
A lda+u study of selected iron compounds 第二章
61
GGAによって研究されたいくつかのd開殻系 http://www.sissa.it/cm/thesis/2002/cococcioni.p df 和訳:@dc1394 A LDA+U study of selected iron compounds 第二章
Transcript of A lda+u study of selected iron compounds 第二章

GGAによって研究されたいくつかのd開殻系 http://www.sissa.it/cm/thesis/2002/cococcioni.pdf 和訳:@dc1394
A LDA+U study of selected iron compounds 第二章

序論

この章の概要
この章では、計算に標準のDFTを使用した、鉄化合物の研究を行う。
すべての選ばれた系が、孤立原子の電子配置で[Ar](3d)6(4s)2である鉄を含んでいる。
鉄の電子配置の最も外側にある、3dと4sの電子の両方が、この元素の化学で重要な役割を果たす。
4s電子は、原子核と最も内部の電子雲で構成されるイオンコアによって弱く結合しており、より高い電気陰性度を有する(例えば酸素)、他の元素の存在で容易に電子を失う。

この章の概要
4s電子より局在化していて、イオンコアとより結合している、3d電子に対しては、違った振る舞いが観測される。
この事実にもかかわらず、3d電子は深くイオンが形成する化学結合にかかわる。
また3d電子は通常、最も高いエネルギーを持つ、占有された状態であり、Fermiエネルギーによって交差する不完全な縮退を表すように、しばしば鉄化合物の伝導特性の原因となる。

この章の概要
鉄化合物の金属的な振る舞いの原因となる電子は、これらの3d状態に主に収容されるという事実にもかかわらず、dレベルは通常、かなり狭いバンドと非常に局在化した電荷密度・磁気モーメントといった原子的な性質を有する。
そして、(開いた)d軌道の電子の局在化が、原子サイト上の電子相関を引き起こし、鉄(と一般に遷移金属)化合物の物理的挙動における非常に重要な役割を果たすかもしれない。
同時にこれは、局在化した強相関電子を扱うように設計されていない、従来のLDAかGGAアプローチへの問題を引き起こすかもしれない。

この章の動機
この章の動機は二つある。
我々は、GGAで正しく記述されるような物性をLDA+U法で研究するが、それはLDA+U法でも、できるだけ良い記述のまま残したい。
また一方で、従来の交換相関汎関数ではよく記述できない場合を考慮する(そして、電子相関のより良い取り扱いが必要となることがわかる)。

この章で扱う物質とその計算法
この章は、3つの異なった物質(バルクの鉄、酸化鉄FeO、Fe2SiO4というフェライトの一種)に対して、3つのセクションで扱っている。
これらの化合物を研究するために使用した計算法は、NLCC擬ポテンシャルのスピン分極GGAである。
これらの系に対して、バンド構造、磁気モーメント配列、そして構造特性(これは全エネルギー計算のMurnaghanフィッティングによって行う)を示す。
そして、これらの結果を実験結果と比較し、(GGAによる)この計算の成功と欠陥を検討する。

バルクの鉄について
バルクの鉄は、第一原理計算によって広範囲にわたって研究されており、そして、NLCC擬ポテンシャルのGGAは、基底状態としてbcc構造の強磁性(FM)相を予測する。
この場合、この標準のDFT(の近似)によって、構造、磁気、そして振動特性を正しく得ることができる。
したがって、Fermi準位の周りのd電子に対する強い相関が、この物質にはそれほど重要でないと予想され、これはこの論文で調査したいと思う。

酸化鉄FeOについて
酸化鉄FeO(と他の多くの遷移金属酸化物)は、GGAで説明が不十分な物質である。
GGAは磁気モーメントと平衡格子間隔に対して、かなり成績が良いが、それは、圧力下(特に[111]方向に沿った変形)のふるまいを、それほど正確に説明しない。
さらに重大なことは、観測されている絶縁性基底状態の再現に失敗することである。

酸化鉄FeOについて
事実、他の遷移金属酸化物のように、FeOは、バンドギャップが実際存在し(これは電子が1つのサイトから別のサイトまで跳ぶことを禁じるオンサイトCoulomb斥力のため)、いわばMott絶縁体のようなものと推定される。
その結果、従来のGGA汎関数の近似は、この化合物の電子物性の良い記述を与えるほど正確ではなく、これがおそらく、本章で示された物性の計算結果が失敗している理由である。

Fe2SiO4について
Fe2SiO4というフェライトの一種は、地球物理学上、興味を持たれている鉱物である。
我々は、この化合物のいくつかの計算(LDAとGGAの両方を用いる)を示す。
そして、磁気および構造特性の計算結果が、実験値とかなり良く一致することを見る(なお、最も良い結果はGGAから得る)。
しかしながら、(実験で)観測された絶縁性のふるまいにもかかわらず、計算結果は金属となる(計算結果からFermiエネルギーで交差する鉄のd-レベルの狭いバンドを得る)。

Fe2SiO4について
この(Fermi)レベルの周りの状態の電荷密度は、鉄部位で非常に局在化されており、それゆえに、これらの電子の強い原子様性質とオンサイト相関が重要である可能性がある。
GGA計算を簡素化されたHubbard模型と比較して、平均したHubbardがUであると本当に荒く見積ると、この化合物のMott-Hubbardのふるまいと一致した、U~2.4eVの値を得ることができるだろう。

バルクの鉄(の計算)

バルクの鉄の計算法
この節で、バルクの鉄の、平衡構造と磁気・電気的な特性に関して、いくつかの結果を報告する。
電子イオン相互作用について説明するために、以前に鉄の構造的・振動的な研究で使用された修正Rappe-Rabe-Kaxiras-Joannopoulos (RRKJ)から作られた、ウルトラソフト擬ポテンシャルを使用する。
LSDAがこの材料の基底状態構造性質に関して間違った結果を与えるのが知られているので、交換と相関汎関数の両方にGGA-PBEフレームワークを採用する。
A. M. Rappe, K. M. Rabe, E Kaxiras, J. D. Joannopoulos, Phys. Rev. B 41, 1227 (1990).

バルクの鉄の計算法
電子擬波動関数について説明するために、35Ryのエネルギーカットオフを選んだ(ただし、電荷密度の補強部分は420Ryまで拡大する)。
LDA+U計算では、鉄に対して、他の研究で使用されるものより大きい波動関数エネルギーカットオフが必要であり、そして、このGGAによる計算と一貫した結果を得るために、同じカットオフ値を採用する。

バルクの鉄の計算法
逆空間積分を実行するために、MonkhorstとPackのスペシャルポイントテクニックを用いる。
そして、メッシュは「スペシャルな」8×8×8ポイント(これは既約Brillouinゾーンのウェッジの中の29非同等ポイントに対応する)を用いれば、非常に正確にBrillouinゾーンをサンプリングすることができるのがわかっている。
MethfesselとPaxtonのsmearingテクニックが、Fermi分布を整えるために使用され、「普通」のDFT計算で必要とされるより小さい0.005Ryの広がりを使用する。
これは最後の章で見るように、LDA+U計算に対して高精度を与え、またこの結果とこの節の結果とを比較する。

バルクの鉄の平衡格子間隔と体積弾性率
バルクの鉄の基底状態はbccの強磁性(FM)であり、この構造特性を研究する。
平衡格子間隔と体積弾性率を見積もるために、体積とユニットセルの値を変えて計算された点をMurnaghan状態式にフィッティングした(図2.1)。
また、これらの量と、各イオンの磁気モーメントに対して、実験結果と計算結果の比較を行った(表2.1)。

図2.1 バルクの鉄の平衡格子間隔と体積弾性率
図2.1 bccのFM鉄のMurnaghan式へのフィッティング。
体積の単位は(a. u.)3、エネルギー-体積曲線の最小値を縦軸のゼロとしている。

表2.1 格子定数、体積弾性率、および磁気モーメントの比較
表2.1 格子定数(a0)、体積弾性率(B0)、および磁気モーメント(μ 0)のGGA(この研究)とLSDA(Corso他)による計算値とMoroni他による実験値の比較
A. Dal Corso, S. de Gironcoli, Phys. Rev. B 62, 273 (2000).
E. G. Moroni, G. Kresse, J. Hafner, J. Furthmüller, Phys. Rev. B 56, 15629 (1997).

平衡格子定数での電子構造
格子定数の計算値の実験との一致はかなり良いが、体積弾性率と磁気モーメントの計算値は実験値とかなり食い違いがある(ただし、LSDAで計算された値より食い違いははるかに小さい)。
図2.2に、平衡格子定数で得られた電子構造が示されている。
また同じ図に、Turner他による光電子放出によって得られたいくつかの実験結果が示されている。
A. M. Turner, A. W. Donoho, J. L. Erskine, Phys. Rev. B 29, 2986 (1984).

図2.2 bcc鉄のバンド構造
図2.2 bcc鉄のバンド構造。多数スピンバンドに対しては実線、少数スピンバンドに対しては点線。エネルギーの0はFermiエネルギーに合わせてある。

図2.2 bcc鉄のバンド構造

電子構造とその実験との比較
二つのスピン分布(これは物質の強磁性の起源となる)の間の有限のスピン分裂が明白であり、そしてそれがFermiレベル付近でより大きいことがわかる。
この計算されたバンド構造と実験を比較すると、H点の周りとΓ -P-H領域ではやや不一致が見られるが、特にΓ -N-P領域でよく一致しているように見える。
この図から、二つのグループの間を明らかに区別することができ、それはまた図2.3の射影状態密度から明らかである。

図2.3 bcc鉄の射影状態密度
図2.3 bcc鉄の射影状態密度。
d状態に対しては赤線、s状態に対しては黒線。多数スピン状態は上側、少数スピン状態は下側。

最初のグループと二番目のグループ
最初のグループは、Fermiエネルギーの周りの領域(およそFermiエネルギーの5eV下から3eV上まで)で広がっていて、鉄の3dレベルによって主に構成されている。
二番目のグループは、はるかに大きい分散を有し、Fermi準位の8eV下~4eV下の間に広がった4s状態があり、それ以上の領域はd状態によって占められている。

遍歴強磁性
二つのスピン分布の間の相対的なシフトによって生成する、イオンの磁化はかなりd状態からきている。
その結果、同じ電子が金属的な挙動の原因となり、また磁気特性をもたらす(遍歴強磁性)。
図2.3からわかるように、s状態は不均衡を生み出す二つのスピン状態の間でわずかに異なった状態密度を有するが、その寄与は非常に小さい(二つの状態密度の積分は、s状態の多数スピンで0.39電子/セル、少数スピンで0.45電子/セルである)。

金属特性の起源
また、Fermi準位の周りの領域では、両方の種類の状態が共存しているが、しかし、s状態の密度は、dレベルが位置している領域では(両方のスピンチャネルに対して)強く減少し、Fermi準位近傍で少なくなっている。
このように、二つの状態の混成がそれほど重要でないと予想されるので、この物質の金属特性は、強く原子的なd状態から主に起こる。

ここまでのGGAアプローチのまとめ
このような、電気・磁気的、構造的な特性に関する研究によって明白なように、GGAアプローチはバルクの鉄に対して非常に良い記述を与える。
しかしながら、物理的挙動に関していくつかの質問がまだ残っている(例えば、原子的な磁気モーメントを導くメカニズム)。
事実、遍歴電子によって生み出された磁気は問題であり、このような背景から、電子相関の重要性は議論のポイントの1つである。

酸化鉄FeO

酸化鉄(FeO)について
酸化鉄(FeO)は最先端の数値計算技術で研究される、Feよりはるかに問題の多い物質である。
ほとんどの遷移金属酸化物において起きることであるが、観測された絶縁性のふるまい(Mottのようなメカニズムによって生み出されると予想される)の再現に、LDAは完全に失敗する。
しかしそれにもかかわらず、常圧・常温状態の実験結果における、この化合物の構造・磁気的特性については、合理的に説明できる。

酸化鉄の計算条件等
この段落では、FeOのσ -GGA研究のいくつかの結果を示し、そして、論文の後の章に示すLDA+U結果と比較するために、構造・電子的特性に注目する。
計算に用いた擬ポテンシャルは、Fe原子については、US GGA-PBE NLCC擬ポテンシャル(バルクの鉄の計算に用いた同じもの)を用い、O原子についてはUS GGA-PBE(非NLCC)擬ポテンシャルを用いた。
またバルクの鉄と同じように、smearingの幅として0.005Ryを取り、Fermi分布を整えるのに使用した。
そして同様に、逆空間積分の実行に十分であることがわかっている、4×4×4メッシュのk-点(これはIBZの中の13独立ベクトルに対応する)を取った。

酸化鉄の計算条件等
電子波動関数については、40Ryのエネルギーカットオフが選ばれている。
ただし、電荷密度への増強部分に対しては、400Ryのカットオフを必要とする。
前節と同じように、これらは「正常な」GGA(またはLDA)計算で必要な値より大きい値であるが、この値が必要であるLDA+Uアプローチで得る結果と、この章で述べるGGAの結果を直接比較するために、この値を選んでいる。

酸化鉄の構造と磁気的特性
ひずみのない相における、FeOのユニットセルには、鉄の磁化から生じる菱面体対称の立方岩塩(B1)型構造を有する。
同じ[111]面に属するイオンに対して、図2.4に示されている基底状態スピン構成は、強磁性である。
間に横たわっている酸素イオンによって仲介される超交換相互作用のため、お互いに、最も近い隣の磁気面は反強磁性配置にある。

図2.4 FeOのユニットセル
図2.4 FeOのユニットセル。赤い球はFe原子、青い球はO原子、矢は磁気イオンのスピン分極を表す。

菱面体晶ストレッチング
常圧下で、Néel値(198K)より下まで温度を下げ、[111]対角線に沿った、結晶構造の菱面体晶の伸びについて述べる。
変形は圧力の負荷で増加することがわかっており、そしてまた、Néel温度も増加するのが観測される。
より高い圧力では、系はここに記述しない他の構造相に変化し、これは最近研究された。

計算の結果
我々は、基底状態菱面体晶AFスピン配位のひずみのない構造を研究する。
計算の結果を図2.5に示す。
この計算された点から、Murnaghanフィッティングされた曲線が描かれており、そしてこの曲線は、この物質に関するいくつかの構造パラメータを取り出すのに使われている。
また、表2.2で、格子パラメータ、体積弾性係数、およびそれぞれの鉄の磁気モーメントを実験結果と比較する。

図2.5 酸化鉄に対するMurnaghan状態式へのフィッティング
図2.5 酸化鉄に対するMurnaghan状態式へのフィッティング。エネルギーのゼロはエネルギー-体積フィッティングの最小値に合わせており、体積の単位は(a.u.)3である。

表2.2 計算値の実験結果との比較
表2.2 計算された格子定数(a0)、体積弾性係数(B0)、および磁気モーメント(μ 0)と、Fangらの論文から取った実験結果との比較
Z. Fang, I. Solovyev, H. Sawada, K. Terakura, Phys. Rev. B 59, 762 (1999).

表2.2について
実験結果における何らかの散乱にもかかわらず、特に格子パラメータと体積弾性係数の値の一致は合理的である(我々は、基本直接格子ベクトルでない図2.4の従来の単位立方格子の側を報告する)。
鉄の磁気モーメントの値は、他よりも大きい食い違いがあるが、これでも他の理論研究(Fangらの研究)よりは良く実験値と一致している。

バンド構造
図2.4に示した、ひずみのない立方構造のAFスピン配位の均衡格子パラメータで酸化鉄のバンド構造を計算した。
結果は図2.6に示す(エネルギーのゼロはFermiエネルギーに合わせてある)。

図2.6 図2.4のスピン配位に対応する酸化鉄のバンド構造
図2.6 図2.4のスピン配位に対応する酸化鉄のバンド構造。実線は多数スピンバンド、点線は少数スピンバンド。少数スピンは完全に縮退している。
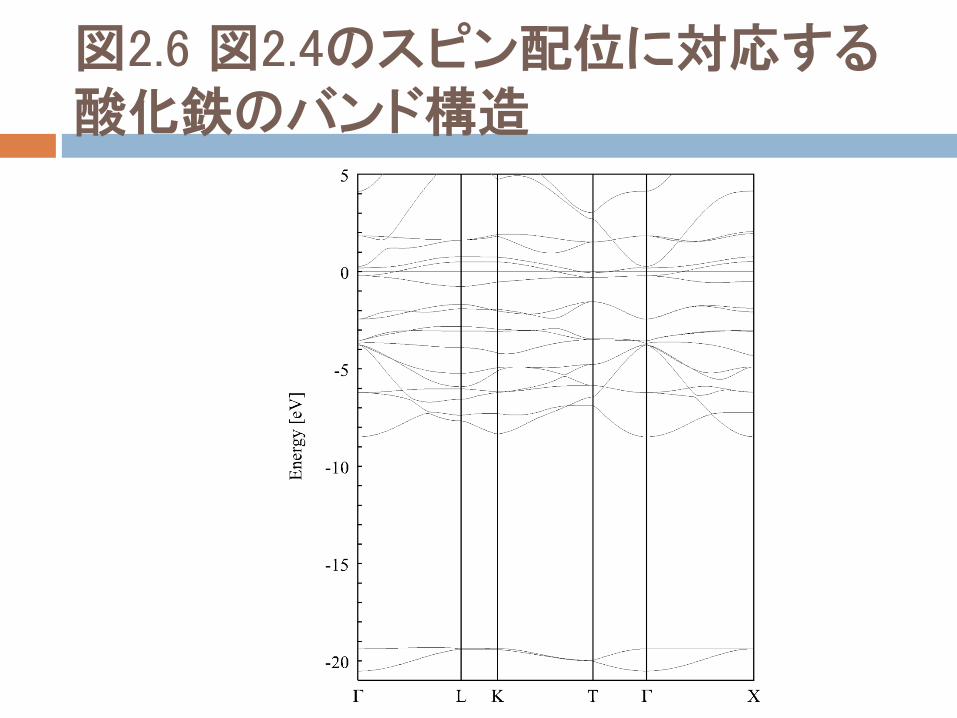
図2.6 図2.4のスピン配位に対応する酸化鉄のバンド構造

図2.6からわかること
図2.6からわかるように、反強磁性の基底状態スピン配位に対して、スピンアップとスピンダウンの間で電子レベルは完全縮退している。
それぞれの鉄の多数スピンd状態は、Fermiエネルギーの4~1eV下に位置している。
また、部分的に占められた少数バンドは、-1~2eVの間のFermi準位の周りで広げられ、またいくつかの点でFermi準位に交差し、その結果金属的な挙動を生じる。

LDA(とGGA)の失敗と成功
この計算結果は、低温低圧で絶縁性基底状態を示しているこの化合物の実験結果と食い違っており、この化合物群について説明する際の、LDA(またはGGA)の最も明確な失敗を示している。
しかしながら、実際に多数と少数スピン状態の間の有限の分裂から生じた非ゼロ磁化とともに、鉄のd電子の遍歴特性は矛盾していない。
したがって、伝導特性の再現に失敗してはいるが、LDAとGGAは各イオンにおける磁化に関して、かなりの結果を与えることができる。

4つのグループ
電子バンド構造において、状態の4つのグループを区別できる。
酸素のs状態は、Fermiエネルギーの約20eV下に存在しており、酸素のp状態は、Fermiエネルギーの9~4eV間に存在している。
鉄のdレベルは、Fermiエネルギー付近の-4から2eVまでに存在する。
鉄のs状態は、Fermiレベルより上の領域にあり、完全に空となっている。

d状態をさらに二つに分割する
完全に立方の環境においては、磁気イオンのd状態をさらに2つのサブグループに分割できた。
t2g(xy, yz, およびxz)レベルは低いエネルギーに存在し、eg(x
2-y2, 3z2-r2)状態はより高いエネルギーにある。
AFスピン配位における鉄によって感じられた菱面体晶配位子場は、立方構造のt2g重なりを上げさせて、A1g対称の1つの状態を生み出す。
このA1g対称は、3Z2-r2の一次結合 に対応し、ここでZは[111]菱面体晶軸に沿って取られる
そして、FM[111]面の上に、Eg対称の他の2つの状態( と )が存在する。

鉄のd電子
この場合、縮退したt2g状態がすべてFermi準位にあり、その結果、1/3充満バンドを生み出すので、菱面体対称なしでは、絶縁性ギャップを実現できなかった。
菱面体対称において生じるギャップのため、 A1g
状態は、t2gトリプレットを起源とする他の二状態に関して、より低いエネルギーとなるべきである。
そして、鉄の6つのd電子は、この場合5つの多数スピン状態と最低エネルギーの少数スピン状態を完全にいっぱいにし、その結果、Fermiエネルギーより上の他の状態を残すだろう。

バンドギャップの問題
その結果、このことは、磁気イオンのd軌道を記述する際に失敗するGGAにおいては、再現できない。
その上、この記述が正しかったとしても、ギャップは、鉄に影響する配位子場によってちょうど生み出されないので(これは、平均場近似のような「普通の」LDAかGGA手法では適切に説明されない電子相関によるためであると予想される)、バンド構造におけるギャップは小さ過ぎるだろう(MnOやNiOで起こるように)。

電荷移動絶縁体
しかしながら、GGA計算でよく説明される場合があり、Fermi準位で開くギャップ(これは再現できないが)の分光的な性質である。
酸化鉄における光電子放出および光学的実験が、混成したO 2p-Fe 3d性質に、この化合物の価電子帯があるのを明らかにしているので、価電子帯と伝導帯の間の遷移状態は酸素から鉄まで跳ぶ電子に関わるべきである(電荷移動絶縁体)。

酸素のp状態と鉄のdレベルの強い混成
したがって、酸素のp状態と鉄のdレベルの間の強い混成は価電子バンドの先で観測されるべきである。
(バンド)ギャップはGGAの計算では得られないが、しかし、これらの状態の混成は我々の計算で(異なった原子の寄与について説明した、射影状態密度をプロットした図2.7のように)明白である。

図2.7 ひずみのないAF酸化鉄の状態密度
図2.7 ひずみのないAF酸化鉄の状態密度。エネルギーのゼロはFermiレベルに合わせてある。

酸素p状態と鉄のd状態の重なり
少数スピンd状態を主に起源とするFermiエネルギーでの状態を無視すると、酸素p状態と多数スピンd状態のかなりの重なりが、Fermi準位の2eV下に近い領域に存在するのを見ることができる。
しかしながら、この重なり領域は、主に鉄の寄与からなっていて、また、Fermi準位の5~9eV下では、酸素p状態と鉄のd状態の、より強い相互作用を観測でき、これは酸素の状態の寄与が支配的である。
つまり、何らかの有限の重なりが存在していても、2つの原子の寄与の中心は、エネルギースペクトルの異なった領域に置かれる。

菱面体晶ひずみ相の不安定性
GGAはひずみのない立方体の構造の格子間隔とFeOの体積弾性率の再現にかなり成功しており、電子物性に対する記述の失敗は、おそらく菱面体晶ひずみ相に対する、量的に間違った結果が理由である。
事実、FeOの立方構造は、[111]立方体対角線に沿う菱面体晶の伸びに関して不安定であることがわかっている。
そしてこの理由は、電子/伝導特性、軌道秩序、および結晶対称性の中の強い相互作用であり、これは非常に効率的な磁気結晶相互作用を発生させる。

Néel温度以下での変形
事実、温度をNéel値(約198K)より下まで下げたとき、大気条件で変形が観察され、圧力下では変形が増加することがわかっている。
FeOの研究に対するGGAアプローチは、歪構造が常圧で基底状態であると正しく予測し(計算はいつもゼロ温度で実行されることに注意)、また、圧力下における菱面体晶の伸びの増大を再現する。

菱面体晶変形に対応するフィッティング曲線
図2.8に、異なった菱面体晶変形に対応するフィッティング曲線を示す(変形パラメタは図2.9に示す2つの表面の対角ベクトルの菱面体晶角度である)。
この図は、圧力が増加すると、変形がますます大きくなるという実際の結果と一致している。
量的な観点から変形の増加を研究するために、我々は、Murnaghan曲線のそれぞれの点に対応する圧力を計算して、0~200kbarsの圧力の範囲について調査した。
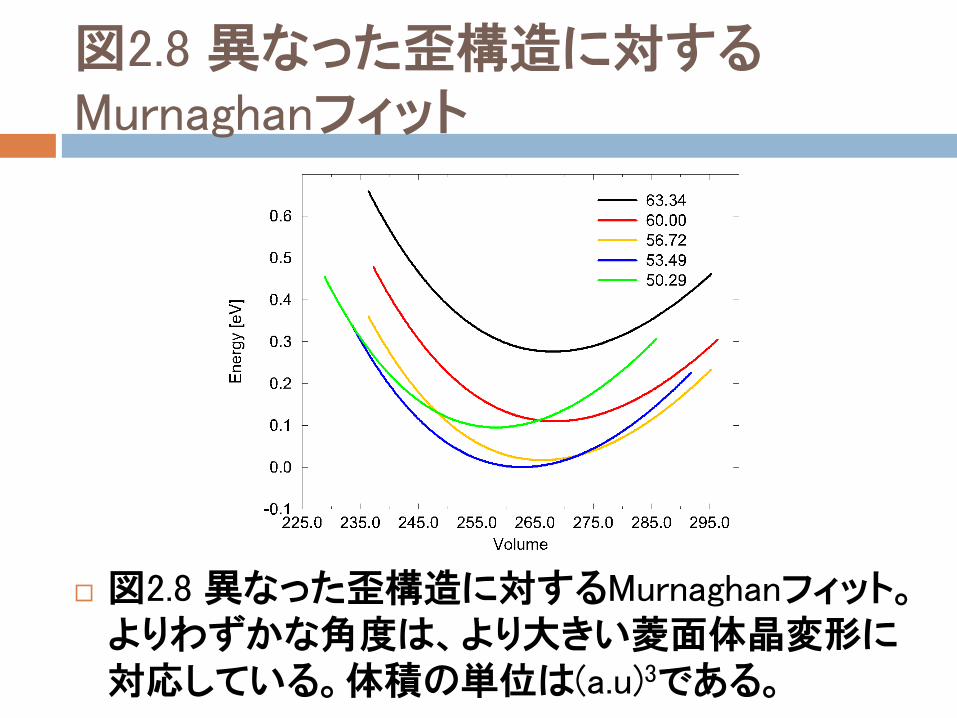
図2.8 異なった歪構造に対するMurnaghanフィット
図2.8 異なった歪構造に対するMurnaghanフィット。よりわずかな角度は、より大きい菱面体晶変形に対応している。体積の単位は(a.u)3である。

図2.9 菱面体角度
図2.9 菱面体晶角度は、2つの表面の対角ベクトルの角度であり、ひずみのない構造では、値は60°である。

菱面体晶変形の増大の明確な傾向
選ばれたそれぞれの圧力について(選ばれた点の間を20の部分に分解した)、系のエンタルピー(E+PV)について計算し、二次曲線でcosα rへの依存をフィッティングして、最終的にエンタルピーの最小値に対応する菱面体晶角度を得た。
この結果は図2.10にあり、菱面体晶変形の増大に向かう明確な傾向が観測される。
同じ図で、実験による2点が示されている。
Willisらの論文から常圧での実験結果を引用し、Yagiらの論文から200kbarの実験結果を引用した。
B. T. M. Willis, H. P. Rooksby, Acta Cryst. 6, 827 (1953).
T.Yagi, T. Suzuki, S. Akimoto, Journ. of Geophys. Res. 90, 8784-8788 (1985)

図2.10 圧力の関数としての菱面体晶角度
図2.10 圧力の関数としての菱面体晶角度。

実験値とGGAによる計算値の比較
実験結果の両方を非化学量論化合物に対して得たので、Willisらの論文の鉄濃度(90K、常圧の実験結果)と、菱面体晶角度の線形依存性を使用することで、同じ量の鉄と酸素に対する構成まで実験結果を外挿した。
200kbarsの点でのイオン密度における同じ線形依存性の使用は、それほど正確でないかもしれない。
両方の場合で、GGAで予測された伸びの量(立方構造値60°からの菱面体晶角度の差)は、実験に用いられたのが非化学量論化合物であることを考慮しても、実験値と比べるとはるかに大きい。

まとめ
Isaakらは、計算と実験結果の一致という観点から、圧力下での菱面体晶変形のふるまいが、GGAで正しく説明されると導き、結論づけた。
我々は、これが蓋し質的な面で本当であると考える。
したがってGGAは、常圧と、より高い圧力領域の両方の基底状態における、この化合物の構造性質について説明しない。
電子構造の不正確な記述に関連するこの失敗を予想するとき、菱面体晶変形が更なる側面になり(もちろん、バンドギャップとその分光性質も)、これはLDA+Uアプローチのときにテストされるだろう。
D. G. Isaak, R. E. Cohen, M. J. Mehl, D. J. Singh, Phys. Rev. B 47, 7720 (1993).





![Generalized Correspondence-LDA Models (GC-LDA) for ... · The GC-LDA and Correspondence-LDA models are extensions of Latent Dirichlet Allocation (LDA) [3]. Several Bayesian methods](https://static.fdocuments.net/doc/165x107/6011a7de37d63b741248406f/generalized-correspondence-lda-models-gc-lda-for-the-gc-lda-and-correspondence-lda.jpg)












