promyelocytic leukemia (APL) and mapping of APL breakpoints within the RARa second intron. Blood 79: I , 1992
62. Chen SJ, Zhu YJ, Tong JH, Dong S , Huang W, Chen Y, Xiang WM, Zhang L, Song Li X, Qian GQ, Wang ZY, Chen Z, Larsen CJ, Berger R Rearrangements in the second intron of the RARa gene are present in a large majority of patients with acute promyelocytic leukemia and are used as molecular marker for reti- noic acid induced leukemic cell differentiation. Blood 78:2696, 1991
63. Tong JH, Dong S, Geng JP, Huang W, Wang ZY, Sun CL, Chen SJ, Chen Z, Larsen CJ, Berger R: Molecular rearrangements of the MYL gene in acute promyelocytic leukemia (APL, M3) de- fine a breakpoint cluster region as well as some molecular variants. Oncogene 7:3 I I , I992
64. Chen Z, Chen SJ, Tong JH, Dong S, Huang W, Chen Y, Xiang WM, Zhang L, Song X, Qian XS, Wang ZY, Chen Z, Larsen CJ, Berger R: The retinoic acid a receptor gene is frequently dis- rupted in its 5’ part in Chinese patients with acute promyelocytic leukemia. Leukemia 5:288, 1991
65. Nagpal S, Saunders M, Kastner P, Durand B, Nakshatri H, Chambon P: Promoter context and response element specificity of the transcriptional activation and modulating function of retinoic acid receptors. Cell 70: 1007, 1992
66. Baniahmad A. Kohne AC, Renkawitz R: A transferable si- lencing domain is present in the thyroid hormone receptor, in v- erbA oncogene product and in the retinoic acid receptor. EMBO J 11:1015, 1992
67. Nervi C, Poindexter CE, Grignani F, Pandolfi PP, Lo Coco F, Avvisati G, Pelicci PG, Jetten AM: Characterization ofthe PML/ RARa chimeric product of the acute promyelocytic leukemia-spe- cific t( 15; 17) translocation. Cancer Res 52:3687, 1992
68. Perez A, Kastner P, Sethi S, Lutz Y, Reibel C, Chambon P: PMLRAR homodimers: Distinct DNA binding properties and heterodimeric interaction with RXR. EMBO J 12:3 17 1, 1993
69. Fagioli M, Alcalay M, Pandolfi PP, Venturini L, Mencarelli A, Simeone A, Acampora D, Grignani F, Pelicci PG: Alternative splicing of PML transcripts predicts coexpression of several carb- oxy-terminally different protein isoforms. Oncogene 7: 1083, 1992
70. Goddard AD, Borrow J, Freemont P, Solomon E: Character- ization of a zinc finger gene disrupted by the t( 15; 17) in acute pro- myelocytic leukemia. Science 254: 1371, 1991
7 1. Freemont PS, Hanson IM, Trowsdale J: A novel cysteine- rich sequence motif. Cell 64:483, I99 I
72. Reddy BA, Etkin LD, Freemont PS: A novel zinc finger coiled-coil domain in a family of nuclear proteins. TIBS I7:344, 1992
73. Lovering R, Hanson IM, Borden KLB, Martin S, OReilly NJ, Evan GI, Rahman D, Pappin DJC, Trowsdale J, Freemont P Identification and preliminary characterization of a protein motif related to the zinc finger. Proc Natl Acad Sci USA 9 0 2 1 12, 1993
74. Daniel MT, Koken M, Romagne 0, Barbey S, Bazarbachi A, Stadler M, Guillemin MC, Degos L, Chomienne C, de Thk H: PML protein expression in hematopoietic and acute promyelocytic leu- kemia cells. Blood 82: 1858, 1993
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

24 GRlGNANl ET AL
75. Forman BM, Samuels HH: Interaction among a subfamily of nuclear hormone receptors: The regulatory zipper model. Mol Endocrinol4:1293,1990
76. Hunter T, Karin M: The regulation oftranscription by phos- phorylation. Cell 70:375, 1992
77. Gaub MP, Lutz Y, Ruberte E, Petkovich M, Brand N, Chambon P: Antibodies specific to the retinoic acid human nuclear receptors a and p. Proc Natl Acad Sci USA 86:3089, 1989
78. Reitsma PH, Rothberg PG, Astrin SM, Trial J, Bar-Shavit Z, Hall A, Teitelbaun S L Regulation of myc gene expression in HL- 60 leukemia cells by a vitamin D metabolite. Nature 306:492, 1983
79. Chang KS, Stass SA, Chu DT, Deaven LL, Trujillo JM, Freireieh El: Characterization ofa fusion cDNA (RARA/myl) tran- scribed from the t( 15; 17) translocation breakpoint in acute promy- elocytic leukemia. Mol Cell Biol 12300, 1992
80. Longo L, Trecca D, Biondi A, Lo Coco F, Grignani F, Mai- olo AT, Pelicci PG, Neri A: Frequency of RAS and p53 mutations in acute promyelocytic leukemias. Leuk Lymphoma 1 I , 1993 (in press)
8 I . Gralnick HR, Sultan C: Acute promyelocytic leukaemia: Haemorrhagic manifestation and morphologic criteria. Br J Haematol29:373, 1975
82. Rodeghiero F, Avvisati G, Castaman G, Barbui T, Mandelli F Early deaths and anti-hemorrhagic treatments in acute promy- elocytic leukemia: A GIMEMA retrospective study in 268 consecu- tive patients. Blood 75:2112, 1990
83. Tallman MS, Kwaan HC: Reassessing the hemostatic disor- der associated with acute promyelocytic leukemia. Blood 79543, 1992
84. Bennet JM, Catovski D, Daniel MT, Randrin G, Galton DAG, Gralnick HR, Sultan C: Proposals for the classification of the acute leukemias. Br J Haematol33:45 1, 1976
85. Golomb HN, Rowley JD, Vardimar JV, Testa JR, Butler A: “Microgranular” acute promyelocytic leukemia: A distinct clinical, ultrastructural and cytogenetic entity. Blood 55:253, 1980
86. Berger R, Bernheim A, Daniel MT. Valensi FT, Flandrin G: Cytological types of mitoses and chromosomal abnormalities in acute leukemia. Leuk Res 7:221, 1983
87. Biondi A, Rambaldi A, Alcalay M, Pandolfi PP. Lo Coco F, Diverio D, Rossi V, Mencarelli A, Longo L, Zangrilli D, Masera G, Barbui T, Mandelli F, Grignani F, Pelicci PG: RARa gene re- arrangements as a genetic marker for diagnosis and monitoring in acute promyelocytic leukaemia. Blood 77: 1418, 199 1
88. Chang KS, Trujillo JM, Ogura T, Castiglione CM, Kidd KK, Zhao SR, Freireich El, Stass SA: Rearrangement ofthe retinoic acid receptor gene in acute promyelocytic leukemia. Leukemia 5:200, 199 1
89. Miller WH Jr, Kakizuka A, Frankel SR, Warrel RP Jr, De- Blasio A, Levine K, Evans RM, Dmitrovsky E: Reverse transcrip- tion polymerase chain reaction for the rearranged retinoic acid re- ceptor a clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci USA 89:2694, 1992
YO. Kantajian HM, Keating MJ, Walters RS: Acute promyelo- cytic leukemia: MD Anderson Hospital experience. Am J Med 80: 789, 1986
9 I . Sanz MA, Jarque I, Martin G, Martinez J, Rafecas J, Sayas MJ. Sanz G, Gomis F: Acute promyelocytic leukemia: Therapy re- sults and prognostic factors. Cancer 6 1 :7, I988
92. Cunningham I, Gee TS, Reich LM, Kempin SJ, Naval AN, Clarkson B D Acute promyelocytic leukemia: Treatment results during a decade at Memorial Hospital. Blood 73: 1 1 16, 1989
93. Fenaux P Management of acute promyelocytic leukemia. Eur J Haematol50:65, 1993
94. Biondi A, Rambaldi A, Pandolfi PP, Rossi V, Giudici G, Al- calay M. LoCoco F, Diverio D, Pogliani EM, Lanzi EM, Mandelli
F, Masera G, Barbui T, Pelicci PG: Molecular monitoring of the myl/retinoic acid receptor-a fusion gene in acute promyelocytic leukemia by polymerase chain reaction. Blood 80:492, 1992
95. Diverio D, Pandolfi PP, Biondi A, Avvisati G, Petti MC, Mandelli F, Pelicci PG, Lo Coco F Blood (in press)
96. Castaigne S, Balitrand N, de The H, Dejean A, Degos L, Cho- mienne C: A PML/retinoic acid receptor a fusion transcript is con- stantly detected by RNA-based polymerase chain reaction in acute promyelocytic leukemia. Blood 79:31 IO, 1992
97. Chen SJ, Chen Z, Chen A, Tong JH, Dong S, Wang ZY, Waxman S, Zelent A: Occurrence of distinct PML-RAR-a fusion gene isoforms in patients with acute promyelocytic leukemia de- tected by reverse transcriptase/polymerase chain reaction. Onco- gene 7:1223, 1992
98. Borrow J, Goddard AD, Gibbons B, Katz F, Swirsky D, Fi- oretos T, Dube I , Winfield DA, Kingston J, Hagemeijer A, Rees JKH, Lister TA, Solomon E: Diagnosis of acute promyelocytic leu- kaemia by RT-PCR: Detection of PML-RARA and RARA/PML fusion transcripts. Br J Haematol82529, 1992
99. Chang KS, Lu J, Wang G, Trujillo JM, Estey E, Cork A, Chu DT, Freireich EJ, Stass SA: The t( 15; 17) breakpoint in acute promyelocytic leukemia clusters within two different sites of the my1 gene: Targets for the detection of minimal residual disease by the polymerase chain reaction. Blood 79554, 1992
100. Lo Coco F, Diverio D, Pandolfi PP, Biondi A, Rossi V, Av- visati G, Rambaldi A, Arcese W, Petti MC, Meloni G, Mandelli F, Grignani F, Masera G, Barbui T, Pelicci PG: Molecular evaluation of residual disease as a predictor of relapse in acute promyelocytic leukaemia. Lancet 340:1437, 1992
I O I . Nilsson B: Probable in vivo induction of differentiation by retinoic acid of promyelocytes in acute promyelocytic leukaemia. Br J Haematol 57:365, 1984
102. Huang W, Sun CL. Li XS, Cao Q, Lu Y, Jang CS, Zhang FQ, Chai JR, Wang ZY, Waxman S, Chen Z, Chen SJ: Acute pro- myelocytic leukemia: Clinical relevance of two major PML-RARa isoforms and detection of minimal residual disease by retrotrascrip- tase/polymerase chain reaction to predict relapse. Blood 82: 1264, 1993
103. Miller WH Jr, Levine K, DeBlasio A, Frankel S, Dmitrov- sky E, Warrel RP Jr: Detection of minimal residual disease in acute promyelocytic leukemia by reverse transcription polymerase chain reaction assay for the PML/RARa fusion mRNA. Blood 82: 1689, 1993
104. Pignon JM, Henni T, Amselem S, Vidaud M, Duquesnoy P, Vernant JP, Kuents M, Cordonnier C, Rochant H, Goossens M: Frequent detection of minimal residual disease by use of the polymerase chain reaction in long-term survivors after bone mar- row transplantation for chronic myeloid leukemia. Leukemia 433. 1990
105. Nucifora G, Larson RA, Rowley JD: Persistence ofthe 8;2 1 translocation in patients with acute myeloid leukemia type M2 in long term remission. Blood 82:712, 1993
106. Rovelli A, Biondi A, Cantu Rajnoldi A, Conter V, Giudici G, Jankovic M, Locasciulli A, Rizzari C, Romitti L, Rossi MR, Scir6 R, Tosi S, Uderzo C, Masera G: Microgranular variant of acute promyelocytic leukemia in children. J Clin Oncol 10:1413. 1992
107. San Miguel JF, Gonzales M, Canizo MC, Anta SP, Zola H, Lopez Burrasca A: Surface marker analysis in acute myeloid leuke- mia and correlation with FAB classification. Br J Haematol64:547, 1986
108. Davey FR, Davis RB, MacCallum JM, Nelson DA, Mayer RJ, Ball ED, Griffin JD, Schiffer CA, Bloomfield CD: Morphologic and cytochemical characteristics of acute promyelocytic leukemia. Am J Hematol30:22 I , 1989
109. Lo Coco F, Avvisati G, Diverio D, Biondi A, Pandolfi PP,
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

APL: FROM GENETICS TO TREATMENT 25
Alcalay M, De Rossi G, Petti MC, Canth-Rajnoldi A, Pasqualetti D, Nanni M, Fenu S, Frontani M, Mandelli F Rearrangements of the RARa gene in acute promyelocytic leukemia: Correlations with morphology and immunophenotype. Br J Haematol78:494,199 1
1 IO. Claxton DF, Reading CL, Nagajan L, Tsujimoto Y, An- dersson BS, Estey E, Cork A, Huh YO, Trujillo J, Deisseroth AB: Correlation of CD2 expression with PML gene breakpoints in pa- tients with acute promyelocytic leukemia. Blood 80582, 1992
1 1 1. Avvisati G, Mandelli F, Petti MC, Vegna ML, Spadea A, Lis0 V, Specchia G, Bernasconi C, Alessandrino EP, Piatti C, Ca- rella AM: Idarubicin (4-demethoxydaunorubicin) as single agent for remission induction of previously untreated acute promyelo- cytic leukemia: A pilot study of the Italian cooperative group GI- MEMA. Eur J Haematol44:257,1990
112. Daenen S, Vellenga E, van Dobbenburgh OA, Halie M R Retinoic acid as antileukemic therapy in a patient with acute pro- myelocytic leukemia and Aspergillus pneumonia. Blood 67:559, 1986
1 13. Chen ZX, Xue YQ, Zhang R, Rei-Fan T, Xue-Ming X, Chun L, Wei W, Wang-Ying Z, Xue-Zen Y, Bao-Yue L: A clinical and experimantal study on all-trans retinoic acid-treated acute pro- myelocytic leukemia patients. Blood 78: 141 3, 199 1
114. Huang ME, Ye YC, Chen SR, Zhao JC, Gu U, Cai JR, Zhan L, Xie JX, Shen ZX, Wang ZY: All-trans retinoic acid with or without low dose cytosine arabinoside in acute promyelocytic leukemia: Report of 6 cases. Chin Med J (Engl) 100:949,1987
I 15. Castaigne S, Chomienne C, Daniel MT, Berger R, Fenaux P, Degos L: All-trans retinoic acid as a differentiating therapy for acute promyelocytic leukemia I. Clinical results. Blood 76: 1704, I990
I 16. Chen ZX, Xue YQ, Zhang R, Tao RF, Xia XM, Li C, Wang W, Zu W, Yao XZ, Ling BJ: A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia pa- tients. Blood 78:1413, 1991
1 17. Bashford J, Szer J, Wiley JS, Buckley M, Carson OM, Van Der Weyden MB: Treatment of acute promyelocytic leukaemia re- lapsing after allogeneic bone marrow transplantation with all-trans- retinoic acid: Suppression of the leukaemic clone. Br J Haematol 79:331, 1991
1 18. Warrel RP Jr, de T h i H, Wang ZY, Degos L Acute promy- elocytic leukemia. N Engl J Med 329: 177, 1993
1 19. LoCoco F, Pelicci PG, DAdamo F, Alimena G, Monte- fusco E, Arcese W, Avvisati G, DeFelice L, Meloni G, Nervi C, Mandelli F, Saglio G: Polyclonal hematopoietic reconstitution in leukemia patients at remission after suppression of specific gene re- arrangements. Blood 82:606, 1993
120. Avvisati G, ten Cate JW, Buller H, Manfelli F Efficacy of tranexamic acid in controlling haemmorhage in patients with acute promyelocytic leukemia: A controlled study. Lancet 2: 122, 1989
121. Castaigne S, Chomienne C, Fenaux P, Daniel MT, Degos L: Hyperleukocytosis during all-trans retinoic acid for acute pro- myelocytic leukemia. Blood 76:260a, 1990 (abstr, suppl)
122. Frankel SR, Eardley A, Lauwers G, Weiss M, Warrell RP Jr: The retinoic acid syndrome in acute promyelocytic leukemia. Ann Intern Med 17:292, 1992
123. Margolin KA, Rayner AA, Hawkins MJ, Atkins MB, Dutcher JP, Fisher RI, Weiss GR, Doroshow JH, Jaffe HS, Roper M, Parkinson DR, Wiernik PH, Creekmore, SP, Boldt DH: In- terleukin-2 and lymphokine-activated killer cell therapy of solid tu- mors: Analysis oftoxicity and management guidelines. J Clin Oncol 7:486, 1989
124. Fenaux P, Castaigne S, Dombret H, Archinbaud E, Duarte M, Morel P, Tilly TH, Guerci A, Maloisel F, Bordessoule D, Sa- doun A, Tiberghien P, Fegueux N, Daniel MT, Chomienne C, Degos L: All-trans-retinoic acid followed by intensive chemother- apy gives a high complete remission rate and may prolong remis- sions in newly diagnosed acute promyelocytic leukemia: A pilot study on 26 cases. Blood 80:2 176, 1992
125. Languino LR, Plescia J, Duperray A, Brian AA, Plow EF, Geltosky JE, Altieri DC: Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM- 1 dependent pathway. Cell 73:1423, 1993
126. Fenaux P, Robert MC, Castaigne S, Archimbald E, Cho- mienne C, Link H, Guerci A, Bowen D, Sanz M, Fey M, Fegueux N, Chastang C, Degos L: A multicenter trial comparing all-trans retinoic acid plus chemotherapy (ATRA + CT) and CT alone in newly diagnosed acute promyelocytic leukemia (APL). Proc Am SOC Clin Oncol 12:300, 1993 (abstr)
127. Chomienne C, Ballerini P, Balitrand N, Daniel MT, Fen- aux P, Castaigne S, Degos L: All-trans retinoic acid in acute promy- elocytic leukemias. 11. In vitro studies: Structure-function relation- ship. Blood 76: I7 10, I990
128. Lo Coco F, Avvisati G, Diverio D, Petti MC, Alcalay M, Pandolfi PP, Zangrilli D, Biondi A, Rambaldi A, Moleti ML, Man- delli F, Pelicci PG: Molecular evaluation of response to all-trans- retinoic acid therapy in patients with acute promyelocytic leuke- mia. Blood 77: 1657, l99 I
129. Dermime S, Grignani Fr, Clerici M, Nervi C, Sozzi G, Ta- lamo GP, Marchesi E, Formelli F, Parmiani G, Pelicci PG, Gambacorti-Passerini C: Occurrence of resistance to retinoic acid in the acute promyelocytic leukemia cell line NB306 is associated with altered expression of the PML/RARa protein. Blood 82: 1573, 1993
130. Hassan HT, Pearson EC, Rees J K Retinoic acid induces granulocytic differentiation of myeloid progenitors in congenital agranulocytosis bone marrow cells. Cell Biol Int Rep 14:247, 1990
13 1. Lefebvre PH, Thomas G, Gourmel B, Agadir A, Castaigne S, Dreux C, Degos C, Degos L, Chomienne C: Pharmacokinetics of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Leukemia 5: 1054, 199 I
132. Gambacorti-Passerini C, Grignani Fr, Arienti F, Pandolfi PP, Pelicci PG, Parmiani G: Human CD4 lymphocytes recognize a peptide representing the fusion region of the hybrid protein PML/ RARa present in acute promyelocytic leukemia cells. Blood 81: 1369, 1993
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom

1994 83: 10-25
and PG PelicciF Grignani, M Fagioli, M Alcalay, L Longo, PP Pandolfi, E Donti, A Biondi, F Lo Coco, F Grignani Acute promyelocytic leukemia: from genetics to treatment
http://www.bloodjournal.org/content/83/1/10.citation.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on January 2, 2019. by guest www.bloodjournal.orgFrom


















