
Accelerating Product Development June 25, 2015 IMDMC REG 101 Pathways To Market.
36
Accelerating Product Development June 25, 2015 IMDMC REG 101 Pathways To Market
-
Upload
lynette-ellis -
Category
Documents
-
view
214 -
download
0
Transcript of Accelerating Product Development June 25, 2015 IMDMC REG 101 Pathways To Market.

Accelerating Product Development
June 25, 2015IMDMC REG 101
Pathways To Market

Pathways To MarketPathways To Market
Classification of Devices
510(k) Requirements
PMA Requirements

Device ClassificationDevice Classification
Class I - low risk, manual toothbrush
Class II – moderate risk, blood pressure monitor
Class III – high risk, heart valve

Classification Tips Classification Tips
Don't think of bucketsThink of ski slopes
◦Light in Class vs HeavyClass I can require clinical dataClass II may not require clinical dataTest to consensus standardsCheck with FDA if not sure

FDA Request for designationFDA Request for designation513(g) process
Costs money
Part of CDRH presubmission process
Follow FDA guidance
Usually takes a couple of months

Class IClass I
Low riskSubject to General ControlsSome may be exemptedCheck regulationsMust register with FDARecall and reporting systemUsually no submission is required

Class IIClass II
Medium RiskSubject to General and Special Controls
◦Performance standards◦Postmarket surveillance◦Patient registries◦Special labeling requirements◦Premarket data requirements◦Guidelines
Some may be exempted510(k) clearance required

Class IIIClass III
Highest risk OR very novelSubject to general and special controlsPre-Market approval requiredFDA inspection requiredClinical data requiredCheck regulations and guidanceTalk to FDA

How to Classify Your ProductHow to Classify Your Product
Define indication for usePick a predicateFDA.gov
◦Classification Database◦Device panel
Revisit IFULook at competitorsConfirm with FDA

ExampleExample
Transcutaneous Electrical Nerve Stimulator (TENS) device
Indications for use: Depression and Pain
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm

Device PanelDevice Panel
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/ucm051530.htm

Panel ListingPanel Listing73 Anesthesiology Part 868
74 Cardiovascular Part 870
75 Chemistry Part 862
76 Dental Part 872
77 Ear, Nose, and Throat Part 874
78 Gastroenterology and Urology Part 876
79 General and Plastic Surgery Part 878
80 General Hospital Part 880
81 Hematology Part 864
82 Immunology Part 866
83 Microbiology Part 866
84 Neurology Part 882
85 Obstetrical and Gynecological Part 884
86 Ophthalmic Part 886
87 Orthopedic Part 888
88 Pathology Part 864
89 Physical Medicine Part 890
90 Radiology Part 892
91 Toxicology Part 862
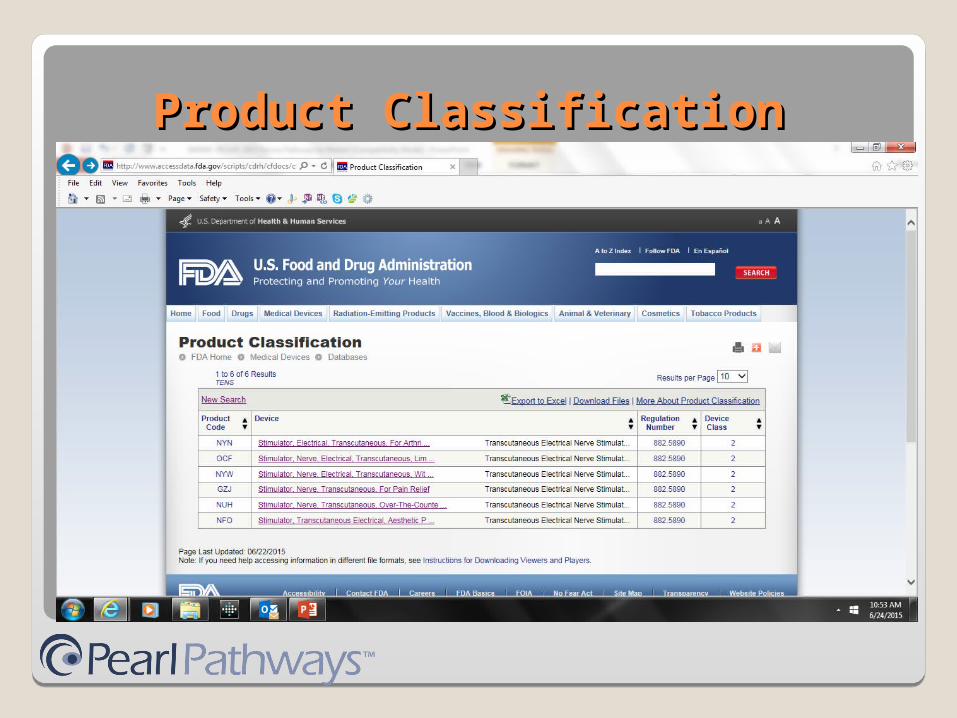
Product ClassificationProduct Classification

RegulationRegulation
TITLE 21--FOOD AND DRUGS
CHAPTER I--FOOD AND DRUG ADMINISTRATIONDEPARTMENT OF HEALTH AND HUMAN SERVICES
SUBCHAPTER H--MEDICAL DEVICESPART 882 -- NEUROLOGICAL DEVICES Subpart F--Neurological Therapeutic Devices Sec. 882.5890 Transcutaneous electrical nerve stimulator for pain relief.
(a) Identification. A transcutaneous electrical nerve stimulator for pain relief is a device used to apply an electrical current to electrodes on a patient's skin to treat pain. (b) Classification. Class II (performance standards).

TENS DeviceTENS Device
Class IICheck guidancehttp://www.fda.gov/downloads/
medicaldevices/deviceregulationandguidance/guidancedocuments/ucm207059.pdf
Guidance applies to specific product codes, not all 6 possible codes


Intended useIntended useWanted: Indications for use of Depression
and PainFound: A transcutaneous electrical nerve
stimulator for pain reliefHow to get a depression claim?
◦Talk to FDA◦Probably run a clinical endpoint◦Is it worth it?

SummarySummary
Multiple classes of devices based on risk, novelty, patient population, intended use, indication for use
First understand market and user requirements
Check FDA resourcesCan pay FDA to tell you (Be careful what
you with for!)

ResourcesResources Product Classification Database
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm 510(k) Database
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm Premarket Approval Database
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pma.cfm Class I and Class II Devices Exempt from 510(k)
Requirementshttp://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm
Device Guidance Documents http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm

Let’s Try it Together!Let’s Try it Together!
510k Requirements510k Requirements
Some class I and most class IIDemonstrates that the new device is
“substantially equivalent” to a predicate device in terms of intended use, technological characteristics, and performance testing, as needed
Follow 21CFR807Use FDA presubmission checklist

Substantial EquivalenceSubstantial Equivalence
Has the same intended use as the predicate; and has the same technological characteristics as the predicate; or
Has the same intended use as the predicate; and has different technological characteristics and the information submitted to FDA; ◦ does not raise new questions of safety and effectiveness;
and◦ demonstrates that the device is at least as safe and
effective as the legally marketed device

FDA Pre-submission ChecklistFDA Pre-submission Checklist
Acceptance Checklist for 510(k)sThe Guidance for Industry and Food and Drug Administration Staff: Refuse to Accept Policy for 510(k)s describes the criteria FDA intends to use in assessing whether a 510(k) submission meets a minimum threshold of acceptability and should be accepted for substantive reviewThe guidance includes acceptance checklists for each type of 510(k) submission:Traditional 510(k) ChecklistAbbreviated 510(k) ChecklistSpecial 510(k) Checklist

Types of 510k SubmissionsTypes of 510k Submissions
Abbreviated – No Brainer
Special – Change that doesn’t alter risk
Traditional – All others

Abbreviated 510KAbbreviated 510K
A guidance documents existSpecial controls are establishedRecognized standards existSummary report
◦Information regarding the efforts to conform with the guidance document/special control(s) & outline any deviations

Not a Special 510kNot a Special 510kModifications that have the potential to alter the
fundamental scientific technology of the device, operating principle(s), mechanism of actionbe submitted as
Examples:◦ surgical instrument that uses a sharpened metal blade to
one that cuts with at a laser ◦ in vitro diagnostic (IVD) device that uses immunoassay
technology to one that uses nucleic acid ◦ Incorporation of a sensing mechanism in a device to
allow the device to function "on demand" rather than continuously

Special 510k examplesSpecial 510k examples
Energy typeEnvironmental specificationsPerformance specificationsErgonomics of the patient-user interfaceDimensional specificationsSoftware or firmwarePackaging or expiration datingSterilizationContact FDA if conducting clinicals

FDA Pre-submission ChecklistFDA Pre-submission Checklist
Checklist for CDRH reviewersUse as a guide for your benefitGood war story

Pre-Market Application (PMA)Pre-Market Application (PMA)
Require FDA approvalProbably not in the classification databaseSupports or sustains human life, is of
substantial importance in preventing impairment of human health, or presents a potential, unreasonable risk of illness or injury
Check for Guidance Documents

Types of PMAsTypes of PMAs
Traditional PMA◦It’s traditional
Modular PMA◦Submitted in pre-defined components◦Project specific
Streamlined PMA◦ FDA guidance document or other published methods for
review which have been evaluated◦ FDA review history dealing with like products (2+)◦ Must be available for the Interactive review process

CBER DevicesCBER Devices
Medical devices associated with the blood collection and processing procedures
Medical devices associated with cellular therapies
InVitro Diagnostics involved with blood testing
Device regulations applyCheck CBER device listFollow CBER guidance documents

PMA Data requirementsPMA Data requirements
Non-clinical data◦ microbiology, toxicology, immunology, biocompatibility,
stress, wear, shelf life, and other laboratory or animal tests
Clinical data◦ study protocols, safety and effectiveness data, adverse
reactions and complications, device failures and replacements, patient information, patient complaints, tabulations of data from all individual subjects, results of statistical analyses, and any other information from the clinical investigations

Companion DiagnosticsCompanion Diagnostics
Any InVitro Diagnostic developed to choose patients for therapy with a specific therapeutic
Must be co-developed with the therapeutic
Starts as a lab developed test and then progresses to an IDE and PMA
PMA and NDA or BLA approved on the same day

FDA Pre-submission ChecklistFDA Pre-submission Checklist
Acceptance and Filing Reviews for Premarket Approval Applications (PMAs) Guidance Document
Have a third party reviewTalk with FDA about Advisory Committee
MeetingMultiple FDA meetings

AM ManufacturingAM ManufacturingWhat class is the Powered wheelchair with
built in monitors?
What questions should you ask about the change in the polymer for the HOA product?
How would you report a change in the data transfer system for the wheelchair

CONTACT USCONTACT US
Diana Caldwell, President & CEO◦[email protected]
Gretchen Bowker, Chief Operating Officer◦[email protected]
Contact us at 317.899.9341Websites: www.pearlpathways.com and
www.pearlirb.com


















