Activation of the Farnesoid X-receptor in breast cancer...
33
This is a repository copy of Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential . White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/118394/ Version: Accepted Version Article: Alasmael, N, Mohan, R, Meira, LB et al. (2 more authors) (2016) Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential. Cancer Letters, 370 (2). pp. 250-259. ISSN 0304-3835 https://doi.org/10.1016/j.canlet.2015.10.031 © 2015, Elsevier. Licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International http://creativecommons.org/licenses/by-nc-nd/4.0/ [email protected] https://eprints.whiterose.ac.uk/ Reuse Unless indicated otherwise, fulltext items are protected by copyright with all rights reserved. The copyright exception in section 29 of the Copyright, Designs and Patents Act 1988 allows the making of a single copy solely for the purpose of non-commercial research or private study within the limits of fair dealing. The publisher or other rights-holder may allow further reproduction and re-use of this version - refer to the White Rose Research Online record for this item. Where records identify the publisher as the copyright holder, users can verify any specific terms of use on the publisher’s website. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request.
Transcript of Activation of the Farnesoid X-receptor in breast cancer...

This is a repository copy of Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential.
White Rose Research Online URL for this paper:http://eprints.whiterose.ac.uk/118394/
Version: Accepted Version
Article:
Alasmael, N, Mohan, R, Meira, LB et al. (2 more authors) (2016) Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential. Cancer Letters, 370 (2). pp. 250-259. ISSN 0304-3835
https://doi.org/10.1016/j.canlet.2015.10.031
© 2015, Elsevier. Licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International http://creativecommons.org/licenses/by-nc-nd/4.0/
[email protected]://eprints.whiterose.ac.uk/
Reuse
Unless indicated otherwise, fulltext items are protected by copyright with all rights reserved. The copyright exception in section 29 of the Copyright, Designs and Patents Act 1988 allows the making of a single copy solely for the purpose of non-commercial research or private study within the limits of fair dealing. The publisher or other rights-holder may allow further reproduction and re-use of this version - refer to the White Rose Research Online record for this item. Where records identify the publisher as the copyright holder, users can verify any specific terms of use on the publisher’s website.
Takedown
If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request.

1
ActivationoftheFarnesoidX-receptorinbreastcancercelllinesresultsincytotoxicitybut
notincreasedmigrationpotential
NouraAlasmael*1,RatiMohan*
1,LisianeB.Meira
1,KarenE.Swales
2andNickJ.Plant
1
1.! SchoolofBiosciencesandMedicine,FacultyofHealthandMedicalSciences,University
ofSurrey,Guildford,Surrey,GU27XH,UK
2.! ClinicalPDBiomarkerGroup,TheInstituteofCancerResearch,Sutton,SM25NG,UK,
*Theseauthorscontributedequallytothemanuscript
AuthorforCorrespondence:DrNickPlant,SchoolofBiosciencesandMedicine,Facultyof
HealthandMedicalSciences,UniversityofSurrey,Guildford,Surrey,GU27XH,UK
Tel:+44(0)1483686412;Fax:+44(0)1483686401;email:[email protected]

2
Abstract
Breastcanceristhecommonestformofcancerinwomen,butsuccessfultreatmentis
confoundedbytheheterogeneousnatureofbreasttumours:Effectivetreatmentsexistfor
hormone-sensitivetumours,buttriple-negativebreastcancerresultsinpoorsurvival.An
areaofincreasinginterestismetabolicreprogramming,wherebydrug-inducedalterationsin
themetaboliclandscapeofatumourslowtumourgrowthand/orincreasesensitivityto
existingtherapeutics.Nuclearreceptorsaretranscriptionfactorscentraltotheexpression
ofmetabolicandtransportproteins,andthusrepresentpotentialtargetsformetabolic
reprogramming.WeshowthatactivationofthenuclearreceptorFXR,eitherbyits
endogenousligandCDCAorthesyntheticGW4064,leadstocelldeathinfourbreastcancer
celllineswithdistinctphenotypes:MCF-10A(normal),MCF-7(receptorpositive),MDA-MB-
231andMDA-MB-468(triplenegative).Furthermore,weshowthatthemechanismofcell
deathispredominantlythroughtheintrinsicapoptoticpathway.Finally,wedemonstrate
thatFXRagonistsdonotstimulatemigrationinbreastcancercelllines,animportant
potentialadverseeffect.Together,ourdatasupportthecontinuedexaminationofFXR
agonistsasanovelclassoftherapeuticsforthetreatmentofbreastcancer.
Keywords:Apoptosis,Autophagy,NuclearReceptor,Triplenegativebreastcancer,Bile
acids,
Abbreviations:CDCA=chenodexoycholicacid;ER=estrogenreceptor;FXR=FarnesoidX-
receptor;Her2=Humanepidermalgrowthfactorreceptor2;PR=progesteronereceptor;
TNBC=Triplenegativebreastcancer

3
1.! Introduction
BreastcancerrepresentsoneofthelargestkillersofwomenintheWesternworld,witha
lifetimeriskofoneineight[1].Inthepastdecadesanumberofsuccessfultherapeutics
targetingbreastcancerhavebeendeveloped,usedinbothsingleandcombination
therapies.However,improvementsinthefiveyearsurvivalratehavenotbeenashighas
hoped.Thisistoalargedegreeduetotheheterogeneousnatureofbreasttumours,with
multiplemolecularlandscapesbeingclassifiedasasingledisease.Thesediversetumour
phenotypesresultinvariedresponsestotherapeuticintervention,oftenleadingtosub-
optimalpatientresponse[2].Toaidoptimisationoftreatmentregimens,breasttumoursare
commonlyclassifiedintothreeclinicallysignificantgroups:hormonesensitive,Her2positive
andtriplenegativebreastcancers(TNBC)[3].BothhormonesensitiveandHer2-positive
tumourshavearangeofgoodtherapeuticoptions,butTNBCtumoursarecharacterizedby
alackofmoleculartargetsandtendencytodevelopdrugresistance.Notsurprisingly,TNBC
representstheleadingcauseofdeathinbreastcancer[4].Itisthusimperativetodevelop
noveltherapeuticoptionsthatwouldexploittumourvulnerabilities,mitigatedrug
resistanceandleadtoimprovedpatientresponse,andespeciallyinthecaseofTNBC
tumours.
Twoareasoftherapeuticinterventionhavereceivedincreasingattentioninthepastfew
years:Syntheticlethalityandmetabolicvulnerability.Theserelatedconceptsexploitthe
greaterunderstandingofnetworkbiologytopredictsynergisticcombinationtherapies[5].
Insyntheticlethality,theconceptof‘rescuepathways’isexploited:Inessence,targetingof
asinglespeciesinabiologicalpathwayisoftennegatedbyre-routingofthebiological
networktoexploitasecondarypathway.Insyntheticlethality,drugcombinationsareused

4
totargetboththeprimaryandsecondarypathways,producingasynergisticcytotoxicity[6].
Inmetabolicvulnerability,thistheoryofnetworktargetingisfurtherexpanded,lookingfor
novelagentsthatwillworkwithexistingtherapeutics.Tumourcellshaveahighmetabolic
load,duetotheirrequirementtomakealltheprecursorsrequiredforconstantcell
proliferation[7].Assuch,thedevelopmentofdrugsthattargetmetabolismmayreducecell
proliferation,andsynergisewithexistingdrugstoproducemoreeffectivecombination
therapies[8].
Membersofthenuclearreceptorfamilyofligand-activatedtranscriptionfactorsgenerally
regulateexpressionofgenesthatencodeproteinsinvolvedinmetabolicandtransport
processes[9].Assuch,theymayrepresentimportanttargetstoinstigatemetabolic
reprogrammingoftumourcells,leadingtoareductioninsupplyofthecomponents
requiredforcellgrowth[10].Nuclearreceptorexpressioniswidespreadthroughoutthe
body[11],withanumberbeing(over)expressedinbreasttumours[12].Indeed,twoofthe
threemajorclassifiersforbreastcancer,theestrogenreceptor(ER;NR3A1)andthe
progesteronereceptor(PR;NR3C3),arenuclearreceptors,anddisruptionoftheirregulatory
actionisasuccessfultreatmentinreceptorpositivetumours[13,14].ThefarnesoidX-
receptor(FXR;NR1H4)isanadoptednuclearreceptorwithoxysterolssuchasprimaryand
secondarybileacidsasitsendogenousligands[15,16].Bileacidsarethemetabolicproducts
ofcholesterol,andcanbetoxictothebodyathighconcentrations.FXRactstoprevent
accumulationofbileacidsbyinducingexpressionofasecondnuclearreceptorsmall
heterodimerpartner(SHP,NR0B2),whichinhibitsexpressionofCYP7A1,therate-limiting
enzymeinbileacidsynthesis[17,18].Themainsiteofbileacidproductionandsecretion
aretheliverandintestine,respectively,andthisismirroredbyhighlevelsofFXRexpression
intheseorgans[19].However,asapproximately70%ofbileacidsarere-absorbedbythe

5
body,theycanreachmicromolarconcentrationsintheplasma,whichmayunderliethe
expressionofFXRinanumberofothertissues[20].FXRhasbeenshowntobeexpressedin
bothnormalbreasttissueandbreasttumours,withitsactivationinvitrostimulates
apoptosisandinhibitsaromatase[21].ThesefeaturessuggestthatFXRagonistsmay
representanovelclassofbreastcancertherapeuticsduetotheirabilitytoaltertheFXR-
regulatedmetabolicnetwork.
Inthepresentwork,wefirstfullydelineatethemolecularmechanismsbywhichFXR
agonistsactivateapoptosisinbreastcancercelllines,demonstratingthistobethroughthe
intrinsicpathway.Next,weexaminetheabilityofFXRagoniststostimulatemigrationin
thesecelllines,concludingthattheydonotpossessthisability.Together,thesedata
supportthefurtherexaminationofFXRagonistsascancerchemotherapeutics.
2.! MaterialsandMethods
2.1.! Materials
ThebreasttumourcelllineMDA-MB-468(ATCC-HTB-132),andthenormalbreastcellline
MCF10A(ATCC-CRL-10317)werepurchasedfromtheATCC(Teddington,UK),whilethe
breasttumourcelllinesMCF-7(ECACC86012803)andMDA-MB-231(ECACC92020424)
werepurchasedfromtheECACC(PortonDown,UK).CDCAandGW4064werepurchased
fromSigmaAldrich(Poole,UK)andTocrisBiosciences(Abingdon,UK),respectively.
2.2.! CellCulture
MCF7,MDA-MB-468andMDA-MB-231cellsweregrowninDulbecco’smodifiedeagle
medium(DMEM)withphenolredandL-glutamine,supplementedwith10%foetalbovine
serum,100units/mlpenicillin,0.1mg/mlstreptomycinsulphate,and0.25µg/ml
amphotericinB.MCF10AcellswereculturedinDulbecco’smodifiedeaglemedium/F12(1:1

6
v/vDMEM/F12)withphenolred,supplementedwith5%horseserum,100ng/mlcholera
toxin,0.5µg/mlhydrocortisone,10µg/mlinsulin,20ng/mlepidermalgrowthfactor,100
µg/mlstreptomycin,and100units/mlpenicillin.
2.3.! ViabilityAssay
Cellviabilitywasassessedbythe3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium
bromide(MTT)assay,aspreviouslydescribed[22].Briefly,breastcancercelllineswere
platedin96-welltissuecultureflasksatadensityof5x104cellsperwell,andallowedto
attachovernight.Forexperimentsusingserum-starvedconditions,cellsweregrownfora
further24hoursinserum-freemedium.Next,cellswereexposedtotestchemicalsforthe
requiredtime;duringthefinal60minofincubation,0.5mg/mLMTTwasadded.Attheend
oftheincubationperiod,theresultantformazansaltwasdissolvedinDMSOandits
absorbancemeasuredat540nm.Resultsareexpressedasapercentageofvehiclecontrol;
eachdatapointrepresentsthemeanofaminimumofthreeindependentexperimentsof6
wellsperexperiment,witherrorbarsrepresentingthestandarderrorofthemean(SEM).
2.4.! CaspaseAnalysis
CaspaseactivitywasmeasuredusingtheCaspase-Gloassaysystem(Promega,UK)as
previouslyreported[23].Briefly,breastcancercelllineswereseededin96-wellplatesata
densityof1x104cellsperwell,allowedtoattachovernight,serum-starvedfor24hours,
andthenexposedtotestchemicals(±specificcaspaseinhibitor)asrequired.Next,Caspase-
Gloreagentwasadded,mixed,incubatedatroomtemperatureforonehour,andthen
luminescencemeasured.Resultsareexpressedasapercentageofvehiclecontrol;eachdata
pointrepresentsthemeanofaminimumofthreeindependentexperimentsof6wellsper
experiment,witherrorbarsrepresentingthestandarderrorofthemean(SEM).
2.5.! ProteinAnalysis

7
Westernblotanalysiswasundertakenaspreviouslydescribed[22].Briefly,totalproteinwas
extractedusingRIPAbuffer,andproteinquantifiedbythemethodofSmith[24].Thirty
microgramsoftotalproteinwasseparatedonprecast6-18%polyacrylamidegels,andthen
transferredtoPVDFmembrane.Proteinweredetectedovernightat4˚Cusingthefollowing
primaryantibodiesanddilutions:FXR1:500(sc13063,SantaCruzBiotechnology);MMP2
1:200(sc-10736,SantaCruzBiotechnology);MMP91:200(sc-10737,SantaCruz
Biotechnology);PARP1:1000(#9542,CellSignalling);cytochromec1:500(sc-7159,Santa
CruzBiotechnology);Bax1:250(sc-493,SantaCruzBiotechnology);Bcl21:250(sc-7382,
SantaCruzBiotechnology);SHP1:250(sc-15283,SantaCruzBiotechnology);LC31:1000
(#2775,CellSignalling);and,B-actin1:2,000(A2228,Sigma-Aldrich).Next,membraneswere
incubatedwithanIRDye800CWgoat,anti-rabbitsecondaryantibody(1:10,000)forone
houratroomtemperature,andimagedusinganOdysseyCLxinfraredimagingsystem(LI-
CORBiosciences).
2.6.! AutophagyAnalysis
InadditiontoexaminationofautophagythroughWesternblottingofLC3cleavage(see
above),theaccumulationofwasp62/SQSTM1inautophagosomeswasquantifiedusingthe
PremoAutophagySensorGFP-p62assay(MolecularProbes,OR,USA)[25].Breastcancer
celllineswereseededin24-wellplatesatadensityof2x105cellsperwell,allowedto
attachovernight,exposedtopGFP-62inductionmixinserum-freemediumfor24hours,
andthenexposedtotestchemicalsasrequired.p62/SQSTM1fluorescencewasquantified
andexpressedasapercentageofvehiclecontrol;eachdatapointrepresentsthemeanofa
minimumofthreeindependentexperiments,witherrorbarsrepresentingthestandard
errorofthemean(SEM).3-methyladenineandrapamycin(Sigma-Aldrich)wereusedas
inhibitorsandactivatorsofautophagy,respectively[26-28].

8
2.7.! CellMigrationAnalysis
Forthetranswellassay,24-welltranswellinsertswithan8μmporesize(CorningInc,USA)
wereused.Breastcancercelllineswereseededintotheupperchamberatadensityof
1x105cellsperwellinserum-freemedium.Plateswereincubated24-72hours,andthen
cellsinthelowerchambercounted;toensurecountingofallmigratedcells,thelower
surfaceofthetranswellwasexposedto1xtrypsin:EDTAfor30minutes.
Forthewound-healingassay,35mmwoundhealing-inserts(Ibidi,Martinsried,Germany)
wereused.Breastcancercelllineswereseededintothechamberatadensityof1x105cells
perwellinserum-freemediumandallowedtoattachovernight.Insertswereremoved,and
gapclosuremeasuredattime0and12hoursafterexposuretotestchemicalusingtScratch
program[29].
2.8.! InhibitionofFXRactivitybyFXRdominant-negativeover-expression
TodemonstratethattheobservedeffectswereFXR-dependent,experimentswere
undertakenusingover-expressionofadominantnegativeFXRmutant[30].MCF-7and
MDA-MB-231cellsweretransfectedwiththeFXR-DNexpressingplasmidusingFugene6
(Promega,UK)accordingtothemanufacturer’sinstruction,andincubatedovernightto
allowproteinexpression.CellswerethenexposedtoGW4064andanalysedaspreviously
described.
2.9.! StatisticalAnalysis
StatisticalanalysiswasundertakenusingGraphPadPrismv6.01(GraphPadSoftwareInc.,La
Jolla,USA).Datasetswerecomparedthrougheitheraone-wayANOVAwithTukey’s
multiplecomparisontestoratwo-wayANOVAwithSidak’smultiplecomparisontest,as
appropriate.Thelevelofstatisticalsignificancewassetaprioriatp<0.05.

9
3.! ResultsandDiscussion
3.1.! ActivationofFXRinducescelldeathinbreastcancercelllines
ThenuclearreceptorFXRisclassicallyassociatedwithbileacidhomeostasisinthebodyand
itstargetgenesimpactonthemetabolicandtransporter-mediatedclearanceofbothbile
acidsandtheirprecursors[15].However,FXRactivationhasalsobeenreportedtoelicita
numberofotherphenotypes,includingcelldeath.Toconfirmthisphenotypeinbreast
cancercelllines,weexaminedtheeffectoftheendogenousligandCDCAandthemore
potentandselectiveartificialligandGW4064[31].Fourcelllineswerechosentorepresent
differentbreastcancerphenotypes:normal(i.e.MCF-10A),receptorpositivetumour(i.e.
MCF-7)andtriplenegativetumours(i.e.MDA-MB-231andMDA-MB-468).Weconfirmed
thatFXRisexpressedinthesecelllines,andforMCF-7andMDA-MB-231cellsthattheFXR-
dependentsignallingcascadeisintact,withactivationofSHPexpressioninresponsetoFXR
activation(supplementalfigureS1).
BothCDCAandGW4064causedaconcentration-dependenttoxicityinallcelllines(Figure
1),withtheeffectgenerallybeingmoremarkedinserum-freeconditions.Thisisconsistent
withtheobservationthatbothCDCAandGW4064arelipophilic,withLogPvaluesof4.15
and8.3,respectively[20].Inserum-containingmedium,thefreeconcentrationofboth
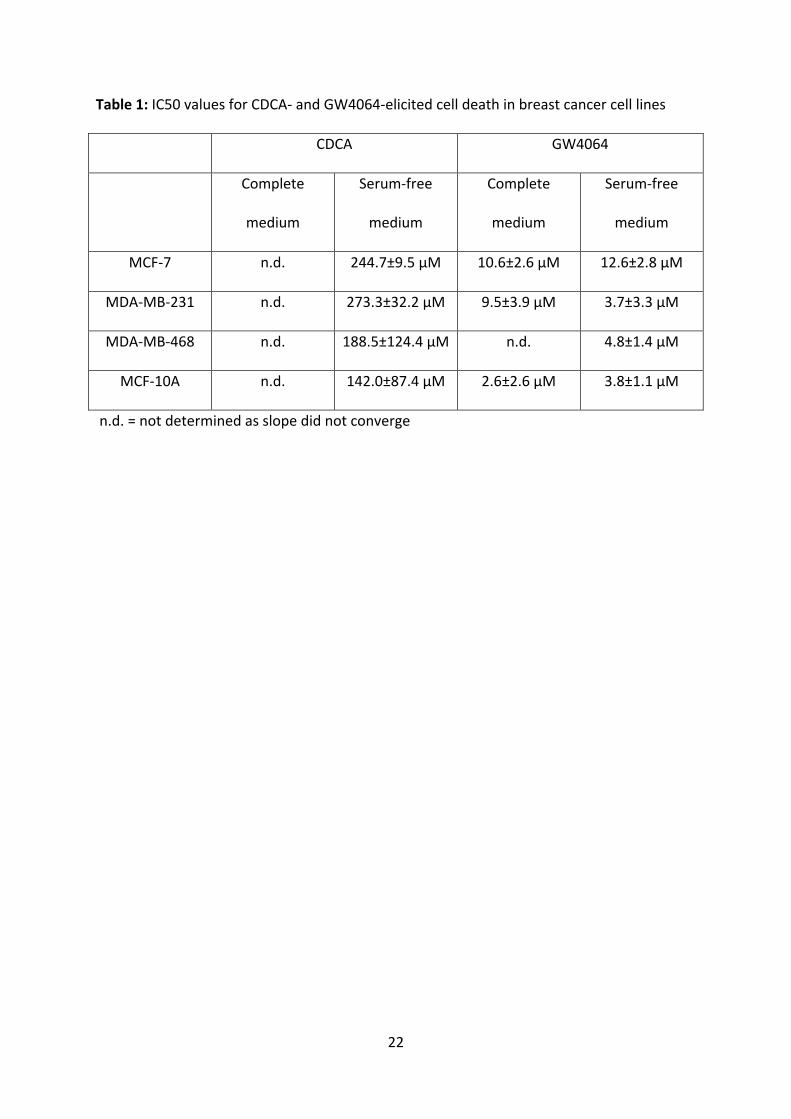
compoundsareexpectedtobereducedduetoproteinbinding[32].IC50valueswere
determinedforallconditionsandarepresentedinTable1.Wenotethat,asexpected,IC50
valuesforGW4064areapproximatelythreeordersofmagnitudelowerthanforCDCA,
consistentwiththehigheraffinityofthisligandforFXR[GW4064Kd=0.01μMversusCDCA
Kd=50μM[15,31]].DataconsistentwiththesevalueswerealsoobtainedwithMTTanalysis
following72hoursofexposureofcelllinestoeachcompound(datanotshown).

10
Despitethediversephenotypesofthefourcelllinesstudied,allrespondedinthesame
mannertoCDCAandGW4064,bothqualitativelyandquantitatively,withnostatistically
significantdifferencesinresponseforeitheragonistbetweencelllines.Thiswouldsuggest
thatFXR-dependentcytotoxicityisageneralcellresponse,andnotdependentofany
emergentphenotypicpropertyofthedifferentgenotypesofeachcellline.
3.2.! FXR-mediatedcytotoxicityismediatedthroughtheintrinsicapoptoticcascade
HavingdeterminedthatFXRagonistsstimulatecytotoxicityinallfourbreastcancercell
lines,wenextexaminedthemolecularmechanismsunderlyingthisresponseinthereceptor
positiveMCF-7andtriplenegativeMDA-MB-231celllines.
ActivationofFXRhasbeenpreviouslyreportedtocauseapoptoticcelldeathinbreast
cancerMCF-7andMDA-MB-468celllines[21].AsdepictedinFigure2,exposureofMCF-7
andMDA-MB-231cellstoGW4064orCDCAfor24hoursresultedinastatisticallysignificant
activationoftheexecutionercaspases3/7,andcleavageofitssubstratepoly(ADP-
ribose)polymerase-1(PARP)[33].ThisconfirmsthedataofSwalesetal,andextendsitto
coveranothertriplenegativecellline.Inaddition,wenoteSwalesetalusedhigh
concentrationsofGW4064(30µM;3,000xKd);hereweusealowerconcentrationthatwill
cause99.9%receptoroccupancywhilereducingtheriskofoff-targeteffects[31].FXR
agonist-mediatedactivationofcaspases3/7,andsubsequentPARPcleavage,ismorepotent
intheMDA-MB-231celllinecomparedtotheMCF-7cellline.Thisisconsistentwithreports
thattheMCF-7celllinelacksexpressionofcaspase3[34,35],meaningonlycaspase7
activityispresent.
HavingconfirmedthatFXRactivationbyGW4064orCDCAcausesapoptosisinbreastcancer
celllines,wenextexaminedthemolecularmechanism(s)underlyingthiseffect.The
apoptoticcascadecanoccurthroughtheintrinsicandextrinsicpathways,witheachbeing

11
composedofdistinctmoleculareventsthatconvergeattheactivationofexecutioner
caspasessuchascaspase3and7[36].Caspase8activationisclassicallyassociatedwiththe
extrinsicapoptoticpathway,whilecaspase9activationisindicativeofapoptosisviathe
intrinsicpathway.ExposureofMCF-7andMDA-MB-231cellstoeither3µMGW4064or
30µMCDCAfor9hourscausedastatisticallysignificantincreaseincaspase9activity,which
couldbepreventedbyco-incubationwiththecaspase-9inhibitorZ-LEHD-FMK(Figure3a).
Foldincreasesincaspase9activitywerethesameorderofmagnitudeasthatobservedwith
thepositivecontrol,1µMstaurosporine.Incontrast,neithercompoundwasabletoactivate
caspase8activityineitherMCF-7orMDA-MB-231celllines(Figure3b);presenceofa
functionalextrinsicpathwaywasdemonstratedthroughtheuseofthepositivecontrol
chemicalFasligand[37].Together,thesedataareconsistentwithactivationofFXRresulting
incelldeaththroughactivationoftheintrinsicpathwayofapoptosis.
ToconfirmtheabilityofGW4064andCDCAtoinduceintrinsicpathway-mediatedapoptosis,
weexaminedseveralothermolecularmarkersforthispathway.Akeystageintheintrinsic
pathway,andonethatdistinguishesitfromtheextrinsicpathway,ismitochondrial
dysfunction.MembersoftheBCL2familyarecentralregulatorsofthisprocess,withthe
relativeexpressionofpro-(e.g.Bcl2)andanti-(e.g.BAX)apoptoticfamilymembers
determiningcellfate[36].Figure4ademonstratesthatFXRactivationresultsinasignificant
increaseinBAX:Bcl2ratio,withGW4064increasingtheratioby2.9±0.3-foldand4.8±1.0-
fold,andCDCAby2.8±0.4-foldand2.5±0.2-foldinMCF-7andMDA-MB-231cells,
respectively.Alterationinthisratioisassociatedwithchangesinmitochondrialmembrane
integrity,leadingtocytochromecreleaseintothecytoplasm,whichcomplexeswithApaf-1
toformtheapoptosome[38].Weobserveatime-dependentreleaseofcytochromec
followingexposureofMCF-7andMDA-MB-231cellstobothGW4064andCDCA(Figure4b).

12
Asnotedabove,theMCF-7celllinemayhavelimitedapoptoticcapacityduetoalackof
expressionofcaspase3[34,35].Undersuchconditions,alternativepathwaysmaybe
initiated.Autophagyisacellrescuepathwayinvolvingdegradationandrecyclingof
damagedcellcomponents[39],andtherelativerolesofautophagyandapoptosisin
determiningcellfateappearstobehighlycontextdependent,withfactorsincludingcellular
background,typeofperturbation,anddegreeofdamage.ConjugationofcytosolicLC3-Ito
phosphatidylethanolamine,formingautophagosomalLC3-IIisanimportantmarkerof
autophagy[40],andinFigure5awepresenttheimpactofGW4064andCDCAontheLC3-
I:LC3-IIratioinbothcelllines.InMCF-7cells,GW4064isobservedtocausea4.4±0.3-fold
increaseinthisratio,consistentwithactivationoftheautophagypathway,butCDCAdoes
notelicitasimilarresponse.InMDA-MB-231cells,neithercompoundwasabletoelicita
significantchangeinLC3-I:LC3-II.Asecondexperimentalapproachwasusedtoconfirmthe
activationofautophagy,namelyaccumulationofp62/SQSTM1,aubiquitin-bindingprotein
thattargetsubiquitinylatedproteinsfordegradationviaautophagy[41].Both3µM
GW4064and30µMCDCAelicitedastatisticallysignificantincreaseinp62-GFPfluorescence
inMCF-7cells,butnotinMDA-MB-231cells.Thisincreasewasreducedbytheadditionof
5mM3-methyladenine,aninhibitorofautophagy.Assayspecificitywasdemonstratedby
thepositivecontrolcompound2.5µMrapamycin[26].
ThesedataareconsistentwithMCF-7cellsusingtheautophagicresponseasacomplement
toapoptosis,whichisnecessaryduetothecompromisedcapacitytoundergoapoptosis
causedbylackofexpressionoftheexecutionercaspase3.Insupportofthis,when
autophagyisinhibitedby3-methyladenine,nosignificantincreaseincaspase3/7activation
isseeninMCF-7cells,suggestingthatthecellsarealreadyutilisingthecaspase-dependent
apoptoticcelldeathpathwayatclosetoitscapacity(Figure5c).

13
3.3.! InductionofapoptosisandautophagyinbreastcancercelllinesisFXR-dependent
Aspreviouslynoted,whileGW4064isahighlyselectiveligandagainstFXR,theactionsof
CDCAaremorepromiscuous.Itisthusimportanttodemonstratethattheobservedeffects
aremediatedviaFXRandnotanalternatemechanism.Toexaminethis,wehaveuseda
dominantnegativeFXRmutant(FXR-DN)tosquelchFXR-mediatedsignalling[21,30,42].As
canbeseenfromfigure6a,transfectionoftheFXR-DNplasmidintoMCF-7andMDA-MB-
231cellseffectivelyblocksFXR-dependentsignalling,asevidencedbytheablationofSHP
induction,aclassicalFXRtargetgene,inresponsetoGW4064.
BlockadeofFXRsignallingbyFXR-DNalsoreducesGW4064andCDCA-dependentcelldeath
(Figure6b),consistentwithpreviousreportsthatthisisFXRdependent[21].Thereduction
incelldeathisduetodecreasedapoptosisinbothcelllines,asevidencebyreduced
activationoftheexecutioncaspases3/7(Figure6c).Aspreviouslydemonstrated,MCF-7
cells,whicharedeficientincaspase-3,arealsostimulatedtoundergoautophagyby
GW4064,andwedemonstratethatthisisalsoFXR-dependent,beingreducedinthe
presenceoftheFXR-DNmutant(figure6d).
Together,thesedatastronglysupportthattheobservedeffectsaremediatedthroughthe
actionofFXR,andnotviaanother,non-specific,mechanism.
3.4.! ActivationofFXRinbreastcancercelllinesdoesnotcauseanincreaseincell
migration
ThedatapresentedhereinsupportsthecytotoxicpotentialofFXRactivationinbreast
cancercelllines.Inaddition,wefurtherelucidatethemolecularmechanismsunderlying
this,whicharethroughtheprogrammedapoptoticandautophagicpathways.Assuch,these
datawouldsupportthepotentialofFXRagonistsascancerchemotherapeuticagents.

14
However,anumberofreportshavesuggestedthatactivationofFXRcanalterthe
migration/invasionphenotypeoftumourcells,althoughthedatainthisareais
controversial.Forexample,Fukaseetal.havereportedapositiveactiononmigrationinthe
Hep3Bhepatomacellline[43],whileStrauchetal.demonstratedasimilareffectin
intestinalepithelialcells[44].Conversely,ZhangetalreportedthatFXRsuppression
promotedproliferationandmigrationofHepG2andHep3Bhepatomacelllines[45],while
Yoyoetal.demonstratedarepressiveeffectonvascularsmoothmusclecellmigration[46].
Anypotentialforenhancedcellmigration/invasionisamajorliabilityinthedevelopmentof
atherapeuticagent,andwethereforeexaminedthepotentialofGW4064andCDCAto
effectmigrationinbreastcancercells.
Inthetranswellmigrationassaywewereabletodetectasignificantnumberofmigrated
cellsfollowing72hoursincubationforbothMCF-7andMDA-MB-231celllines,importantas
MCF-7areconsideredpoorlyinvasive[47].Exposureofcellsintheupperchambertoeither
3µMGW4064or30µMCDCAthroughouttheincubationperioddidnotresultinany
increaseinmigrationrates(Figure7a).Inaddition,wenotethatextensionoftheincubation
periodtoincludea1-hourpre-incubationwithFXRagonistalsodidnotimpactupon
migrationrates(datanotshown).Asecondexperimentalapproachwasalsousedto
examinemigration,thescratchassay.Datafromthisassaywasconsistentwiththetranswell
assay;whilesignificantmigrationcouldbeobservedoverthe24hourincubationperiod,
therewasnosignificantincreasewhenMCF-7orMDA-MB-231cellswereexposedtoFXR
agonists(Figure7b).
ThisdataappearsinconflictwiththatofSilvaetal,whodemonstratedthatthesecondary
bileaciddeoxycholatecouldinducemigrationofMDA-MB-231,butnotMCF-7cells[48].
However,themethodologyusedbySilvaetalwassignificantlydifferentfromthepresent

15
work,andonlythesecondarybileaciddeoxycholatewasexamined.Thisbileacidisa
productofbacterialmetabolismofCDCA,andattheconcentrationusedlikelytocauseboth
FXR-dependentandFXR-independenteffects[49].
Giventheimportantnegativeliabilityassociatedwithanypro-migratoryeffect,andthe
conflictingdatainthisarea,wealsoexaminedMCF10AandMDA-MB-468cellsfortheir
migratoryresponseuponFXRactivation.AspresentedinsupplementaryfigureS2,no
evidencewasfoundtosupportapro-migratoryphenotypeineitherofthesecelllines.As
such,thesedatasupportthenotionthatFXRactivationinbreastcancercelllinesdoesnot
enhancemigratorypotential.ThissuggeststhatanyinteractionbetweenFXRandcell
migration/invasioniscomplex,andalmostcertainlycontext-dependent.
Conflictofinterest
KESisanemployeeoftheInstituteofCancerResearch.NA,RM,LMandNJPdeclarethat
theyhavenoconflictsofinterest
Acknowledgements
NAwasfundedbytheKingdomofSaudiArabiaMinistryofEducation.
References
[1]J.Ferlay,I.Soerjomataram,R.Dikshit,S.Eser,C.Mathers,M.Rebelo,D.M.Parkin,D.
Forman,F.Bray,Cancerincidenceandmortalityworldwide:Sources,methodsandmajor
patternsinGLOBOCAN2012,Int.J.Cancer,136(2015)E359-E386.

16
[2]B.Weigelt,A.Mackay,R.A'Hern,R.Natrajan,D.S.P.Tan,M.Dowsett,A.Ashworth,J.S.
Reis-Filho,Breastcancermolecularprofilingwithsinglesamplepredictors:aretrospective
analysis,LancetOncology,11(2010)339-349.
[3]J.S.Reis-Filho,L.Pusztai,BreastCancer2Geneexpressionprofilinginbreastcancer:
classification,prognostication,andprediction,Lancet,378(2011)1812-1823.
[4]J.Crown,J.O'Shaughnessy,G.Gullo,Emergingtargetedtherapiesintriple-negative
breastcancer,Ann.Oncol.,23(2012)56-65.
[5]A.Kolodkin,F.C.Boogerd,N.Plant,F.J.Bruggeman,V.Goncharuk,J.Lunshof,R.Moreno-
Sanchez,N.Yilmaz,B.M.Bakker,J.L.Snoep,R.Balling,H.V.Westerhoff,Emergenceofthe
siliconhumanandnetworktargetingdrugs,EurJPharmSci,46(2012)190-197.
[6]W.G.Kaelin,Theconceptofsyntheticlethalityinthecontextofanticancertherapy,Nat.
Rev.Cancer,5(2005)689-698.
[7]D.Hanahan,R.A.Weinberg,Thehallmarksofcancer,Cell,100(2000)57-70.
[8]B.Al-Lazikani,U.Banerji,P.Workman,Combinatorialdrugtherapyforcancerinthepost-
genomicera,NatureBiotechnology,30(2012)679-691.
[9]N.Plant,S.Aouabdi,Nuclearreceptors:thecontrollingforceindrugmetabolismofthe
liver?,Xenobiotica,39(2009)597-605.
[10]H.Gronemeyer,J.A.Gustafsson,V.Laudet,Principlesformodulationofthenuclear
receptorsuperfamily,Nat.Rev.DrugDiscov.,3(2004)950-964.
[11]A.L.Bookout,Y.Jeong,M.Downes,R.T.Yu,R.M.Evans,D.J.Mangelsdorf,Anatomical
profilingofnuclearreceptorexpressionrevealsahierarchicaltranscriptionalnetwork,Cell,
126(2006)789-799.
[12]M.Uhlen,L.Fagerberg,B.M.Hallstroem,C.Lindskog,P.Oksvold,A.Mardinoglu,A.
Sivertsson,C.Kampf,E.Sjoestedt,A.Asplund,I.Olsson,K.Edlund,E.Lundberg,S.Navani,

17
C.A.-K.Szigyarto,J.Odeberg,D.Djureinovic,J.O.Takanen,S.Hober,T.Alm,P.-H.Edqvist,H.
Berling,H.Tegel,J.Mulder,J.Rockberg,P.Nilsson,J.M.Schwenk,M.Hamsten,K.von
Feilitzen,M.Forsberg,L.Persson,F.Johansson,M.Zwahlen,G.vonHeijne,J.Nielsen,F.
Ponten,Tissue-basedmapofthehumanproteome,Science,347(2015)394-+.
[13]N.E.Hynes,H.A.Lane,ERBBreceptorsandcancer:Thecomplexityoftargeted
inhibitors,Nat.Rev.Cancer,5(2005)341-354.
[14]X.J.Cui,R.Schiff,G.Arpino,C.K.Osborne,A.V.Lee,Biologyofprogesteronereceptor
lossinbreastcanceranditsimplicationsforendocrinetherapy,J.Clin.Oncol.,23(2005)
7721-7735.
[15]M.Makishima,A.Y.Okamoto,J.J.Repa,H.Tu,R.M.Learned,A.Luk,M.V.Hull,K.D.
Lustig,D.J.Mangelsdorf,B.Shan,Identificationofanuclearreceptorforbileacids,Science,
284(1999)1362-1365.
[16]D.J.Parks,S.G.Blanchard,R.K.Bledsoe,G.Chandra,T.G.Consler,S.A.Kliewer,J.B.
Stimmel,T.M.Willson,A.M.Zavacki,D.D.Moore,J.M.Lehmann,Bileacids:Naturalligands
foranorphannuclearreceptor,Science,284(1999)1365-1368.
[17]B.Goodwin,S.A.Jones,R.R.Price,M.A.Watson,D.D.McKee,L.B.Moore,C.Galardi,
J.G.Wilson,M.C.Lewis,M.E.Roth,P.R.Maloney,T.M.Willson,S.A.Kliewer,Aregulatory
cascadeofthenuclearreceptorsFXR,SHP-1,andLRH-1repressesbileacidbiosynthesis,
MolecularCell,6(2000)517-526.
[18]G.Rizzo,B.Renga,A.Mencarelli,R.Pellicciari,S.Fiorucci,RoleofFXRinregulatingbile
acidhomeostasisandrelevanceforhumandiseases.,CurrentDrugTargetsforImmune,
EndocrinologyandMetabolicDisorders,5(2005)289-303.
[19]G.R.Mishra,M.Suresh,K.Kumaran,N.Kannabiran,S.Suresh,P.Bala,K.Shivakumar,N.
Anuradha,R.Reddy,T.M.Raghavan,S.Menon,G.Hanumanthu,M.Gupta,S.Upendran,S.

18
Gupta,M.Mahesh,B.Jacob,P.Mathew,P.Chatterjee,K.S.Arun,S.Sharma,K.N.Chandrika,
N.Deshpande,K.Palvankar,R.Raghavnath,R.Krishnakanth,H.Karathia,B.Rekha,R.Nayak,
G.Vishnupriya,H.G.M.Kumar,M.Nagini,G.S.S.Kumar,R.Jose,P.Deepthi,S.S.Mohan,
T.K.B.Gandhi,H.C.Harsha,K.S.Deshpande,M.Sarker,T.S.K.Prasad,A.Pandey,Human
proteinreferencedatabase-2006update,NucleicAcidsRes.,34(2006)D411-D414.
[20]D.S.Wishart,T.Jewison,A.C.Guo,M.Wilson,C.Knox,Y.Liu,Y.Djoumbou,R.Mandal,
F.Aziat,E.Dong,S.Bouatra,I.Sinelnikov,D.Arndt,J.Xia,P.Liu,F.Yallou,T.Bjorndahl,R.
Perez-Pineiro,R.Eisner,F.Allen,V.Neveu,R.Greiner,A.Scalbert,HMDB3.0-TheHuman
MetabolomeDatabasein2013,NucleicAcidsRes.,41(2013)D801-D807.
[21]K.E.Swales,M.Korbonits,R.Carpenter,D.T.Walsh,T.D.Warner,B.-B.D.,Thefarnesoid
Xreceptorisexpressedinbreastcancerandregulatesapoptosisandaromataseexpression.,
CancerRes.,66(2006)10120-10126.
[22]R.H.Gee,J.N.Spinks,J.M.Malia,J.D.Johnston,N.J.Plant,K.E.Plant,Inhibitionof
prenyltransferaseactivitybystatinsinbothliverandmusclecelllinesisnotcausativeof
cytotoxicity,Toxicology,329(2015)40-48.
[23]K.E.Plant,E.Anderson,N.Simecek,R.Brown,S.Forster,J.Spinks,N.Toms,G.G.
Gibson,J.Lyon,N.Plant,Theneuroprotectiveactionofthemoodstabilizingdrugslithium
chlorideandsodiumvalproateismediatedthroughtheup-regulationofthehomeodomain
proteinSix1,Toxicol.Appl.Pharmacol.,235(2009)124-134.
[24]P.K.Smith,R.I.Krohn,G.T.Hermanson,A.K.Mallia,F.H.Gartner,M.D.Provenzano,E.K.
Fujimoto,N.M.Goeke,B.J.Olson,D.C.Klenk,Measurementofproteinusingbicinchoninic
acid,AnalyticalBiochemistry,150(1985)76-85.

19
[25]N.J.Dolman,K.M.Chambers,B.Mandavilli,R.H.Batchelor,M.S.Janes,Toolsand
techniquestomeasuremitophagyusingfluorescencemicroscopy,Autophagy,9(2013)
1653-1662.
[26]Y.-p.Yang,L.-f.Hu,H.-f.Zheng,C.-j.Mao,W.-d.Hu,K.-p.Xiong,F.Wang,C.-f.Liu,
Applicationandinterpretationofcurrentautophagyinhibitorsandactivators,Acta
Pharmacol.Sin.,34(2013)625-635.
[27]Y.Sheng,B.Sun,W.-T.Guo,Y.-H.Zhang,X.Liu,Y.Xing,D.-L.Dong,3-Methyladenine
inducescelldeathanditsinteractionwithchemotherapeuticdrugsisindependentof
autophagy,Biochem.Biophys.Res.Commun.,432(2013)5-9.
[28]K.A.Tekirdag,G.Korkmaz,D.G.Ozturk,R.Agami,D.Gozuacik,MIR181Aregulates
starvation-andrapamycin-inducedautophagythroughtargetingofATG5,Autophagy,9
(2013)374-385.
[29]T.Gebaeck,M.M.P.Schulz,P.Koumoutsakos,M.Detmar,TScratch:anovelandsimple
softwaretoolforautomatedanalysisofmonolayerwoundhealingassays,Biotechniques,46
(2009)265-+.
[30]T.A.Kocarek,S.D.Shenoy,N.A.Mercer-Haines,M.Runge-Morris,Useofdominant
negativenuclearreceptorstostudyxenobiotic-induciblegeneexpressioninprimary
culturedhepatocytes,J.Pharmacol.Toxicol.Methods,47(2002)177-187.
[31]P.R.Maloney,D.J.Parks,C.D.Haffner,A.M.Fivush,G.Chandra,K.D.Plunket,K.L.
Creech,L.B.Moore,J.G.Wilson,M.C.Lewis,S.A.Jones,T.M.Willson,Identificationofa
chemicaltoolfortheorphannuclearreceptorFXR,JournalofMedicinalChemistry,43
(2000)2971-2974.
[32]T.Ghafourian,Z.Amin,QSARmodelsforthepredictionofplasmaproteinbinding,
BioImpacts:BI,3(2013)21-27.

20
[33]S.H.Kaufmann,S.Desnoyers,Y.Ottaviano,N.E.Davidson,G.G.Poirier,Specific
proteolyticcleavageofpoly(adp-ribose)polymerase-anearlymarkerofchemotherapy-
inducedapoptosis,CancerRes.,53(1993)3976-3985.
[34]M.J.Abedin,D.Wang,M.A.McDonnell,U.Lehmann,A.Kelekar,Autophagydelays
apoptoticdeathinbreastcancercellsfollowingDNAdamage,CellDeathandDifferentiation,
14(2007)500-510.
[35]R.U.Jaenicke,MCF-7breastcarcinomacellsdonotexpresscaspase-3,BreastCancer
ResearchandTreatment,117(2009)219-221.
[36]S.Elmore,Apoptosis:Areviewofprogrammedcelldeath,ToxicologicPathology,35
(2007)495-516.
[37]S.Nagata,P.Golstein,TheFASdeathfactor,Science,267(1995)1449-1456.
[38]H.Zou,Y.C.Li,H.S.Liu,X.D.Wang,AnAPAF-1centerdotcytochromecmultimeric
complexisafunctionalapoptosomethatactivatesprocaspase-9,J.Biol.Chem.,274(1999)
11549-11556.
[39]D.J.Klionsky,S.D.Emr,Cellbiology-Autophagyasaregulatedpathwayofcellular
degradation,Science,290(2000)1717-1721.
[40]Y.Kabeya,N.Mizushima,T.Uero,A.Yamamoto,T.Kirisako,T.Noda,E.Kominami,Y.
Ohsumi,T.Yoshimori,LC3,amammalianhomologueofyeastApg8p,islocalizedin
autophagosomemembranesafterprocessing,EmboJournal,19(2000)5720-5728.
[41]S.Barth,D.Glick,K.F.Macleod,Autophagy:assaysandartifacts,JournalofPathology,
221(2010)117-124.
[42]F.T.He,J.Li,Y.Mu,R.Kuruba,Z.Ma,A.Wilson,S.Alber,Y.Jiang,T.Stevens,S.Watkins,
B.Pitt,W.Xie,S.Li,Downregulationofendothelin-1byfarnesoidXreceptorinvascular
endothelialcells,CirculationResearch,98(2006)192-199.

21
[43]K.Fukase,H.Ohtsuka,T.Onogawa,H.Oshio,T.Ii,M.Mutoh,Y.Katayose,T.Rikiyama,
M.Oikawa,F.Motoi,S.Egawa,T.Abe,M.Unno,BileacidsrepressE-cadherinthroughthe
inductionofSnailandincreasecancerinvasivenessinhumanhepatobiliarycarcinoma,
CancerScience,99(2008)1785-1792.
[44]E.D.Strauch,J.Yamaguchi,B.L.Bass,J.Y.Wang,Bilesaltsregulateintestinalepithelial
cellmigrationbynuclearfactor-kappaB-inducedexpressionoftransforminggrowthfactor-
beta,JournaloftheAmericanCollegeofSurgeons,197(2003)974-984.
[45]Y.Zhang,W.Gong,S.Dai,G.Huang,X.Shen,M.Gao,Z.Xu,Y.Zeng,F.He,
DownregulationofHumanFarnesoidXReceptorbymiR-421PromotesProliferationand
MigrationofHepatocellularCarcinomaCells,MolecularCancerResearch,10(2012)516-
522.
[46]Y.T.Y.Li,K.E.Swales,G.J.Thomas,T.D.Warner,D.Bishop-Bailey,FarnesoidXreceptor
ligandsinhibitvascularsmoothmusclecellinflammationandmigration,Arteriosclerosis
ThrombosisandVascularBiology,27(2007)2606-2611.
[47]D.A.Zajchowski,M.F.Bartholdi,Y.Gong,L.Webster,H.L.Liu,A.Munishkin,C.
Beauheim,S.Harvey,S.P.Ethier,P.H.Johnson,Identificationofgeneexpressionprofilesthat
predicttheaggressivebehaviorofbreastcancercells,CancerRes.,61(2001)5168-5178.
[48]J.Silva,S.Dasgupta,G.H.Wang,K.Krishnamurthy,E.Ritter,E.Bieberich,Lipidsisolated
fromboneinducethemigrationofhumanbreastcancercells,JournalofLipidResearch,47
(2006)724-733.
[49]P.Lefebvre,B.Cariou,F.Lien,F.Kuipers,B.Staels,RoleofBileAcidsandBileAcid
ReceptorsinMetabolicRegulation,Physiol.Rev.,89(2009)147-191.
22
Table1:IC50valuesforCDCA-andGW4064-elicitedcelldeathinbreastcancercelllines
CDCA GW4064
Complete
medium
Serum-free
medium
Complete
medium
Serum-free
medium
MCF-7 n.d. 244.7±9.5µM 10.6±2.6µM 12.6±2.8µM
MDA-MB-231 n.d. 273.3±32.2µM 9.5±3.9µM 3.7±3.3µM
MDA-MB-468 n.d. 188.5±124.4µM n.d. 4.8±1.4µM
MCF-10A n.d. 142.0±87.4µM 2.6±2.6µM 3.8±1.1µM
n.d.=notdeterminedasslopedidnotconverge

23
Figure1:TheFXRagonistsGW4064andCDCAcauseconcentration-dependenttoxicityin
breastcancercelllines.MCF-10A(A),MCF-7(B),MDA-MB-231(C)andMDA-MB-468(D)
cellswereexposedtovaryingconcentrationsofGW4064orCDCAincompleteorserum-free
medium.Following48or72hoursofexposure,cellviabilitywasassessedbyMTT.Eachdata
pointrepresentsthemeanofthreeindependentexperiments,whileerrorbarsrepresent
thestandarderrorofthemean(SEM).*=p<0.05,**=p<0.01,***=p<0.001

24
Figure2:TheFXRagonistsGW4064andCDCAcauseconcentration-dependentactivation
ofcaspase3andcaspase7intheMCF-7andMDA-MB-231celllines.(A)MCF-7andMDA-
MB-231cellswereexposedto3µMGW4064,30µMCDCA,or1µMstaurosporineinserum-
freemedium,inthepresenceorabsenceofspecificcaspaseinhibitorsfor24hours,and
thenCaspase3andcaspase7activitymeasured.(B)MCF-7andMDA-MB-231cellswere
exposedtoindicatedconcentrationsofGW4064,CDCAorstaurosporineinserum-free
mediumfor24hoursandthenpoly(ADP-ribose)polymerase-1(PARP)cleavageassessedby
Westernblot.Eachdatapointrepresentsthemeanofthreeindependentexperiments,

25
whileerrorbarsrepresentthestandarderrorofthemean(SEM).*=p<0.05,**=p<0.01,
***=p<0.001
Figure3:TheFXRagonistsGW4064andCDCAactivatecaspase9,butnotcaspase8,
activityintheMCF-7andMDA-MB-231celllines.MCF-7andMDA-MB-231cellswere
exposedto3µMGW4064,30µMCDCAor1µMstaurosporineinserum-freemediumfor24
hours,andthenCaspase8andcaspase9activitymeasured.Eachdatapointrepresentsthe
meanofthreeindependentexperiments,whileerrorbarsrepresentthestandarderrorof
themean(SEM).*=p<0.05,**=p<0.01,***=p<0.001

26
Figure4:TheFXRagonistsGW4064andCDCAstimulateapoptosisthroughtheintrinsic
pathwayinMCF-7andMDA-MB-231celllines.MCF-7andMDA-MB-231cellswere
exposedto3µMGW4064,30µMCDCAor1µMstaurosporineinserum-freemedium.(A)
Subcellularlocalisationofcytochromecwasexaminedfollowing0,4,6and8hoursof
exposure,withLDHasamarkerforthecytosolicfraction.(B)BaxandBcl2proteinlevels
weremeasuredintotalproteinfollowing24hoursofexposure.Eachdatapointrepresents
themeanofthreeindependentexperiments,whileerrorbarsrepresentthestandarderror
ofthemean(SEM).*=p<0.05,**=p<0.01,***=p<0.001

27
Figure5:TheFXRagonistGW4064stimulatesautophagyinMCF-7cells.MCF-7andMDA-
MB-231cellswereexposedto3µMGW4064or30µMCDCAinserum-freemediumfor24
hoursandautophagyassessedby(A)LC3cleavageand(B)p62/SQSTM1accumulation.5mM
3-methyladenineand2.5µMrapamycinwereincludedwhereindicatedasaninhibitorand
activatorofautophagy,respectively.Eachdatapointrepresentsthemeanofthree
independentexperiments,whileerrorbarsrepresentthestandarderrorofthemean(SEM).
*=p<0.05,***=p<0.001

28

29
Figure6:InductionofapoptosisandautophagyinbreastcancercelllinesbyGW6064and
CDCAisFXR-dependent.MCF-7andMDA-MB-231cellswereexposedtoGW4064orCDCA
inserum-freemediumfor24hoursinthepresenceorabsenceofadominantnegativeFXR
protein(FXR-DN).(A)Cellswereexposedto3µMGW4064or30µMCDCAandexpression
oftheFXRtargetgeneSHPmonitoredbyWesternblotting.(B)CellswereexposedtoIC50
concentrationofGW4064orCDCAandcellviabilityassessedbyMTTassay.(C)Cellswere
exposedto3µMGW4064or30µMCDCAandapoptosisassessedbycaspase3/7activity
assay.(D)Cellswereexposedto3µMGW4064or30µMCDCAandautophagyassessedby
p62/SQSTM1accumulation.Eachdatapointrepresentsthemeanofthreeindependent
experiments,whileerrorbarsrepresentthestandarderrorofthemean(SEM).*=p<0.05,
**=p<0.01,***=p<0.001

30
Figure7:TheFXRagonistsGW4064andCDCAdonotstimulatemigrationofMCF-7or
MDA-MB-231celllines.(A)MCF-7andMDA-MB-231cellswereseededintranswellplates
andexposedto3µMGW4064or30µMCDCAinserum-freemediumfor72hours.The
numberofcellsmigratingintothebottomchamberwasquantified.(B)MCF-7andMDA-
MB-231cellswereseededinawound-healingassayformatandexposedto3µMGW4064or
30µMCDCAinserum-freemediumfor24hours.Cellmigrationwasquantifiedbyclosureof
thescratchareausingthetScatchprogram.Eachdatapointrepresentsthemeanofthree
independentexperiments,whileerrorbarsrepresentthestandarderrorofthemean(SEM).

31
SupplementaryFigures
ActivationoftheFarnesoidX-receptorinbreastcancercelllinesresultsincytotoxicitybut
notincreasedmigrationpotential
NouraAlasmael,RatiMohan,LisianeB.Meira,KarenE.SwalesandNickJ.Plant
SupplementaryFigure1:BreastcancercelllinesexpressafunctionalFXRgeneregulatory
network
FigureS1:BreastcancercelllinesexpressafunctionalFXRgeneregulatorynetwork.(A)
MCF-10A,MCF-7,MDA-MB-231andMDA-MB-468cellswereexposedto3µMGW4064,
30µMCDCAorvehiclecontrolinserum-freemediumfor24hours,andthenFarnesoidX-
receptor(FXR)expressionassessedbyWesternblot.(B)MCF-7,MDA-MB-231orHepG2
(positivecontrol)cellswereexposedto3µMGW4064,30µMCDCAorvehiclecontrolin
serum-freemediumfor24hours,andthensmallheterodimerpartner(SHP)expression
assessedbyWesternblot.Eachdatapointrepresentsthemeanofthreeindependent
experiments,whileerrorbarsrepresentthestandarderrorofthemean(SEM).**=p<0.01,
***=p<0.001

32
SupplementaryFigureS2:TheFXRagonistsGW4064andCDCAdonotstimulatemigration
ofMCF-10AorMDA-MB-468celllines
FigureS2:TheFXRagonistsGW4064andCDCAdonotstimulatemigrationofMCF-10Aor
MDA-MB-468celllines.(A)MCF-10AandMDA-MB-468cellswereseededintranswell
platesandexposedto3µMGW4064or30µMCDCAinserum-freemediumfor72hours.
Thenumberofcellsmigratingintothebottomchamberwasquantified.(B)MCF-7and
MDA-MB-231cellswereseededinawound-healingassayformatandexposedto3µM
GW4064or30µMCDCAinserum-freemediumfor24hours.Cellmigrationwasquantified
byclosureofthescratchareausingthetScatchprogram.Eachdatapointrepresentsthe
meanofthreeindependentexperiments,whileerrorbarsrepresentthestandarderrorof
themean(SEM).