
Acute Liver Failure Including Acetaminophen Overdose...Amanita poisoning Recent mushroom ingestion,...
34
Acute Liver Failure Including Acetaminophen Overdose Robert J. Fontana, MD Division of Gastroenterology, Department of Internal Medicine, University of Michigan Medical School, University of Michigan Medical Center, Ann Arbor, MI 48109-0362, USA Acute liver failure (ALF) is an uncommon, but dramatic, clinical syn- drome defined by the onset of coagulopathy (international normalized ratio [INR] O 1.5), and mental status changes within 8 to 26 weeks of presenta- tion [1,2]. The cause of ALF usually is established rapidly by patient history, laboratory tests, and imaging studies but remains unknown in up to 20% of cases [3]. Acetaminophen overdose is the most common cause of ALF in Western countries, and its incidence seems to be increasing. Fortunately, most patients who have acetaminophen overdose recover with early N-acetylcysteine (NAC) therapy and supportive care, but regulatory actions are needed to prevent future cases. Many patients who have ALF develop infectious, cardiopulmonary, or renal complications that can progress to multiorgan failure. In addition, cerebral edema is notoriously difficult to diagnose and treat and can lead to irreversible brain ischemia and even- tual death. Emergency liver transplantation is associated with a 70% 1-year patient survival, but less than 10% of patients who have ALF are listed, and up to 20% of listed patients die awaiting liver transplantation. Therefore, early referral of patients who have a poor prognosis to a liver transplant center is essential to optimize clinical outcomes. Etiology in the United States The low annual incidence of ALF in the United States, estimated at 2800 cases per annum, makes it difficult to collect reliable data on the causes, risk factors, and outcomes of this clinical syndrome [2,4]. This low annual incidence also can lead to referral bias, selection bias, and ascertainment The author was supported in part by NIH grant (UO1 DK058389-07) as a participant in the Acute Liver Failure Study Group. E-mail address: [email protected] 0025-7125/08/$ - see front matter Ó 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.mcna.2008.03.005 medical.theclinics.com Med Clin N Am 92 (2008) 761–794
Transcript of Acute Liver Failure Including Acetaminophen Overdose...Amanita poisoning Recent mushroom ingestion,...

Acute Liver Failure IncludingAcetaminophen Overdose
Robert J. Fontana, MDDivision of Gastroenterology, Department of Internal Medicine, University of Michigan
Medical School, University of Michigan Medical Center, Ann Arbor, MI 48109-0362, USA
Acute liver failure (ALF) is an uncommon, but dramatic, clinical syn-drome defined by the onset of coagulopathy (international normalized ratio[INR] O 1.5), and mental status changes within 8 to 26 weeks of presenta-tion [1,2]. The cause of ALF usually is established rapidly by patient history,laboratory tests, and imaging studies but remains unknown in up to 20% ofcases [3]. Acetaminophen overdose is the most common cause of ALF inWestern countries, and its incidence seems to be increasing. Fortunately,most patients who have acetaminophen overdose recover with earlyN-acetylcysteine (NAC) therapy and supportive care, but regulatory actionsare needed to prevent future cases. Many patients who have ALF developinfectious, cardiopulmonary, or renal complications that can progress tomultiorgan failure. In addition, cerebral edema is notoriously difficultto diagnose and treat and can lead to irreversible brain ischemia and even-tual death. Emergency liver transplantation is associated with a 70% 1-yearpatient survival, but less than 10% of patients who have ALF are listed, andup to 20% of listed patients die awaiting liver transplantation. Therefore,early referral of patients who have a poor prognosis to a liver transplantcenter is essential to optimize clinical outcomes.
Med Clin N Am 92 (2008) 761–794
Etiology in the United States
The low annual incidence of ALF in the United States, estimated at2800 cases per annum, makes it difficult to collect reliable data on the causes,risk factors, and outcomes of this clinical syndrome [2,4]. This low annualincidence also can lead to referral bias, selection bias, and ascertainment
The author was supported in part by NIH grant (UO1 DK058389-07) as a participant in
the Acute Liver Failure Study Group.
E-mail address: [email protected]
0025-7125/08/$ - see front matter � 2008 Elsevier Inc. All rights reserved.
doi:10.1016/j.mcna.2008.03.005 medical.theclinics.com

762 FONTANA
bias in single-center reports. ALF occurs in patients of all ages, but the causesand prognosis in adults and in infants and children differ markedly [2,5]. Inaddition, for reasons that are unclear, a predominance of female patients hasbeen reported consistently for nearly all causes of ALF (Table 1).
The United States Acute Liver Failure Study Group (ALFSG) is a net-work of 23 tertiary care centers that have studied the causes and outcomesof ALF prospectively since 1998 [3]. A recent analysis of 1033 consecutiveadult patients enrolled through July 2007 demonstrates that acetaminophenoverdose accounts for 46% of cases, followed by indeterminate ALF (15%)and idiosyncratic drug reactions (12%) (Fig. 1). During the past 8 years, anincreasing frequency of acetaminophen overdose cases and a decreasing fre-quency of indeterminate cases and cases caused by hepatitis A virus (HAV)have been noted [6,7]. Other identifiable causes of ALF include acute hepa-titis B virus (HBV) infection (7%), autoimmune hepatitis (5%), ischemichepatitis (4%), and various other causes (5%). Overall survival was 67%at 3 weeks after presentation, with 46% of patients improving spontane-ously and 25% requiring emergency liver transplantation [3]. The likelihoodof spontaneous recovery was highest in patients who had acetaminophenoverdose, HAV, and pregnancy (58%–64%). Patients who had Wilson’sdisease, indeterminate ALF, and drug reactions had the worst prognosis(0–30%).
Table 1
Clinical features in 1033 consecutive adults with who had acute liver failure in the United States
(1998–2007)
Patient
characteristics
Acetaminophen
N ¼ 475
Drug
N ¼ 119
Indeterminate
N ¼ 151
Hepatitis
A virus
N ¼ 31
Hepatitis
B virus
N ¼ 75
Other
N ¼ 182
Presenting features
Median age (years) 36 43 37 47 41 42
Female gender (%) 74 67 56 45 44 76
Jaundice (days) 0 10 10 3 7 7
Median serum
alanine
transferase
level (IU/L)
4149 571 851 2404 1601 677
Median bilirubin
level (mg/dL)
4.5 21.6 23.0 11.9 20.8 15.2
Outcomes at 3 weeks
Transplant (%) 9 40 42 29 47 35
Spontaneous
survival (%)
64 26 27 58 24 30
Overall
survival (%)
71 63 65 84 64 60
Data courtesy ofWilliam Lee, MD, and the United States Acute Liver Failure Study Group,
July 2007.

151
50
8917
4355
31
75
119
475
0
50
100
150
200
250
300
350
400
450
500
AC
M
Dru
g
Hep
B
Hep
A
Au
to
im
m
Isch
em
ic
Wilso
n's
Bu
dd
-C
hiari
Preg
nan
cy
Oth
er
In
deter
n
Fig. 1. Causes of acute liver failure in the United States. Among the 1033 adult patients who
had acute liver failure (ALF) enrolled in the Acute Liver Failure Study Group registry from
1998 through July 2007, acetaminophen overdose was the most common cause (45%), followed
by indeterminate ALF (15%) and idiosyncratic drug reactions (12%). Spontaneous survival at
3 weeks was greatest in patients who had acetaminophen overdose (63%). Patients who had
malignancy and Wilson’s disease had the lowest survival (0%) ACM, acetaminophen; Hep
A, hepatitis A; Hep B, hepatitis B. (Unpublished data, courtesy ofWilliam Lee, MD, University
of Texas Southwestern, October, 2007).
763ACUTE LIVER FAILURE
Initial evaluation
The diagnosis of ALF is based on the physical examination (altered men-tal status) and laboratory findings (INR O 1.5). The initial evaluationshould include rapid identification of the underlying cause, with an empha-sis on treatable conditions (Table 2). In addition to serologic testing, a urinetoxicology screen, and liver imaging, a careful review of all ingested medica-tions is important. ALF occasionally is confused with other clinical entitiessuch as sepsis, systemic disorders with hepatic and brain involvement (eg,systemic lupus erythematosus, thrombotic thrombocytopenic purpura),and acute decompensation of chronic liver disease. Alcoholic hepatitis orflares of chronic HBV may be mistaken for ALF, but a careful review of thepatient’s medical history, laboratory tests, and imaging studies should differ-entiate these conditions. Septic patients who have intrahepatic cholestasisand disseminated intravascular coagulation typically have low factorVIII levels, whereas patients who have ALF typically have normal factorVIII levels but low factor V levels [8].
ALF usually presents initially with nonspecific symptoms such as nausea,vomiting, and malaise. Severe acute liver injury often leads to impaired elim-ination of bilirubin, manifesting as jaundice immediately before or shortlyafter presentation. In addition, the depressed synthesis and excessive con-sumption of clotting factors results in a complex coagulopathy. A dimin-ished synthesis of glucose, increased intracellular lactate production, and

Table 2
Treatable causes of acute liver failure
Cause Etiology Evaluation Treatment
Viruses Hepatitis B HBsAg, anti-HBc IgM
HBV-DNA by PCR
Lamivudine,
entecavir
Hepatitis D HDV-RNA, anti-HDV
IgM, HDV antigen
Lamivudine,
entecavir
Cytomegalovirus CMV-DNA PCR,
CMV-IgM, biopsy
Ganciclovir,
valganciclovir
Epstein-Barr virus EBV-DNA PCR,
serology, biopsy
Steroids, acyclovir
Herpes simplex virus HSV-DNA PCR,
anti-HSV IgM, biopsy
Acyclovir
Metabolic Wilson’s disease Ceruloplasmin, urinary
and hepatic copper,
slit lamp examination
Chelating agents?
plasmapharesis
Acute fatty liver of
pregnancy, HELLP
syndrome
Preeclampsia findings
(hypertension, edema,
proteinuria)
Emergency delivery
of infant
Autoimmune hepatitis ANA, ASMA, IgG,
IgM, IgA liver biopsy
Corticosteroids
Infiltrative Metastatic malignancy Imaging, liver biopsy Chemotherapy
Acute leukemia/lymphoma Bone marrow aspiration,
liver biopsy
Chemotherapy
Drugs/Toxins Acetaminophen
toxicity
Medication history,
serum acetaminophen
level, acetaminophen-
cysteine adducts (?)
N-acetylcysteine
Idiosyncratic drug
reaction
Temporal relationship Withdraw suspect
medication
Amanita poisoning Recent mushroom
ingestion, severe
gastrointestinal
symptoms
Gastric lavage,
charcoal,
penicillin G,
silymarin,
hemodialysis
Vascular Budd-Chiari
syndrome
Liver ultrasound with
Doppler, angiogram
Heparin, low
molecular
weight heparin
Ischemic hepatitis Systemic hypotension
(cardiogenic shock,
pulmonary embolism,
hypovolemia)
Reversal of
hypotension,
inotropes
Abbreviations: ANA, antinuclear antibodies; anti-HGc, anti-hepatitis B core; ASMA, anti-
smooth muscle antibody; CMV, cytomegalovirus; EBV, Epstein-Barr virus; HbsAg, hepatitis
B surface antigen; HDV, hepatitis D virus; HELLP, hemolysis, elevated liver enzymes, and
low platelet count; HSV, herpes simplex virus; PCR, polymerase chain reaction.
764 FONTANA
reduced hepatic uptake of lactate can lead to hypoglycemia and metabolicacidosis. Thirty percent to 40% of patients who have ALF present with im-paired renal function and associated azotemia and oliguria [9].
Mental status changes or encephalopathy are defining criteria of ALF.They are believed to be caused by cerebral edema, particularly in patients

765ACUTE LIVER FAILURE
who have rapid-onset ALF, whereas portosystemic shunting of toxins is im-plicated in patients who have subacute ALF. The complications of ALF,such as hypoglycemia, sepsis, fever, and hypoxia/hypotension, also contrib-ute to neurologic abnormalities. The West Haven criteria for encephalopa-thy frequently are applied to patients who have ALF, although the Glasgowcoma score is more useful for intubated patients. Patients who have grade1 encephalopathy have only subtle changes in affect, altered sleep patterns,or difficulties in concentration. Patients who have stage 2 encephalopathyhave drowsiness, disorientation, and confusion. Stage 3 is marked by som-nolence and incoherence. Patients who have stage 4 have frank coma withminimal (4A) or no (4B) responses to noxious stimuli. Patients who haveALF often have asterixis or tremors in stages 1 or 2 and hyperreflexia, clo-nus, or muscular rigidity in stages 3 or 4. Although worrisome, these uppermotor neuron signs are reversible with hepatic recovery. Patients progress-ing to stage 3 or 4 encephalopathy have a poorer outcome than those whohave a maximum of stage of 1 or 2 encephalopathy (70% survival in patientswho have stage 2 encephalopathy versus 20% survival in patients who havestage 4 encephalopathy) [10].
Diagnosis of acetaminophen hepatotoxicity
Acetaminophen overdose is the leading cause of ALF in the UnitedStates and in other Western countries and recently has been increasing[3,6]. There are an estimated 60,000 cases of acetaminophen overdose annu-ally, most of which are intentional suicide gestures [11]. Nearly 26,000 pa-tients who have acetaminophen overdose are hospitalized each year; anestimated 1% of these patients develops severe coagulopathy or encephalop-athy. The mortality attributed to acetaminophen overdose is 500 per annum,and at least 20% of these deaths occur in patients who have unintentionalacetaminophen overdose [11]. Nearly half of acetaminophen-related casesof ALF are therapeutic misadventures [6]. The increasing incidence ofacetaminophen-induced ALF may, in part, reflect a shift from aspirin toacetaminophen-based products to treat acute febrile illnesses, the presenceof acetaminophen in numerous over-the-counter and prescription medica-tions, and underappreciation of its hepatotoxicity [12].
Acetaminophen is a dose-dependent hepatotoxin that can cause severeacute hepatocellular injury. The injury leads to a characteristic pattern ofpericentral necrosis because of the cytochrome P-450–mediated oxidativemetabolism of acetaminophen to the highly reactive intermediate metabo-lite, N-acetyl-p-benzoquinone imine (NAPQI) (Fig. 2) [13]. Although thereare intracellular mechanisms to detoxify NAPQI, excessive production candeplete intrahepatic glutathione stores and bind to intracellular proteins,leading to hepatocellular necrosis. Chronic consumption of alcohol caninduce cytochrome P-450 2E1 (CYP2E1) activity and increase the rate ofNAPQI formation with therapeutic dosing [14]. Short-term studies of

Glucuronyltransferases
sulfotransferases
Acetaminophen Stable metabolites,
excretion
CYP2E1,
CYP3A4,CYP1A2Gluthathione transferases
NAPQI
Covalent binding,oxidative stress
Hepatocyte damage
Fig. 2. Metabolism of acetaminophen. Acetaminophen is metabolized by hepatic glucuronyl
transferases and sulfotransferases to conjugatedmetabolites that are excreted in the urine.A small
fraction also can be metabolized oxidatively to a reactive intermediate, N-acetyl-p-benzoquinone
imine (NAPQI). If excessive doses of acetaminophen are ingested (ie, O 4 g/d), NAPQI can bind
covalently to intracellular proteins and lead to hepatocyte necrosis. In addition, if cytochrome
P-450 enzyme activity is induced, the high rate of NAPQI formation may deplete intrahepatic
glutathione stores, resulting in liver toxicity. Finally, depletion of glutathione stores by prolonged
fasting also may increase the risk of acetaminophen hepatotoxicity, but supporting evidence in
humans is lacking. CYP2E1, cytochrome P-450 2E1 CYP1A2, cytochrome P-450 1A2;
CYP3A4, cytochrome P-450 3A4.
766 FONTANA
therapeutic doses of acetaminophen in recently abstinent alcoholics have notdemonstrated hepatotoxicity, however [15]. Ingestion of other cytochromeP-450 inducers, such as phenytoin and isoniazid, can lower the thresholdfor acetaminophen hepatotoxicity [16]. Many patients who have uninten-tional acetaminophen overdose report short-term fasting and/or poor nutri-tional status immediately preceding the event, but prospective studies ofhepatic glutathione stores at presentation with ALF are unavailable [17].
The hallmark of acetaminophen hepatotoxicity is the presence of elevatedserum aminotransferase levels (up to 400 times the upper limit of normal)with concomitant hypoprothrombinemia, metabolic acidosis, and renal fail-ure. Most patients have normal or minimally elevated serum bilirubin levelsat presentation because of the acuity of the liver injury. The diagnosis ofacetaminophen hepatotoxicity requires a high index of suspicion (Box 1).Some patients present with unexplained nausea and vomiting and are notedto have only mild aminotransferase elevations, metabolic acidosis, or iso-lated hypoprothrombinemia. Others present with obtundation followinga witnessed or unwitnessed overdose. The minimal dose of acetaminophen

767ACUTE LIVER FAILURE
that produces liver injury varies from 4 to 10 g. Recent prospective studiesdemonstrate evidence of mild biochemical liver injury with therapeuticdosing of 1 g of acetaminophen every 6 hours in healthy volunteers [18].Acetaminophen hepatotoxicity therefore should be considered wheneverthe dose exceeds 4 g/d.
Patients who have unintentional acetaminophen overdose often presentwith 2 to 3 days of nonspecific symptoms superimposed on the acute orchronic medical condition for which they were taking an acetaminophen-based analgesic [6,19]. Patients who have taken an unintentional overdosegenerally have been exposed over several days, have low or undetectableserum acetaminophen levels, and have more advanced encephalopathy atpresentation. In addition to a serum and urine toxicology screen for illicitsubstances, a careful review of all prescription and over-the-counter medica-tions is critical (Tables 3, 4).
Serum acetaminophen levels can help estimate the risk of liver injury fol-lowing a single ingestion [13,20]. A low serum level does not exclude signif-icant overdose, however, and repeated serum samples at 4 to 12 hours maybe needed to define the hepatic risks. Serum bilirubin levels exceeding 10 to15 mg/dL can lead to false-positive acetaminophen levels with some colori-metric assays [21]. Given these limitations, detection of serum acetamino-phen-cysteine protein adducts that emanate from the liver may prove tobe a more sensitive and specific biomarker [22,23]. Although the diagnosticand prognostic significance of adduct levels is still evolving, this assay mayprove particularly useful in patients unable to provide a medication historyor in patients presenting after multiple ingestions over time. Furthermore,detection of adducts in patients who have virally mediated ALF, in whichacetaminophen may be a toxic cofactor, could permit more rapid adminis-tration of NAC.
Management of acetaminophen overdose
Standard medical therapy of known or suspected acetaminophen over-dose includes induction of emesis by ipecac syrup, gastric lavage of pillfragments, and administration of activated charcoal to reduce absorption(see Box 1) [24]. In patients who have a single ingestion, the likelihood ofsubsequent hepatotoxicity is estimated by the Rumack nomogram.
Patients who have known or suspected intentional acetaminophen over-dose should be hospitalized to assess their suicidal risk. Patients who haveunstable hemodynamics, renal failure, or altered mental status should bemonitored in an ICU and transferred to a liver transplant center early, ifdeemed potential transplant candidates. NAC should be administered im-mediately to patients at risk for hepatotoxicity based on the initial serumacetaminophen level, elevated serum aminotransferase level, or INR level.Oral NAC is given as a loading dose of 140 mg/kg followed by a mainte-nance dose of 70 mg/kg for up to 72 hours or until the INR has become

Box 1. Diagnosis and management of acetaminophen overdose
Diagnosis� Ingestion of a toxic dose of acetaminophen-containing
product(s)� Review intake of all over-the-counter and prescription
medications� Intake of more than 4 g (usually > 10 g) of acetaminophen
in 24 hours� Consider in all patients who have an unexplained
serum alanine aminotransferase levels higher than1000 IU/mL� Obtain serum acetaminophen level for single-dose ingestion
(Rumack nomogram)� Bilirubin level higher than 10 mg/dL may lead to
false-positive serum acetaminophen levels� Check urine toxicology screen for other toxins/illicit
substances� Exclude acute hepatitis A virus, hepatitis B virus,
and ischemia
Management and treatment� Within 4 hours of ingestion administer� Ipecac syrup/nasogastric lavage� Activated charcoal, 1 g/kg
� Admit to hospital if there is potential for hepatotoxicity,coagulopathy, altered mentation, or intentional overdosewith suicide attempt.
� Admit to ICU if there is encephalopathy, metabolic acidosis,renal failure.� Arrange early transfer to transplant center if there
is grade 2 encephalopathy or other adverse prognosticcriteria
� Obtain serum liver biochemistries, arterial blood gas andlactate, prothrombin time/international normalized ratio,and factor V levels at admission and every 12 hours
� Administer oral N-acetylcysteine� Loading dose: 140 mg/kg� Maintenance dose: 70 mg/kg every 4 hours for 17 doses
or until international normalized ratio is less than 1.5� Nausea and vomiting are seen in 20% of patients.� Mix N-acetylcysteine with carbonated beverage to improve
gastrointestinal tolerance.
768 FONTANA

� Intravenous N-acetylcysteine is approved by the Foodand Drug administration for acetaminophen overdose.� Indications: gastrointestinal intolerance of oral
N-acetylcysteine, ileus, pancreatitis, bowel obstruction,short gut syndrome, and pregnancy� Contraindications: known sulfa allergy� Loading dose: 150 mg/kg in 250 mL dextrose 5% over
1 hour� Maintenance dose: 50 mg/kg in 500 mL dextrose 5% over
4 hours; then 125 mg/kg in 1000 mL dextrose 5% over19 hours; 100 mg/kg in 1000 mL dextrose 5% over 24 hoursfor 2 days or until the international normalized ratio is lessthan 1.5
� Telemetry monitoring is needed during infusion.� Hypersensitivity/anaphylactoid reactions occur in 3% of
patients.� For a mild hypersensitivity reaction, reduce the infusion
rate by 50% and consider intravenous diphenhydramineor corticosteroids
769ACUTE LIVER FAILURE
lower than 1.5 [13]. Most patients tolerate oral NAC, with the coadministra-tion of antiemetics, but an intravenous formulation is available for patientswho cannot tolerate oral NAC [25]. Most experts recommend continuousintravenous infusion of NAC until the INR is less than 1.5. This formula-tion is particularly useful in pregnant women, patients who have a shortgut, or patients who have an ileus. This drug should be administered ina monitored unit, because up to 3% of patients receiving intravenousNAC develop a hypersensitivity reaction. Patients who experience a mildto moderate infusion reaction should have the infusion rate decreased by50% and receive antihistamines and/or corticosteroids.
Unintentional acetaminophen overdose
The ALFSG recently demonstrated that nearly 50% of acetaminophen-related ALF occurs without an overt suicide intent [6]. In most of thesepatients, the amount of acetaminophen ingested exceeded the maximal dailyrecommended dose of 4 g/d. Nearly 50% of these patients, however,reported ingesting only 4 to 10 g/d, and 38% of patients ingested a multitudeof products. Contrary to earlier reports, these patients were not more likelyto be taking antidepressants or to have a history of alcohol abuse [14,19].Nonetheless, patients who had an unintentional overdose had more ad-vanced encephalopathy at presentation, presumably because of frequentnarcotic administration. Fortunately, 95% of these patients received

Table 3
Acetaminophen content of selected narcotic analgesicsa
Prescription analgesics
Product (active ingredients) Acetaminophen per dose (mg)b
Anexsia (hydrocodone bitartrate) 325–660 mg
Capital with Codeine Suspension
(codeine phosphate)
120 mg/5 ml
Darvocet-N50 (propoxyphene napsylate) 325 mg
Darvocet-N100 (propoxyphene napsylate) 650 mg
Darvocet-A500 (propoxyphene napsylate) 500 mg
Endocet (oxycodone hydrochloride) 325–650 mg
Esgic Plus (butalbital/caffeine) 500 mg
Fioricet (butalbital/caffeine) 325 mg
Fioricet with Codeine (butalbital/caffeine/
codeine phosphate)
325 mg
Lorcet (hydrocodone bitartrate) 325–750 mg, 500 mg/15 ml
Lortab (hydrocodone bitartrate) 325–500 mg 500 mg/15 ml
Maxidone (hydrocodone bitartrate) 750 mg
Norco (hydrocodone bitartrate) 325 mg
Panadol #3 and #4 (codeine phosphate) 300 mg
Percocet/Oxycet (oxycodone hydrochloride) 325–650 mg
Phenaphen with Codeine (codeine phosphate) 325–650 mg
Roxicet (oxycodone hydrochloride) 325–500 mg, 325 mg/5 ml
Sedapap (butalbital) 650 mg
Talacen (pentazocine hydrochloride) 650 mg
Tylenol #2, #3, and #4 (codeine phosphate) 300 mg
Tylox (oxycodone hydrochloride) 500 mg
Ultracet (tramadol hydrochloride) 325 mg
Vicodin (hydrocodone bitartrate) 500 mg
Vicodin ES (hydrocodone bitartrate) 750 mg
Vicodin HP (hydrocodone bitartrate) 660 mg
Wygesic (propoxyphene napsylate) 650 mg
Zydone (hydrocodone bitartrate) 400 mg
a This list does not contain all acetaminophen-containing prescription products.b Contact Poison Control Center for exact dosage of other constituents.
770 FONTANA
NAC, and their rate of spontaneous survival was similar to that of patientswho had taken an intentional overdose (64% versus 66%).
Because acetaminophen hepatotoxicity is the leading cause of ALF but iscompletely preventable, some experts have recommended regulatory changesregarding the labeling and dispensation of acetaminophen-containing prod-ucts [12,26]. In the United Kingdom, blister packaging and restrictions on thedispensation of acetaminophen tablets have led to a reduction in the numberof patients taking an intentional overdose and in those referred for livertransplantation [27,28]. The Food and Drug Administration (FDA) recentlyproposed changes in the labeling of all over-the-counter products that con-tain acetaminophen as well as nonsteroidal anti-inflammatory drugs [29].Additional limitations on the dispensing of prescription acetaminophen-narcotic congeners and on reducing or eliminating the acetaminophen

Table 4
Acetaminophen content of selected over-the-counter medicationsa
Over-the-counter medications
Product (active ingredients) Acetaminophen per dose (mg)
Actifed products (triprolidine/
pseudoephedrine)
325–500 mg
Alka Seltzer products (sodium bicarbonate) 250–325 mg
Allerest products (naphazoline) 325–500 mg
Anacin, Anacin-3 (aspirin/caffeine) 80–500 mg, 100 mg/ml, 160 mg/5 ml
Arthritis Foundation Aspirin-Free
(acetaminophen)b500 mg
Benadryl Allergy/Cold Tablets
(diphenhydramine hydrochloride)
500 mg
Children’s Tylenol products (acetaminophen)b 80–160 mg, 160 mg/5 ml
Comtrex products (pseudoephedrine,
chlorpheniramine)
325–1000 mg, 500 mg/5 ml, 500 mg/15 ml,
650 mg/oz, 1000 mg/oz, 1000 mg/5ml
Datril Extra (acetaminophen)b 325–500 mg, 130 mg/5 ml
Drixoral products (dexbrompheniramine,
pseudoephedrine)
325–500 mg
Excedrin Migraine Products
(aspirin/caffeine)
250–500 mg
Goody’s Extra Strength Headache Powder
(aspirin/caffeine)
130–500 mg, 260 mg per powder paper
Liquiprin (acetaminophen)b 80 mg/0.8 ml, 80 mg/1.66 ml, 80 mg/2.5 ml
Midrin (dichloralphenazone,
isometheptene mucate)
325 mg
NyQuil (dextromethorphan, pseudoephedrine,
doxylamine)
250 mg, 167 mg/5 ml, 1000 mg/packet,
1000 mg/30 ml
Pamprin (ibuprofen) 250–500 mg, 650 mg/packet
Panadol (acetaminophen)b 80–500 mg, 60 mg/0.6 ml, 80 mg/0.5 ml
Percogesic (phenyltoloxamine) 325–500 mg
Sine-Aid Sinus Medicine (ibuprofen,
pseudoephedrine)
500 mg
Sinutab products (pseudoephedrine,
chlorpheniramine)
325–500 mg, 1000 mg/oz
Sominex Pain Relief Formula
(diphenhydramine)
500 mg
St. Joseph’s Aspirin Free Products
(acetaminophen)b80–500 mg, 60 mg/0.6 ml, 80 mg/0.8 ml,
80 mg/2.5 ml, 120 mg/5 ml, 160 mg/5 ml
Sudafed Sinus Products
(pseudoephedrine hydrochloride)
325–500 mg
Tempra products (acetaminophen) 80, 160 mg/5 ml
TheraFlu products (dextromethorphan,
pseudoephedrine)
325–650 mg, 650–1000 mg/packet
Tylenol products (acetaminophen)b 325–650 mg, 250 mg/5 ml, 650 mg/30 ml,
650–1000 mg/packet, 1000 mg/30 ml
Vanquish products (aspirin, caffeine) 194 mg
a This list does not contain all over-the-counter products that contain acetaminophen.b Only active ingredient is acetaminophen.
771ACUTE LIVER FAILURE

772 FONTANA
component of these products have been suggested but not yet implemented[12]. In the interim, health care providers and pharmacies should be awareof the acetaminophen content in many compound medications.
Acute liver failure related to viral hepatitis
Severe acute HAV, HBV, and hepatitis E virus (HEV) infections occa-sionally produce ALF. The diagnosis of HAV-related ALF depends onthe detection of anti-HAV IgM. Young children, persons more than 50 yearsold, and individuals who have underlying liver disease may be more prone todevelop severe acute HAV. The overall incidence of ALF from acute HAVinfection is less than 1% [7,30]. A recent analysis of the United Network forOrgan Sharing (UNOS) transplant database and the ALFSG confirmeda significant decline in the incidence of fulminant HAV in the United Statesbetween 1998 and 2005 [7]. This decline presumably results from more wide-spread HAV vaccination and the reduced incidence of sporadic acute infec-tion. Of note, the Centers for Disease Control and Prevention liberalizedrecommendations for HAV and HBV vaccination in 2007 so that nowany individual is eligible for vaccination [31].
Fulminant HBV infection occurs in less than 1% of acutely infectedindividuals. It is diagnosed by the presence of detectable hepatitis B surfaceantigen (HBsAg) and/or anti-hepatitis B core (HBc) IgM antibody. Some pa-tients who have chronic HBV may develop transiently detectable anti-HBcIgM during a disease flare, however [32,33]. Patients who have fulminantHBV occasionally have hepatitis D virus (HDV) coinfection or superinfec-tion as confirmed by detection of anti-HDV antibodies. Although earlystudies suggested that pre-core and core-promoter variants of HBV wereassociated with ALF, recent studies have failed to demonstrate this associa-tion [32,33]. The role of HBV genotypes and host factors in determining sus-ceptibility to HBV-related ALF is unclear. Patients who have HBV-relatedALF have only a 30% likelihood of survival [33,34]. Although fulminantHBV is believed to be caused by an overwhelming immune response to in-fected hepatocytes, the use of oral antiviral agents such as lamivudine orentecavir has been proposed [34]. A recent randomized, controlled trial oflamivudine in patients in India who had severe acute HBV failed to demon-strate any clinical benefit, however [35]. A retrospective review of the ALFSGexperience from 1995 to 2006 also failed to demonstrate any benefit fromantiviral therapy in 76 patients who had HBV-related ALF [36]. Nonetheless,many experts use oral antiviral agents for fulminant HBV because of theirrelative safety [2].
Severe acute HEV infection is a leading cause of ALF in tropical coun-tries. It occurs most commonly in pregnant women [37,38]. It is diagnosedby detection of anti-HEV IgM antibody. The treatment is supportive. Re-cently, a recombinant protein vaccine for HEV was shown to be safe andeffective in preventing acute infection in a high-risk population from Nepal

773ACUTE LIVER FAILURE
[39]. Rarely, other nonhepatotropic viruses including Epstein-Barr virus, cy-tomegalovirus, herpes simplex virus (HSV), varicella zoster virus, humanherpes virus-6, and parvovirus B-19 can cause ALF [40–42]. Whether theserare causes of ALF are caused by viral variants or an aberrant host immuneresponse is unclear. Diagnosing ALF caused by one of these nonhepato-tropic viruses frequently is difficult and often requires histologic confirma-tion as well as polymerase chain reaction testing. In particular, mostpatients who have HSV-related ALF have no skin lesions at presentation[43,44]. Severe acute Epstein-Barr virus, cytomegalovirus, or HSV shouldbe considered as causes of ALF because they can be treated successfullywith antiviral therapy (see Table 2).
Idiosyncratic drug reactions
Drug-induced liver injury (DILI) is a leading cause for the discontinua-tion of drugs in development and for regulatory actions on previouslyapproved drugs [45]. DILI is rare (1 in 10,000 to 1 in 1,000,000 patient years)and is thought to be caused by host metabolic idiosyncrasy [46,47]. Most pa-tients who have severe DILI experience acute hepatocellular injury resultingin jaundice, but some patients develop severe DILI from severe cholestatichepatic injury [48,49]. Multiple case series demonstrate a preponderanceof women in patients who have DILI and in patients progressing to ALF[3,48]. Whether women are more susceptible to idiosyncratic drug-inducedALF because of differences in body weight, drug dosing, or metabolizing/detoxification enzyme activity is unknown.
Idiosyncratic drug reactions are characterized by variable latency afterinitial administration but usually occur within 12 months of drug initiation.Genetically determined variability in host toxification, detoxification, andregeneration pathways is implicated in the pathogenesis and outcome ofidiosyncratic DILI, but supportive data are limited [47]. The roles of medi-cation dose, drug–drug interactions, alcohol consumption, host immune re-sponse, and other environmental cofactors are largely unknown [50,51]. TheDrug Induced Liver Injury Network should provide insight into theetiologies and mechanisms of DILI by prospectively collecting biologic sam-ples from well-phenotyped cases (see http://dilin.dcri.duke.edu for addi-tional information) [52].
The primary treatment of drug-induced ALF is discontinuing the sus-pected drug to avoid further hepatic injury [53]. A recent uncontrolledseries suggested a potential role for corticosteroids in some patients whohave severe DILI, but this approach is controversial [54]. In addition, cor-ticosteroids were not beneficial in large, randomized controlled studies ofpatients who had ALF [55]. DILI is notoriously difficult to diagnose be-cause patients usually lack immunologic or allergic features at presenta-tion, often are taking multiple drugs, and a confirmatory laboratory testis not available. Liver histology in severe DILI usually is not beneficial

774 FONTANA
except for excluding other treatable causes. Therefore, DILI is a diagnosisof exclusion that requires causality assessment instruments that have sub-stantial limitations [56,57].
In addition to prescription drugs, a careful history of herbal, complemen-tary, and alternative medicines is needed in patients who have unexplainedALF. For example, green tea, ephedra, and various weight-loss agents havebeen associated with ALF [58–60]. Unfortunately, herbal products are notregulated closely during development, manufacturing, or marketing, andin many mixtures the specific hepatotoxic ingredient(s) cannot be identified.
Development of jaundice in combination with high serum aminotransfer-ase levels in patients who have DILI has an estimated mortality rate of 10%(Hy’s rule) [61]. A recent retrospective review of 784 Swedish DILI casesconfirmed that serum aspartate aminotransferase and bilirubin levels at pre-sentation are the most important predictors of mortality or liver transplan-tation in severe hepatocellular DILI [48]. A recent review of 95 JapaneseDILI cases also identified a high serum bilirubin level at presentation anda prolonged latency period to be risk factors for mortality [62]. A reviewof the UNOS liver transplantation database from 1990 to 2002 highlightedthe causes and outcomes of 270 adult liver transplant recipients who haddrug-induced ALF [63]. A striking female predominance was reported(76%), the mean age was 35 years, and acetaminophen was the culprit in49% of cases. Commonly implicated medications in the idiosyncraticDILI group included isoniazid (17.5%), propylthiouracil (9.5%), phenytoin(7.3%), and valproate (7.3%) [63]. In the US ALFSG, two thirds of thepatients who had DILI were female. Most presented with high serum biliru-bin levels (median, 22 mg/dL) and had symptoms for an average of 10 daysbefore presentation (see Table 1). Implicated medications included antitu-berculosis drugs (20%), sulfa compounds (12%), phenytoin (10%), andvarious herbs (10%) (personal communication, WM Lee, MD, 2007). Over-all, patients who had idiosyncratic DILI resulting in ALF had a poor prog-nosis, with a spontaneous survival rate of only 26% at 3 weeks. Therefore,any patient who develops jaundice with coagulopathy or encephalopathyfrom suspected DILI should be referred urgently to a liver transplant center.
Other identifiable causes of acute liver failure
Autoimmune hepatitis rarely causes ALF [64]. Autoimmune serologiesand liver biopsy can aid in the diagnosis, but many of these patients havelow titer or undetectable autoantibodies. The benefit of corticosteroids in ful-minant autoimmune hepatitis is unclear. Early identification of ALF causedby autoimmune hepatitis is important, however, because of the low rate ofspontaneous survival (Fig. 1). ALF is a well-known complication of severalpregnancy-related liver diseases including acute fatty liver of pregnancy(AFLP) and the syndrome of hemolysis, elevated liver enzyme levels andlow platelet count (HELLP) [65–67]. Treatment of these conditions is directed

775ACUTE LIVER FAILURE
toward prompt delivery of the fetus. The hallmark of AFLP is the rapiddevelopment of microvesicular steatosis in the third trimester with resultantmitochondrial dysfunction, metabolic acidosis, and coagulopathy, withonly mild to moderate serum aminotransferase elevations. Women whohave long-chain fatty acid metabolic defects are at increased risk of develop-ing AFLP, but only 25% of women who have AFLP exhibit an identifiablemutation [68]. Although most women who have AFLP or HELLP improvewith prompt delivery, some require emergency liver transplantation. Severeacute viral hepatitis and HSV hepatitis should also be considered in pregnantwomen who have ALF, particularly in the third trimester, because these con-ditions are associated with a poor prognosis even with prompt delivery.
Sudden hepatic outflowobstruction caused by occlusion of all three hepaticveins (Budd-Chiari syndrome) is a rare but potentially treatable cause of ALF[69]. Patients usually present with recent onset of abdominal pain, hepatomeg-aly, and ascites.More than 80%of patients have an identifiable thrombophiliathat may be treated with anticoagulation, but many require liver transplanta-tion [70]. Arterial hypoperfusion to the liver caused by cardiogenic shock orhypovolemia can lead to ischemic hepatitis and may progress to ALF [71].The outcome in these patients is determined primarily by the underlying car-diopulmonarydisease, and liver transplantation rarely is requiredor indicated.
Amanita phalloides mushroom poisoning is a rare cause of ALF thatoften presents with severe gastrointestinal symptoms and diarrhea. Assaysfor amanita toxin are unavailable. Patients can be treated with successfullyintravenous penicillin G, silymarin, and dialysis, although many requireliver transplantation [72,73].
Metabolic and infiltrative diseases
Wilson’s disease is a hereditary disorder of impaired biliary excretion ofcopper that presents as ALF in up to 25% of adolescent or young-adult pa-tients [74]. Clues to fulminant Wilson’s disease include the presence ofKayser-Fleischer rings on slit-lamp examination in up to 50% of cases,low serum alkaline phosphatase levels, hemolytic anemia with hyperbilirubi-nemia, and low serum ceruloplasmin levels (although these levels are normalin 15% of patients) [75]. Elevated serum and urinary copper levels oftenoccur, but these tests may not be feasible because of the frequent presenceof concomitant renal failure. A transjugular liver biopsy can establish thediagnosis definitively by detecting elevated quantitative hepatic copper levelsand advanced hepatic fibrosis, but this biopsy is not always feasible. Becausefulminant Wilson’s disease has 100% mortality in the absence of liver trans-plantation, all these patients should be listed quickly for transplantation.
Acute Hodgkin’s and non-Hodgkin’s lymphoma, metastatic carcinoma(eg, lung, breast, melanoma), and several variants of leukemia are rare infil-trative causes of ALF [76,77]. Although a diagnosis of fulminant malig-nancy may be suspected based on history, laboratory tests, or imaging,

776 FONTANA
liver biopsy frequently is required for confirmation. These patients havea poor prognosis and are not candidates for liver transplantation [3].
Indeterminate acute liver failure
No cause is identified in up to 20% of adult patients who have ALF and in50% of children who have ALF [3,5,78]. Prior studies failed to demonstrateoccult infection with HBV, HEV, parvovirus B-19, HSV, or SEN virus in USALFSG adult patients who had indeterminate ALF [33,43,79,80]. Other pro-posed causes include occult autoimmune hepatitis, undiagnosed acetamino-phen hepatotoxicity, or DILI [23]. In the ALFSG, 19% of the patients whohad indeterminate ALF had detectable serum acetaminophen-cysteine ad-ducts; these patients tended to have higher serum aminotransferase levelsand lower bilirubin levels at presentation than adduct-negative patientswho had indeterminate ALF [23]. Whether acetaminophen was the primarycause of ALF or merely a cofactor in these cases is unclear, however. Patientswho have indeterminate ALF have a poor likelihood of spontaneous recov-ery and should be evaluated rapidly for liver transplantation.
Management of acute liver failure
A key principle in management is the unpredictable and rapid manner inwhich patients who have ALF can deteriorate. Therefore, patients who haveALF should be monitored in an ICU for frequent neurologic and hemody-namic assessment [2]. If the prognosis is poor, early transfer to a liver trans-plant center is recommended.
General management measures
A rapid evaluation for treatable causes of ALF allows the initiation ofspecific, appropriate therapy (see Table 2). Except for liver transplantation,however, no single medical intervention has been shown to be beneficial forall patients who have ALF. Corticosteroids or intravenous prostaglandin E1infusions failed to decrease morbidity or mortality in randomized, con-trolled trials [55,81,82]. NAC is of proven benefit in patients who have acet-aminophen hepatotoxicity [13,20]. Some physiologic studies suggest thatNAC may be beneficial in non-acetaminophen ALF, possibly because of im-proved tissue oxygenation [83,84]. Preliminary results from the ALFSGmulticenter, double-blind study of NAC in non-acetaminophen ALFrecently demonstrated a survival benefit only in patients who had grade1 or 2 encephalopathy [85]. Therefore, this simple and widely available ther-apy may be useful in some patients who have ALF.
An experienced hepatologist, transplant surgeon, and intensivist shouldwork as a team to direct the management of patients who have ALF. Eval-uation for liver transplantation includes obtaining diagnostic serologies,

777ACUTE LIVER FAILURE
a chest roentgenogram, a bedside echocardiogram, and psychosocial evalu-ation. Placement of central venous access and arterial lines can allow fluidresuscitation, infusion of medications, frequent laboratory monitoring,and titration of acid/base status. Routine laboratory tests including seriallactate, factor V, INR, and liver biochemistries should be obtained at leastevery 8 to 12 hours. Glucose levels should be monitored hourly and supple-mented as needed.
Neurologic features
Continuous assessment of neurologic status is critical. Classical signs ofintracranial hypertension, such as papilledema, loss of pupillary reflexes,and clonus, do not correlate reliably with intracranial pressure (ICP) mea-surements or grade of encephalopathy (Box 2). Similarly, head CT findingsof cerebral edema frequently occur late and are not adequately sensitive orreliable to detect intracranial hypertension (Fig. 3) [86,87]. Moreover, thescanning time and transportation logistics usually preclude use of MR im-aging in critically ill patients who have ALF. The pathogenesis of cerebraledema in patients who have ALF may involve the ‘‘glutamine hypothesis,’’wherein detoxification of ammonia by astrocytes leads to the conversion ofglutamate to glutamine that can increase tissue osmolarity and cause edema[88]. Alternatively, cerebral edema may develop from failure of intracerebralvascular autoregulation with resultant increases in brain water and brainvolume, particularly in patients who have advanced encephalopathy [89,90].
Although invasive, ICP monitoring is the most reliable means to monitorchanges in ICP in patients who have ALF [91]. Information from an ICPmonitor helps guide management decisions regarding the use of mannitoland paralytic agents. Controversy exists, however, about whether ICP mon-itoring should be used only in liver transplant candidates, only in patientsenrolled in clinical trials, or in all patients who have grade 3 or grade 4 en-cephalopathy. Sedation should be withheld for at least 2 to 4 hours in intu-bated patients who have ALF who are being considered for placement of anICP monitor to assess brain function. A preoperative head CT is recommen-ded to exclude spontaneous hemorrhage. Although parenchymal cathetershave a greater risk of intracranial bleeding, they provide more reliable pres-sure readings [91,92]. ICP measurements can help intensivists maintain anadequate cerebral perfusion pressure (CPP) (ie, O 50 mm Hg) by the intro-duction of vasopressors to raise the mean arterial pressure (MAP) ormaneuvers to lower the elevated ICP (ie, CPP ¼ MAP � ICP).
To prevent exacerbation of cerebral edema, the head of the bed should beelevated more than 30� from horizontal in all patients who have ALF [93].Vigorous suctioning or other Valsalva maneuvers should be avoided to pre-vent surges in ICP. Prophylactic intravenous lidocaine may be of value[5,93]. Mechanical ventilation with high levels of positive end-expiratorypressure should be avoided. Cooling blankets can be used to keep the

Box 2. Management of cerebral edema in acute liver failure
Grade 1 or 2 encephalopathy� Grade 1: Mild changes in mood and speech, disordered sleep� Grade 2: Inappropriate behavior, mild irritability, agitation,
or somnolence� Hyperreflexia, clonus, asterixis (may or may not be present)
� Transfer patient to ICU for frequent monitoring and neurologicchecks.� Maintain a quiet environment with minimal environmental
stimuli.� Avoid sedatives/hypnotics.
� Administer dextrose 10% drip with hourly blood glucosemonitoring.
� Lactulose may be of benefit in patients who have subacuteacute liver failure (see text).� Toxicities: megacolon, volume depletion, hypernatremia
Grade 3 or 4 encephalopathy� Grade 3: Patient is somnolent but arousable to verbal
command and demonstrates marked confusion, incoherentspeech.
� Grade 4: Patient is not arousable by painful stimuli.� Avoid medications with sedative properties (eg, narcotics,
benzodiazepines) unless patient is intubated� Elevate head of bed to 30� from horizontal.� Avoid Valsalva maneuvers, vigorous straining, or suctioning.� Use cooling blankets to keep core temperature at 37�C or
lower.� Consider intubation to protect airway, hypoxia, respiratory
failure.� If intubated, propofol or midazolam are preferred for
sedation.� Obtain a head CT to rule out intracranial hemorrhage.� Consider placement of an intracranial pressure monitor.� Correct coagulopathy (international normalized ratio < 1.5)
with fresh frozen plasma or recombinant factor VIIa.� Balance risk of procedure versus benefit of accurate data
(eg, epidural versus subdural versus parenchymal) inselecting type of intracranial pressure catheter used.
Measures for elevated intracranial pressure� Maintain cerebral perfusion pressure above 50 mm Hg
(cerebral perfusion pressure = mean arterialpressure � intracranial pressure).
778 FONTANA

� Hyperventilate to PCO2 of approximately w28 to 30 mm Hg.� If intracranial pressure is greater than 20 mm Hg for more than
5 minutes, administer a mannitol bolus (0.5–1.0 mg/kg) over5 minutes.� Monitor serum osmolarity and osmolar gap.
� If intracranial pressure is persistently elevated, administera bolus pentobarbital infusion (100–150 mg) over 15 minutesfollowed by continuous infusion at 1 to 3 mg/kg/h.� Pressors may be needed if pentobarbital is used or cerebral
perfusion pressure is lower than 50 mm Hg.� Intravenous infusion of dopamine or norepinephrine
is preferred.� Avoid vasopressin because of adverse effect on cerebral
blood flow.� Moderate hypothermia (33�–35�C) is investigational
for refractory cerebral edema in acute liver failure.� Use a paralytic agent (atracurium) or propofol to prevent
shivering� Perform a brain perfusion scan to exclude brain death
if increase in intracranial pressure is prolonged.
779ACUTE LIVER FAILURE
patient’s core temperature below 37.0�C. Sedative medications, especiallylong-acting benzodiazepines, narcotics, and diphenhydramine, should beavoided in nonintubated patients because they can obscure neurologicchanges. If sedation is required for patient comfort and safety, agentswith a short half-life, such as midazolam or propofol, are preferred.
If a patient deteriorates clinically or has an ICP exceeding 20 mm Hg formore than 5 to 10 minutes, several measures should be undertaken. Initially,hyperventilation of intubated patients to a PCO2 of 28 to 30 mm Hg is rec-ommended to induce cerebral vasoconstriction [94]. Lactulose can helplower systemic ammonia levels in cirrhotic patients by its osmotic activityand acidification of stool, but lactulose has not been tested prospectivelyin patients who have ALF. In addition, lactulose raises concerns aboutfree water depletion and potential abdominal distention with associatedbowel ischemia. Nonetheless, many centers use lactulose, particularly forpatients who have subacute liver failure who have evidence of portosystemicshunting [2].
Mannitol (0.5–1.0 g/kg) is a first-line therapy for management of ICPsurges exceeding 20 mm Hg that do not respond to hyperventilation [95].Mannitol reduces intracranial volume by drawing fluid into the intravascu-lar space. Mannitol infusions should be withheld in patients who have renalfailure or fluid overload until these problems have been addressed. Monitor-ing of serum osmolarity also is recommended to avoid a hyperosmolar state.

Fig. 3. Head CT findings in a patient who has acute liver failure with cerebral edema. A
39-year-old man ingested an unknown quantity of acetaminophen, zolpidem, and lamotrigine
in a suicide attempt 48 hours before presentation. His initial acetaminophen level was 87 mg/mL,
his alanine aminotransferase level was above 9000 IU/L, his international normalized ratio
was 8.9, and his factor V level was less than 15%. He also had a severe lactic acidosis with
hypotension requiring the use of pressors and intubation. (A) At the time he was list for liver trans-
plantation, a head CT showed loss of the gray–white matter interface, but an intracranial
pressure monitor revealed an opening pressure of only 12 to 15 mm Hg. After an uneventful
liver transplantation, the patient did not wake up. (B) A follow-up head CT showed diffuse
changes of worsening cerebral edema and the patient was pronounced brain dead after the
absence of intracranial blood flow was determined on a Technitium-99 albumin scan.
780 FONTANA
Recent data suggest that hypertonic saline infusion, with a target serumsodium level of 145 to 155 mmol/L, may reduce the incidence and severityof intracranial hypertension, but further studies are needed because of thenarrow therapeutic index [96].
Thiopental and pentobarbital are centrally acting hypnotics that reducebrain oxygen use. They represent a second-line therapy for severe intracranialhypertension [97]. Pentobarbital, administered as a 100- to150-mg bolus over15minutes followed by continuous infusion at 1 to 3mg/kg/h, should bemon-itored to maintain serum drug levels at 20 to 35 mg/L. Because barbiturateinfusions can cause systemic hypotension, dopamine may be required tomaintain an adequate CPP. Propofol has been used to reduce ICP. It maybe advantageous because of its low risk of systemic hemodynamic effects [98].
Moderate hypothermia can reduce cerebral hyperemia and decrease ICPin patients who have ALF refractory to medical therapy [99–101]. Reducingthe core body temperature to 33� or 35�C reduces cerebral oxygen use andblood flow. Whole-body hypothermia can be achieved by external coolingblankets, intravascular cooling devices, and body suits with core body tem-peratures monitored by a rectal or intravascular thermometer. Sedationwith a paralytic agent such as atracurium may be needed to prevent reflexive

781ACUTE LIVER FAILURE
shivering, but propofol or deep sedation also may be effective. The optimalmeans to rewarm hypothermic patients who have ALF safely have not beenestablished. Because of the potential risks of hypothermia, including cardiacarrhythmias, worsening coagulopathy, hypotension, and impaired liverregeneration, randomized, controlled trials of therapeutic and prophylactichypothermia with ICP monitoring are needed before this investigationaltherapy can be recommended routinely.
Seizures in patients who have ALF may be difficult to detect, particularlyin patients receiving deep sedation or barbiturates. In one study of 42 intu-bated patients who had ALF, 31% had subclinical seizure activity. Theincidence was lower in patients who received prophylactic phenytoin[102]. Some experts recommend continuous or intermittent electroencepha-logram monitoring for patients who have grade 3 or grade 4 coma, but thispractice has not been adopted widely. Hypoglycemia and electrolyte distur-bances should be excluded as precipitating factors for seizure developmentand treated aggressively if detected. Phenytoin, infused as an 18-mg/kg load-ing dose over 30 minutes followed by 100 mg every 8 hours, is the first-linetreatment for seizures; pentobarbital, 3 mg /kg, is reserved for refractoryseizures [102].
Infections
Infectious complications are common in patients who have ALF and area leading cause of mortality. Eighty percent of patients who have ALF de-velop bacterial infections, and 20% to 30% develop fungal infections duringtheir hospitalization [103]. Therefore, daily surveillance cultures of blood,urine, and sputum are recommended on admission to the ICU [103]. A di-agnostic paracentesis should be performed on all patients who have ALF atpresentation with ascites, unexplained fever, or leukocytosis. Patients com-monly are infected with staphylococcal species, streptococcal species, orgram-negative rods [2,103]. Coverage with broad-spectrum antibioticsshould be initiated if the patient develops fever, leukocytosis, or unexplaineddeterioration in clinical status. Frequently a quinolone or third-generationcephalosporin is used. Vancomycin can be added for patients suspected ofhaving line infection or further deterioration. Enteral decontaminationwith poorly absorbed orally administered antibiotics does not seem to alterthe outcome of patients who have ALF who receive parenteral antibiotics.Fluconazole or amphotericin should be added for suspected or proven fun-gal infection.
Renal failure and fluid management
Acute renal failure in ALF usually is multifactorial with components ofacute tubular necrosis (ATN), hypovolemia, and even hepatorenal syn-drome. Renal failure is particularly common in patients who have acetamin-ophen toxicity and portends a poor prognosis [104]. In addition to

782 FONTANA
monitoring of central pressures, a urinalysis and urine electrolytes can helpdistinguish ATN from hepatorenal or prerenal causes of renal failure. Lacticacidosis is a common complication of ALF that can be worsened by hypo-volemia, infection, and poor perfusion pressures [105]. Infusion of normalsaline or other colloids may help in the management of hypovolemia.Avoidance of nephrotoxic agents, including aminoglycosides, nonsteroidalanti-inflammatory drugs, and intravenous contrast dye, is critical in patientswho have ALF. Enteral feedings are preferred to parenteral nutritionbecause of the high rate of infectious and metabolic complications withthe latter method. Hyponatremia is a poor prognostic sign. In addition,serum sodium levels below 125 mmol/L should be avoided because hypona-tremia can exacerbate cerebral edema.
If progressive renal failure ensues with oliguria, azotemia, or fluid over-load, continuous venovenous hemofiltration is preferred to standard hemo-dialysis because of the less dramatic fluid shifts and higher perfusionpressures [106]. Citrate anticoagulation may be preferred to heparin inpatients who have liver disease, but randomized, controlled trials have notbeen completed.
Hemodynamic monitoring and inotropes
ALF is characterized by a hyperdynamic circulation with high cardiacoutput, low MAP, and low systemic vascular resistance. Following fluidresuscitation, dopamine or norepinephrine may be used to maintain anadequate MAP and a CPP higher than 50 mm Hg [2,88]. Vasopressin andits analogue terlipressin should be avoided, because they produce cerebralvasodilation and increased cerebral blood flow leading to worsening intra-cranial hypertension [107]. Placement of a Swan-Ganz catheter is helpfulwhen inotropes or ICP monitors are used. Surges in systemic hypertensionand bradycardia (Cushing’s reflex) may herald impending uncal herniation.In terminal ALF, patients can become refractory to inotropes and diefrom circulatory failure. A Technitium-99 albumin scan can document theabsence of blood flow in patients who have ALF and refractory cerebraledema so that these patients can be removed from the liver transplant wait-ing list.
Coagulopathy and bleeding
Serial INR and factor V levels provide useful prognostic information.Routine correction of elevated INR levels with fresh frozen plasma (FFP)is not recommended unless there is evidence of active bleeding or an invasiveprocedure is planned (Box 3). A therapeutic trial of vitamin K, 10 mg, sub-cutaneously for 3 consecutive days is recommended at presentation, becausemany patients who have ALF are vitamin K deficient. Before an invasiveprocedure, such as placement of a central line or an ICP monitor or liverbiopsy, FFP is infused in an attempt to decrease the INR below 1.5. The

783ACUTE LIVER FAILURE
volume of infused FFP needs to be monitored carefully to avoid exacerba-tion of fluid overload and cerebral edema. Similarly, immediately beforeinvasive procedures the platelet count should be maintained above50,000 platelets/mL by means of platelet infusions [108]. Cryoprecipitatecan be administered if the fibrinogen level is less than 100 mg/dL. Acid sup-pression with a proton-pump inhibitor rather than sucralfate or histamine-2receptor blockers is used to prevent upper gastrointestinal bleeding in intu-bated patients who have ALF [2,109].
Recombinant activated factor VII (rFVIIa) has been used before invasiveprocedures in patients who have severe coagulopathy, but it is not FDAapproved for this indication [110,111]. The goal of rFVIIa infusion is to pro-mote localized clot formation in areas of tissue factor release. Because of itscost and risks, most centers reserve rFVIIa infusion for patients who havean INR higher than 1.5 despite infusion of at least 4 units of FFP who re-quire an invasive procedure. Typically a single dose of 80 ug/kg is infusedrapidly to enhance clot formation and to normalize the INR for 2 to12 hours. Contraindications include Budd-Chiari syndrome; known or sus-pected malignancy; a history of deep venous thrombosis, pulmonary embo-lism, or thrombophilia; pregnancy; and hypersensitivity to vitamin K. Themedication should be administered immediately before invasive procedures.Repeating coagulation parameters immediately thereafter is not recommen-ded because of its short half-life. It is unclear whether additional doses orcontinuous infusions of rFVIIa prevent spontaneous bleeding in patientswho have ALF.
Prognosis in acute liver failure
Before the widespread availability of liver transplantation, the reportedsurvival of patients who had ALF was 3% to 18% [2,112]. Later studiesreported survival of 14% to 25% without liver transplantation and 41%to 49% with liver transplantation [113]. Among transplant recipients, the1-year patient survival rate now varies between 60% and 80% [114,115].The severity of encephalopathy and coagulopathy correlate inversely withsurvival [116,117]. Numerous prognostic scales have been proposed to iden-tify patients who have the greatest need for liver transplantation.
King’s College criteria
The King’s College criteria were developed from a retrospective cohort of588 medically managed patients who had ALF and then were validated pro-spectively in an additional 175 patients who had ALF [118]. Readilyobtained clinical and laboratory parameters were selected to enhance theirclinical usefulness in patients who had acetaminophen and non-acetamino-phen–related ALF. In the acetaminophen cohort, an arterial pH below 7.3or INR higher than 6.5, a serum creatinine level higher than 3.4 mg/dL, and

Box 3. Management of coagulopathy in acute liver failure
Multifactorial causes� Hypoprothrombinemia caused by reduced hepatic synthesis
of coagulation factors and disseminated intravascularcoagulation/hypofibrinogenemia
� Thrombocytopenia caused by reduced hepatic thrombopoietinproduction, consumption, acute portal hypertension, andreduced marrow production (eg, aplastic anemia, acute viralillness)
� Vitamin K deficiency caused by poor oral intake and jaundice/cholestasis
Assessment� Obtain prothrombin time/international normalized ratio, partial
thromboplastin time, complete blood cell and platelet count,and fibrinogen every 12 hours.� Serial international normalized ratio and factor V levels have
prognostic value.� Approximately10% of patients who have acute liver failure
have clinically significant bleeding.� Mucocutaneous hemorrhage, gastrointestinal bleeding,
and bleeding at insertion sites
Management� Prophylaxis for gastrointestinal bleeding is recommended in all
patients treated with a proton-pump inhibitor or histamine 2blocker.
� Vitamin K (10 mg) subcutaneously for 3 days is recommendedfor all patients.
� Prophylactic fresh frozen plasma infusions are notrecommended in the absence of active bleeding.� Concerns about volume overload/worsening cerebral edema� Lose prognostic value of international normalized ratio
� If there is active bleeding or a planned procedure, administer� Fresh frozen plasma to maintain international normalized
ratio below 1.5� Platelet infusion to maintain a level higher than
50,000 platelets/mL� Cryoprecipitate to maintain fibrinogen level higher than
100 mg/dL� Consider recombinant factor VIIa only if an invasive procedure
such as placement of an intracranial pressure monitor will beperformed and the international normalized ratio is below 1.5after 4 units of fresh frozen plasmaa
784 FONTANA

� Mechanism: Enhances clot formation at areas of tissue factorrelease
� Contraindications: Budd-Chiari syndrome, malignancy,history of deep vein thrombosis/pulmonary embolism,pregnancy, thrombophilia
� Dose: Administer recombinant factor VIIa as bolus, 80 mg/kg,intravenously over 2 to 5 minutes
� Therapeutic window: Half-life is 2 to 12 hours forinterventions
a Not approved by the Food and Drug Administration for use in this setting.
785ACUTE LIVER FAILURE
grade 3 or grade 4 encephalopathy had prognostic significance. In the non-acetaminophen cohort, an INR higher than 6.5 or three or more of the fol-lowing five parameters were independent predictors of poor outcome [118]:
1. Unfavorable causes (non-A, non-B hepatitis, DILI)2. Jaundice for more than 7 days before encephalopathy3. Age under 10 years or over 40 years4. INR higher than 3.55. Serum bilirubin level higher than17.5 mg/dL
The positive predictive value of these criteria for mortality was 84% inthe acetaminophen cohort and 98% in the non-acetaminophen cohort; thenegative predictive values were 86% and 82%, respectively [118]. This prog-nostic model has been tested in other patient cohorts, and lower positivepredictive values and negative predictive values were found [119,120]. Re-cently, the value of early arterial lactic acid levels in conjunction with thestandard King’s College criteria have been studied in patients who haveacetaminophen-induced ALF. A postresuscitation arterial lactate levelhigher than 3.0 mmol/L and an ‘‘early’’ level higher than 3.5 mmol/L hadnegative predictive values of 97% and 99%, respectively, but had positivepredictive values of only 79% and 74%, respectively [105].
Other prognostic models
The Model for End Stage Liver Disease (MELD) score, consisting ofserum creatinine level, total bilirubin level, and INR, was shown to predict3-month survival better than the Child-Turcotte-Pugh score in liver trans-plant candidates who had cirrhosis [121]. The MELD score has becomethe means by which liver allografts are allocated to cirrhotic patients inthe United States. In patients who have non-acetaminophen ALF, MELDscores identified patients who have the worst prognosis; other patientshad a high rate of spontaneous recovery independent of MELD score [122].

786 FONTANA
Global assessment models have been proposed to predict outcome [123].Larson and colleagues [6] recently showed that admission APACHE IIscores were superior to the King’s College criteria or MELD scores in pre-dicting outcomes in patients who had acetaminophen-induced ALF. Otherpotential prognostic laboratory markers include serum phosphate levels,which decline in patients who have rapid hepatic regeneration [124]. Simi-larly, serum alpha-fetoprotein levels increase in patients undergoing rapidliver regeneration, and increasing levels indicate a better prognosis[125,126]. Although these biochemical parameters probably have inadequatepredictive power independently, they may be useful clinically in combina-tion with other prognostic variables. Abdominal CT scanning to assess livervolume and liver biopsy to assess hepatic histopathology also have been pro-posed, but both methods have limited sensitivity, specificity, and feasibility[127,128]. Because the cause of ALF is an important, consistent predictor ofoutcome, disease-specific prognostic models may prove useful for non-acetaminophen as well as acetaminophen-related ALF [7].
Liver transplantation
Emergency liver transplantation is the only intervention with knownsurvival benefit in patients who have ALF carrying a poor prognosis[114]. Outcome after liver transplantation is linked closely to the severityof the pretransplant illness and the nature of the graft used. Currently,the 1-year survival of patients undergoing transplantation for ALF is lowerthan that of patients undergoing transplantation for chronic liver failure(70% versus 85%), probably because of the emergent nature of the surgery,concomitant organ failure, and higher incidence of immunologically medi-ated graft dysfunction.
Rapid medical and surgical evaluation is required for all ALF transplantcandidates before listing to exclude significant cardiopulmonary disease,malignancy, or other conditions that negatively affect patient outcomes[2]. In addition, a comprehensive psychosocial evaluation of patient compli-ance, family support, and substance abuse is extremely important in patientswho have acetaminophen overdose. Ongoing evaluation of the need and thesuitability of patients listed for transplantation because of ALF is necessarybecause of the unstable nature of this patient population and the potentialdevelopment of contraindications. The frequent delay in securing a suitabledonor liver renders medical decisions complex and difficult. Most centersconsider refractory systemic hypotension or intracranial hypertension, un-controlled sepsis, or progressive multiorgan failure to be contraindicationsto transplantation.
To facilitate rapid distribution of livers for life-saving transplantation,the UNOS developed a special status 1 designation for patients who havea high short-term risk of death. Patients eligible for status 1 designationinclude patients who have ALF with onset of illness in the prior 8 weeks,

787ACUTE LIVER FAILURE
patients who have fulminant Wilson’s disease, and transplant recipients whohave primary graft nonfunction or early hepatic artery thromboses. Status 1patients move ahead of all other listed patients who have chronic liver fail-ure. Grafts are allocated to status 1 patients based on blood type, geogra-phy, and waiting time [122,129]. In August 2005, the status 1 categorywas modified to include specific clinical and laboratory criteria for fulminanthepatic failure, hepatic artery thromboses, and primary graft nonfunction.Donor livers are offered initially to status 1A adults or children; a status1B category was developed for pediatric patients who had chronic liver dis-ease requiring intensive care, nonmetastatic hepatoblastoma, or metabolicdisease. In calendar years 2004 and 2005, 1529 patients were listed asUNOS status 1 [130]. Fifteen days after listing, 54.2% of patients had under-gone transplantation, 16.1% were dying or too sick to undergo transplanta-tion, 11.6% had recovered, 8.7% were still listed for transplantation, and9.1% had been delisted. Extrapolating these data to the overall population,less than 10% of the 2800 patients who have ALF receive a liver transplanteach year in the United States.
Artificial and bioartificial liver devices
Artificial and bioartificial liver-support devices are under development forpatients who have acute and acute on chronic liver failure. These devices maybe ideally suited for patients who have ALF as a bridge to spontaneous recov-ery during native liver regeneration. The design of a clinical trial design is dif-ficult, however, because of variable spontaneous recovery rates and variableavailability of liver transplantation. The ideal liver replacement device shouldperform normal hepatocyte functions including detoxification, metabolism,and synthesis of critical proteins. Early attempts at artificial liver detoxifica-tion included hemodialysis, hemofiltration, exchange transfusion, plasma ex-change, and resin hemoperfusion, but none of these interventions improvedoutcome [131,132]. Newer artificial detoxification devices, such as the Molec-ularAbsorbent Recirculating System, use charcoal or other adherent particlesin an extracorporeal circuit [132,133]. These artificial devices provide only thefiltration function, however. The need for arterial and venous cannulation, an-ticoagulation, and extracorporeal perfusion can cause complications.
Bioartificial liver support devices use human or other mammalian-de-rived hepatocytes in an extracorporeal circuit [132]. In theory, these systemscan synthesize proteins and metabolize xenobiotics in addition toperforming filtration and detoxification. Maintaining viable, sterile hepato-cytes for continuous extracorporeal use is a formidable challenge, however.Concerns have been raised regarding transmission of hepatocytes to thehost, transmission of zoonoses, activation of the clotting cascade, and im-munologic reactions with development of xenoantibodies [134]. In the larg-est randomized, controlled trial, 171 patients who had fulminant orsubfulminant liver failure or primary graft nonfunction following liver

788 FONTANA
transplantation were assigned randomly to receive a daily 6-hour treatmentwith the a device that contains 100 g of porcine hepatocytes loaded in a di-alysis cartridge in series with charcoal filters, versus standard care [135,136].Overall, 30-day survival was similar in both treatment groups (71% in thegroup treated with the porcine hepatocyte device versus 62% in the controlgroup; P¼ .26). This landmark study highlights the difficulties of performingclinical trials in ALF and the need for appropriate patient inclusion criteriaand clinically relevant end points. Further refinements of device componentsand perfusion circuitry and an improved understanding of liver regenerationare needed to improve these devices for use in patients who have ALF.
Summary
ALF remains a dramatic and highly unpredictable clinical syndrome. Stud-ies of its causes and natural history are hampered by its low incidence, variableterminology, and variable clinical management. In the United States, acet-aminophen is the leading cause of ALF, and the incidence of unintentionalacetaminophen overdose seems to be increasing. ALF is a clinical syndromeof coagulopathy and encephalopathy ensuing from a multitude of infectious,immunologic, vascular, infiltrative, and metabolic diseases (see Table 2).Proven treatments for specific causes ofALF includeNAC for acetaminophenoverdose and delivery in pregnancy-relatedALF. In addition, because of theirgenerally safe medication profile, antiviral agents frequently are recommen-ded for HBV- and HSV-related ALF. Intravenous NAC for non-acetamino-phen–related ALF seems promising, but the available data indicate thatcorticosteroids should not be used in indeterminate ALF or DILI . Cerebraledema is a hallmark of ALF that requires specializedmanagement by a groupof experienced intensivists, hepatologists, and transplant surgeons. A lowthreshold for broad-spectrum antibiotics is recommended, because patientswho have ALF are at high risk of bacterial and fungal infections. Patientswho have advanced encephalopathy or an otherwise unfavorable prognosisshould be referred promptly to a liver transplant center for further evaluation.Emergency liver transplantation can have a favorable outcome but requirescoordinated intensive care and constant reassessment.
Acknowledgments
The author would like to acknowledge the contributions and mentorshipprovided by Dr. William Lee of the University of Texas Southwestern, whois the principal investigator of the Acute Liver Failure Study Group.
References
[1] Trey C, Davidson CS. The management of fulminant hepatic failure. In: Popper H,
Schaffner F, editors. Progress in Liver Disease. New York: Grune & Stratton; 1970.
p. 282–98.

789ACUTE LIVER FAILURE
[2] Polson JP, Lee WM. American Association for the Study of Liver Disease position paper:
the management of acute liver failure. Hepatology 2005;41:1179–97.
[3] OstapowiczGA,FontanaRJ, Schiodt FV, et al. Results of a prospective study of acute liver
failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947–54.
[4] Hoofnagle JH, Carithers RL, Shapiro C, et al. Fulminant hepatic failure: summary of
a workshop. Hepatology 1995;21:240–53.
[5] Squires RH, Shneider BL, Bucuvalas J, et al. Acute liver failure in children: the first
348 patients in the pediatric acute liver failure study group. J Pediatr 2006;148:652–8.
[6] LarsonAM,Polson J, FontanaRJ, et al. Acetaminophen-induced acute liver failure: results
of a United States multicenter, prospective study. Hepatology 2005;42:1364–72.
[7] Taylor RM, Davern T, Munoz S, et al. Fulminant hepatitis A virus in the United States:
incidence, prognosis, and outcomes. Hepatology 2006;44:1589–97.
[8] Lisman T, Leebeek FWG, DeGroot PG. Haemostatic abnormalities in patients with liver
disease. J Hepatol 2002;37:280–7.
[9] Ring-Larsen H, Palazzo U. Renal failure in fulminant hepatic failure and terminal cirrho-
sis: a comparison between incidence, types, and prognosis. Gut 1981;22:585–91.
[10] Vaquero J, Polson J, Chung C, et al. Infection and the progression of encephalopathy in
acute liver failure. Gastroenterology 2003;125:755–64.
[11] Nourjah P, Ahmad SR, Karwoski C, et al. Estimates of acetaminophen (Paracetamol)-
associated overdoses in the United States. Pharmacoepidemiol Drug Saf 2006;15:
398–405.
[12] Fontana RJ, Adams PC. ‘‘Unintentional’’ acetaminophen overdose on the rise: who is
responsible? Can J Gastroenterol 2006;20:319–24.
[13] Smilkstein MJ, Knapp GL, Kulig KW, et al. Efficacy of oral N-acetylcysteine in
the treatment of acetaminophen overdose. Analysis of the national multicenter study
(1976–1985). N Engl J Med 1988;319:1557–62.
[14] Zimmerman HJ, Maddrey WC. Acetaminophen hepatotoxicity with regular intake of
alcohol: analysis of instances of therapeutic misadventure. Hepatology 1995;22:767–73.
[15] Kuffner EK, Dart RC, Bogdan GM, et al. Effect of maximal daily doses of acetaminophen
on the liver of alcoholic patients: a randomized, double-blind, placebo-controlled trial.
Arch Intern Med 2001;161:2247–52.
[16] Nolan CM, Sandblom RE, Thummel KE, et al. Hepatotoxicity associated with acetamin-
ophen usage in patients receiving multiple drug therapy for tuberculosis. Chest 1994;105:
408–11.
[17] Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and
alcohol use. JAMA 1994;272:1845–50.
[18] Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults
receiving 4 grams of acetaminophen daily. JAMA 2006;296:87–93.
[19] Schiodt FV, Rochling FA, Casey DL, et al. Acetaminophen toxicity in an urban county
hospital. N Engl J Med 1997;337:1112–7.
[20] Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol
2002;40:3–20.
[21] Polson J, Orsulak PJ, Wians F, et al. Elevated bilirubin may cause false positive acetamin-
ophen levels in hepatitis patients [abstract]. Hepatology 2004;40:496A.
[22] James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab
Dispos 2003;31:1499–506.
[23] Davern TJ, James LP, Hinson J, et al. Measurement of serum acetaminophen-protein
adducts in patients with acute liver failure. Gastroenterology 2006;130:687–94.
[24] Sato RL, Wong JJ, Sumida SM, et al. Efficacy of superactivated charcoal administration
late (3 hours) after acetaminophen overdose. Am J Emerg Med 2003;21:189–91.
[25] Kao LW, Kirk MA, Furbee RB, et al. What is the rate of adverse events after oral
N-acetylcysteine administered by the intravenous route to patients with suspected
acetaminophen poisoning? Ann Emerg Med 2003;42:741–50.

790 FONTANA
[26] Kaplowitz N. Acetaminophen hepatotoxicity: what do we know, what we don’t know, and
what do we do next? Hepatology 2004;40:24–6.
[27] Hawton K, Townsend E, Deeks J, et al. Effects of legislation restricting pack sizes of para-
cetamol and salicylate on self poisoning in the United Kingdom: before and after study.
BMJ 2001;322:1–7.
[28] Hawton K, Simkin S, Deeks J, et al. UK legislation in analgesic packs: before and after
study of long term effect on poisonings. BMJ 2004;329:1076–9.
[29] Food andDrug Administration, 21 CRF Parts 201 and 343. Internal analgesic, antipyretic,
and antirheumatic drug products for over the counter human use: proposed amendment
of the tentative final monograph, required warnings and other labeling. Federal Register
2006;71(247):77314–52.
[30] Rezende G, Roque-Afonso AM, Samuel D, et al. Viral and clinical factors associated with
the fulminant course of hepatitis A infection. Hepatology 2003;38:613–8.
[31] Recommended adult immunization scheduledUnited States October 2006–September
2007. MMWR 2006;55:2430–3.
[32] Wai CT, Fontana RJ, Polson J, et al. Clinical outcome and virological characteristics
of hepatitis B related acute liver failure in the United States. J Viral Hepat 2005;12:
192–8.
[33] Teo EK, Ostapowicz G, Hussain M, et al. Hepatitis B infection in patients with acute liver
failure in the United States. Hepatology 2001;33:972–6.
[34] Schmilovitz-Weiss H, Ben-Ari Z, Sikular E, et al. Lamivudine treatment for acute severe
hepatitis B: a pilot study. Liver Int 2004;24:547–51.
[35] Kumar M, Satapathy S, Monga R, et al. A randomized controlled trial of lamivudine to
treat acute hepatitis B. Hepatology 2007;45:97–101.
[36] SerembaE, Sanders C, JainM, et al. Use of nucleoside analogues inHBV related acute liver
failure [abstract]. Hepatology 2007;46(Suppl 1):79.
[37] Kharoo MS, Kamili S. Aetiology and prognostic factors in acute liver failure in India.
J Viral Hepat 2003;10:224–31.
[38] Pal R, Aggarwal R, Naik SR, et al. Immunological alterations in pregnant women with
acute hepatitis E. J Gastroenterol Hepatol 2005;20:1094–101.
[39] ShresthaMP,McNair ScottR, JoshiDM, et al. Safety and efficacy of recombinant hepatitis
E vaccine. N Engl J Med 2007;356:895–903.
[40] Kang AH, Graves CR. Herpes simplex hepatitis in pregnancy: a case report and review of
the literature. Obstet Gynecol Surv 1999;54:463–8.
[41] Peters DJ, Greene WH, Ruggiero F, et al. Herpes simplex induced fulminant hepatitis in
adults: a call for empiric therapy. Dig Dis Sci 2000;45:2399–404.
[42] Dits H, Frans E,Wilmer A, et al. Varicella-zoster virus infection associated with acute liver
failure. Clin Infect Dis 1998;27:209–10.
[43] Levitsky J, ThadareddyA, Lakeman FD, et al. Measurement of herpes simplexDNA-emia
before and after liver transplantation. Liver Transpl 2008, in press.
[44] Ichai P, Alfonso AM, Sebagh M, et al. Herpes simplex virus-associated acute liver failure:
a difficult diagnosis with a poor prognosis. Liver Transpl 2005;11:1550–5.
[45] Lee WM, Senior JR. Recognizing drug induced liver injury: current problems, possible
solutions. Toxicol Pathol 2005;33:155–64.
[46] Sgro C, Clinard F, Ouazir L, et al. Incidence of drug-induced hepatic injuries: a French
population-based study. Hepatology 2002;36:451–5.
[47] Watkins PB, Seeff LB.Drug induced liver injury: summary of a single topic clinical research
conference. Hepatology 2006;43:618–31.
[48] Bjornsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver
disease. Hepatology 2005;42:481–9.
[49] Andrade RJ, Lucena MI, Fernandez MC, et al. Drug-induced liver injury: an analysis of
461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology
2005;129:512–21.

791ACUTE LIVER FAILURE
[50] Verma A, Lilienfeld DE. The need for a population-based surveillance system for liver
disease in the United States. Pharmacoepidemiol Drug Saf 2004;13:821–4.
[51] Lee WM. Drug-induced hepatotoxicity. N Engl J Med 2003;349:474–85.
[52] Hoofnagle JH. Drug induced liver injury network (DILIN). Hepatology 2004;40:773.
[53] Aithal PG, Day CP. The natural history of histologically proved drug induced liver disease.
Gut 1999;44:731–5.
[54] DecheneA, Treichel U, GerkenG, et al. Effectiveness of a steroid and ursodeoxycholic acid
combination therapy with drug induced subacute liver failure [abstract]. Hepatology 2005;
42:358A.
[55] Rakela J, Mosley JW, Edwards VM, et al. A double-blinded randomized trial of hydrocor-
tisone in acute hepatic failure. Dig Dis Sci 1991;36:1223–8.
[56] Danan G, Benichou C. Causality assessment of adverse reactionsda novel method based
on conclusions of international consensus meetings: application to drug-induced liver
injuries. J Clin Epidemiol 1993;46:1323–30.
[57] LucenaMI, Camargo R, Andrade RJ, et al. Comparison of two clinical scales for causality
assessment in hepatotoxicity. Hepatology 2001;33:123–30.
[58] Favreau JT, FyuML, Braunstein G, et al. Severe hepatotoxicity associated with the dietary
supplement LipoKinetix. Ann Intern Med 2002;136:590–5.
[59] Estes JD, Stolpman D, Olyaei A, et al. High prevalence of potentially hepatotoxic herbal
supplement use in patients with fulminant hepatic failure. Arch Surg 2003;138:852–8.
[60] Watt K, Molinari M, Kruszyna T, et al. Acute liver failure induced by green tea extracts:
case report and review of the literature. Liver Transpl 2006;12:1892–5.
[61] Shapiro MA, Lewis JH. Causality assessment of drug-induced hepatotoxicity: promises
and pitfalls. Clin Liver Dis 2007;11:477–505.
[62] Ohmori S, Shiraki K, Inoue H, et al. Clinical characteristics and prognostic indicators of
drug induced fulminant hepatic failure. Hepatogastroenterology 2003;50:1531–4.
[63] Russo MW, Galanko JA, Shrestha R, et al. Liver transplantation for acute liver failure
from drug induced liver injury in the United States. Liver Transpl 2004;10:1018–23.
[64] KesslerWR, Cummings OQ, EckertG, et al. Fulminant hepatic failure as the initial presen-
tation of acute autoimmune hepatitis. Clin Gastroenterol Hepatol 2004;2:625–31.
[65] Rolfes DB, IshakKG. Acute fatty liver of pregnancy: a clinicopathologic study of 35 cases.
Hepatology 1985;5:1149–58.
[66] Weinstein L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count:
a severe consequence of hypertension in pregnancy. Am J Obstet Gynecol 1982;142:
159–67.
[67] Pereira SP, O’Donohue J, Wendon J, et al. Maternal and perinatal outcome in severe
pregnancy-related liver disease. Hepatology 1997;26:1258–62.
[68] Ibdah JA, Bennett MJ, Rinaldo P, et al. A fetal fatty-acid oxidation disorder as a cause of
liver disease in pregnant women. N Engl J Med 1999;340:1723–31.
[69] Fickert P, Ramschak H, Kenner L, et al. Acute Budd-Chiari syndrome with fulminant
hepatic failure in a pregnant woman with factor V Leiden mutation. Gastroenterology
1996;111:1670–3.
[70] Olzinski AT, Sanyal AJ. Treating Budd-Chiari syndrome: making rational choices from
a myriad of options. J Clin Gastroenterol 2000;30:155–61.
[71] Taylor RM, Fontana RJ, Shakil AO, et al. Acute liver failure due to ischemic hepatitis:
natural history and predictors of outcome in a prospective, multi-center U.S. study
[abstract]. Gastroenterology 2005;128(4 Supp 2):A-706.
[72] Broussard CN,Aggarwal A, Lacey SR, et al.Mushroom poisoningdfrom diarrhea to liver
transplantation. Am J Gastroenterol 2001;96:3195–8.
[73] Klein AS, Hart J, Brems JJ, et al. Amanita poisoning: treatment and the role of liver trans-
plantation. Am J Med 1989;86:187–93.
[74] Roberts EA, Schilsky ML. American Association for the Study of Liver Disease practice
guidelines: a practice guideline on Wilson disease. Hepatology 2003;37:1475–92.

792 FONTANA
[75] Nazer H, Ede RJ, Mowat AP, et al. Wilson’s disease: clinical presentation and use of
prognostic index. Gut 1986;27:1377–81.
[76] Woolf GM, Petrovic LM,Rojter SE, et al. Acute liver failure due to lymphoma. A diagnos-
tic concern when considering liver transplantation. Dig Dis Sci 1994;391:1351–8.
[77] Shehab TM, Kaminski MS, Lok ASF. Case report: acute liver failure due to hepatic
involvement by hematologic malignancy. Dig Dis Sci 1997;42(7):1400–5.
[78] Wigg AJ, Gunson BK,Mutimer DJ. Outcomes following liver transplantation for seroneg-
ative acute liver failure: experience during a 12-year period with more than 100 patients.
Liver Transpl 2005;11:27–34.
[79] Lee WM, Brown KE, Young NS, et al. Brief report: no evidence of parvovirus B19 or
hepatitis E virus as causes of acute liver failure. Dig Dis Sci 2006;51:1712–5.
[80] Umemura T, Tanaka E, Ostapowicz G, et al. Investigation of SEN virus infection in
patients with cryptogenic acute liver failure, hepatitis-associated aplastic anemia, or acute
and chronic non-A-E hepatitis. J Infect Dis 2003;188:1545–52.
[81] Ware AJ, Jones RE, Shorey JW, et al. A controlled trial of steroid therapy in massive
hepatic necrosis. Am J Gastroenterol 1974;62:130–3.
[82] Sterling RK, Luketic VA, Sanyal AJ, et al. Treatment of fulminant hepatic failure with
intravenous prostaglandin E1. Liver Transpl Surg 1998;4:424–31.
[83] Harrison PM, Wendon JA, Gimson ES, et al. Improvement by acetylcysteine of
hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med 1991;
324:1852–7.
[84] Sklar GE, Subramaniam M. Acetylcysteine treatment for non-acetaminophen-induced
acute liver failure. Ann Pharmacother 2004;38:498–501.
[85] Lee WM, Rossaro L, Fontana RJ, et al. Intravenous N-acetylcysteine improves spontane-
ous survival in early stage non-acetaminophen acute liver failure [abstract]. Hepatology
2007;46(Suppl 1):79.
[86] Wijdicks EFM, Plevak DJ, Rakela J, et al. Clinical and radiologic features of cerebral
edema in fulminant hepatic failure. Mayo Clin Proc 1995;70:119–24.
[87] Itai Y, SekiyamaK, Ahmadi T, et al. Fulminant hepatic failure: observation with serial CT.
Radiology 1997;202:379–82.
[88] Jalan R. Pathophysiological basis of therapy of raised intracranial pressure in acute liver
failure. Neurochem Int 2005;47:78–83.
[89] Clemmesen JO, Larsen FS, Kondrup J, et al. Cerebral herniation in patients with acute
liver failure is correlated with arterial ammonia concentration. Hepatology 1999;29:
648–53.
[90] Larsen FS, Ejlersen E, Hansen BA, et al. Functional loss of cerebral blood flow autoregu-
lation in patients with fulminant hepatic failure. J Hepatol 1995;23:212–7.
[91] Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure
monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl
2005;11:1581–9.
[92] Durward QJ, Amacher AL, Del Maestro RF, et al. Cerebral and cardiovascular responses
to changes in head elevation in patients with intracranial hypertension. J Neurosurg 1983;
59:938–44.
[93] Yano M, Nishiyama H, Yokota H, et al. Effect of lidocaine on ICP response to endotra-
cheal suctioning. Anesthesiology 1986;64:651–3.
[94] Strauss G, Hansen BA, Knudsen GM, et al. Hyperventilation restores cerebral blood flow
autoregulation in patients with acute liver failure. J Hepatol 1998;28:199–203.
[95] Canalese J, GimsonAE,Davis C, et al. Controlled trial of dexamethasone andmannitol for
the cerebral oedema of fulminant hepatic failure. Gut 1982;23:625–9.
[96] Murphy N, Auzinger G, Bernal W, et al. The effect of hypertonic sodium chloride on
intracranial pressure in patients with acute liver failure. Hepatology 2004;39:464–70.
[97] Forbes A, Alexander GJ, O’Grady JG, et al. Thiopental infusion in the treatment of
intracranialhypertension complicating fulminanthepatic failure.Hepatology1989;10:306–10.

793ACUTE LIVER FAILURE
[98] Wijkicks EFM, Nyberg SL. Propofol to control intracranial pressure in fulminant hepatic
failure. Transplant Proc 2002;34:1220–2.
[99] Jalan R, Olde Damink SW, Deutz NE, et al. Restoration of cerebral blood flow
autoregulation and reactivity to carbon dioxide in acute liver failure bymoderate hypother-
mia. Hepatology 2001;34:50–4.
[100] JalanR,Damink SW,DeutzNE, et al.Moderate hypothermia prevents cerebral hyperemia
and increase in intracranial pressure in patients undergoing liver transplantation for acute
liver failure. Transplantation 2003;75:2034–9.
[101] Jalan R, Olde Damink SW, Deutz NE, et al. Moderate hypothermia in patients with acute
liver failure and uncontrolled intracranial hypertension. Gastroenterology 2004;127:
1338–46.
[102] Ellis AJ, Wendon JA, Williams R. Subclinical seizure activity and prophylactic phenyt-
oin infusion in acute liver failure: a controlled clinical trial. Hepatology 2000;32:
536–41.
[103] Wade J, RolandoN, Philpott-Howard J, et al. Timing and etiology of bacterial infections in
a liver intensive care unit. J Hosp Infect 2003;53:144–6.
[104] Cobden I, Record CO, Ward MK, et al. Paracetamol-induced acute renal failure in the
absence of fulminant liver damage. Br Med J 1982;284:21–2.
[105] BernalW,DonaldsonN,Wyncoll D, et al. Blood lactate as an early predictor of outcome in
paracetamol-induced acute liver failure: a cohort study. Lancet 2002;359:558–63.
[106] Davenport A, Will EJ, Davidson AM. Improved cardiovascular stability during continu-
ous modes of renal replacement therapy in critically ill patients with acute hepatic and renal
failure. Crit Care Med 1993;21:328–38.
[107] Shawcross DL, Davies NA, Mookerjee RP, et al. Worsening of cerebral hyperemia by the
administration of terlipressin in acute liver failure with severe encephalopathy. Hepatology
2004;39:471–5.
[108] Drews R, Weinberger S. Thrombocytopenic disorders in critically ill patients. Am J Respir
Crit Care Med 2000;162:347–51.
[109] Cook D, Guyatt G, Marshall J, et al. A comparison of sucralfate and ranitidine for the
prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation.
N Engl J Med 1998;338:l791–7.
[110] Shami VM, Caldwell SH, Hespenheide EE, et al. Recombinant activated factor VII for
coagulopathy in fulminant hepatic failure compared with conventional therapy. Liver
Transpl 2003;9:138–43.
[111] Caldwell SH, ChangC,Macik BG.Recombinant activated factor VII as a hemostatic agent
in liver disease: a break from convention in need of controlled trials. Hepatology 2004;39:
592–8.
[112] Ritt DJ, Whelan G, Werner DJ, et al. Acute hepatic necrosis with stupor or coma. An
analysis of 31 patients. Medicine 1969;48:151–72.
[113] Schiodt FV,Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute
liver failure in the United States. Liver Transpl Surg 1999;5:29–34.
[114] Higgins PD, Fontana RJ. Liver transplantation in acute liver failure. Panminerva Med
2002;52:93–7.
[115] McDiarmid SV, Goodrich NP, Harper AM, et al. Liver transplantation for status 1: the
consequences of good intentions. Liver Transpl 2007;13:699–707.
[116] Neuberger J. Prediction of survival for patients with fulminant hepatic failure. Hepatology
2005;41:19–21.
[117] O’Grady J. Attempting to predict the unpredictable in acute liver injury. JHepatol 2005;42:
5–6.
[118] O’Grady JG, Alexander GJ, Hayllar KM, et al. Early indicators of prognosis in fulminant
hepatic failure. Gastroenterology 1989;97:439–55.
[119] Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant
hepatic failure: an assessment of the King’s criteria. J Hepatol 1997;26:62–8.

794 FONTANA
[120] Pauwels A, Mostefa-Kara N, Florent C, et al. Emergency liver transplantation for acute
liver failure: evaluation of London and Clichy criteria. J Hepatol 1993;17:124–7.
[121] Weisner R, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and
allocation of donor livers. Gastroenterology 2003;124:91–6.
[122] KremersWK, IjperenMV,KimWR, et al.MELD score as a predictor of pretransplant and
posttransplant survival in OPTN/UNOS status 1 patients. Hepatology 2004;39:764–9.
[123] Mitchell I, Bihari D, ChangR, et al. Earlier identification of patients at risk from acetamin-
ophen-induced acute liver failure. Crit Care Med 1998;26:279–84.
[124] Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe
acetaminophen-induced hepatotoxicity. Hepatology 2002;36:659–65.
[125] Schiodt FV, OstapowiczG,MurrayN, et al. Alpha-fetoprotein and prognosis in acute liver
failure. Liver Transpl 2006;12:1776–81.
[126] Schmidt LE, Dalhoff K. Alpha-fetoprotein is a predictor of outcome in acetaminophen-
induced liver injury. Hepatology 2005;41:26–31.
[127] Shakil AO, Jones BC, Lee RG, et al. Prognostic value of abdominal CT scanning and
hepatic histopathology in patients with acute liver failure. Dig Dis Sci 2000;45:334–9.
[128] Hanau C, Munoz SJ, Rubin R. Histopathological heterogeneity in fulminant hepatic
failure. Hepatology 1995;21:345–51.
[129] Wiesner RH. MELD/PELD and the allocation of deceased donor livers for status 1 recip-
ients with acute fulminant hepatic failure, primary nonfunction, hepatic artery thrombosis,
and acute Wilson’s disease. Liver Transpl 2004;10:S17–22.
[130] 2006 organ procurement and transplantation network/scientific registry of transplant recip-
ients annual report, Table 9.2a. Available at: http://www.optn.org/AR2006902a_lihtm.
Accessed September 25, 2007.
[131] O’Grady JG, Gimson AE, O’Brien CJ, et al. Controlled trials of charcoal hemoperfusion
and prognostic factors in fulminant hepatic failure. Gastroenterology 1988;94:1186–92.
[132] Redeker AG, YamahiroHS. Controlled trial of exchange-transfusion therapy in fulminant
hepatitis. Lancet 1973;1:3–6.
[133] Sen S, Williams R. New liver support devices in acute liver failure: a critical evaluation.
Semin Liver Dis 2003;23:283–94.
[134] Rifai K, Ernst T, KretschmerU, et al. The Prometheus device for extracorporeal support of
combined liver and renal failure. Blood Purif 2005;23:298–302.
[135] Demetriou AA. Hepatic assist devices. Panminerva Med 2005;47:31–7.
[136] Demetriou AA, Brown RS Jr, Busuttil RW, et al. Prospective, randomized, multicenter
controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg 2004;239:
660–7.




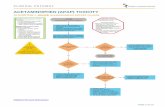








![Paracetamol(acetaminophen)ornon-steroidalanti ... (acetaminophen) … · [Intervention Review] Paracetamol (acetaminophen) or non-steroidal anti-inflammatory drugs, alone or combined,](https://static.fdocuments.net/doc/165x107/5f387cbd43d51a2eb45648f8/paracetamolacetaminophenornon-steroidalanti-acetaminophen-intervention.jpg)




